 Xing Niu
Xing Niu Jingping Xu
Jingping Xu Jinwen Liu2
Jinwen Liu2 Ming Zhong
Ming Zhong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 14 October 2020
Sec. Head and Neck Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.556497
Objective: To comprehensively analyze the global N6-methyladenosine (m6A) modification pattern in ameloblastoma.
Methods: m6A peaks in ameloblastoma and normal oral tissues were detected by MeRIP-seq. Differentially methylated m6A sites within messenger RNAs (mRNAs), long no-coding RNA (lncRNAs) and circular RNA (circRNAs) were identified, followed by functional enrichment analysis. By comprehensively analyzing MeRIP-seq and RNA-seq data, differentially expressed mRNAs, lncRNAs and circRNAs containing differentially methylated sites were identified. RNA binding proteins (RBPs) were then identified for differentially methylated m6A sites.
Results: In total, 3,673 differentially methylated m6A sites within coding genes were detected, of which 16.2% (704/3,673) were significantly upmethylated sites in ameloblastoma compared to normal oral tissues. Furthermore, 4,975 differentially methylated m6A sites within lncRNAs were identified, of which 29.4% (1,465/4,975) were upmethylated sites in ameloblastoma. We also found 364 differentially methylated m6A sites within circRNAs, of which 22.5% (82/364) were upmethylated sites in ameloblastoma. Differentially methylated m6A was most often harbored in the CDS (54.10%), followed by 5’UTR (21.71%). Functional enrichment analysis revealed that m6A modification could be involved in the development of ameloblastoma by organism developmental processes. A total of 158 RBPs within differentially methylated m6A sites were identified, which were significantly involved in mRNA metabolic process, mRNA processing, RNA processing, RNA splicing and RNA transport.
Conclusion: Our findings for the first time provide m6A landscape of human ameloblastoma, which expand the understanding of m6A modifications and uncover regulation of lncRNAs and circRNAs through m6A modification in ameloblastoma.
Ameloblastoma is the most common epithelial odontogenic neoplasm globally, accounting for approximately 36% of all odontogenic tumors (1). Ameloblastoma has a high risk of recurrence (90%) and even occurs distant metastasis (2). Surgery resection is the main treatment option for ameloblastoma, but it often leads to facial deformity, masticatory function loss, and psychological burden (3). Thus, it is of importance to develop new treatment strategies for ameloblastoma. To date, most molecular studies on ameloblastoma have focused on exploring markers and genetic variation, which help to ensure diagnosis and better determine patients’ prognosis (4–6).
m6A is one of the most popular RNA modifications, which is widely found in mRNA and non-coding RNA like lncRNA (7) and circRNA (8). Specific methylation of these RNA molecules regulates RNA structure and protein-RNA interactions, which may affect RNA metabolism, cell signaling, cell survival and differentiation (9, 10). m6A modification provides a critical transcriptomic mechanism, which regulates RNA metabolism and function. Increasing studies have confirmed that dysregulation of RNA modification like m6A is in association with severe human diseases including tumors (11, 12). However, no studies have reported m6A modification in ameloblastoma.
LncRNAs are transcribed RNA molecules with more than 200 nucleotides (13). Growing evidence suggests that lncRNAs are key regulators of gene expression, which participate in a variety of physiological and pathological processes during the occurrence and development of tumors, including ameloblastoma (14). The roles and potential mechanisms of several lncRNAs in ameloblastoma have been reported (15). The key role of m6A modified lncRNAs has been emphasized in cancer. For example, m6A modified lncRNA THOR regulates the proliferation of tumor cells (16). m6A reader YTHDF3 negatively mediates lncRNA GAS5 in colorectal cancer. However, the specific functions of lncRNAs with m6A modification remain unclear in ameloblastoma.
CircRNAs, a class of ncRNAs, have emerged as mediators of gene expression. To date, more than 100,000 circRNAs have been identified, which are expressed in specific tissues or cells and are associated with various physiological and pathological conditions (17). The biogenesis of circRNAs is regulated by various molecular factors such as RNA-binding proteins (RBPs), splicing components, proteins that affect transcriptional elongation, and the presence of reverse RNA repeats (18). CircRNAs have been shown to be functional and can affect gene expression patterns by acting as a sponge for microRNAs (miRNAs) and RBPs (18). For instance, Yang et al. found that the most abundant RNA modification motif m6A was enriched in the circRNA population (19). It has been demonstrated that m6A-modified circNSUN2 may facilitate cytoplasmic export and stabilize HMGA2, thereby accelerating colorectal liver metastasis (20). Thus, m6A modified circRNAs could be promising therapeutic targets for tumors. However, little is known about m6A modification of circRNAs in ameloblastoma. In this study, we for the first time comprehensively analyzed m6A modification patterns and uncovered m6A modified lncRNAs and circRNAs in ameloblastoma.
