 Maryam Najafi1,2Dor Mohammad Kordi Tamandani2*Anoush Azarfar3Zeineb Bakey1,4Farkhondeh Behjati5Dinu Antony1,4Isabel Schüle4Simin Sadeghi-Bojd6Ehsan Ghayoor Karimiani7,8
Maryam Najafi1,2Dor Mohammad Kordi Tamandani2*Anoush Azarfar3Zeineb Bakey1,4Farkhondeh Behjati5Dinu Antony1,4Isabel Schüle4Simin Sadeghi-Bojd6Ehsan Ghayoor Karimiani7,8 Miriam Schmidts1,4*
Miriam Schmidts1,4*- 1Genome Research Division, Human Genetics Department, Radboud University Medical Center Nijmegen and Radboud Institute for Molecular Life Sciences, Nijmegen, Netherlands
- 2Department of Biology, University of Sistan and Baluchestan, Zahedan, Iran
- 3Department of Pediatrics, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
- 4Center for Pediatrics and Adolescent Medicine, Faculty of Medicine, University Hospital Freiburg, Freiburg University, Freiburg, Germany
- 5Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
- 6Children and Adolescents Health Research Center, Resistant Tuberculosis Institute, Zahedan University of Medical Sciences, Zahedan, Iran
- 7Next Generation Genetic Polyclinic, Mashhad, Iran
- 8Razavi Cancer Research, Razavi Hospital, Imam Reza International University, Mashhad, Iran
Background: Nephropathic Cystinosis, the most common cause of renal Fanconi syndrome, is a lysosomal transport disorder with an autosomal recessive inheritance pattern. A large number of mutations in CTNS have been identified as causative to date. A 57 kb deletion encompassing parts of CTNS is most commonly identified in Caucasians but this allele has not been identified in individuals of Eastern Mediterranean, Middle Eastern, Persian, or Arab origin to date.
Methods and Results: Implementing whole exome sequencing (WES) in a consanguineous Iranian family, we identified this large deletion affecting CTNS in a patient initially presenting with hypokalemic metabolic alkalosis symptoms and considerable proteinuria.
Conclusion: We show WES is a cost and time efficient genetic diagnostics modality to identify the underlying molecular pathology in Cystinosis individuals and provide a summary of all previously reported CTNS alleles in the Middle east population. Our work also highlights the importance to consider the 57-kb deletion as underlying genetic cause in non-European populations, including the Middle East. Limited diagnostic modalities for Cystinosis in developing countries could account for the lack of previously reported cases in these populations carrying this allele. Further, our findings emphasize the utility of WES to define genetic causes in clinically poorly defined phenotypes and demonstrate the requirement of Copy number variation (CNV) analysis of WES data.
Introduction
Cystinosis is an autosomal recessive lysosomal storage disorder (general incidence of 1:100,000–200,000 live births) caused by mutation of the CTNS gene (1, 2). CTNS locates to chromosome 17p13, contains 12 exons of which ten exons, exon 3–12, encode for Cystinosin, a protein facilitating cysteine-proton co-transport out of lysosomes (3, 4). Impaired Cystinosin function leads to the build-up of intralysosomal cystine crystals in cells of many different organs, notably the bone marrow, lymph nodes, kidney, cornea, thyroid, liver and spleen (5). Based on the age at onset and the severity of clinical disease, three cystinosis subtypes are distinguished: the most severe subtype is nephropathic cystinosis (OMIM#219800) which is also known as infantile onset renal tubular fanconi syndrome that affects proximal renal tubular function and results in failure to thrive, polyuria, and polydipsia with a disease onset between age 6 and 18 months on average. This is also the most frequent manifestation of cystinosis accounting for up to 95% of all cases (6). End-stage renal disease (ESRD) occurs often around 10 years of age if untreated (1). The two other milder subtypes, the nephropathic juvenile (late onset) subtype (OMIM# 219900) and the ocular non-nephropathic adult subtype (OMIM#219750) are rarely diagnosed before adult-hood (7, 8). Genotype-phenotype correlations suggest that milder adult-onset phenotypes may occur as a result of at least one milder mutation in comparison to mutations identified to cause the early-onset phenotype (9). Early diagnosis is important especially in nephropathic cystinosis as treatment with aminothiol cysteamine can slow down progression of the renal phenotype (10).
