 Jose Antonio Tavares de Albuquerque1,2
Jose Antonio Tavares de Albuquerque1,2 Edgar Borges de Oliveira Junior1,2
Edgar Borges de Oliveira Junior1,2 Nuria Bengala Zurro3Paola Vendramini3Edson Kiyotaka Ishizuka1,2Daniela de Souza Paiva Borgli4Monica Soares de Souza4
Nuria Bengala Zurro3Paola Vendramini3Edson Kiyotaka Ishizuka1,2Daniela de Souza Paiva Borgli4Monica Soares de Souza4 Antonio Condino-Neto3*
Antonio Condino-Neto3*- 1Immunogenic Assessoria e Diagnóstico em Saúde LTDA, São Paulo, Brazil
- 2PENSI Institute, José Luiz Egídio Setúbal Foundation, Sabará Hospital, São Paulo, Brazil
- 3Department of Immunology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil
- 4Hospital Federal dos Servidores do Estado, Rio de Janeiro, Brazil
Chronic granulomatous disease (CGD) is an innate immune deficiency of phagocytic cells caused by mutations that affect components of the NADPH oxidase system, with resulting impairment in reactive oxygen species production. Patients with CGD are susceptible to recurrent infections and hyperinflammatory responses. Mutations in CYBB lead to the X-linked form of CGD and are responsible for ~ 70% of cases. In this study, we report the case of a 2.5-year-old male patient with recurrent pneumonia and Bacillus Calmette-Guérin infection (BCGitis). As his first clinical manifestation, he presented with bullous impetigo at 18 days of age, which was followed by recurrent pneumonia and regional BCGitis. Genetic analysis revealed a de novo mutation in exon 5 of the CYBB gene: a single-nucleotide substitution, c.376T > C, leading to a C126R change.
Introduction
The production of reactive oxygen species (ROS) is fundamental for the ability of phagocytes to eliminate microorganisms. One of the key producers of ROS in these cells is NADPH oxidase, which is composed of membrane and cytoplasmic subunits and generates superoxide by transferring electrons from NADPH to oxygen inside phagolysosome (1–3). Of these components, flavocytochrome b558, which is located at the membrane, consists of gp91phox and p22phox; p47phox, p67phox, and p40phox are located in the cytoplasm (4). The genes encoding gp91phox, p22phox, p47phox, p67phox, and p40phox are CYBB, CYBA, NCF1, NCF2, and NCF4, respectively. When activated, the cytoplasmic subunits translocate to the membrane, where the NADPH oxidase complex catalyzes the conversion of molecular oxygen (O2) to superoxide anion () and other reactive oxygen intermediates (5).
Chronic granulomatous disease (CGD) is an inherited immunodeficiency disease caused by defects in NADPH oxidase. Mutations in all five structural genes of the NADPH oxidase complex have been implicated in CGD, affecting 1 in 250,000 live births in the USA. Mutations in CYBB, which cause X-linked CGD (XL-CGD), are responsible for ~ 70% of CGD cases in Latin America (2). The hallmark of this immunodeficiency is the development of chronic inflammatory granulomas, hyperinflammatory responses, and recurrent infections (1, 3, 4). Indeed, lung infections are prevalent in CGD patients and may be complicated by granulomatous inflammation, persistent hilar or mediastinal lymphadenopathy, and pulmonary fibrosis (1, 2, 6).
Common pathogenic agents in these cases include Staphylococcus aureus, Escherichia coli, Aspergillus spp, Candida spp, Klebsiella spp, and Burkholderia cepacia. Other symptomatic occurrences involve colitis/enteritis or granulomatous obstruction of either the gastric outlet or urinary tract (1, 2, 6). Approximately 30% of patients in Latin America have adverse reactions to Bacillus Calmette-Guérin (BCG) vaccinations (2).
In this study, we report a male patient with X-linked CGD with recurrent pneumonia, regional BCG vaccine dissemination and a de novo mutation in CYBB that results in the single-amino acid substitution C126R in the gp91phox protein.
Case Report
The proband (2.5 years old) was a male child from a non-consanguineous family with no immunodeficiency history. The mother reported two abortions prior to the birth of the child by cesarean section at 37 weeks. The patient was discharged from the hospital after 48 h without any clinical manifestations. The first clinical manifestation was bullous impetigo at 18 days of age, which was treated with oxacillin and amikacin for 28 days.