A total of three patients with ameloblastoma were involved in our study. Tumor tissue specimens and corresponding normal oral specimens were collected during the operation from the Department of Maxillofacial Surgery, Stomatological Hospital of China Medical University. All specimens were immediately stored at −80°C before RNA isolation. Our research was approved by the Ethics Committee of School and Hospital of Stomatology, China Medical University (2019012). All participants signed written informed consents.
Total RNA was extracted from tissue specimens using TRIzol reagent (Life Technologies, Carlsbad, CA) following the manufacturer’s instructions. RNA quantification and quality were determined using NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA). RNA integrity and gDNA contamination were evaluated via denaturing agarose gel electrophoresis.
MeRIP-seq and RNA-seq were performed by Cloudseq Biotech, Inc. (Shanghai, China), in accordance with a previously reported procedure (21). Briefly, m6A RNA immunoprecipitation was performed with the GenSeqTM m6A RNA IP Kit (GenSeq Inc., China) in line with the manufacturer’s instructions. Both the input sample without immunoprecipitation and the m6A IP samples were used for RNA-seq library generation with NEBNext® Ultra II Directional RNA Library Prep Kit (New England Biolabs, Inc., USA). The library quality was evaluated with BioAnalyzer 2100 system (Agilent Technologies, Inc., USA). Library sequencing was performed on Illumina Hiseq instrument with 150bp paired-end reads. MeRIP-seq and RNA-seq data have been uploaded to the Gene Expression Omnibus (GEO) database (accession number: GSE156886).
Paired-end reads were collected from Illumina HiSeq 4000 sequencer, followed by quality control via Q30. Then, 3’ adaptor-trimming and low-quality reads were removed using cutadapt software (version: 1.9.3). After that, the clean reads of all libraries were aligned to the reference genome (UCSC HG19) by Hisat2 software (version: 2.0.4). CircRNAs were identified by DCC software according to STAR alignment results. Interested genes were directly visualized in the Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/; version: 2.4.10) (22). For MeRIP-seq, m6A sites on RNAs (peaks) were analyzed by overlapping three pairs of ameloblastoma and adjacent normal oral tissues using MACS software. diffReps was used to identify differentially methylated m6A sites between ameloblastoma tissues and adjacent normal oral tissues with the threshold of |log2 fold change (FC)|>1 and p-value<0.05. m6A methylation peaks that were overlapping with transcript exons were figured out and chosen by home-made scripts. For RNA-seq, raw counts were obtained based on HTSeq software (version 0.9.1), followed by normalization using the edgeR software. Differentially expressed mRNAs, lncRNAs and circRNAs were identified according to p-value and fold change.
The Gene ontology (GO) project provides a controlled vocabulary to describe gene and gene product attributes in any organism (http://www.geneontology.org). The ontology covers three domains: biological process (BP), cellular component (CC) and molecular function (MF). Pathway analysis is a functional analysis mapping gene to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. In this study, functional enrichment analysis was presented for the differentially methylated mRNAs. P-value ≤ 0.05 was considered significantly enriched.
Ameloblastoma tissues and adjacent normal oral tissues from three patients were used for MeRIP-seq analysis. Venn diagram showed that 16,477 m6A peaks within mRNAs (Figure 1A), 5,895 m6A peaks within lncRNAs (Figure 1B) and 2,808 m6A peaks within circRNAs (Figure 1C) were overlapped between ameloblastoma tissues and adjacent normal oral tissues. Noticeably, there were 6,870 nonoverlapping m6A peaks within mRNAs (Figure 1A), 4,558 nonoverlapping m6A peaks within lncRNAs (Figure 1B), and 1,091 nonoverlapping m6A peaks within circRNAs (Figure 1B) in ameloblastoma tissues compared to adjacent normal oral tissues. The high nonoverlapping percentages of m6A peaks within mRNAs, lncRNAs and circRNAs suggest the differences in the m6A modification patterns between the two groups. Furthermore, 8,881 m6A-modified mRNAs (Figure 1D), 5,675 m6A-modified lncRNAs (Figure 1E) and 1,844 m6A-modified circRNAs (Figure 1E) were found within both the two groups.