Commonly followed diagnostic approaches for cystinosis include measurement of leukocyte cystine, detection of corneal cystine crystals by the slit lamp examination and molecular testing of the relatively small CTNS gene (11). Nonetheless, in some developing countries, cystinosis diagnosis is still significantly delayed or may not happen due to limited access to healthcare and likewise limited diagnostic resources. In most instances, a cystinosis diagnosis in these countries is made due to (often repetitive) ophthalmological examinations showing accumulation of cystine crystals in the cornea. To date, ~120 causative CTNS mutations including small deletions or insertions, missense, nonsense, and splice site mutations in the coding regions or adjacent intronic regions as well as in the CTNS gene promoter have been described (www.hgmd.cf.ac.uk). Of note, the most common mutation is a large deletion, affecting the first 10 exons of the CTNS gene in either homozygous or heterozygous form. This variant is particularly frequent in Caucasians, accounting for up to 76% of all cases in this group of individuals (12). The breakpoints of this 57,257 base pair deletion locate in exon 10 of CTNS and intron 2/3 of TRPV1, resulting in deletion of large parts of CTNS, all of CARKL/SHPK and the two non-coding exons of TRPV1 (13). However, in other populations, this deletion appears a lot less frequent and previous reports did not identify this allele in cystinosis cases from Middle Eastern countries including Egypt, Iran, Jordan, Turkey, and Saudi Arabia (14).
In the present study, we used Whole Exome Sequencing (WES) to identify the underlying molecular cause in an Iranian subject with metabolic alkalosis. Interestingly, we identified the previously described 57 kB deletion encompassing CTNS exon 1–10.
Methods
DNA Samples
Consent forms were obtained from all the participants of this study. Ethical approval was obtained from the Ethics committee of Mashhad University of medical (IR.MUMS.REC.1395.534) and samples were processed in Nijmegen under the Diagnostic Innovation Program. Written informed consent from the parents or legal guardians of the patients/participants for the publication of their data. Genomic DNA was extracted from EDTA blood using standard salting out method. The concentration of DNA was measured by Qubit 2.0 (life technologies, Carlsbad, CA, USA).
Whole Exome Sequencing
Two micrograms of DNA from the index case was subjected to WES at Novogene, Hongkong. Illumina HiSeq 2500. Exome capture was performed using Agilent SureSelect Human All Exon V6 Kit; sequencing was undertaken on an Illumina HiSeq 2500 Genome Analyzer machine. Paired-end sequencing resulting in sequences of 150 bases from each end of the fragments with a fraction_of_target_covered_with_at_least_10x of 99.5%, of at least 20x of 98.1%, of at least 50x of 87.7% and of 100x of 44.3%. UCSC hg19 was used as a reference genome. VarScan version 2.2.5 and MuTec and GATK Somatic Indel Detector were used to detect SNV and InDels, respectively. Data was filtered for MAF < 1% in public control databases such as dbSNP, ExAc, and gnomAD (gnomAD, http://gnomad.broadinstitute.org). subsequently, variants occurring with MAF > 1% in the Iranome (contains 800 healthy individuals) (http://www.iranome.ir) were also excluded. The remaining variants were filtered for known disease causing genes with emphasis on diseases compatible with the patient phenotype (renal tubular disorders). We prioritized homozygous variants due to the autosomal recessive inheritance pattern of disease and known consanguinity in the family. This did not reveal any variant in a gene previously associated with a tubular disorder. We therefore proceeded to visual inspection of the BAM file for known tubuluopathy genes and detected a large homozygous deletion encompassing most of CTNS.