Thereafter, the child was hospitalized with pneumonia, with chest X-ray showing a hypotransparent lesion in the lower right lobe (Figure 1A). The patient was treated with antibiotics for 35 days (5 days of clavulanate, 10 days of cefuroxime, and 20 days of cefepime and vancomycin). Due to his bullous impetigo and pneumonia, with a delayed response to treatment and low weight development, primary immunodeficiency was suspected, and the patient was discharged from the hospital with sulfamethoxazole-trimethoprim (TMP-SMX) and itraconazole prophylaxis at 3 months of age. At 5 months of age, he developed axillary lymphadenitis caused by a BCG vaccination reaction. His regional BCG infection (BCGitis) improved after isoniazid and ethambutol treatment for 45 days, with a normal BCG scar and without BCG dissemination.
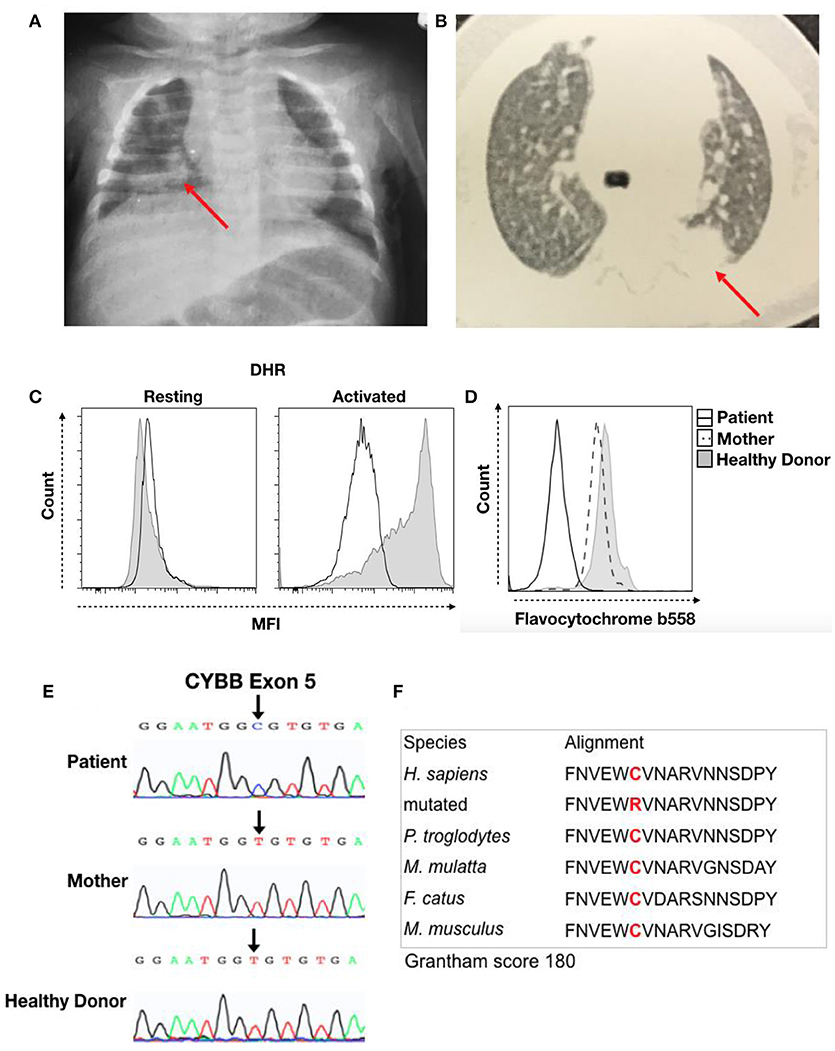
Figure 1. Clinical record of the patient with CGD. (A) Chest X-ray from the first hospitalization at 3 months of age and (B) thorax computed tomography at 5 months of age. The red arrow indicates the local lesion. (C) DHR of granulocytes in resting or activated states from the patient (Black Line) and a healthy donor (Solid Gray). (D) Flavocytochrome b558 expression in granulocytes from the patient (Black Line), his mother (Dash Line), and a healthy donor (Solid Gray). (E) Sequencing results for the CYBB gene. Chromatographs for the patient (Top), his mother (Middle), and a healthy donor (Bottom). The black arrow shows a single-nucleotide substitution, c.376T > C, that results in a C126R change. (F) We performed sequence analysis using MutationTaster to observe amino acid conservation in alignment with other species and obtain the Grantham matrix score.