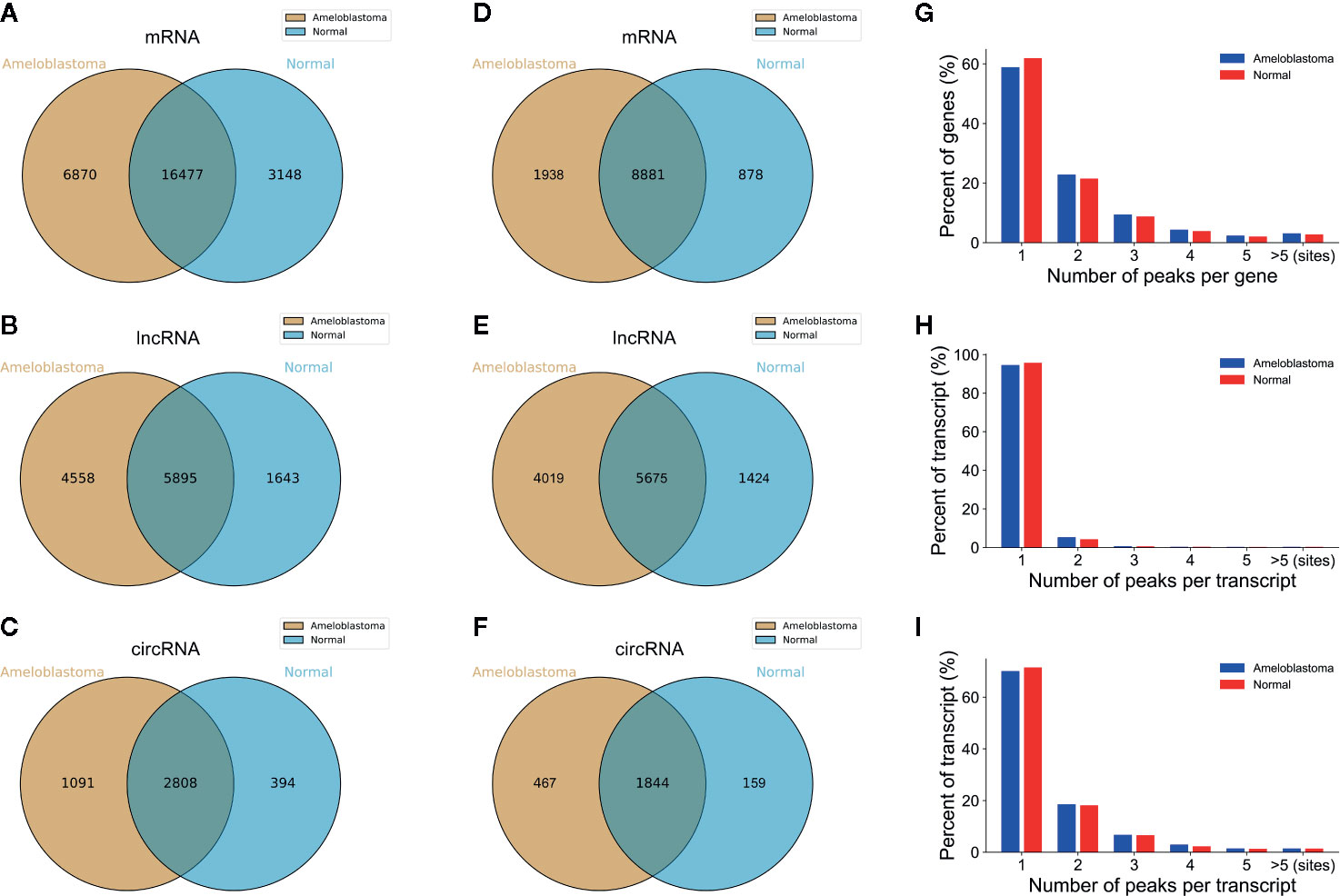
Figure 1 Overview of m6A-modified peaks within mRNAs, lncRNAs and circRNAs in ameloblastoma and adjacent normal oral tissues. (A–C) Venn diagram depicting the overlapped and non-overlapped m6A peaks within mRNAs, lncRNAs and circRNAs between the two groups. (D–F) Venn diagram showing the differences and overlaps in m6A-modified mRNAs, lncRNAs and circRNAs between the two groups. (G-I) The number of m6A peaks per mRNA, lncRNA and circRNA between the two groups.
