 George C. Gabriel
George C. Gabriel Gregory J. Pazour2
Gregory J. Pazour2 Cecilia W. Lo
Cecilia W. Lo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pediatr., 15 June 2018
Sec. Pediatric Nephrology
Volume 6 - 2018 | https://doi.org/10.3389/fped.2018.00175
This article is part of the Research TopicGenetic Kidney Diseases of ChildhoodView all 27 articles
Congenital heart disease (CHD) is one of the most common birth defects, and recent studies indicate cilia-related mutations play a central role in the genetic etiology of CHD. As cilia are also known to have important roles in kidney development and disease, it is not surprising that renal anomalies were found to be enriched among CHD mutant mice recovered in a large-scale mouse forward genetic screen. Indeed 42% of mutations identified to cause both CHD and renal anomalies were cilia-related. Many of these cilia mutations comprise cilia transition zone or inversin compartment components, consistent with the known role of these cilia proteins in a wide variety of ciliopathies. The high prevalence of CHD with congenital anomalies of the kidney and urinary tract (CAKUT) observed in mice was also corroborated with clinical studies that showed 20–30% of CHD patients have renal anomalies. Together these findings suggest CHD patients may benefit from early screening for renal anomalies to allow early diagnosis and intervention to improve outcome for this vulnerable patient population.
Congenital heart disease (CHD) is the most common birth defect, occurring in up to 1% of live births (1). Interestingly, CHD often presents in combination with extra cardiac defects including congenital anomalies of the kidney and urinary tract (CAKUT) (2–4). In fact, renal or urinary system defects are associated with 23.1% of congenital heart defects (5). These findings suggest an overlapping genetic etiology for CHD and CAKUT. Consistent with this is the fact that several previously reported mouse models (not individually discussed in this review) have found genetic disruption of a single gene can affect both heart and kidney development, and that many genetic syndromes can present with both cardiac and renal anomalies (6–9). Hence, insights into the genetic etiology of CHD may help elucidate the etiology of CAKUT. Relevant to this is the recent unexpected finding from a large-scale forward genetic screen in mice indicating a significant role for cilia-related mutations in the pathogenesis of CHD (10). As cilia mutations are well-described in the context of CAKUT, this would suggest cilia defects may have a central role in mediating both CHD and CAKUT phenotypes. Below, we briefly discuss CAKUT and its association with CHD, and its shared genetic etiology involving cilia-related mutations.
CAKUT represent a broad range of kidney and urinary tract defects. This can include abnormalities in the shape, size, or structure of the kidney including kidney agenesis, kidney hypoplasia or dysplasia, horseshoe/fused kidney, cystic kidneys, or duplex/multiplex kidney, and multiple collecting ducts or ureters (11). These defects can occur in combination such as with the finding of cystic dysplastic kidneys. These structural abnormalities can result in vesicoureteral reflux, ureteropelvic junction obstruction, hydroureter, and hydronephrosis (10, 12). In humans, monogenic mutations in several genes have been implicated in CAKUT development; however, these mutations explain only 5–20% of patient disease (13). Additional causes of CAKUT phenotypes include genetic syndromes, which can present with a spectrum of phenotypes often involving defects in many organs including both the heart and the kidney.
In DiGeorge syndrome, heterozygous deletion of chromosome 22q11.2 is associated with multiple organ defects, including the heart, the kidney, the thymus, and the nervous system. It has an estimated prevalence of 1:3,000 to 1:4,000 live births, though prenatal chromosomal analysis suggests this may be an underestimate, and affected patients characteristically present with CHD, thymus hypoplasia or agenesis, and craniofacial defects (14–16). Approximately 20–30% of DiGeorge syndrome patients also exhibit CAKUT (17). Haploinsufficiency of TBX1, a transcription factor within the T-box family, has been shown to contribute to the cardiac phenotypes associated with this disorder (18, 19). More recently, another gene within the 22q11.2 locus, CRKL, an adaptor protein that regulates tyrosine kinase signaling, was found to contribute to developmental kidney defects in the context of DiGeorge syndrome (20, 21).
Another syndrome known as VACTERL association also presents with both congenital heart defects and renal abnormalities. Occurring in 1:10,000 to 1:40,000 live births, VACTERL association is a birth defect associated with at least 3 of the following phenotypes: vertebral defects, anal atresia, cardiac defects, trachea-esophageal fistula, renal defects, and limb defects (22). In a large study of patients diagnosed with VACTERL association, 80% exhibited kidney defects and 48% exhibited cardiac defects (23). In humans, several genes have been related to VACTERL association including FGF8, FOXF1, HOXD13, LPP, TRAP1, and ZIC3, all of which have been implicated in kidney developmental anomalies, with ZIC3 being an X-linked gene also shown to cause heterotaxy (24).