The patient then developed a persistent subcutaneous nodule in his upper limb at 6 months of age. Biopsy showed granulomatous dermatitis with epithelioid histiocytes, few lymphocytes and no giant cells, and it was negative for BK and fungi. At 9 months of age, a new episode of pneumonia developed. Thorax computed tomography (CT) revealed a hypotransparent lesion in the left upper lobe that improved with cefuroxime treatment. However, chest X-ray confirmed a consolidation lesion in the left upper lobes (Figure 1B), and subcutaneous nodular lesion biopsy showed nonspecific granular cells. The pulmonary lesions improved with treatment. His prophylaxis treatment was suspended at 10 months of age.
The patient developed bacterial cervical adenitis, which was treated with amoxicillin and clavulanate for 21 days, with total resolution when he was 1 year old. However, 1 month later, he started to present intermittent fever and received a new diagnosis of pneumonia and large consolidation lesions. The pediatric service indicated an oncology study, which revealed a normal myelogram. There was no evidence of neoplasm. Pulmonary biopsy showed a granuloma lesion with areas of central necrosis that was not caseous and was negative for BK and fungi; abdominal ultrasound was normal, and PCR was negative for BK in gastric lavage and culture. Immunological tests revealed hypergammaglobulinemia and a subpopulation of CD4, CD8, and CD19 lymphocytes above the 90th percentile. According to chest X-ray, the parents had no contact with BK. His lesion improved markedly after prolonged antibiotic therapy, with the presence of a hypotransparent lesion.
The hypothesis for primary immunodeficiency in phagocytes was investigated using the dihydrorhodamine (DHR) assay, and the abnormal result of ROS production in granulocytes after stimulation was suggestive of CGD (Figure 1C). We then performed flavocytochrome b558 staining to observe gp91phox and p22phox expression at the membrane by flow cytometry. The lack of membrane flavocytochrome b558 in this patient in comparison to his mother and a healthy donor suggested gp91phox and p22phox expression failure (Figure 1D). In addition, we sequenced the CYBB gene to confirm CGD diagnosis. The sequence data were analyzed using NCBI (www.ncbi.nlm.nih.gov) and Ensembl SNP (https://www.ensembl.org/index.html) databases. This analysis confirmed a missense mutation (c.376T > C) in exon 5 that leads to a C126R substitution in the gp91phox protein; conversely, the mother and healthy donor presented the normal sequence, confirming a diagnosis of CGD caused by a de novo mutation (Figure 1E). Cysteine 126 is conserved, and this C126R substitution has a 180 score according to the Grantham matrix in MutationTaster (http://mutationtaster.org) (Figure 1F). At follow-up visits, the patient showed improvement with long-term sulfamethoxazole and itraconazole treatment for CGD prophylaxis.
Discussion
CGD is a rare, primary genetic immunodeficiency resulting from a defect in phagocytic cells that leads to increased susceptibility to bacterial and fungal infections, such as abscess, pneumonia, skin ulcers, sepsis, and osteomyelitis (1–3, 7). A common clinical manifestation in patients with CGD is several infections by catalase-producing microorganisms (2, 4). The most commonly reported invasive infectious agents are Staphylococcus, Serratia ssp, and Aspergillus ssp. S. aureus is the major cause of infection and subcutaneous/liver abscess formation in these patients (1, 8).
Skin manifestations in CGD patients are reported with systemic or deep infections that affect periorbital and perioral skin as well as the nares, scalp, neck, and retroauricular fold. In most cases, atopic dermatitis is prevalent, whereas bullous impetigo is rarely reported (9, 10).
Lung infections are frequent, occurring in more than 80% of CGD patients (2, 11). The high incidence of pneumonia in CGD is caused by many bacteria and fungi and may be complicated by abscess formation or pleural empyema in up to 20% of patients (6, 7, 11, 12). Thus, respiratory complications are a significant cause of mortality and morbidity (4). Our patient had been developing recurrent pneumonia since he was 3 months old, and it was the main cause of his hospitalization. Although no bacteria or fungi were isolated from this patient, antibiotic treatment and TMP-SMX and itraconazole prophylactic treatment helped the patient recover in all episodes and decreased his number of infections.