Most of m6A-motified mRNAs (Figure 1G), lncRNAs (Figure 1H) and circRNAs (Figure 1I) contained only one m6A peak, while a small number of them contained two or more, which was consistent with previous studies such as clear cell renal cell carcinoma (23). The top ten hypermethylated and hypomethylated m6A-modified peaks for ameloblastoma tissues are listed in Tables 1, 2.
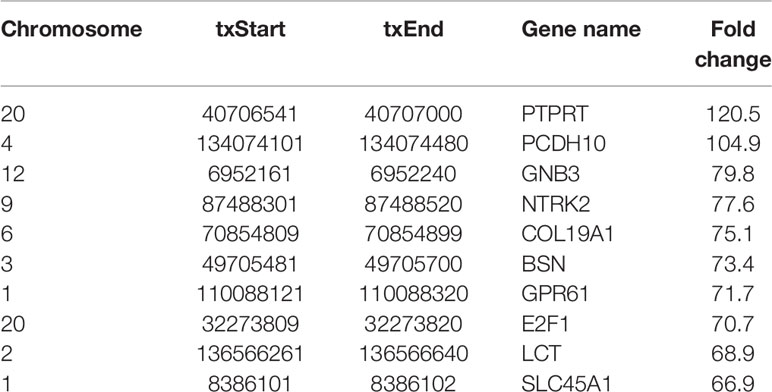
Table 1 The top ten hypermethylated m6A-modified peaks in ameloblastoma tissues compared to normal oral tissues.
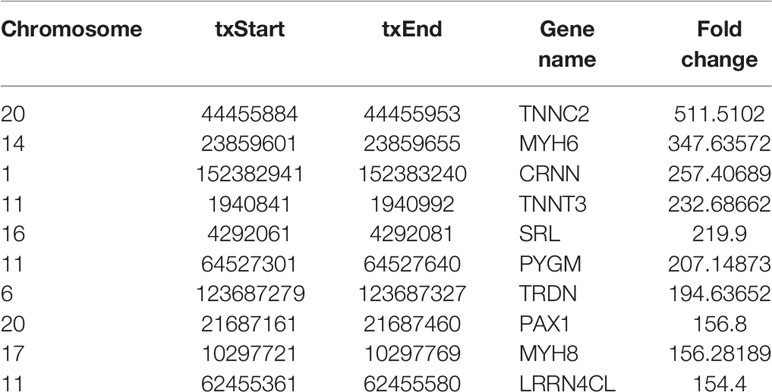
Table 2 The top ten hypomethylated m6A-modified peaks in ameloblastoma tissues compared to normal oral tissues.
Motif analysis was performed by DREME software (version: 5.0.4) (24). Compared to adjacent normal oral samples, the top consensus motifs in the m6A peaks within mRNAs, lncRNAs and circRNAs were respectively AAACU, GAACU and AAACC in ameloblastoma samples (Figure 2A). The distribution of m6A was further investigated in the whole transcriptome of ameloblastoma tissues and adjacent normal oral tissues. The m6A peaks were mainly assigned into 5’UTR, coding sequence (CDS), start codon, stop codon and 3’UTR. As shown in Figure 2B, m6A peaks were especially enriched in the stop codon both for ameloblastoma and normal oral samples. Moreover, there was a higher peak density in the CDS region for normal oral samples compared to ameloblastoma samples. For all m6A peaks both in ameloblastoma and normal oral samples, the CDS was most often harbored (Figure 2C). The distribution of m6A peaks was consistent with previous m6A results (25).