A rarer genetic disorder known as Fraser syndrome can also cause both CHD and genitourinary abnormalities. It is a recessive disorder with a prevalence of only 1 in 200,000 (25). Patients with Fraser syndrome can present with eye defects, syndactyly, kidney defects, most commonly kidney agenesis, and heart defects (25). Fraser syndrome is associated with mutations in FRAS1, FREM2, or GRIP1, all of which are known to interact to form a FRAS/FREM complex that is localized to the basement membrane (26, 27). Mutations in these genes cause loss of the FRAS/FREM complex that likely drives the disease phenotypes observed in Fraser syndrome, although the precise disease mechanism remains unclear (27, 28).
The close association of renal anomalies with CHD suggests insights into the genetics of CHD can yield significant insights into the mechanism of disease pathogenesis for CAKUT. Interestingly, the pursuit of a large-scale forward genetic screen in mice for mutations causing CHD yielded a large preponderance of mutations in cilia-related genes (29). From screening more than 100,000 ethylnitrosourea (ENU) mutagenized mice using in utero fetal ultrasound phenotyping, we recovered over 200 mouse lines with a wide spectrum of CHD. Using whole exome sequencing nearly 100 CHD-causing mutations were recovered in 61 genes, 34 (55.7%) of which were cilia-related (29). As this is a phenotype driven mutagenesis screen in which fetal echocardiography was used to identify mutants with CHD, these findings point to cilia as playing a central role in the pathogenesis of CHD. This is corroborated with the completion of the screen, showing over 50 genes are cilia-related among 100 genes recovered causing CHD. Moreover, analysis of de novo mutations identified in whole exome sequencing analysis from CHD patients also identified similar mutations contributing to CHD pathogenesis (30).
Cilia are microtubule-based organelles that have well-described compartments including an axoneme that projects from the apical cell surface, and a basal body base with an overlying transition zone and inversin compartment that regulate protein trafficking into and out of the cilium (Figure 1) (32). Cilia can be motile with the expression of motor dyneins that drive ciliary motility, or nonmotile, termed primary cilia, which extend from almost all cell types in the body, including cells of the developing heart, and are known to serve important cell signaling functions by mediating various signal transduction processes (33–35). Motile cilia are required for left-right patterning as well as in mediating sperm motility, mucociliary clearance in the airway, and also cerebral spinal fluid flow in the brain. Importantly, it is now appreciated that cilia mutations can cause a wide spectrum of human diseases collectively known as ciliopathies (31). Many of these ciliopathies can present with renal anomalies, such as in nephronepthesis, Meckel-Gruber syndrome (MKS), Bardet-Biedl syndrome (BBS), Joubert syndrome, Alstrom syndrome and polycystic kidney disease among others (36). The renal defects observed may include CAKUT, polycystic kidney disease or progressive renal dysfunction. Other structural birth defects observed in the ciliopathies include CHD, cardiomyopathy, skeletal malformations, brain abnormalities, blindness, and obesity (31).
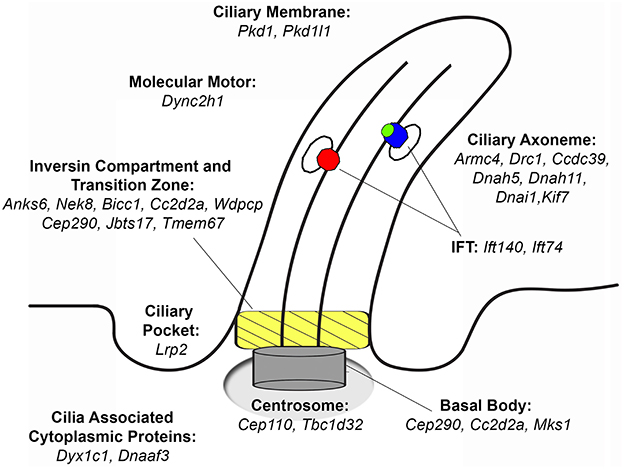
Figure 1. Ciliary localization of proteins that when mutated cause congenital heart disease recovered from a large-scale screen. Mutations in several of these proteins can also cause CAKUT. Adapted from Damerla et al. (31).