Prophylaxis with TMP-SMX for bacterial infections and with itraconazole for fungal infections has been routinely used since the 1970s (13). Prophylaxis treatment decreases the incidence of non-fungal infections in patients with autosomal and X-linked CGD without increasing the incidence of fungal infections. More than 40% of CGD cases remain infection free for more than 1 year with TMP-SMX prophylaxis (14).
The median age at onset of symptoms in CGD patients, with episodes of infection or noninfectious complications, is 4 months of age. However, tuberculosis-endemic countries provide BCG vaccination within the first month of life, and patients with immunodeficiency may quickly develop adverse reactions to BCG vaccination (6). Indeed, studies have shown that ~ 30% of CGD patients suffer from BCG reactions after vaccination (2). Furthermore, patients with any form of CGD can fail to develop a protective immune response against Mycobacterium species and may develop tuberculosis later in life. Clinically, BCG infection has in some cases been the first manifestation of CGD in Brazil. Nevertheless, our patient developed only regional BCGitis at 5 months of age, which was successfully treated with antibiotics, and BCG was not disseminated after the episodes of bullous impetigo and pneumonia.
Mutations in all five genes encoding components of the NADPH enzyme complex have been reported. The most affected gene is CYBB; mutations in this gene lead to an absence of or nonfunctional gp91phox, which is associated with a lack of ROS production by NADPH oxidase in phagocytes (2, 5). An international XL-CGD database includes over 680 mutations in the CYBB gene (15). In the present study, we reported a de novo mutation leading to X-linked CGD in a male patient. The mutation in exon 5 of CYBB leads to a C126R missense mutation in gp91phox that was not found in the patient's mother but has been reported in other CGD patients (16). This cysteine residue is responsible for forming disulfide bonds, which are important for protein folding. Therefore, this missense mutation may abolish protein expression, as observed for the flavocytochrome b558 histogram, and may result in abnormal DHR results. Additionally, patients with mutations in CYBB have been reported to be predisposed to tuberculous mycobacterial disease, corroborating our case report (17).
In summary, we report a de novo mutation, C126R, in gp91phox that led to X-linked CGD in a patient with recurrent pneumonia and regional BCGitis who had no family history, suggesting primary immunodeficiency. Clinicians should consider the possibility of a de novo mutation under such circumstances, and the results of this study highlight the relevance of genetic diagnosis for definitive genetic counseling for families with children who do not have any indications regarding inherited disease.
Ethics Statement
This study was approved by the Ethics Committee for Research in Humans at the Institute of Biomedical Sciences of the University of Sao Paulo in accordance with the Declaration of Helsinki, with written informed consent from the mother of the pediatric patient. The patient, his mother, and the healthy donor provided written informed consent before the study began. Written and informed consent for publication of this case report was obtained from the mother of the patient.
Author Contributions
JdA, EdOJ, and NZ designed and conducted the experiments and wrote the manuscript. EI and PV conducted the experiments. DB and MdS performed clinical and laboratory data collection and analysis. DB, MdS, and AC-N designed the experiments and wrote and reviewed the manuscript.
Funding
This work was supported by the PENSI Institute at the Ministério da Saúde do Brasil (PRONAS/PDC 2015, 25000.077928/2015-06, JdA, EdOJ, EI, AC-N) and the Jeffrey Modell Diagnostic Center São Paulo (AC-N).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our patient and his family for their participation in this study. We thank Instituto PENSI for administrative support.