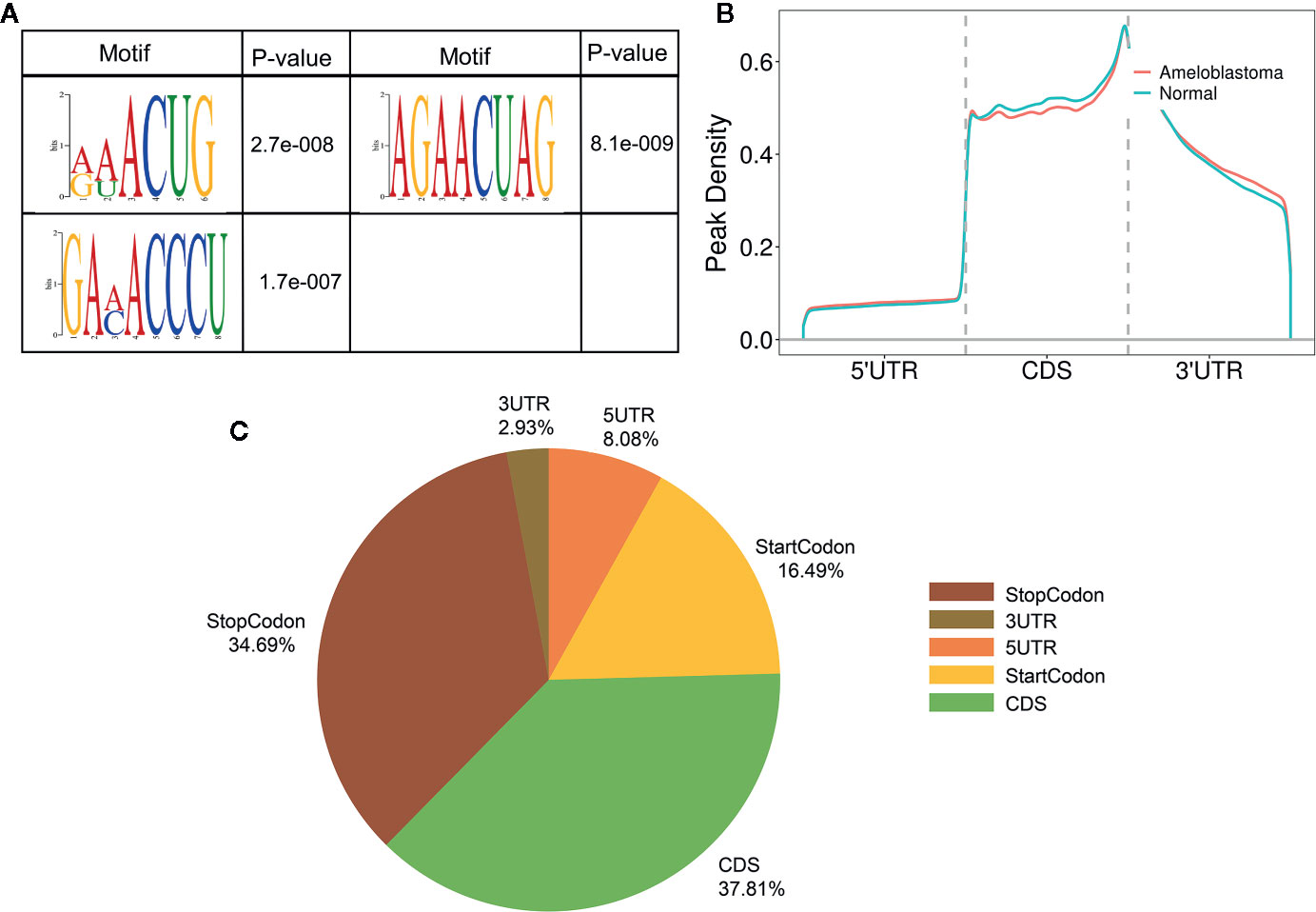
Figure 2 Characteristics of m6A peaks within mRNAs, lncRNAs and circRNAs in ameloblastoma and adjacent normal oral tissues. (A) Top motifs enriched from all identified m6A peaks within mRNAs, lncRNAs and circRNAs in ameloblastoma tissues compared to adjacent normal oral tissues. (B) Difference in the density of m6A peaks in the indicated regions between ameloblastoma and adjacent normal oral tissues. (C) Pie diagram showing the distributions of m6A peaks in the whole transcriptome of ameloblastoma tissues and adjacent normal oral tissues.
