 Rebeca Silveira Grasel1
Rebeca Silveira Grasel1 Paula Silva Felicio1
Paula Silva Felicio1 André Escremim de Paula2
André Escremim de Paula2 Natalia Campacci1
Natalia Campacci1 Felipe Antônio de Oliveira Garcia1
Felipe Antônio de Oliveira Garcia1 Edilene Santos de Andrade1
Edilene Santos de Andrade1 Adriane Feijó Evangelista1
Adriane Feijó Evangelista1 Gabriela Carvalho Fernandes2
Gabriela Carvalho Fernandes2 Cristina da Silva Sabato2
Cristina da Silva Sabato2 Pedro De Marchi3,4
Pedro De Marchi3,4 Cristiano de Pádua Souza3,5
Cristiano de Pádua Souza3,5 Cláudia Alessandra Andrade de Paula6Giovana Tardin Torrezan7
Cláudia Alessandra Andrade de Paula6Giovana Tardin Torrezan7 Henrique de Campos Reis Galvão5
Henrique de Campos Reis Galvão5 Dirce Maria Carraro6,7Edenir Inêz Palmero1,2,8,9*
Dirce Maria Carraro6,7Edenir Inêz Palmero1,2,8,9*- 1Molecular Oncology Research Center, Barretos Cancer Hospital, São Paulo, Brazil
- 2Center of Molecular Diagnosis, Barretos Cancer Hospital, São Paulo, Brazil
- 3Department of Medical Oncology, Barretos Cancer Hospital, São Paulo, Brazil
- 4Oncoclinicas, Rio de Janeiro, Brazil
- 5Department of Oncogenetics, Barretos Cancer Hospital, São Paulo, Brazil
- 6Genomic Diagnostic Center, AC Camargo Cancer Center, São Paulo, Brazil
- 7Genomics and Molecular Biology Group, CIPE - A. C. Camargo Cancer Center, São Paulo, Brazil
- 8Pele Little Prince Research Institute, Curitiba, Brazil
- 9Faculdades Pequeno Príncipe, Curitiba, Brazil
The use of gene panels introduces a new dilemma in the genetics field due to the high frequency of variants of uncertain significance (VUS). The objective of this study was to provide evidence that may help in the classification of these germline variants in terms of their clinical impact and association with the disease in question. A total of 52 unrelated women at-risk for HBOC and negative for BRCA1/BRCA2 pathogenic variants were evaluated through a gene panel comprising 14 breast and/or ovarian cancer susceptibility genes. Of the 453 germline variants identified, 15 variants (classes 3, 4, and 5) in the ATM, BRIP1, CHEK2, MRE11A, MUTHY, PALB2, RAD50, and RAD51C genes were evaluated via databases, co-segregation studies and loss of heterozygosity in the tumor. The co-segregation analysis allowed the establishment of an association with the presence of variants and the risk of cancer for variant c.316C>T in the BRIP1 gene. Four variants of uncertain significance showed loss of heterozygosity in the tumor (ATM c.4709T>C; CHEK2 c.1036C>T; PALB2 c.1001A>G, and RAD50 c.281T>C), which is an indication of pathogenicity. Thus, the present study provides novel evidence that favors the association of variants in moderate-risk genes with the development of hereditary breast cancer.
Introduction
Germline pathogenic BRCA1 or BRCA2 variants are responsible for ~15–25% of the hereditary breast/ovarian cancer (HBOC) cases (1, 2). Other than BRCA1/BRCA2, several high and moderate cancer genes have been associated with HBOC. The wide use of gene panels has proven to be extremely useful in clinical practice due to the optimization of both time and cost, and to the identification and monitoring of high-risk families for hereditary breast cancer, harboring pathogenic germline variants in genes other than BRCA1 and BRCA2. However, the use of gene panels introduces a new dilemma: a high frequency of variants of uncertain significance (VUS).
In order to assist in the classification of variants, the American College of Medical Genetics and Genomics (ACMG) published, in 2015, a report with the Association for Molecular Pathology (AMP) and College of American Pathologists (CAP) experts' describing the recommendations for germline variants classification, identified through genetic testing (3). This guideline describes the classification of variants based on evidences, such as population, in silico, functional and segregation analysis. Among all the criteria, it is worth noting that co-segregation and loss of heterozygosity analysis have been widely used and have an important role in the classification of variants of tumor suppressor genes identified in high risk families (4–7).
In this context, the objective of this study was to evaluate the mutational profile of high- and moderate-risk genes in patients with a personal and/or family history suggestive of HBOC and negative for mutations in the BRCA1 and BRCA2 genes and also to provide evidence that may help to classify the identified germline variants in terms of their clinical impact and their association with the disease in question.
Materials and Methods
Patients
A retrospective cohort study of 52 unrelated women over 18 years of age with a personal and/or family history (FH) suggestive of HBOC were included. These women were referred to the Department of Cancer Genetics of the Barretos Cancer Hospital (BCH) and tested negative for pathogenic germline variants of the BRCA1, BRCA2, and TP53 genes [as described by Fernandes et al. (8)]. Clinical-pathological data and family history data were collected from the patients' clinical records and the family records in the Department of Cancer Genetics of BCH.
Molecular Analyses
Genomic DNA obtained from peripheral blood was extracted using the QIAamp Blood DNA Mini Kit (Qiagen) according to the manufacturer's instructions. The extracted samples were quantified by fluorimetry (Qubit Fluorometer, Thermo Fisher Scientific). The libraries were prepared using the Ion Ampliseq Library 2.0 kit (Applied Biosystems) according to the manufacturer's protocol. The gene panel used in this study consisted of 14 genes (Supplementary Table 1) related to increased risk (moderate to high) of hereditary breast and/or ovarian cancer. The coding region, along with the immediately adjacent intronic regions (5 bases at the beginning and end of each exon) of each gene, were sequenced using the Ion Torrent PGM platform (Applied Biosystems) following the manufacturer's instructions.