PCR and Sanger Sequencing
The visually identified large homozygous deletion encompassing CARKL/SHPK and the first 10 exons of CTNS found in the BAM file of the index case was confirmed using PCR and subsequent Sanger sequencing. Primers were designed such as the forward primer located in TRPV1 and the reverse primer located in intron 10 of CTNS gene. AmpliTaq mastermix (Applied Biosystems) was used according to the manufacturer's protocol. A standard amplification program was used (initial denaturation at 95°C for 10min; followed by 35 cycles of denaturing at 94°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 3 min and final extension at 72°C for 7 min). PCR products were cleaned using ExoSAP-IT® (USB, Cleveland, Ohio, USA) and subsequently submitted for bidirectional Sanger sequencing using a 3730XL DNA analyzer (ABI, Foster City). Primers sequences are available on request.
Results
Our index case was a 1-year-old boy who was the third child of consanguineous healthy parents of Persian ethnicity from Iran. He was born after term gestation with birth weight of 3,200 g (P25-50) and a length of 49.2 cm (P25-50); the pregnancy and delivery were uncomplicated. At the age of nearly 5 months, he was hospitalized due to recurrent vomiting, polydipsia, and polyuria, failure to thrive was noted since the 4th month of life with a length of 62.2 cm (<P3.) weighing 6,800 g (<P3.) at the age of 8 months. On admission, urine analysis (dip stick) revealed a specific weight of 1,004, urine pH of 5, glucosuria (+++), proteinuria (+), and ketonuria (+) with no bilirubin, nitrite, or urobilinogene present. Urine microscopy did not reveal significant hematuria or leucocytes in the urine. Chemical urine analysis, for example for protein quantification and protein subtype identification could not be performed in the admitting center. Venous blood pH was 7.48 (normal range 7.35–7.45), HCO3 16.7 mmol/L (normal range 22–26 mmol/L), pCO2 22.6 mmHg (normal range 35–45 mmhg), pO2 55.3 mmHg (normal range >30 mmHg), and O2 saturation was found to be 91.1%. Serum sodium was within normal ranges (140 mmol/L) but hypokalemia (potassium 2.4 mmol/L, normal range 3.6–5.4 mmol/L) was noted. Creatinine, ferritine, vitamin D, and TSH were within the normal range. Abdominal ultrasound did not reveal any abnormalities in the liver, gallbladder, pancreas, spleen, kidneys, or bladder. The biochemical findings of mild hypokalemic alkalosis (possibly with a respiratory component indicated by low CO2 e.g., due to screaming child) together with severe growth retardation, polydipsia and polyuria as well as the urine findings suggested a renal tubulopathy such as Bartter syndrome or Fanconi syndrome. After 1 month of receiving Polycitra, the patient gained 400 g of weight (P3.) and his serum potassium level increased to 3 mmol/L while the blood pH decreased to pH 7.39. However, HCO3 was measured 14.1 mmol/L, urine pH was 6 and glucosuria, proteinuria, and ketonuria persisted. After 6 months of treatment, serum potassium was still below the normal range with 3.2 mmol/L. The patient showed mild hypophosphatemia (3.5 mg/dl) (normal range 4–7 mg/dl).
An older sibling who passed away previously at the age of 10 years had a medical history of renal transplant rejection, however the underlying cause of ESRD had not been clearly defined. An 8 years old sister did not show any kidney or eye problems (Figure 1). We therefore proceeded to perform WES to establish a molecular diagnosis to optimize the therapeutic approach in knowledge of the underlying condition as well as in light of being able to offer genetic counseling for the family.