Abnormal m6A-modified mRNAs, lncRNAs and circRNAs were identified between ameloblastoma tissues and adjacent normal oral tissues. With the threshold of |log2FC|>1 and p-value<0.05, volcano plots depicted 6,429 hypermethylated and 9,225 hypomethylated mRNAs (Figure 3A), 15,512 hypermethylated and 15,052 hypomethylated lncRNAs (Figure 3B), 1,135 hypermethylated and 12,313 hypomethylated circRNAs (Figure 3C) in ameloblastoma tissues compared to adjacent normal oral tissues. Based on |log2FC|>1 and p-value<0.0001, we identified 1,032 hypermethylated and 3,274 hypomethylated mRNAs (Figure 3D), 2,012 hypermethylated and 3,774 hypomethylated lncRNAs (Figure 3E), 148 hypermethylated and 325 hypomethylated circRNAs (Figure 3F) for ameloblastoma. Then, hierarchical clustering analysis results suggested that there were significant differences in the m6A methylation patterns within mRNAs (Figure 3G), lncRNAs (Figure 3H) and circRNAs (Figure 3I) between ameloblastoma tissues and adjacent normal oral tissues.
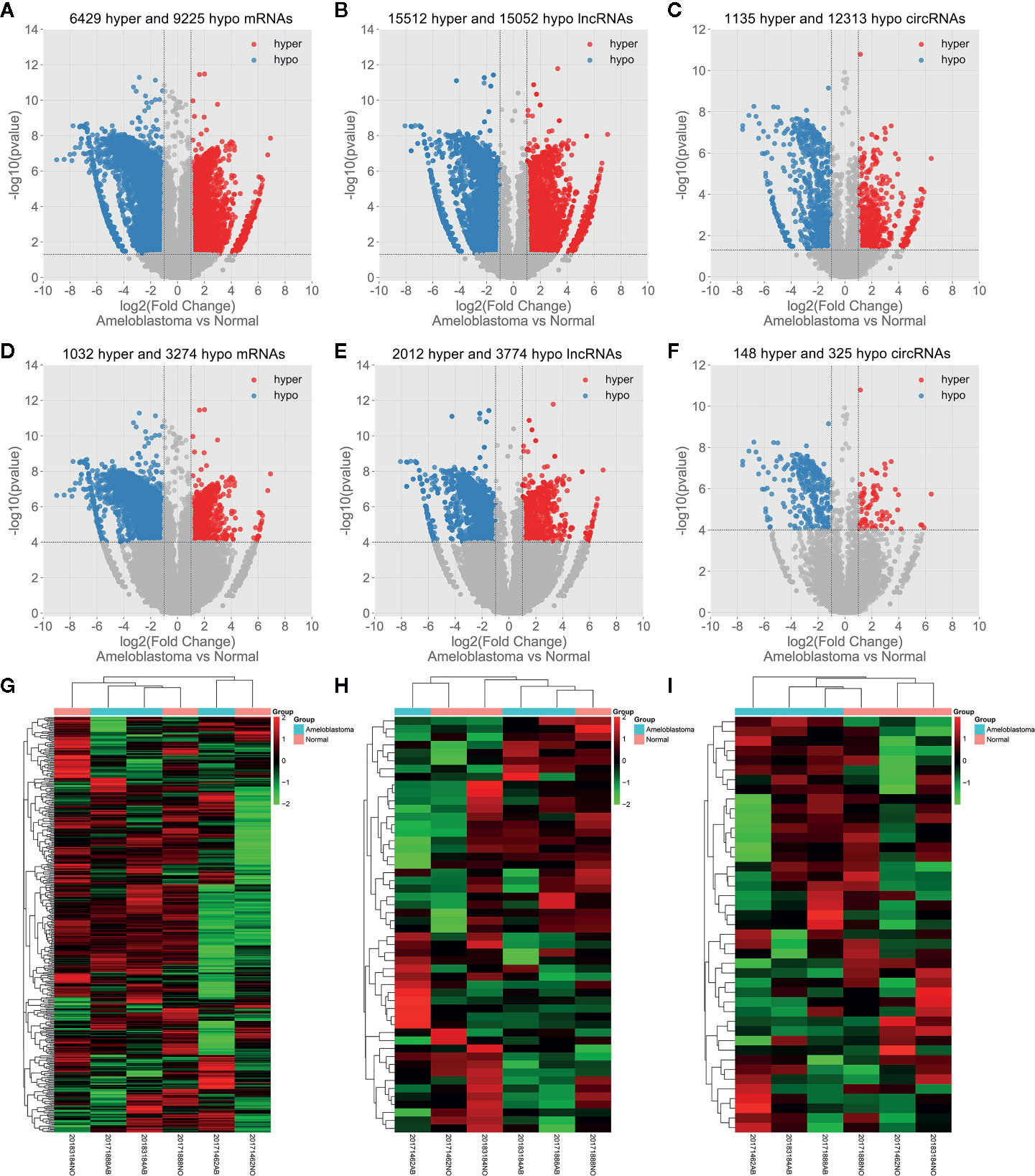
Figure 3 Abnormal m6A-modied mRNAs, lncRNAs and circRNAs in ameloblastoma tissues compared to adjacent normal oral tissues. Volcano plots showing differentially m6A-modified mRNAs (A), lncRNAs (B) and circRNAs (C) based on |log2FC|>1 and p-value<0.05. In volcano plots, red blots represent hypermethylation and blue blots represent hypomethylation. Differentially m6A-modified mRNAs (D), lncRNAs (E) and circRNAs (F) according to |log2 FC|>1 and p-value<0.0001. (G–I) Hierarchical clustering analysis results showing the differences in m6A modification patterns within mRNAs, lncRNAs and circRNAs between ameloblastoma tissues compared to adjacent normal oral tissues according to |log2FC|>1 and p-value<0.0001. In heat maps, red suggests hypermethylation and green suggests hypomethylation.