The primary analysis (signal processing, base calling, sequence filtering and mapping) of the generated data was performed using the Torrent suite software, v2.2. The sequences generated were mapped to the reference genome hg19 (Homo sapiens). The variants were called and annotated using the Ion Reporter v5.6 software, and the standard parameters suggested by the manufacturer were applied. The first filters for the selection of variants of the gene panel were as follows: exclusion of variants with coverage below 50 X; intronic variants with distance higher than ten base pairs upstream and downstream of exons; variants located in non-coding UTR regions and synonymous variants located in regions not associated with splicing. Copy Number Variations (CNVs) were not assessed by our gene panel NGS approach.
Data Ansalysis
Besides, after application of the quality filters, all variants were evaluated for their frequency in international population databases [gnomAD (9)] and in a Brazilian database (10) that were used for manually excluding population-specific variants (MAF ≤ 0.01 were maintained) (3). The identified genetic variants were evaluated for their pathogenicity in the ClinVar database (11) and the Human Gene Mutation Database (HGMD) (12). Furthermore, the variants were classified according to the criteria recommended by the ACMG (Varsome web platform) (3). Additionally, the following in silico prediction tools were used: PolyPhen-2 (13); MutationTaster (14); Align GVGD (15); SIFT (16); Panther (17); Human Splice Finder (18); Revel (19) CADD (20) and PROVEAN (21).
Confirmation and Validation of Results
Conventional sequencing: All variants classified by both ClinVar and ACMG as VUS (class 3), likely pathogenic (class 4), and pathogenic (class 5) were confirmed by conventional sequencing (sanger). For this validation, the genomic DNAs were amplified by PCR, purified with the enzyme Exosap-IT (USB) and Big Dye X terminator kit (Applied Biosystems) and sequenced bi-directionally using the 3500XL platform (Applied Biosystems).
Segregation analysis: For the co-segregation analysis, all families with class 3 germline variants according to the ACMG or ClinVar classification and confirmed by conventional sequencing were invited to participate. All relatives of the index patient with or without cancer at any age who were willing to participate in the study were tested. Co-segregation with disease was considered positive when the variant was present in all affected relatives, and absent in all unaffected individuals (only individuals with diagnosed breast or ovarian cancer were considered as affected).
Loss of heterozygosity (LOH): Variants classified as class 3 and that had available tumor material were subjected to LOH analysis. LOH analysis was conducted using NGS sequencing. Amplicon libraries were prepared by PCR and sequenced using Ion Torrent Proton Platform according to manufacturer's recommended protocol. Germline variant NGS sequencing allows the identification of possible LOH events, taking into consideration the purity of tumor cells and VAF. Data analysis was performed using Torrent Suite 5.10.1 software, and variants of interest were manually inspected with Integrative Genomics Viewer (IGV) visualization. LOH of a wild-type allele was considered when the variant allele had a frequency of >60%. The VAF cut-off value above 60% is based on the model VAF = 100/(% tumor cells + 2X % normal cells), which considers that the deletion of the wild-type allele in hereditary syndromes is an early event, thus present in most/all tumor cells.
Statistical Analysis
Statistical analysis was performed descriptively. SPSS v.21.0 software was used for data storage and statistical analysis.
Ethical Aspects
The study was approved by the institution's Ethics Committee (40814115.4.0000.5437). All patients included in the study signed the study's consent form.
Results
Characterization of the Samples
A detailed clinical-pathological characterization of the 52 patients included in the study is shown in Table 1. In summary, the mean age at first diagnosis of cancer was 41.9 years (ranging from 21 to 61 years). Forty-four patients had breast cancer only (84.6%), three patients had ovarian cancer (5.8%), two patients had breast and ovarian cancer (3.8%), and, three index cases had a personal diagnosis of breast and another tumor type (5.8%). Seventeen percent of the patients were diagnosed with more than one tumor, and one patient had three primary tumors (kidney, breast and thyroid cancer), the first of which was diagnosed when the patient was 31 years old.
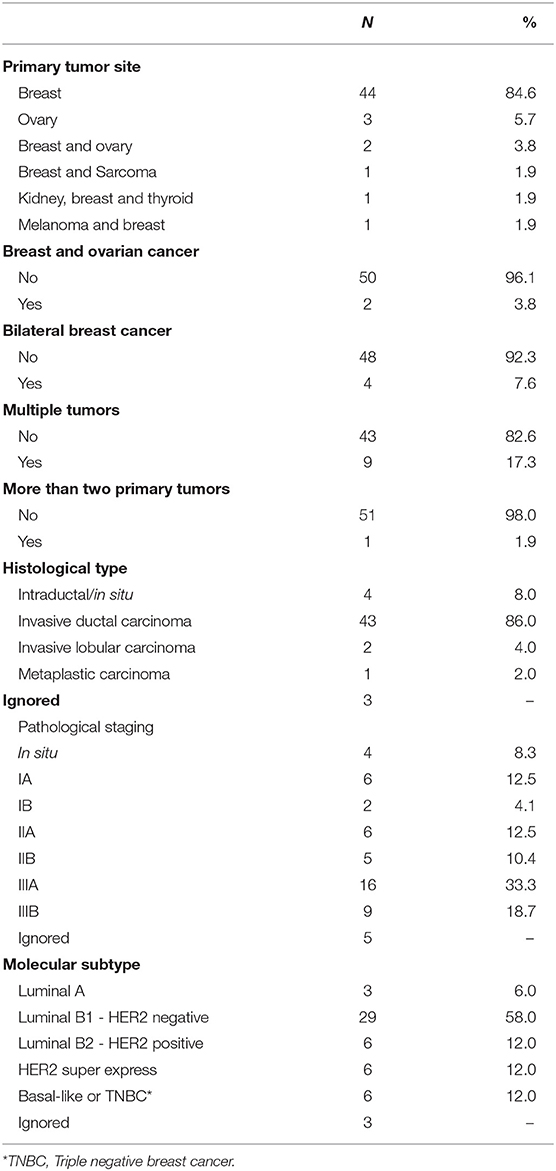
Table 1. Clinicopathological characteristics of the 52 patients included in the study.