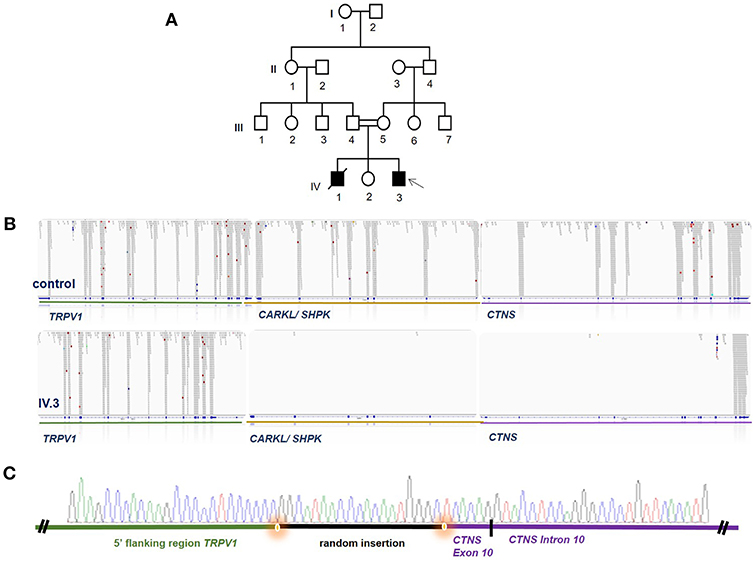
Figure 1. WES identifies a homozygous deletion encompassing large parts of CTNS. (A) Pedigree of the Iranian family, DNA of individual IV3 was analyzed by WES (arrow) while no DNA of other family members was available. Females are marked with circle, males with square, solid filling marks affected individuals, crossed symbols marks a deceased individual, double lines between individuals mark a consang. marriage and single lines mark non-consang.-marriages. (B) WES revealed a large genomic deletion for individual IV.3 encompassing parts of TRPV1, all of CARKL/SHPK and large parts of CTNS as visualized in the BAM file generated from WES data. Read alignment of a control person is shown above for comparison. (C) Breakpoint confirmation by Sanger sequencing confirms presence of the homozygous deletion and reveals a random insertion shown in black.
WES data filtering was performed as described in the Methods section of this manuscript. No promising homozygous variant in known tubulopathy genes were identified amongst the SNVs or INDELs remaining after filtering. We therefore proceeded to inspect the BAM file for homozygous deletions in known tubulopathy genes and, somewhat surprisingly, detected a large homozygous deletion encompassing the CTNS gene (Figure 1). Sanger sequencing confirmed this deletion which spans from exon 10 of the CTNS gene to intron 2/3 of TRPV1, encompassing CARKL/SHP but sparing any coding sequence of TRPV1 gene (Figure 1). This deletion has been previously reported as European founder allele but has not been detected in Middle-Eastern cystinosis patients before. Unfortunately, no DNA of the parents of the index case or the deceased sibling was available for genetic testing.
Discussion
We describe a case clinically presenting with proximal renal tubular dysfunction as suggested by proteinuria and glucosuria as well as polydipsia, polyuria, vomiting, failure to thrive, and mild hypokalemia. However, blood gas analysis revealed very mild alkalosis, likely with a respiratory component possibly due to screaming of the child during blood draw as indicated by low CO2 in combination with low HCO3–. This caused confusion amongst the admitting local clinicians with regards to the precise underlying condition of the tubular disore as (metabolic) alkalosis would rather be compatible with Bartter Syndrome while metabolic acidosis would be expected in nephropathic cystinosis. Mild hypophosphatemia was noted at a repeat hospital visit in Iran but the patient never developed a clinical picture of rickets. In combination with limited clinical information of his older sibling who passed away after he developed ESRD, we decided to perform WES as first line diagnostic test instead of single gene analyses of known Bartter and renal fanconi syndrome genes. This resulted in the identification of the previously described homozygous 57-kb deletion comprising nearly the entire CTNS gene and led to the clinical diagnosis of infantile nephropathic cystinosis in the family. This allele is very frequent in Northern European/Northern American populations of European ancestry due to a German founder mutation originating around 500 AD (15). In other populations this allele is less often identified and previously published reports suggested it doesn't contribute to Cystinosis in the Middle East. However, largely unavailable genetic testing and incomplete clinical data in developing countries creates a significant gap in the rate of cystinosis diagnosis between developed and developing countries (16).