Many of the mutations known to cause renal anomalies within the ciliopathy spectrum are associated with proteins localized in the cilia transition zone or inversin compartment. Thus, among the 61 genes recovered causing CHD, half (N = 34) were cilia-related, including 11 affecting transition zone or inversin compartment components (29). While these are mainly thought to disrupt primary cilia function, recent studies showed ciliopathies generally considered to involve primary cilia defects may also impact motile cilia function. This is indicated by the observation of airway clearance defects in patients diagnosed with ciliopathies such as Leber congenital amaurosis or Sensenbrenner syndrome (37, 38). This is not unexpected given over 70% of proteins required for primary cilia function are also expressed in motile cilia (39). Indeed, airway clearance defects are the hallmark of a ciliopathy known as primary ciliary dyskinesia (PCD).
PCD patients have severe sinopulmonary disease and typically this is associated with mutations in genes required for motile cilia function, such as motor dyneins (40–42). It is interesting to note some isolated reports of bronchiectasis associated with polycystic kidney disease, although the connection with airway clearance defects is unknown (43). PCD patients also can exhibit laterality defects, a reflection of the requirement for motile cilia in the specification of the left-right body axis (44, 45). Consistent with this, PCD patients can display a spectrum of phenotypes including situs solitus (normal visceral organ situs), situs inversus totalis also known as Kartagener's syndrome (reversed visceral organ situs), and heterotaxy, or the randomization of visceral organ situs. In this context, CHD is often associated with heterotaxy as a result of defects in motile cilia function.
Interestingly, some ciliopathies thought to affect only primary cilia function also can be associated with laterality defects, one prominent example being MKS, a ciliopathy that causes severe birth defects and is usually prenatal or neonatal lethal. Studies of Mks1 mutant mice showed defects involving not only the primary cilia, but also laterality defects associated with the loss of motile cilia function in the embryonic node (46). Together, these findings suggest that the distinction of ciliopathies as either affecting motile or primary cilia can be problematic, given the promiscuity of cilia genes in having functional roles in both primary and motile cilia.
Intriguingly, primary cilia proteins can be found in other cellular locations, suggesting that they may have other non-ciliary functions (47). In fact, ciliary proteins are also involved in a wide range of cellular processes including cell cycle progression, cytoskeletal organization, and vesicular trafficking (48–50). Further, cilia themselves are involved in several cell signaling pathways including the sonic hedgehog, TGFβ, wnt, and notch pathways, and defects can arise in these pathways through defective cilia or pathway components outside of the cilium (51). Thus, future research will be important to determine the precise roles cilia and cilia associated proteins play in CHD and CAKUT development.
Extra-cardiac phenotypes, including renal anomalies, were recovered in many CHD mutant mice even though the screen was entirely focused on CHD phenotyping. This included a spectrum of kidney phenotypes including duplex kidney, multiplex kidney, hydronephrosis, kidney agenesis, and cystic kidney (Figure 2), with the most commonly observed renal phenotype being duplex kidneys (10). Among the 39 mutant mouse lines recovered with congenital kidney abnormalities, the most common CHD phenotype is double outlet right ventricle (DORV), occurring in 19/39 lines (48.7%). This is followed by atrioventricular septal defect, which occurred in 15/39 lines (38.5%; Table 1). Other CHD phenotypes associated with kidney defects included transposition of the great arteries (TGA), persistent truncus arteriosus (PTA), ventricular septal defect (VSD), aortic arch anomalies, and biventricular hypertrophy (Table 1).