Most of the patients included in the study were from the state of São Paulo (Southeast Region 64.2%), were white (86.3%), married (66.7%), and had completed high school (35.3%)—(Supplementary Table 2). Most participants were pre-menopausal (76.7%) and had first-degree relatives with breast cancer (56.9%); and among the postmenopausal/perimenopausal women, none was undergoing hormone replacement therapy (Supplementary Table 3).
Among breast tumors, the mean age at diagnosis was 42.6 years of age. The predominant histology was invasive ductal carcinoma (86.0%), followed by carcinoma in situ (8.0%). Four of them had bilateral breast cancer (7.7%). There was a predominance of clinical and pathological stage IIIA tumors, with 33.3% for both. Regarding molecular subtypes, there was a predominance of luminal B1 tumors (58%), while triple negative and Her2-overexpressing tumors together represented ~24% of cases (Table 1).
Among the five women with a personal history of ovarian cancer, the mean age at diagnosis of ovarian cancer was 43.4 years, ranging from 21 to 60 years. Among the five ovarian cases, the histology was available for four cases (all of them were serous ovarian carcinomas), and the predominant pathological stage was IIIC.
Regarding the family history of cancer, the presence of breast cancer in mother-daughter pairs was observed in 41.2% of cases (cancer at any age). When breast cancer in first-degree relatives before age 50 was evaluated, it was observed that 51% of patients had at least one case in their family. In addition, 23.5% of the patients had at least one family member with multiple tumors (Supplementary Table 4).
Molecular Characterization
Sequencing of the 14 genes identified 453 variants. After the application of the quality filters, 50 variants were maintained for the following classification phases. Subsequently, other 35 variants were excluded: (1) variants with a population frequency ≥1% and (2) variants reported as Benign/Probably Benign by ClinVar or according to the ACMG classification. The flowchart shown in Figure 1 details the filters applied to germline variants prioritization employed in this study.
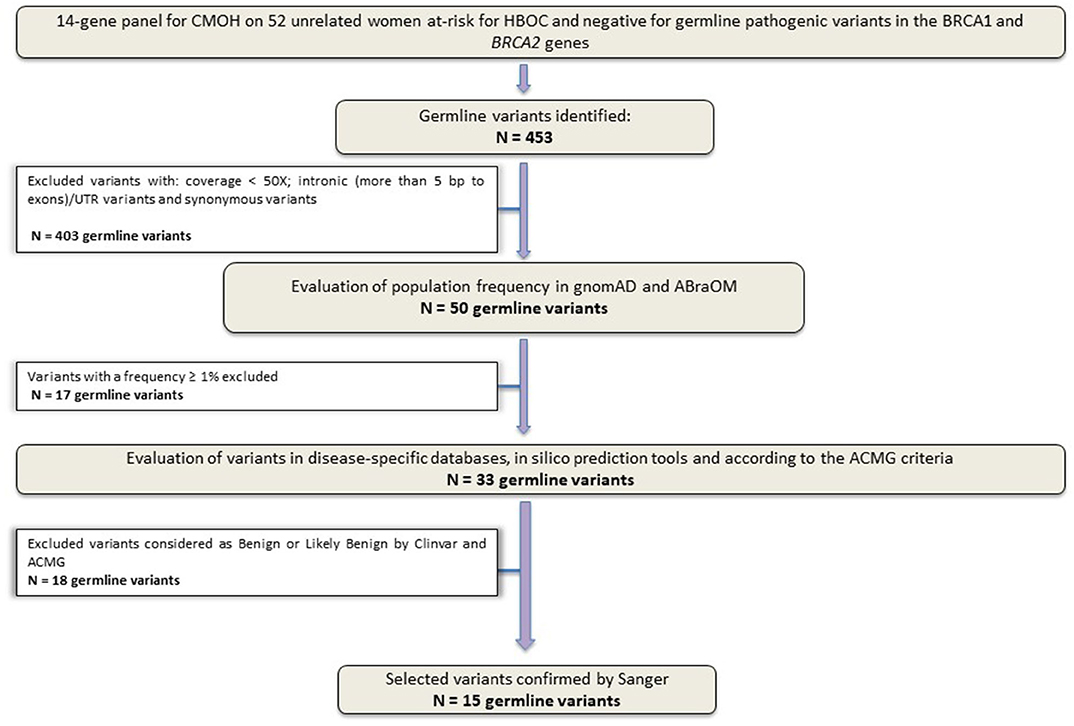
Figure 1. Study design flowchart. Variants selection workflow. Panel sequencing data from 52 unrelated Brazilian women at-risk for HBOC, without germline pathogenic variants in BRCA1 and BRCA2 genes.
After applying all filters described above, a total of 15 unique variants were identified. Two loss of function variants were considered Pathogenic/Probably Pathogenic by the ACMG and ClinVar: a frameshift variant in MUTYH (in heterozygosis): c.1147delC and the splicing variant c.315-2A>G in the MRE11A gene, respectively. VUS were considered to be all those classified by ClinVar or by the ACMG as class 3, and 13 variants were found in 11 women (21%). Table 2 contains all information from the databases used and the in silico predictions of the 15 germline variants identified.