CTNS mutation surveys of Cystinosis in the Middle East population including Iran, Turkey, Saudi Arabia, Egypt, and Jordan have been summarized in Table 1 with mutation localization to CTNS on protein level displayed in Figure 2. The most commonly identified CTNS allele in Iranian individuals was the c.681G>A (p.Glu227*) variant and together with the c.922G>A (p.Gly308Arg), c.681G>A (p.Glu227*) is also the most common variant in the Saudi Arabia population. In Jordan, c.890G>A, p.Trp297* has been identified in more than one family. c.451A>G, p.Arg151Gly, c.681G>A (p.Glu227*) and 0.834_842del, p. Val279_Tyr281del are the most common alleles in the Turkish population while in Egypt, c.829dup, p.Thr277Asnfs*19 has been reported in 4 different cases. Interestingly, c.681G>A (p.Glu227*) is the overall most frequently identified allele detected in all Middle eastern cohorts and most variant represent stop or frameshift alleles (Table 1) (17–22, 26). Studies investigating CTNS mutations in Cystinosis patients from the Far East have likewise reported several alleles including compound heterozygous mutation c.329G>C, c.329+2T>C in Japan, c.926G>A, c.850C>T, c.18-21del/c and c.969C>G in Thailand, and compound heterozygous mutations c.329+1del andc.463_464del in China while none of the reported cases carried the 57-kb deletion (27–29) but a smaller deletion encompassing ~20 kb of genomic DNA extending from CARKL intron 1 to CTNS intron 6, c.771–793del, and a c.1515G>A variant have been reported in Tunisian nephrotic patients (30). The 57-kb deletion has however been previously detected in patients from African American ethnicity (31).

Table 1. Summary of previously published CTNS causing variants in patients from the Middle-East.
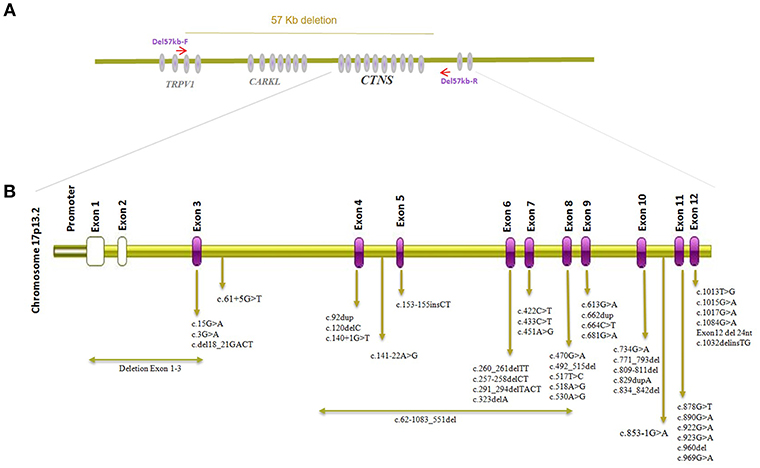
Figure 2. CTNS gene structure and visualization of alleles detected in individuals of Middle-Eastern ethnical origin. (A) Visualization of the genomic location of CTNS and genomic breakpoints of the 57 kb deletion. (B) Localizations of CTNS alleles described in cystinosis patients from the Middle East.
Currently, in Iran as well as many other developing countries, cystinosis is most often diagnosed by slit lamp examination, however corneal crystals may not develop until 1–1.5 years of age. There is therefore a possibility that renal disease may be present before corneal abnormalities can be noted and a critical treatment window is left unused. Renal cystinosis symptoms can closely resemble findings in other tubulopathic diseases such as Dent‘s disease, Lowe’s syndrome, Bartter syndrome and nephrogenic diabetes insipidus and without corneal findings, the correct diagnosis of cystinosis may not be achieved (32). Therefore, additional easy to access diagnostic tests are required, including genetic evaluation. Our study highlights the importance to specifically investigate this allele in non-European populations, including subjects from Iran. Exome data offers great possibilities to detect homozygous alleles by visual BAM file inspection, however heterozygous alleles will be missed by this method and require CNVs analysis. Nevertheless, slit lamp examination of the eyes should be used as initial diagnostic test in patients with clinical suspicion of cystinosis.