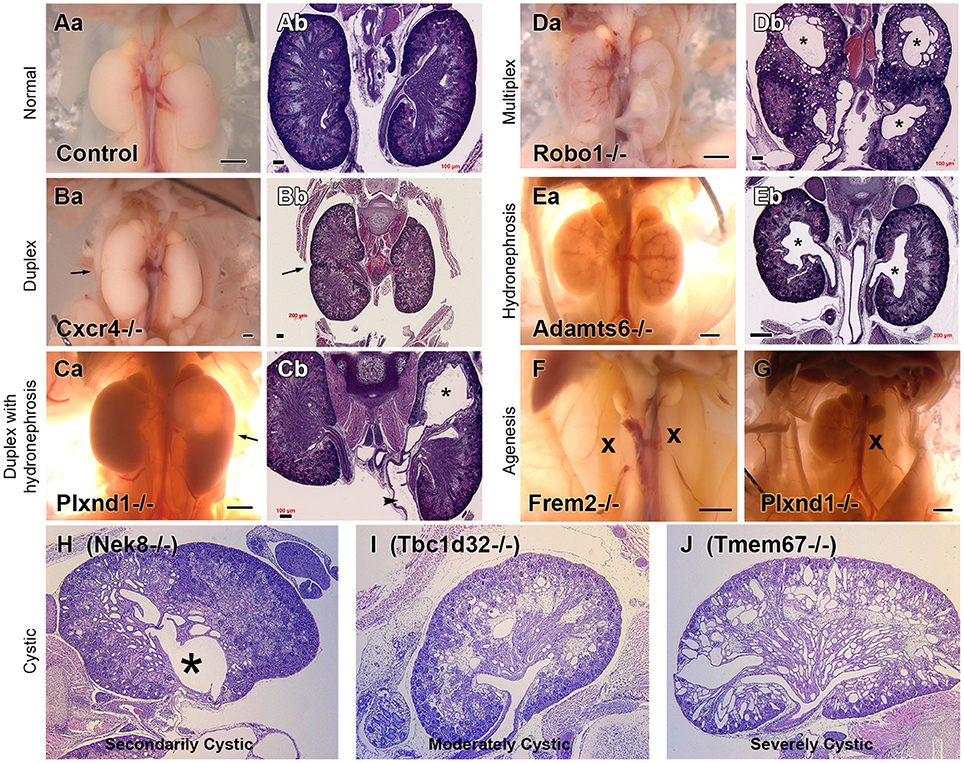
Figure 2. Kidney Phenotypes Observed in Mice with Congenital Heart Disease. Representative examples of the kidney phenotypes observed in mice with congenital heart disease recovered from a large scale forward genetic screen. Phenotypes can be observed at necropsy and upon H&E staining. Adapted from San Agustin et al. (10). Control necropsy (Aa) and control H&E stained kidneys (Ab) can be compared with mutant necropsy (Ba,Ca,Da,Ea,F,G) and mutant H&E stained kidneys (Bb,Cb,Db,Eb, H–J). *represents hydronephrosis.
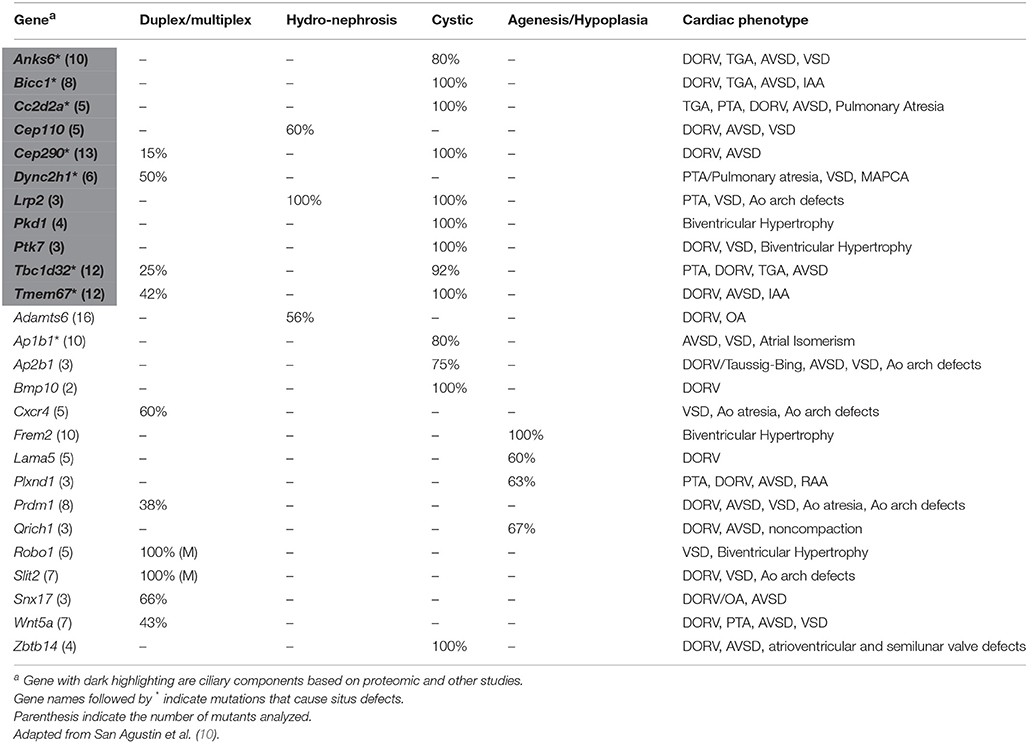
Table 1. Renal anomalies associated with genes causing congenital heart defects.