References
1. Labrosse R, Abou-Diab J, Blincoe A, Cros G, Luu TM, Deslandres C, et al. Very early-onset inflammatory manifestations of x-linked chronic granulomatous disease. Front Immunol. (2017) 8:1167. doi: 10.3389/fimmu.2017.01167
2. de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and genotypic spectrum of chronic granulomatous disease in 71 Latin American patients: first report from the LASID registry. Pediatr Blood Cancer (2015) 62:2101–7. doi: 10.1002/pbc.25674
3. Vignesh P, Bhattad S, Shandilya JK, Vyas S, Garg R, Rawat A. Vertebral osteomyelitis and Acinetobacter Spp. paravertebral soft tissue infection in a 4-year-old boy with X-linked chronic granulomatous disease. Pediatr Infect Dis J. (2016) 35:1043–5. doi: 10.1097/INF.0000000000001221
4. Dunogue B, Pilmis B, Mahlaoui N, Elie C, Coignard-Biehler H, Amazzough K, et al. Chronic granulomatous disease in patients reaching adulthood: a nationwide study in France. Clin Infect Dis. (2017) 64:767–75. doi: 10.1093/cid/ciw837
5. Beghin A, Comini M, Soresina A, Imberti L, Zucchi M, Plebani A, et al. Chronic granulomatous disease in children: a single center experience. Clin Immunol. (2018) 188:12–19. doi: 10.1016/j.clim.2017.11.016
6. Zhou Q, Hui X, Ying W, Hou J, Wang W, Liu D, et al. A Cohort of 169 chronic granulomatous disease patients exposed to BCG vaccination: a retrospective study from a single center in Shanghai, China (2004-2017). J Clin Immunol. (2018) 38:260–72. doi: 10.1007/s10875-018-0486-y
7. Magnani A, Brosselin P, Beaute J, de Vergnes N, Mouy R, Debre M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. (2014) 134:655–62 e658. doi: 10.1016/j.jaci.2014.04.014
8. Bazregari S, Azizi G, Tavakol M, Asgardoon MH, Kiaee F, Tavakolinia N, et al. Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Cent Eur J Immunol. (2017) 42:336–41. doi: 10.5114/ceji.2017.72825
9. Dohil M, Prendiville JS, Crawford RI, Speert DP. Cutaneous manifestations of chronic granulomatous disease. a report of four cases and review of the literature. J Am Acad Dermatol. (1997) 36:899–907. doi: 10.1016/S0190-9622(97)80269-1
10. Low LC, Manson AL, Hardman C, Carton J, Seneviratne SL, Ninis N. Autosomal recessive chronic granulomatous disease presenting with cutaneous dermatoses and ocular infection. Clin Exp Dermatol. (2013) 38:270–3. doi: 10.1111/j.1365-2230.2012.04392.x
11. Khanna G, Kao SC, Kirby P, Sato Y. Imaging of chronic granulomatous disease in children. Radiographics (2005) 25:1183–95. doi: 10.1148/rg.255055011
12. Khan TA, Cabral-Marques O, Schimke LF, de Oliveira EB Jr, Amaral EP, D'Imperio Lima MR, et al. Tuberculosis in an autosomal recessive case of chronic granulomatous disease due to mutation of the NCF1 gene. Allergol Immunopathol. (2016) 44:276–9. doi: 10.1016/j.aller.2015.04.003
13. Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. (2003) 348:2416–22. doi: 10.1056/NEJMoa021931
14. Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. (1990) 162:723–6.
15. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger-Achituv S, Stauber T, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. (2017) 92:28–36. doi: 10.1002/ajh.24573
16. Wu J, Wang WF, Zhang YD, Chen TX. Clinical features and genetic analysis of 48 patients with chronic granulomatous disease in a single center study from Shanghai, China (2005-2015): new studies and a literature review. J Immunol Res. (2017) 2017:8745254. doi: 10.1155/2017/8745254
Keywords: chronic granulomatous disease (CGD), NADPH oxidase, CYBB gene, novel mutation, pneumonia
Citation: de Albuquerque JAT, de Oliveira Junior EB, Zurro NB, Vendramini P, Ishizuka EK, Borgli DdSP, de Souza MS and Condino-Neto A (2018) A C126R de novo Mutation in CYBB Leads to X-linked Chronic Granulomatous Disease With Recurrent Pneumonia and BCGitis. Front. Pediatr. 6:248. doi: 10.3389/fped.2018.00248
Received: 16 May 2018; Accepted: 20 August 2018;
Published: 11 September 2018.
Edited by:
Ivan K. Chinn, Baylor College of Medicine, United StatesReviewed by:
Beatriz Elena Marciano, National Institutes of Health (NIH), United StatesSaul Oswaldo Lugo Reyes, Instituto Nacional de Pediatria, Mexico
Copyright © 2018 de Albuquerque, de Oliveira Junior, Zurro, Vendramini, Ishizuka, Borgli, de Souza and Condino-Neto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Condino-Neto, YW50b25pb2NvbmRpbm9AZ21haWwuY29t