As the 57-kb deletion encompasses CARKL/SHPK encoding sedoheptulokinas enzyme, a recent study has also suggested measuring activity of the encoded sedoheptulokinas enzyme if cystinosis is suspected. This test however is not applicable for patients with other CTNS mutations (33) and given the rarity of the phenotype, may only be available in large centers. Genetic workup therefore remains currently the most reliable and feasible diagnostic method with whole exome sequencing costs below 500 Euros per sample and available through a number of commercial companies and Sanger sequencing being even cheaper.
In summary, while previous studies suggested that the 57.kb deletion does not play a role in the Middle East, in fact this allele also needs to be taken into consideration when screening CTNS, particularly in Iranian patient with Persian ethnicity. As corneal findings may occur after the onset of renal symptoms which in turn share common characteristics with other tubulopathies, we suggest to perform WES or NGS gene panel sequencing as a cost- and time-efficient genetic workup to secure the diagnosis and enable genetic counseling and prenatal diagnosis.
Ethics Statement
Genetic analysis was conducted under the Innovative Diagnostics Program Human Genetics Department, Radboudumc Nijmegen, The Netherlands.
Author Contributions
MN and MS wrote the manuscript. MN performed WES analysis and Sanger sequencing. ZB and DA were involved in WES logistics and sample preparation. IS was involved in assessment of clinical data. DMKT, AA, FB, SS-B, and EGK were involved in patient recruitment and clinical care. MS supervised the work and provided funding. MS, MN, and DMKT designed the study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MS acknowledges funding from Radboudumc and RIMLS Nijmegen (Hypatia tenure track fellowship), the Deutsche Forschungsgemeinschaft (DFG CRC1140 KIDGEM), and the European Research Council (ERC StG TREATCilia, grant No. 716344).
References
1. Gahl WA. Cystinosis. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, editors. Pediatric Nephrology. Berlin; Heidelberg: Springer (2009). p. 1019–38. doi: 10.1007/978-3-540-76341-3_41
2. Levy M. Feingold J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. (2000) 58:925–43. doi: 10.1046/j.1523-1755.2000.00250.x
3. Kalatzis V, Nevo N, Cherqui S, Gasnier B, Antignac C. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet. (2004) 13:1361–71. doi: 10.1093/hmg/ddh152
4. McDowell GA, Gahl WA, Stephenson LA, Schneider JA, Weissenbach J, Polymeropoulos MH. Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat Genet. (1995) 10:246–8. doi: 10.1038/ng0695-246
5. Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol. (2017) 13:115–31. doi: 10.1038/nrneph.2016.182
6. Emma F, Nesterova G, Langman C, Labbé A, Cherqui S, Goodyer P, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. (2014) 29(Suppl 4):iv87–94. doi: 10.1093/ndt/gfu090
7. Goldman H, Scriver CR, Aaron K, Delvin E, Canlas Z. Adolescent cystinosis: comparisons with infantile and adult forms. Pediatrics. (1971) 47:979–88.
