
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr., 31 January 2019
Sec. Pediatric Immunology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00429
This article is part of the Research TopicMeaningful Cases of Primary Immunodeficiencies: Volume 2View all 21 articles
 Puja Sood Rajani
Puja Sood Rajani Maria A. Slack*
Maria A. Slack*Here we describe two term male infants diagnosed with X-linked CGD who present, in addition to frequent infection, with a unique papulopustular skin rash. CGD is caused by a number of genetic defects that impair phagocyte function. This disease results in recurrent infections and granuloma formation. Rarely do patients develop cutaneous symptoms, unless associated with autoimmune disorders such as systemic erythematous lupus (1). Each male infant mentioned here was diagnosed with CGD based on abnormal DHR testing and confirmatory genetic testing. The presenting papulopustular dermatitis was initially characterized as non-classic appearing eczema and subsequently found to be refractory to usual eczema treatment and antibiotics. After obtaining written informed consent from both families, we have documented photographs of the development of a characteristic rash in two newly diagnosed infants with CGD. One infant underwent cutaneous biopsy with histologic evaluation and negative cultures. The dermatitis for both infants was refractory to topical and systemic therapies, and resolved after bone marrow transplantation. Our objective was to characterize cutaneous findings in X-linked CGD and emphasize the importance of considering further immune workup in patients who present with unusual cutaneous findings that do not fit with common infant rashes in conjunction with concerning features for primary immunodeficiency.
Chronic granulomatous disease (CGD) is caused by genetic defects which impair phagocyte function. This disease results in recurrent infections and granulomatous inflammation. Rarely do patients present with systemic rash as a feature of X-linked CGD, unless associated with autoimmune disorders such as systemic erythematous lupus (1, 2). Previously described cutaneous findings include granulomas, abscesses, photosensitivity, malar rash, discoid lupus, vasculitis, and rarely vesicular rashes due to infection (1–5). Here we describe two term infants diagnosed with X-linked CGD who present, in addition to frequent infection, with a unique papulopustular skin rash initially diagnosed as non-classic appearing eczema refractory to usual eczema treatment and antibiotics.
Our first patient presented at 8 months of age with a known history of reflux born full term. His presenting symptoms included episodic fever and chronic leukocytosis, iron deficiency anemia, thrombocytosis, elevated inflammatory markers, blood in the stool concerning for proctocolitis for the past month with elevated calprotectin, and a rash present since 4 months of age. He was on no medications. Lab work showed an elevated WBC to 29.4 × 103/μL (5.0–17.0 × 103/μL), decreased hemoglobin down to 8.6 g/dL (11.5–14.0 g/dL) (thought to be due to blood in the stool), elevated platelet count to 621 × 103/μL (150–330 × 103/μL), ESR max to >130 mm/h, elevated absolute neutrophil count to 13,500 and elevated absolute lymphocyte count to 14,400. Blood cultures were negative, and T, B, and NK lymphocyte subsets were normal. Esophagogastroduodenoscopy with flexible sigmoidoscopy was performed due to his history of reflux and his proctocolitis. It showed no signs of inflammatory bowel disease, the left colon had macroscopically raised erythematous lesions but was microscopically normal with no signs of active colitis. He was initially diagnosed with food protein-inducted enterocolitis syndrome to dairy, given his upper (reflux) and lower (proctocolitis) symptoms, and eczema due to his rash with this allergic profile in the setting of diagnosis of various infections (otitis, upper respiratory illness). The skin rash was described as non-pruritic, generalized pink-purple papulopustular lesions with an erythematous base, most prominent on upper and lower extremities, mostly sparing the trunk (Figure 1). Skin cultures were negative. Skin biopsy was performed and histopathology revealed an essentially unremarkable appearing epidermis and a superficial perivascular lymphohistiocytic infiltrate within the dermis. Eosinophils and plasma cells were not abundant. The histologic changes were thought to be non-specific, but could be seen in a drug reaction, urticaria, or viral exanthem. Initial immunologic evaluation revealed normal levels of IgA, IgG, IgM, IgE and T, B, and NK lymphocyte subsets. Outside of medications and infection, these findings have not been described in CGD related pathology. Giemsa stain and a stain for mast cell tryptase revealed normal numbers of mast cells in the biopsy specimen. He was not on any systemic medications at the time and did not respond to treatment with topical moisturizers, oral antihistamines, topical steroids, dietary avoidances, or any standard for eczema and urticarial care. He had a maternal uncle who passed away in infancy from infection. Heightened clinical suspicion for primary immunodeficiency with this family history and concern that his rash was related to cutaneous inflammatory condition led to a neutrophil oxidative burst assay being obtained. This demonstrated complete loss of neutrophil oxidative function, which was consistent with CGD and genetic testing, thus being confirmatory for X-linked CGD. Pathogenic mutation nucleic acid change was c. 469c>T; Hemizygous; Amino acid alteration was p.157Arg>X within the CYBB gene locus. He had no other features of auto-immune disease; a negative anti-nuclear antibody (ANA) result was obtained later in his clinical course and his proctocolitis was thought to be CGD-associated colitis which is well described in CGD. His rash did not respond to systemic treatments for his colitis, with sulfasalazine, oral antibiotics for both gram positive and gram negative organisms, or IFN-gamma, and he solely improved after hematopoietic stem cell transplantation (HSCT) by day 35 post-transplant. The patient was 18 months of age at the time of transplantation with reduced intensity myeloablative conditioning and has been without recurrence of the rash over the past 18 months. His colitis also improved after transplantation.