In total, 3,673 differentially methylated m6A sites within mRNAs were identified, of which 16.2% (704/3,673) were significantly hypermethylated in ameloblastoma tissues compared to adjacent normal oral tissues. Furthermore, 4,975 differentially methylated m6A sites within lncRNAs were identified in ameloblastoma, of which 29.4% (1,465/4,975) were hypermethylated. We also identified 364 differentially methylated m6A sites within circRNAs, of which 22.5% (82/364) were hypermethylated in ameloblastoma. All differentially methylated m6A sites within mRNAs, lncRNAs and circRNAs were mapped to chromosomes to obtain their distribution profiles. The top five chromosomes harboring the most hypermethylated and hypomethylated m6A sites within mRNAs were 1 (76), 2 (73), 11 (48), 3 (43) and X (40); 1 (338), 3 (195), 19 (189), 17 (179) and 2 (174) in Figures 4A–C. The top five chromosomes harboring the most hypermethylated and hypomethylated m6A sites within lncRNAs were 1 (144), 3 (120), 2 (105), 16 (96) and 12 (92); 1 (437), 2 (333), 15 (243), 17 (233) and 11 (209) in Figures 4D–F. Moreover, the top five chromosomes harboring the most hypermethylated and hypomethylated m6A sites within circRNAs were 3 (11), 15 (8), 11 (7), 1 (6) and 14 (6); 2 (36), 1 (26), 3 (22), 6 (20) and 7 (20) in Figures 4G–I.
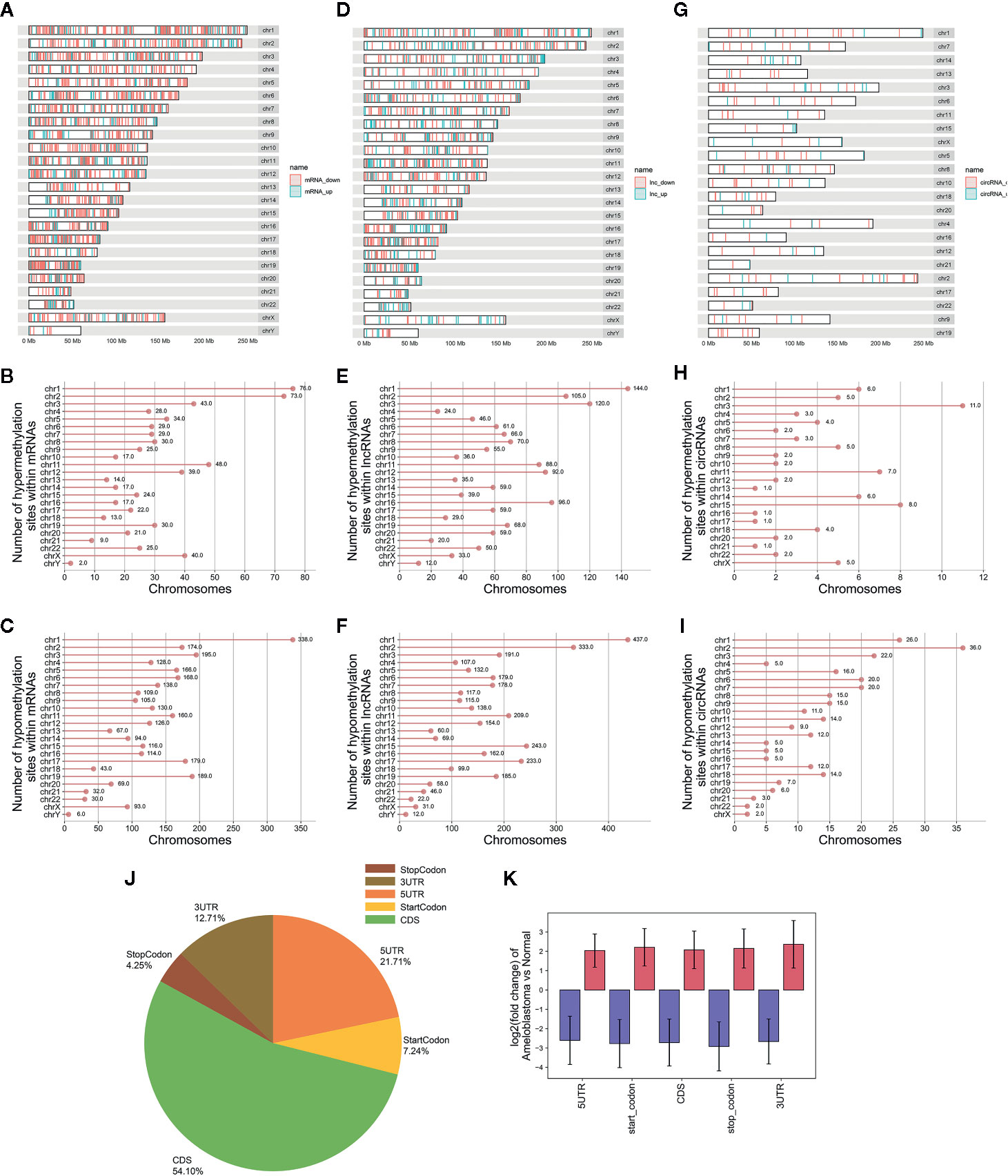
Figure 4 Differences in distribution of differentially methylated m6A sites between ameloblastoma and adjacent normal oral tissues. Chromosomal distribution of differentially methylated m6A sites within mRNAs (A–C); lncRNAs (D–F); circRNAs (G–I). (J) Pie diagram showing the distribution of differentially methylated m6A sites in the whole transcriptome of ameloblastoma tissues and adjacent normal oral tissues. (K) The fold change of differentially methylated m6A sites in five regions between ameloblastoma and adjacent normal oral tissues. Red represents high methylation and purple represents low methylation.