8. Cogan DG, Kuwabara T, Kinoshita J, Sheehan L, Merola L. Cystinosis in an adult. J Am Med Assoc. (1957) 164:394–6. doi: 10.1001/jama.1957.02980040034009
9. Servais A, Morinière V, Grünfeld JP, Noël LH, Goujon JM, Chadefaux-Vekemans B, et al. Late-onset nephropathic cystinosis: clinical presentation, outcome, and genotyping. Clin J Am Soc Nephrol. (2008) 3:27–35. doi: 10.2215/CJN.01740407
10. Thoene J, Oshima RG, Crawhall JC, Olson DL, Schneider JA. Cystinosis. Intracellular cystine depletion by aminothiols in vitro and in vivo. J Clin Invest. (1976) 58:180–9. doi: 10.1172/JCI108448
11. Wilmer MJ, Schoeber JP, van den Heuvel LP, Levtchenko EN. Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol. (2011) 26:205–15. doi: 10.1007/s00467-010-1627-6
12. Forestier L, Jean G, Attard M, Cherqui S, Lewis C, van't Hoff W, et al. Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. Am J Hum Genet. (1999) 65:353–9. doi: 10.1086/302509
13. Anikster Y, Lucero C, Touchman JW, Huizing M, McDowell G, Shotelersuk V, et al. Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS). Molecul Genet Metabol. (1999) 66:111–6. doi: 10.1006/mgme.1998.2790
14. Topaloglu R, Gulhan B, Inözü M, Canpolat N, Yilmaz A, Noyan A, et al. The clinical and mutational spectrum of Turkish patients with cystinosis. Clin J Am Soc Nephrol. (2017) 9:CJN.00180117. doi: 10.2215/CJN.00180117
15. Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. (2002) 347:111–21. doi: 10.1056/NEJMra020552
16. Bertholet-Thomas A, Berthiller J, Tasic V, Kassai B, Otukesh H, Greco M, et al. Worldwide view of nephropathic cystinosis: results from a survey from 30 countries. BMC Nephrol. (2017) 18:210. doi: 10.1186/s12882-017-0633-3
17. Jaradat S, Al-Rababah B, Hazza I, Akl K, Saca E, Al-Younis D. Molecular analysis of the CTNS gene in Jordanian families with nephropathic cystinosis. Nefrologia. (2015) 35:547–53. doi: 10.1016/j.nefro.2015.09.009
18. Shahkarami S, Galehdari H, Ahmadzadeh A, Babaahmadi M, Pedram M, Shahkarami S, et al. The first molecular genetics analysis of individuals suffering from nephropatic cystinosis in the Southwestern Iran. Nefrologia. (2013) 33:308–15. doi: 10.3265/Nefrologia.pre2012.Sep.11558
19. Sadeghipour F, Basiratnia M, Derakhshan A, Fardaei M. Mutation analysis of the CTNS gene in Iranian patients with infantile nephropathic cystinosis: identification of two novel mutations. Hum Genome Var. (2017) 4:17038. doi: 10.1038/hgv.2017.38
20. Ghazi F, Hosseini R, Akouchekian M, Teimourian S, Ataei Kachoei Z, Otukesh H, et al. CTNS molecular genetics profile in a Persian nephropathic cystinosis population. Nefrologia. (2017) 37:301–10. doi: 10.1016/j.nefro.2016.11.024
21. Soliman NA, Elmonem MA, van den Heuvel L, Abdel Hamid RH, Gamal M, Bongaers I, et al. Mutational spectrum of the CTNS gene in Egyptian patients with nephropathic cystinosis. JIMD Rep. (2014) 14:87–97. doi: 10.1007/8904_2013_288
22. Topaloglu R, Vilboux T, Coskun T, Ozaltin F, Tinloy B, Gunay-Aygun M, et al. Genetic basis of cystinosis in Turkish patients: a single-center experience. Pediatr Nephrol. (2012) 27:115–21. doi: 10.1007/s00467-011-1942-6
23. Önenli-Mungan N, Kör D, Karabay-Bayazit A, Cengiz N, Yavuz S, Noyan A, et al. Genotypic and phenotypic features of the cystinosis patients from the South Eastern part of Turkey. Turk J Pediatr. (2016) 58:362–70. doi: 10.24953/turkjped.2016.04.003
24. Doneray H, Aldahmesh M, Yilmaz G, Cinici E, Orbak Z. Infantile nephropathic cystinosis: a novel CTNS mutation. Eur J Med. (2017) 49:148–51. doi: 10.5152/eurasianjmed.2017.17039
25. Bastug F, Nalcacioglu H, Ozaltin F, Korkmaz E, Yel S. Nephropathic cystinosis mimicking bartter syndrome: a novel mutation. Iran J Kidney Dis. (2018) 12:61–3.