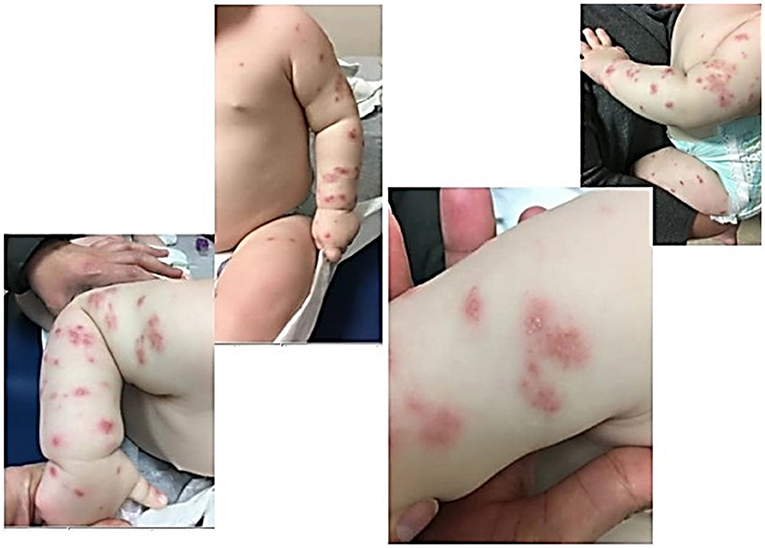
Figure 1. Patient One demonstrates papulopustlar lesions with an erythematous base scattered on upper and lower extremities.
Our second patient presented at 14 months of age with no significant past medical history born full term. He had a paternal history of autoimmune gastrointestinal disease from his father. His presenting symptoms included persistent fevers, inguinal lymphadenopathy, leukocytosis, elevated inflammatory markers, non-bloody diarrhea and rash. He presented to the hospital with a fever of unknown origin with concern for infection, atypical Kawasaki's, or drug rash. His rash was present since 9 months of age and described as scattered, erythematous, papulopustular lesions most prominent on the upper arms but also present on lower arms, legs, and chest, faint lesions on the face (Figure 2). There was some induration and thickness to the rash in some areas (confluent on the arms with more erythema and induration) and more faint pink papules (scattered on legs) elsewhere. Lab work showed an elevated WBC to 20.5 × 103/μL, decreased hemoglobin down to 9.2 g/dL, elevated platelets to 433 × 103/μL, ESR max to 73 mm/h, increased CRP to 104 and elevated ALC to 13,900. Blood cultures were negative, and T, B, and NK subsets were normal. Unfortunately, a skin biopsy of the rash was not performed. Initial differential diagnosis included fever of unknown origin, infection, atypical Kawasaki's, drug rash (he had been on trimethoprim/sulfamethoxazole), eczema flare, malignancy, Kikuchi Fujimoto disease, and tularemia. A biopsy of his inguinal lymph node showed “granulomatous lymphadenitis with neutrophilic abscess formation,” and culture was positive for Serratia marcescens, increasing suspicion for CGD. Neutrophil oxidative burst assay showing complete loss of oxidative function was consistent with CGD, and genetic testing was confirmatory for X-linked CGD, due to the presence of a mutation within the CYBB gene. Pathogenic nucleic acid change was c. 141 + 5G>A; Hemizygous; Amino acid alteration was p.del Exon 2. The rash remained unchanged with treatment for infection, initiation of antifungal and bacterial prophylaxis, along with topical steroid therapy and resolved solely with HSCT by day 24 post-transplant. The patient was 18 months at the time of bone marrow transplantation, and after receiving reduced intensity myeloablative conditioning, the rash has not recurred over the following 6 months.