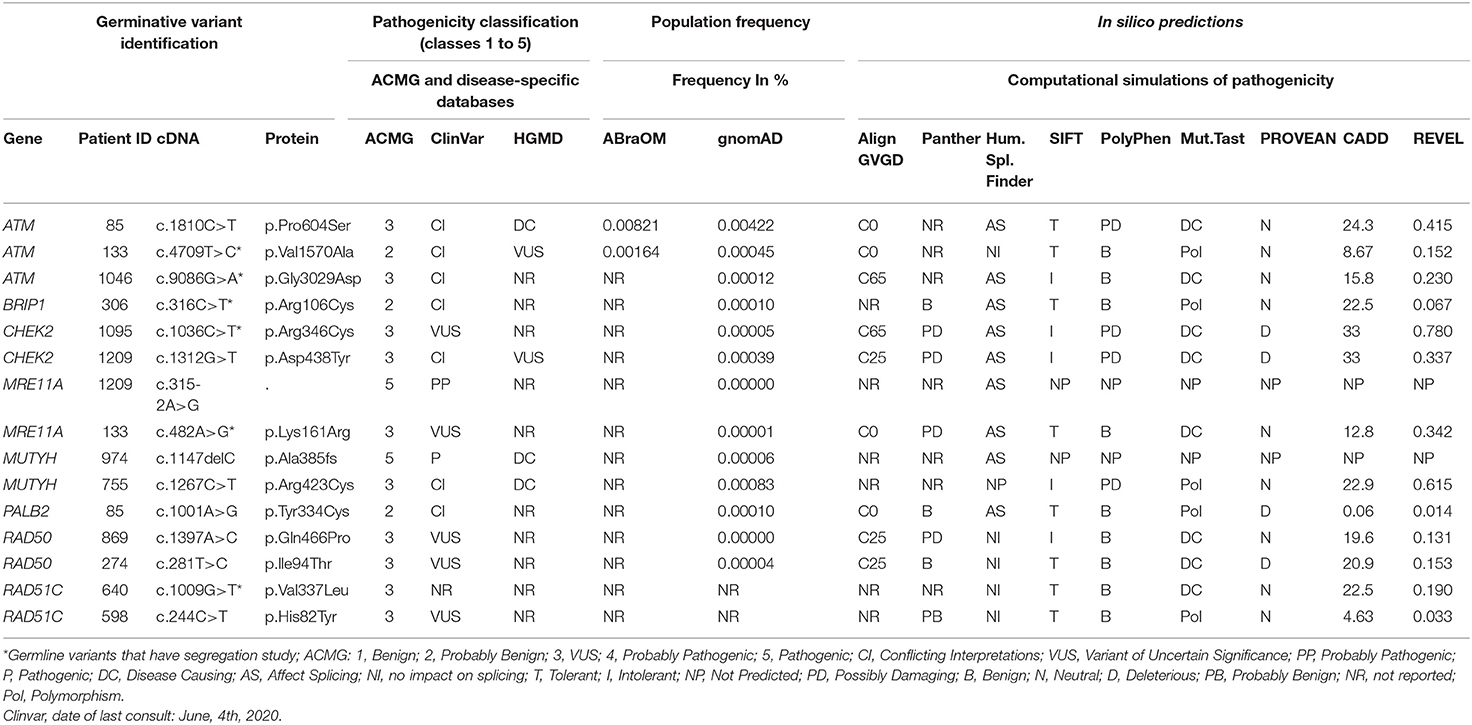
Table 2. Description of the 15 variants identified in the 14-gene panel.
Regarding the personal history of cancer of the 12 probands (with identified class 3, 4, or 5 variants), as shown in Figure 2, seven patients (58.3%) were diagnosed with cancer before the age of 45 and three were diagnosed before the age of 30. Two patients, ID306 (presence of the variant in the BRIP1 gene, c.316C>T) and ID869 (presence of variant in the RAD50 c.1397A>C gene) had two primary tumors: melanoma and breast, and breast and ovary, respectively. The patients with pathogenic germline variants in the MUTYH and MRE11A genes had unilateral breast tumors diagnosed after 46 years of age.
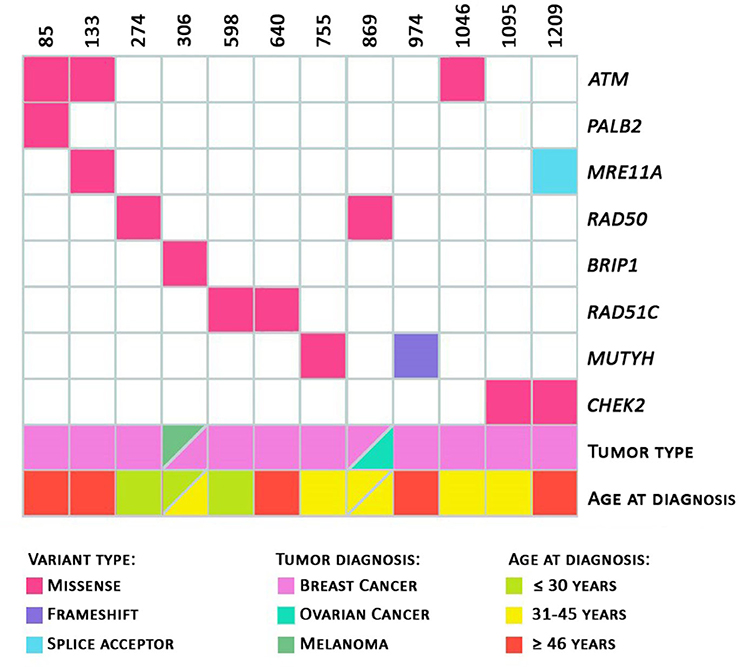
Figure 2. Heatmap. Total number of variants per patient. Variants identified with information regarding the tumor diagnosed and the patient's age at diagnosis; and age at diagnosis (in green: diagnosis ≤ 30 years of age; in yellow: 31 to 45 years of age; in red: ≥ 46 years of age) are represented.