Previous attempts to recover CAKUT genes have been confounded by incomplete penetrance and the probable involvement of more complex genetics. The few CAKUT genes previously identified are largely those that mediate the early steps in kidney development and patterning. Other than Pax2, which causes renal coloboma syndrome, and Hnf1b, which causes renal cystic disease and diabetes, most genes identified to cause CAKUT are associated with disease in only a few patients and hence, the evidence supporting pathogenesis is limited (52). Among the 135 CHD mouse lines recovered, 39 (28.8%) had kidney defects caused by mutations in 26 genes, 11 (42%) being known cilia genes (Table 1, Figure 1) (10). Remarkably, this included four cilia genes that encoded known direct protein-protein interactors: Anks6, Nek8, Bicc1, and Wwtr1 (53–56). These mutations were found in four independent CHD mouse lines, with mutation in each gene causing a similar spectrum of kidney and heart defect phenotypes comprising double outlet right ventricle and cystic kidney disease (Figure 3).
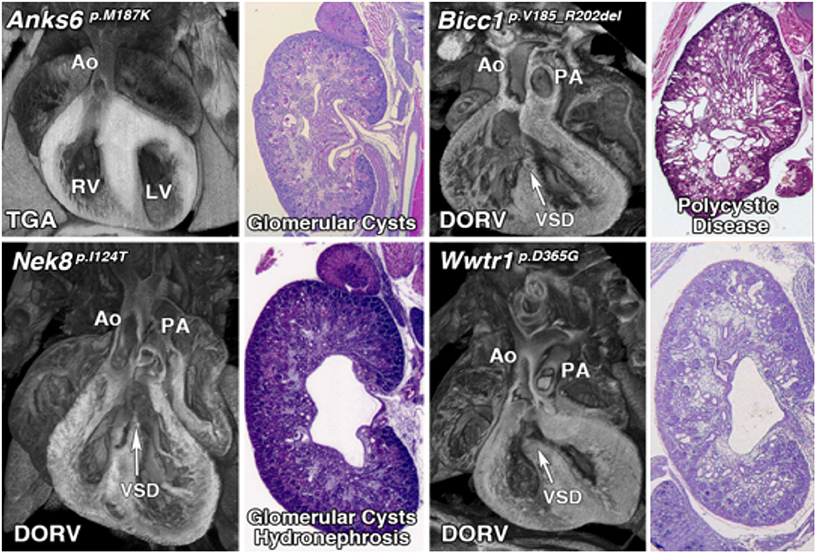
Figure 3. Mutations in interacting proteins cause a similar spectrum of both heart and kidney phenotypes. Mutations in four interacting proteins, Anks6, Bicc1, Nek8, and Wwtr1 cause similar phenotypes including both congenital heart disease and cystic kidney disease (Unpublished data).
The relevance of CAKUT abnormalities in CHD was confirmed with a clinical study of 77 CHD patients recruited from the Children's Hospital of Pittsburgh. Retrospective review of the medical records of these 77 patients revealed 23 (30%) also had some form of renal defect such as renal cysts, kidney agenesis, cystic dysplastic kidneys, and horseshoe kidney (Figure 4, Table 2) (10). These findings are in agreement with a previous epidemiological study in the Atlanta metro area, which showed 23% of 8,000 subjects with CHD also had renal abnormalities (1). Thus, both the mouse and human studies point to a significant overlap in the genetic etiology of CHD and kidney abnormalities. This probably reflects the conservation of developmental pathways and cell signaling mechanisms that regulate cardiovascular and renal development, including a central role for cilia in the pathogenesis of CHD and renal birth defects. Together these findings suggest the routine evaluation of CHD patients for renal anomalies with simple non-invasive renal ultrasound may be warranted. This may allow early diagnosis and early therapeutic intervention to treat and manage any renal dysfunction that could negatively impact the long-term outcome of this high-risk patient population.