26. Aldahmesh MA, Humeidan A, Almojalli HA, Khan AO, Rajab M, AL-Abbad AA, et al. Characterization of CTNS mutations in Arab patients with cystinosis. Ophthal Genet. (2009) 30:185–9. doi: 10.3109/13816810903200953
27. Higashi S, Matsunoshita N, Otani M, Tokuhiro E, Nozu K, Ito S. Diagnostic challenge in a patient with nephropathic juvenile cystinosis: a case report. BMC Nephrol. (2017) 18:300. doi: 10.1186/s12882-017-0721-4
28. Yeetong P, Tongkobpetch S, Kingwatanakul P, Deekajorndech T, Bernardini IM, Suphapeetiporn K, et al. Two novel CTNS mutations in cystinosis patients in Thailand. Gene. (2012) 499:323–5. doi: 10.1016/j.gene.2012.03.047
29. Yang YJ, Hu Y, Zhao R, He X, Zhao L, Tu M, et al. First report of CTNS mutations in a Chinese family with infantile cystinosis. Sci World J. (2015) 2015:309410. doi: 10.1155/2015/309410
30. Chkioua L, Khedhiri S, Grissa O, Aloui C, Turkia HB, Ferchichi S, et al. Genetic basis of cystinosis in Tunisian patients: identification of novel mutation in CTNS gene. MetaGene. (2015) 5:144–9. doi: 10.1016/j.mgene.2015.07.003
31. Kleta R, Anikster Y, Lucero C, Shotelersuk V, Huizing M, Bernardini I, et al. CTNS mutations in African American patients with cystinosis. Mol Genet Metab. (2001) 74:332–7. doi: 10.1006/mgme.2001.3218
32. Elmonem MA, Veys KR, Soliman NA, van Dyck M, van den Heuvel LP, Levtchenko E. Elmonem MA, et al. Cystinosis: a review. Orphanet J Rare Dis. (2016) 11:47. doi: 10.1186/s13023-016-0426-y
33. Wamelink MM, Struys EA, Jansen EE, Blom HJ, Vilboux T, Gahl WA, et al. Elevated concentrations of sedoheptulose in bloodspots of patients with cystinosis caused by the 57-kb deletion: implications for diagnostics and neonatal screening. Mol Genet Metab. (2011) 102:339–42. doi: 10.1016/j.ymgme.2010.12.002
Keywords: Cystinosis, CTNS deletion, Middle East population, Iran, tubulopathy
Citation: Najafi M, Tamandani DMK, Azarfar A, Bakey Z, Behjati F, Antony D, Schüle I, Sadeghi-Bojd S, Karimiani EG and Schmidts M (2019) A 57 kB Genomic Deletion Causing CTNS Loss of Function Contributes to the CTNS Mutational Spectrum in the Middle East. Front. Pediatr. 7:89. doi: 10.3389/fped.2019.00089
Received: 06 November 2018; Accepted: 28 February 2019;
Published: 21 March 2019.
Edited by:
Douglas Marc Silverstein, United States Food and Drug Administration, United StatesReviewed by:
Fernando Santos, Universidad de Oviedo, SpainShashi Kumar Nagaraj, Duke University Medical Center, United States
Copyright © 2019 Najafi, Tamandani, Azarfar, Bakey, Behjati, Antony, Schüle, Sadeghi-Bojd, Karimiani and Schmidts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dor Mohammad Kordi Tamandani, ZG9yX2tvcmRpQHlhaG9vLmNvbQ==
Miriam Schmidts, bWlyaWFtLnNjaG1pZHRzQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==