Segregation and LOH Analysis
Of the 15 germline variants resulting from the gene panel, it was possible to perform a more in-depth analysis of the biological-clinical impact in 9 of them. For six variants (five families), it was possible to perform co-segregation analysis. In addition, the LOH analysis was performed for seven VUS, and both analyses—co-segregation and LOH—were performed for four variants (Table 3, Supplementary Figure 1). The variant c.4709C>T in the ATM gene (class 2 according to the ACMG and conflicting data according to ClinVar) was included in the analysis because it was present in the same patient (ID133) as one MRE11A class 3 variant. Pedigree of index cases, where segregation and/or LOH analysis were performed, are depicted in Supplementary Figures 2–7 and in Figure 3.
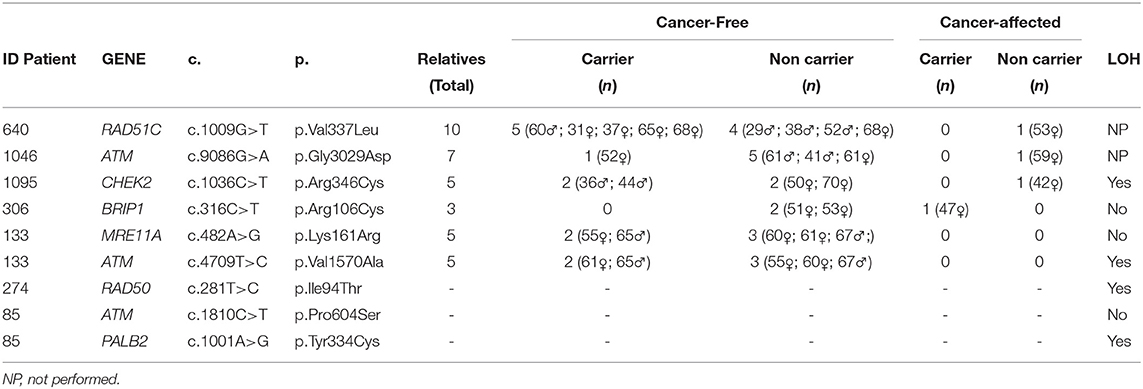
Table 3. Segregation and LOH analysis.
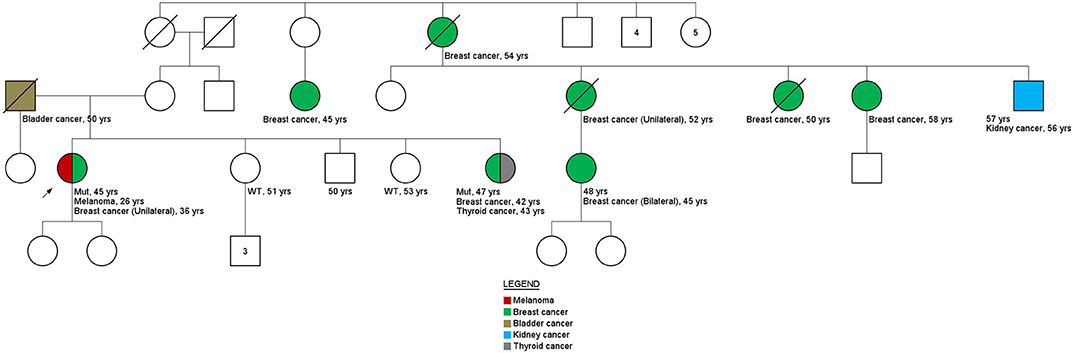
Figure 3. Family history. Pedigree family ID306. Variant c.316C>T in the BRIP1 gene.
For all variants evaluated through segregation analysis, the sex and the age of the relatives are detailed in Table 3. Given the fact that no cancer affected relatives of family ID133 were available for testing, segregation analysis was not possible. Variants under evaluation were not identified in the cancer-affected relatives tested (1 per family) of ID640, ID1046, and ID1095 families, but were carried by one (ID1046), two (ID1095), and five (ID640) cancer-free relatives (Table 3).
Interestingly, results obtained for variant c.316C>T in BRIP1 gene of ID306 patient's family (Figure 3) shown that, only the sister's patient with cancer history (breast cancer at 42 years of age and thyroid cancer at 53 years of age) carried the c.316C>T variant (two other siblings without cancer history were tested). These results are an indicative of the of the co-segregation of the variant with the disease.
Regarding the LOH analyses, among the seven VUS evaluated, the loss of the normal allele was identified in four of them: the variants c.1036C>T in the CHEK2 gene; c.4709T>C variant (ATM gene); c.281T>C in the RAD50 gene, and c.1001A>G in the PALB2 gene (Supplementary Figure 1).
Discussion
The patients included in the study were women considered to be at high risk for HBOC. Fifteen germline variants classified as VUS, probably pathogenic and pathogenic were identified in moderate-risk genes (22–24). The frequency of pathogenic germline variants in patients who previously tested negative for pathogenic variants in BRCA1/BRCA2 was 3.8%. Although it is quite difficult to compare mutation frequencies among studies, due to differences in inclusion criteria and/or ethnicity, our mutation frequency corroborates other studies based on family cancer history inclusion criteria (25–32).
Regarding the pathogenic variants identified, one of them is located in the MRE11A gene, which is considered a partner of the ATM gene in terms of signaling damage to and repair of double-stranded DNA (33, 34). This gene was described in the first studies using NGS as a gene conferring a moderate risk of HBOC (OR = 2.88, P = 0.0090) and therefore was included in panels (34). However, more recent studies with significant sample sizes (case-control) revealed a low risk associated with the MRE11A gene. In the classification by Lee et al. (24), which evaluated the clinical validity of genes usually included in panels, the MRE11A is classified as “disputed,” i.e., there are conflicting data and/or divergent opinions about its association to HBOC. In our study the index patient who carried the pathogenic germline variant in the MRE11A gene was diagnosed with breast cancer at 58 years of age, and her mother had an ovarian tumor diagnosed after 50 years of age. Her family had three generations affected by cancer and a total of 11 individuals with cancer. In addition to the pathogenic germline variant in the MRE11A gene, this patient also had a VUS in the CHEK2 c.1312G>T gene. Southey et al. (35) showed evidence of the relationship of this VUS (CHEK2 c.1312G>T) with risk of prostate cancer, with OR 2.21 (95% CI 1.06 to 4.63, p = 0.030), in European men. However, no case of prostate cancer was reported in our family.