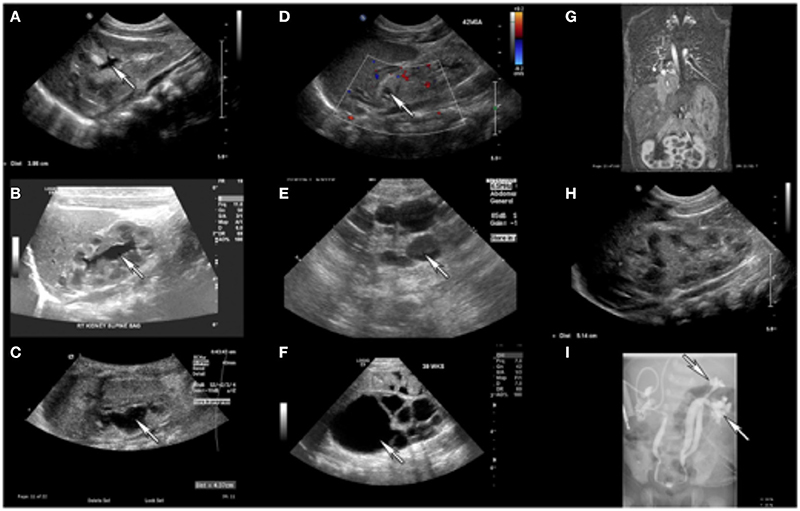
Figure 4. Kidney Defects Observed in Patients with CHD. Renal ultrasound of patients with congenital heart disease unveils a spectrum of renal abnormalities including intrarenal collecting system dilation (A–C), cystic-dysplastic abnormalities (D–F), horseshoe kidney (G), and duplicated collecting system depicted with renal ultrasound (H), and voiding cystourethrogram (I). Adapted from San Agustin et al. (10).

Table 2. Clinical characteristics of congenital heart disease patients.
Studies in both CHD mutant mice and CHD patients showed CAKUT is highly associated with CHD. This is supported by the finding of CAKUT and CHD in various genetic syndromes, and the finding that many mutations identified to cause CHD can also cause CAKUT phenotypes. Among genes identified to cause both CHD and CAKUT there is a significant enrichment of cilia genes, indicating a central role for cilia in the pathogenesis of CHD and CAKUT phenotypes. However, as half of the mutations identified to cause CHD and renal anomalies are not known to be cilia-related, the association of CHD with renal anomalies likely extend beyond the role of cilia in heart and kidney development. The clinical relevance of these findings in mice were shown with the observation that 23–30% of CHD patients also exhibited renal anomalies. This would suggest CHD patients may benefit from routine evaluation for renal anomalies. This might improve the long-term outcome of this high-risk patient population by reducing potential renal complications with early diagnosis and therapeutic intervention.
All authors listed contributed intellectually to this work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by NIH grants HL098180, 1S10 OD010340, HL132024, and GM104412 to CL and GM060992 and DK103632 to GP.
1. Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J Pediatr. (2008) 153:807–13. doi: 10.1016/j.jpeds.2008.05.059
2. Nedima Atić HB, Ibrahimović J, Zulić S, Osmanović E. Cardiac evaluation of children with congenital anomalies of the kidneys and urinary tract. Paediatr Tdy (2016) 12:169–75. doi: 10.5457/p2005-114.153
3. Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. (2014) 7:86–91. doi: 10.4103/0974-2069.132474
4. Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science (2015) 350:1262–6. doi: 10.1126/science.aac9396
5. Miller A, Riehle-Colarusso T, Alverson CJ, Frias JL, Correa A. Congenital heart defects and major structural noncardiac anomalies, Atlanta, Georgia, 1968 to 2005. J Pediatr. (2011) 159:70–8.e2. doi: 10.1016/j.jpeds.2010.12.051
6. McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, et al. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development (2001) 128:491–502.