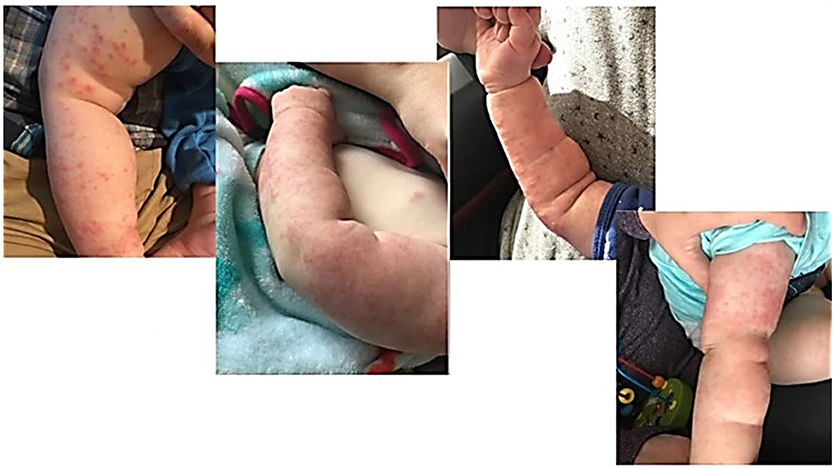
Figure 2. Patient Two demonstrates papulopustular lesions found diffusely scattered on the upper and lower extremities with areas of confluence.
These cases emphasize the importance of heighted awareness for primary immunodeficiency in patients who present with frequent infections and who have unusual cutaneous findings that do not fit with common infant rashes. Our patients presented with a papulopustular dermatitis that was scattered throughout the extremities, refractory to routine therapies, and non-diagnostic on skin biopsy in conjunction with a characteristic infection associated with CGD. Afrough et al. described a somewhat similar vesiculopustular rash in a 24 day old male infant of the periorbita, genitalia, foot, and sacroiliac regions. Although these cases share a descriptive visual resemblance, the patient described by Afrouh et al. was quite sick and acquired multiple severe infections early in life, including osteomyelitis. Their patient's skin cultures were positive for Staphylococcus aureus and skin biopsies showed “necrotizing granulomatous tissue reaction with infectious etiology,” which aided in early diagnosis of CGD. In our first case, there was no identifiable skin infection or disease specific inflammation on skin biopsy to suggest a particular primary immunodeficiency. The resolution of the rash after HSCT in both cases highlights the relationship of this cutaneous finding with the underlying diagnosis of CGD. Specifically, our findings stress the need for considering CGD in a male infant presenting with generalized papulopustular rash in the right clinical context. Differential for generalized papulopustular rashes can include eczema, rosacea, acne, folliculitis, scabies, candida infection, cutaneous lupus, and other disorders. The need to assess infection history, other comorbidities, family history, and skin biopsies for rashes that are not improving with standard care, is essential to aid in diagnosis and work up of rare disease. Based on our findings, we would recommend consideration of dihydrorhodamine (DHR) analysis in children presenting with refractory papulopustular dermatitis with suggestive family and/or infectious history consistent with CGD. Further, it is important to note in such patients, HSCT led to resolution of these CGD associated symptoms, specifically papulopustular rash and colitis in our patients.
The above was a case report involving human subjects who provided consent.
PR made significant contributions to the manuscript by drafting. MS was involved in drafting the manuscript and revising it critically for important intellectual content. PR and MS have given final approval of the version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The above cases were previously published in a condensed format in abstract in the Journal of Clinical Immunology by Springer Nature and permission was obtained (License 4435380742170 obtained Sep 24, 2018 by Springer Nature/Journal of Clinical Immunology). We thank the families of our patients for their steadfastness and willingness to share their experience to aid others in better understanding this complex disease and written consent was obtained for permission to publish this case report along with pictures of the rash.
1. Agarwal S. Chronic granulomatous disease. J Clin Diagn Res. (2015) 9:SD01–02. doi: 10.7860/JCDR/2015/12139.5945
2. Ben Abdallah Chabchoub R, Turki H, Mahfoudh A. [Systemic lupus erythematosus in a boy with chronic granulomatous disease: case report and review of the literature]. Arch Pediatr. (2014) 21:1364–6. doi: 10.1016/j.arcped.2014.09.011
3. Wolpowitz D, Wise EM. Lupus-like rash of chronic granulomatous disease effectively treated with hydroxychloroquine. Cutis (2015) 95:E25–27.
4. Afrough R, Mohseni SS, Sagheb S. An uncommon feature of chronic granulomatous disease in a neonate. Case Rep Infect Dis. (2016) 2016:5943783. doi: 10.1155/2016/5943783
Keywords: rash, papulopustlar lesions, chronic granulomatous disease (CGD), immune deficiency, immunodeficiency-primary, dermatitis, papulopustular
Citation: Rajani PS and Slack MA (2019) Papulopustular Dermatitis in X-Linked Chronic Granulomatous Disease. Front. Pediatr. 6:429. doi: 10.3389/fped.2018.00429
Received: 15 September 2018; Accepted: 24 December 2018;
Published: 31 January 2019.
Edited by:
Ivan K. Chinn, Baylor College of Medicine, United StatesReviewed by:
Nicholas L. Rider, Baylor College of Medicine, United StatesCopyright © 2019 Rajani and Slack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria A. Slack, bWFyaWFfc2xhY2tAdXJtYy5yb2NoZXN0ZXIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.