The second pathogenic mutation has been identified in the tumor suppressor gene MUTYH. Recent studies point to a low risk of cancer in carriers of monoallelic pathogenic variants in MUTYH in HBOC (22, 36). In the present study, the heterozygous MUTYH mutation carrier was diagnosed with breast carcinoma at 46 years of age. Additionally, eight cases of breast cancer and two cases of prostate cancer were present in the family history.
Thirteen germline variants of uncertain significance were identified, corresponding to a VUS frequency of 21%. The percentage of VUS, although relatively high, is comparable to that reported in other studies that included moderate and low-risk genes, such as the one published in 2018 by Xie et al. (37), which evaluated 100 women and found a VUS rate of 34.25% in 10 genes associated with susceptibility to breast cancer (excluding BRCA1 and BRCA2).
Among the variants in which the co-segregation analysis could be performed, evidence of an association was found for the germline missense variant c.316C>T in the BRIP1 gene, present in ID306. Although only three relatives were tested, the family member with a previous diagnosis of breast and thyroid tumors had the variant in question. Nevertheless, it is worth noting that the index patient had two primary tumors: melanoma, diagnosed at 26 years of age, and breast cancer, at 36 years old. The family history suggests that the c.316C>T germline variant in the BRIP1 gene may be involved in the development of cancer in this family, although the analysis of the tumor material of the index patient did not show LOH. However, loss of the normal allele can also be caused by other genetic and epigenetic processes, such as occurrence of a pathogenic point mutation at the functional allele and promoter hypermethylation.
The presence of pathogenic germline variants in BRIP1 has been specifically related to ovarian tumors (38, 39). However, other studies have found a moderate association with the risk of breast tumors. A case-control study conducted by Couch et al. (40) showed an OR of 1.27 for breast cancer and 1.67 when both breast cancer and ovarian cancer were included. In the study by Tung and collaborators (36), among 488 patients with breast cancer, four women (0.81%) had a pathogenic variant in the BRIP1 gene. In the family of patient ID306, no cases of ovarian cancer were reported by the index patient. This fact corroborated the studies by Tung et al. (36) (out of the four patients analyzed, only one had a history of ovarian cancer) and Slavin et al. (23), which identified BRIP1 mutations in families with other tumors then ovarian. The melanoma phenotype present in patient ID306 and the bladder phenotype present in the patient's father are noteworthy because both were also present in the study by Tung et al. (36). These results suggest that in-depth analyses of the identified variant should continue, together with a close family monitoring and screening.
Regarding the other families in which co-segregation analysis was performed, there was no co-segregation between the variants in ATM c.9086G>A, CHEK2 c.1036C>T, RAD51C c.1009G>T, and the disease. However, taking into consideration that only one cancer-affected individual was tested in each family, these findings need to be confirmed in a larger number of relatives. Besides, result needs to be interpreted with caution given the incomplete penetrance of these genes and the moderate risk of cancer associated with them (23, 26, 36, 41–43).
In the family where the co-segregation of the CHEK2 variant was analyzed, although co-segregation could not be observed, it is worthy to reinforce that this “negative” result needs to be interpreted with caution given the incomplete penetrance of this gene and the fact that LOH was observed in the tumor tissue, suggesting a pathogenic behavior for this variant. In the clinical classification of the relationship of the CHEK2 gene with HBOC, the association is considered “definitive” for breast tumors (24). Regarding specifically the variant c.1036C>T, it was previously identified by Southey et al. (35) in a case-control study involving breast cancer (42,671 cases and 42,164 controls), prostate cancer (22,301 cases and 22,320 controls), and ovarian cancer (14,542 cases and 23,491 controls), and the results showed strong evidence of an association with the risk of development of breast cancer, with an OR of 5.06 (95% CI 1.09 to 23.5) (35).
For some variants, co-segregation analysis was not possible, but the LOH analysis was performed. The VUS in the gene PALB2 c.1001A>G, identified in ID85, showed LOH in the tumor. The PALB2 gene is considered a high-risk gene (40) for hereditary breast cancer. In the study by Slavin et al. (23), the PALB2 gene had a significant OR of 6.95 (CI 3.71 to 12.70), and in the study by Couch et al. (40), the OR was 7.46 (CI 5,12–11,19). Additionally, in addition to breast cancer, new phenotypes are being associated with the gene, such as pancreatic cancer (42) and gastric cancer (44) (ID85 had two cases of gastric cancer in her family history). Preventive measures and risk management already exist for carriers of the pathogenic germline variant of this gene (https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf, 2019), which some authors are currently considering “BRCA3” (40).
We also identified LOH in the tumor specimen of the patient with the variant c.281T>C (RAD50 gene). In the most recent studies, the RAD50 gene demonstrated a low OR for breast cancer. In the study by Couch et al. (40), an OR = 0.77 was observed (CI 0.52-1.61), and in the study by Slavin et al. (23) only one woman had a pathogenic germline variant in this gene. Regarding the “gene-disease” evaluation in the study by Lee et al. (24), it was observed that the classification is considered “limited” for breast tumors and “disputed” for ovarian tumors, once again suggesting that the risk of cancer associated with the gene is still uncertain.
Although there are conflicting reports in the literature regarding the management of patients with class 3 germline variants, the guidelines of the Brazilian Society of Medical Genetics (45) and NCCN (46) recommend that individuals with VUS should be managed based on the risk of the gene along with the personal and family history of cancer. Discussing genetic results with patients, especially those with VUS-type variants or pathogenic variants in genes whose risk is not well-founded, is not easy. It is a challenge for clinicians and for their patients, who face decisions about screening and prevention strategies in the case of different outcomes.