7. Riazi AM, Arsdell GV, Buchwald M. Transgenic expression of CECR1 adenosine deaminase in mice results in abnormal development of heart and kidney. Transgen Res. (2005) 14:333–6. doi: 10.1007/s11248-005-1174-2
8. Martinez-Estrada OM, Lettice LA, Essafi A, Guadix JA, Slight J, Velecela V, et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat Genet. (2010) 42:89–93. doi: 10.1038/ng.494
9. Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci USA. (2001) 98:12174–9. doi: 10.1073/pnas.211191098
10. San Agustin JT, Klena N, Granath K, Panigrahy A, Stewart E, Devine W, et al. Genetic link between renal birth defects and congenital heart disease. Nat Commun. (2016) 7:11103. doi: 10.1038/ncomms11103
11. Renkema KY, Winyard PJ, Skovorodkin IN, Levtchenko E, Hindryckx A, Jeanpierre C, et al. Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol Dial Transplant. (2011) 26:3843–51. doi: 10.1093/ndt/gfr655
12. Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. (2014) 85:1429–33. doi: 10.1038/ki.2013.508
13. van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. (2018) 29:36–50. doi: 10.1681/ASN.2017050561
14. Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Childhood (1998) 79:348–51. doi: 10.1136/adc.79.4.348
15. Leana-Cox J, Pangkanon S, Eanet KR, Curtin MS, Wulfsberg EA. Familial DiGeorge/velocardiofacial syndrome with deletions of chromosome area22q11.2: report of five families with a review of the literature. Am J Med Genet. (1996) 65:309–16. doi: 10.1002/(sici)1096-8628(19961111)65:4<309::Aid-ajmg12>3.0.Co;2-y
16. Grati FR, Molina Gomes D, Ferreira JC, Dupont C, Alesi V, Gouas L, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. (2015) 35:801–9. doi: 10.1002/pd.4613
17. Burnside RD. 22q11.21 deletion syndromes: a review of proximal, central, and distal deletions and their associated features. Cytogenet Genome Res. (2015) 146:89–99. doi: 10.1159/000438708
18. Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature (2001) 410:97–101. doi: 10.1038/35065105
19. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet (2003) 362:1366–73. doi: 10.1016/S0140-6736(03)14632-6
20. Haller M, Mo Q, Imamoto A, Lamb DJ. Murine model indicates 22q11.2 signaling adaptor CRKL is a dosage-sensitive regulator of genitourinary development. Proc Natl Acad Sci USA. (2017) 114:4981–6. doi: 10.1073/pnas.1619523114
21. Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, et al. Genetic drivers of kidney defects in the DiGeorge syndrome. N Engl J Med. (2017) 376:742–54. doi: 10.1056/NEJMoa1609009
22. Hilger A, Schramm C, Pennimpede T, Wittler L, Dworschak GC, Bartels E, et al. De novo microduplications at 1q41, 2q37.3, and 8q24.3 in patients with VATER/VACTERL association. Eur J Hum Genet. (2013) 21:1377–82. doi: 10.1038/ejhg.2013.58
23. Botto LD, Khoury MJ, Mastroiacovo P, Castilla EE, Moore CA, Skjaerven R, et al. The spectrum of congenital anomalies of the VATER association: an international study. Am J. Med. Genet. (1997) 71:8–15. doi: 10.1002/(sici)1096-8628(19970711)71:1<8::aid-ajmg2>3.0.co;2-v
24. Reutter H, Hilger AC, Hildebrandt F, Ludwig M. Underlying genetic factors of the VATER/VACTERL association with special emphasis on the “Renal” phenotype. Pediatr Nephrol. (2016) 31:2025–33. doi: 10.1007/s00467-016-3335-3
25. Barisic I, Odak L, Loane M, Garne E, Wellesley D, Calzolari E, et al. Fraser syndrome: epidemiological study in a European population. Am J Med Genet A (2013) 161A:1012–8. doi: 10.1002/ajmg.a.35839
26. van Haelst MM, Maiburg M, Baujat G, Jadeja S, Monti E, Bland E, et al. Molecular study of 33 families with Fraser syndrome new data and mutation review. Am J Med Genet A (2008) 146A:2252–7. doi: 10.1002/ajmg.a.32440
27. Vogel MJ, van Zon P, Brueton L, Gijzen M, van Tuil MC, Cox P, et al. Mutations in GRIP1 cause Fraser syndrome. J Med Genet. (2012) 49:303–6. doi: 10.1136/jmedgenet-2011-100590
28. McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. (2003) 34:203–8. doi: 10.1038/ng1142
29. Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature (2015) 521:520–4. doi: 10.1038/nature14269
30. Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature (2013) 498:220–3. doi: 10.1038/nature12141
31. Rao Damerla R, Gabriel GC, Li Y, Klena NT, Liu X, Chen Y, et al. Role of cilia in structural birth defects: insights from ciliopathy mutant mouse models. Birth Defects Res C Embryo Tdy (2014) 102:115–25. doi: 10.1002/bdrc.21067
32. Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. (2007) 69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236
33. Pala R, Alomari N, Nauli SM. Primary cilium-dependent signaling mechanisms. Int J Mol Sci. (2017) 18:E2272. doi: 10.3390/ijms18112272
34. Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. (2017) 18:533–47. doi: 10.1038/nrm.2017.60
35. Slough J, Cooney L, Brueckner M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev Dyn. (2008) 237:2304–14. doi: 10.1002/dvdy.21669
36. Mitchison HM, Valente EM. Motile and non-motile cilia in human pathology: from function to phenotypes. J Pathol. (2017) 241:294–309. doi: 10.1002/path.4843
37. Papon JF, Perrault I, Coste A, Louis B, Gerard X, Hanein S, et al. Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations. J Med Genet. (2010) 47:829–34. doi: 10.1136/jmg.2010.077883
38. Li Y, Garrod AS, Madan-Khetarpal S, Sreedher G, McGuire M, Yagi H, et al. Respiratory motile cilia dysfunction in a patient with cranioectodermal dysplasia. Am J Med Genet A (2015) 167A:2188–96. doi: 10.1002/ajmg.a.37133
39. Ishikawa H, Thompson J, Yates JR III, Marshall WF. Proteomic analysis of mammalian primary cilia. Curr Biol. (2012) 22:414–9. doi: 10.1016/j.cub.2012.01.031
40. Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. (2004) 169:459–67. doi: 10.1164/rccm.200303-365OC
41. Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev. (2016) 18:18–24. doi: 10.1016/j.prrv.2015.09.001
42. Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature (1997) 389:963–6. doi: 10.1038/40140
43. Driscoll JA, Bhalla S, Liapis H, Ibricevic A, Brody SL. Autosomal dominant polycystic kidney disease is associated with an increased prevalence of radiographic bronchiectasis. Chest (2008) 133:1181–8. doi: 10.1378/chest.07-2147
44. Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, Davis SD, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. (2009) 11:473–87. doi: 10.1097/GIM.0b013e3181a53562
45. McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell (2003) 114:61–73. doi: 10.1016/s0092-8674(03)00511-7
46. Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, et al. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech. (2011) 4:43–56. doi: 10.1242/dmm.006262
47. Hua K, Ferland RJ. Primary cilia proteins: ciliary and extraciliary sites and functions. Cell Mol Life Sci. (2018) 75:1521–40. doi: 10.1007/s00018-017-2740-5
48. Vertii A, Bright A, Delaval B, Hehnly H, Doxsey S. New frontiers: discovering cilia-independent functions of cilia proteins. EMBO Rep. (2015) 16:1275–87. doi: 10.15252/embr.201540632
49. McClure-Begley TD, Klymkowsky MW. Nuclear roles for cilia-associated proteins. Cilia (2017) 6:8. doi: 10.1186/s13630-017-0052-x
50. Yuan S, Sun Z. Expanding horizons: ciliary proteins reach beyond cilia. Annu Rev Genet. (2013) 47:353–76. doi: 10.1146/annurev-genet-111212-133243
51. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. (2010) 11:331–44. doi: 10.1038/nrg2774
52. Nicolaou N, Renkema KY, Bongers EM, Giles RH, Knoers NV. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. (2015) 11:720–31. doi: 10.1038/nrneph.2015.140
53. Hoff S, Halbritter J, Epting D, Frank V, Nguyen TM, van Reeuwijk J, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet. (2013) 45:951–6. doi: 10.1038/ng.2681
54. Stagner EE, Bouvrette DJ, Cheng J, Bryda EC. The polycystic kidney disease-related proteins Bicc1 and SamCystin interact. Biochem Biophys Res Commun. (2009) 383:16–21. doi: 10.1016/j.bbrc.2009.03.113
55. Czarnecki PG, Gabriel GC, Manning DK, Sergeev M, Lemke K, Klena NT, et al. ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat Commun. (2015) 6:6023. doi: 10.1038/ncomms7023
Keywords: cilia, CAKUT, congenital heart disease, congenital abnormalities, ciliopathies, genetic syndromes
Citation: Gabriel GC, Pazour GJ and Lo CW (2018) Congenital Heart Defects and Ciliopathies Associated With Renal Phenotypes. Front. Pediatr. 6:175. doi: 10.3389/fped.2018.00175
Received: 29 December 2017; Accepted: 29 May 2018;
Published: 15 June 2018.
Edited by:
Miriam Schmidts, Radboud University Nijmegen, NetherlandsReviewed by:
Kathryn Hentges, University of Manchester, United KingdomCopyright © 2018 Gabriel, Pazour and Lo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia W. Lo, Y2VsMzZAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.