The results presented in this study can contribute to the definition of the pathogenicity of VUS in breast/ovarian susceptibility genes, especially for the variants in which some indicative of pathogenicity has been found (co-segregation with the disease and/or LOH). Further validation could be obtained by functional analyses. The present study had several limitations, which may contribute to the relatively low frequency of pathogenic variants identified. Among these, it is worth noting that the number of patients tested was relatively low compared to other recently published studies. Besides, although relatively rare, the presence of CNVs were not evaluated. In addition, as the understanding of the role of moderate-penetrance genes on breast/ovarian cancer susceptibility is constantly being updated, some genes that were recently reported as HBOC-associated were not included in the panel. Finally, due to a limitation in the access to relatives with cancer and to tumor material, the segregation and LOH analysis were not possible in all families with identified VUS.
Conclusion
Of the 52 unrelated women with a personal and family history suggestive of breast and/or ovarian cancer, two pathogenic germline variants and 13 germline VUS were identified through the use of a gene panel containing 14 genes associated with high- and moderate-risks of breast and ovarian cancer.
The co-segregation analysis in 5 families allowed the establishment of an association with the presence of the variant and the risk of cancer for variant c.316C>T in the BRIP1 gene. Four variants showed LOH in the tumor specimen (ATM c.4709T>C; CHEK2 c.1036C>T; PALB2 c.1001A>G, and RAD50 c.281T>C), which is an indication of pathogenicity. Thus, the present study provides novel evidence that favors the association of variants in moderate-risk genes with the development of hereditary breast cancer.
Data Availability Statement
The datasets presented in this article are not readily available due to privacy and ethical restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comitê de Ética em Pesquisa, Hospital de Câncer de Barretos. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
RG: methodology, formal analysis, investigation, data analysis, data curation, and writing—original draft. PF: formal analysis, investigation, data curation, and writing—original draft. AE, GF, and CS: methodology, investigation, data curation, and writing—review and editing. NC: patient recruitment, data curation, and writing—review and editing. FG, EA, and AE: data analysis; data curation, and writing—review and editing. PD: data analysis; data curation, and writing—original draft. CP and HG: patient recruitment; data curation, and writing—review and editing. CAAP and GT: LOH analysis, data analysis, and writing—review and editing. DC: conceptualization, LOH analysis, data analysis, and writing—review and editing. EP: conceptualization, writing—review and editing, supervision, project administration, and funding acquisition. All authors: contributed to the article and approved the submitted version.
Funding
This project was funded through grants from FAPESP (2013/24633-2), PRONON (25000.056766/2015-64), and Coordination for the Improvement of Higher Education Personnel (CAPES/PROSUP). EP and DC are recipients of CNPq Productivity Grants.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.571330/full#supplementary-material
References
1. Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention. Science. (2014) 343:1466–70. doi: 10.1126/science.1251827
2. Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. (2016) 16:599–612. doi: 10.1038/nrc.2016.72
3. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
4. Gracia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, de la Hoya M, et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS ONE. (2013) 8:e55681. doi: 10.1371/journal.pone.0055681
5. Diaz-Gay M, Franch-Exposito S, Arnau-Collell C, Park S, Supek F, Munoz J, et al. Integrated analysis of germline and tumor DNA identifies new candidate genes involved in familial colorectal cancer. Cancers. (2019) 11:3. doi: 10.3390/cancers11030362
6. Duran-Lozano L, Montalban G, Bonache S, Moles-Fernandez A, Tenes A, Castroviejo-Bermejo M, et al. Alternative transcript imbalance underlying breast cancer susceptibility in a family carrying PALB2 c.3201+5G>T. Breast Cancer Res Treat. (2019) 174:543–50. doi: 10.1007/s10549-018-05094-8
7. Pastorino L, Andreotti V, Dalmasso B, Vanni I, Ciccarese G, Mandala M, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers. (2020) 12:4. doi: 10.3390/cancers12041007
8. Fernandes GC, Michelli RA, Galvao HC, Paula AE, Pereira R, Andrade CE, et al. Prevalence of BRCA1/BRCA2 mutations in a Brazilian population sample at-risk for hereditary breast cancer and characterization of its genetic ancestry. Oncotarget. (2016) 7:80465–81. doi: 10.18632/oncotarget.12610
9. Monkol SL, Konrad JK, Eric VM, Kaitlin ES, Eric B, Timothy F. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Available online at: http://gnomad.broadinstitute.org/
10. AbraOM. Available online at: http://abraom.ib.usp.br/
11. COSMIC Catalogue of Somatic Mutations In Cancer. Available online at: www.Sanger.ac.uk/genetics/CGP/cosmic
12. HGMD Human Gene Mutation Database. Available online at: www.hgmd.cf.ac.uk
13. PolyPhen-2 PolyPhen-2. Available online at: www.genetics.bwh.harvard.edu/pph2
14. MutationTaster. Available online at: www.mutationtaster.org
15. AlignGVGD Align GVGD. Available online at: http://agvgd.hci.utah.edu/agvgd_input.php
16. SIFT Sorting Intolerant from Tolerant. Available online at: http://sift.bii.a-star.edu.sg/
17. Panther. Available online at: http://www.pantherdb.org/tips/tips_csnpScores.jsp
18. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. (2009) 37:e67. doi: 10.1093/nar/gkp215
19. Ioannidis NM, Pejaver RJ, Middha V, McDonnell S, Baheti SK, Musolf S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
20. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
21. Choi SY, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
22. Slavin TP, Niell-Swiller M, Solomon I, Nehoray B, Rybak C, Blazer KR, et al. Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front Oncol. (2015) 5:208. doi: 10.3389/fonc.2015.00271
23. Slavin TP, Maxwell KN, Lilyquist J, Vijai J, Neuhausen SL, Hart SN, et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer. (2017) 3:22. doi: 10.1038/s41523-017-0046-2
24. Lee K, Seifert BA, Shimelis H, Ghosh R, Crowley SB, Carter NJ, et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet Med. (2018). doi: 10.1038/s41436-018-0361-5
25. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. (2015) 121:25–33. doi: 10.1002/cncr.29010
26. Thompson ER, Rowley SM, Li N, McInerny S, Devereux L, Wong-Brown MW, et al. Panel testing for familial breast cancer: calibrating the tension between research and clinical care. J Clin Oncol. (2016) 34:1455–9. doi: 10.1200/JCO.2015.63.7454
27. Moran O, Nikitina D, Royer R, Poll A, Metcalfe K, Narod SA, et al. Revisiting breast cancer patients who previously tested negative for BRCA mutations using a 12-gene panel. Breast Cancer Res Treat. (2017) 161:135–42. doi: 10.1007/s10549-016-4038-y
28. Weitzel JN, Neuhausen SL, Adamson A, Tao S, Ricker C, Maoz A, et al. Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA-negative Hispanics with breast cancer. Cancer. (2019) 125:2829–36. doi: 10.1002/cncr.32083
29. Kaneyasu T, Mori S, Yamauchi H, Ohsumi S, Ohno S, Aoki D, et al. Prevalence of disease-causing genes in Japanese patients with BRCA1/2-wildtype hereditary breast and ovarian cancer syndrome. NPJ Breast Cancer. (2020) 6:25. doi: 10.1038/s41523-020-0163-1
30. Lerner-Ellis J, Sopik V, Wong A, Lazaro C, Narod SA, Charames GS. Retesting of women who are negative for a BRCA1 and BRCA2 mutation using a 20-gene panel. J Med Genet. (2020) 57:380–4. doi: 10.1136/jmedgenet-2019-106403
31. Velazquez C, Esteban-Cardenosa KL, Avila Cobos EM, Lastra E, Abella LE, et al. Germline genetic findings which may impact therapeutic decisions in families with a presumed predisposition for hereditary breast and ovarian cancer. Cancers. (2020) 2:8. doi: 10.3390/cancers12082151
32. Yoo J, Lee GD, Kim JH, Lee SN, Chae H, Han E, et al. Clinical validity of next-generation sequencing multi-gene panel testing for detecting pathogenic variants in patients with hereditary breast-ovarian cancer syndrome. Ann Lab Med. (2020) 40:148–54. doi: 10.3343/alm.2020.40.2.148
33. Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. (2007) 26:7749–58. doi: 10.1038/sj.onc.1210880
34. Damiola F, Pertesi M, Oliver J, Le Calvez-Kelm F, Voegele C, Young EL, et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. (2014) 16:R58. doi: 10.1186/bcr3669
35. Southey MC, Goldgar DE, Winqvist R, Pylkas K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. (2016) 53:800–11. doi: 10.1136/jmedgenet-2016-103839
36. Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol. (2016) 34:1460–8. doi: 10.1200/JCO.2015.65.0747
37. Xie Y, Li G, Chen M, Guo X, Tang L, Luo X, et al. Mutation screening of 10 cancer susceptibility genes in unselected breast cancer patients. Clin Genet. (2018) 93:41–51. doi: 10.1111/cge.13063
38. Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. (2011) 43:1104–7. doi: 10.1038/ng.955
39. Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. (2015) 107:11. doi: 10.1093/jnci/djv214
40. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. (2017) 3:1190–6. doi: 10.1001/jamaoncol.2017.0424
41. Cybulski C, Lubinski J, Wokolorczyk D, Kuzniak W, Kashyap A, Sopik V, et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin Genet. (2015) 88:366–70. doi: 10.1111/cge.12524
42. Hu C, Hart SN, Bamlet WR, Moore RM, Nandakumar K, Eckloff BW, et al. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol Biomarkers Prev. (2016) 25:207–11. doi: 10.1158/1055-9965.EPI-15-0455
43. Schubert S, van Luttikhuizen JL, Auber B, Schmidt G, Hofmann W, Penkert J, et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int J Cancer. (2018) 144:2683–94. doi: 10.1002/ijc.31992
44. Fewings E, Larionov A, Redman J, Goldgraben MA, Scarth J, Richardson S, et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol Hepatol. (2018) 3:489–98. doi: 10.1016/S2468-1253(18)30079-7
45. Sociedade Brasileira de Genética Médica. (2019). Available online at: https://diretrizes.amb.org.br/_BibliotecaAntiga/cancer-familial.pdf
46. NCCN Guidelines. (2019). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf
Keywords: genetics, hereditary breast and ovarian cancer predisposition syndrome, hereditary cancer, variant of unknown significance, segregation analysis
Citation: Grasel RS, Felicio PS, de Paula AE, Campacci N, Garcia FAdO, Andrade ES, Evangelista AF, Fernandes GC, Sabato CdS, De Marchi P, de Padua Souza C, de Paula CAA, Torrezan GT, Galvão HdCR, Carraro DM and Palmero EI (2020) Using Co-segregation and Loss of Heterozygosity Analysis to Define the Pathogenicity of Unclassified Variants in Hereditary Breast Cancer Patients. Front. Oncol. 10:571330. doi: 10.3389/fonc.2020.571330
Received: 10 June 2020; Accepted: 28 August 2020;
Published: 02 October 2020.
Edited by:
Laura Ottini, Sapienza University of Rome, ItalyReviewed by:
Florentia Fostira, National Centre of Scientific Research Demokritos, GreeceJiaqi Liu, National Cancer Center of China, China
Maria Adelaide Caligo, Pisana University Hospital, Italy
Copyright © 2020 Grasel, Felicio, de Paula, Campacci, Garcia, de Andrade, Evangelista, Fernandes, Sabato, De Marchi, Souza, de Paula, Torrezan, Galvão, Carraro and Palmero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edenir Inêz Palmero, ZWRlbmlyaXBAeWFob28uY29tLmJy