The distribution of differentially methylated m6A peaks was analyzed in ameloblastoma. In Figure 4J, differentially methylated m6A sites were most often harbored in the CDS (54.10%), followed by 5’UTR (21.71%). Then, these differentially methylated m6A sites were classified according to the five regions. As shown in Figure 4K, there were distinct differences in m6A levels within the five regions between ameloblastoma tissues and adjacent normal oral tissues, indicating abnormal m6A patterns could be involved in the development of ameloblastoma.
To uncover the potential functions of protein coding genes harboring differentially methylated m6A sites in ameloblastoma, differentially methylated m6A sites-contained mRNAs-, lncRNAs- and circRNAs-associated genes were selected for functional enrichment analyses. For the BP term, we found that mRNAs harboring differentially methylated m6A sites were significantly enriched in organism developmental processes such as multicellular organismal development and system development (Figure 5A). For the CC term, these mRNAs harboring upmethylated m6A sites in ameloblastoma were mainly enriched in intrinsic/integral component of plasma membrane, postsynaptic membrane and synaptic membrane, while those harboring downmethylated m6A sites were highly involved in myofibril, contractile fiber and contractile fiber part (Figure 5B). For the MF term, these mRNAs were significantly correlated with DNA binding and protein binding (Figure 5C). Furthermore, these mRNAs were found to be significantly enriched in cancer-associated pathways, such as pathways in cancer, cGMP-PKG signaling pathway and oxytocin signaling pathway (Figure 5D).
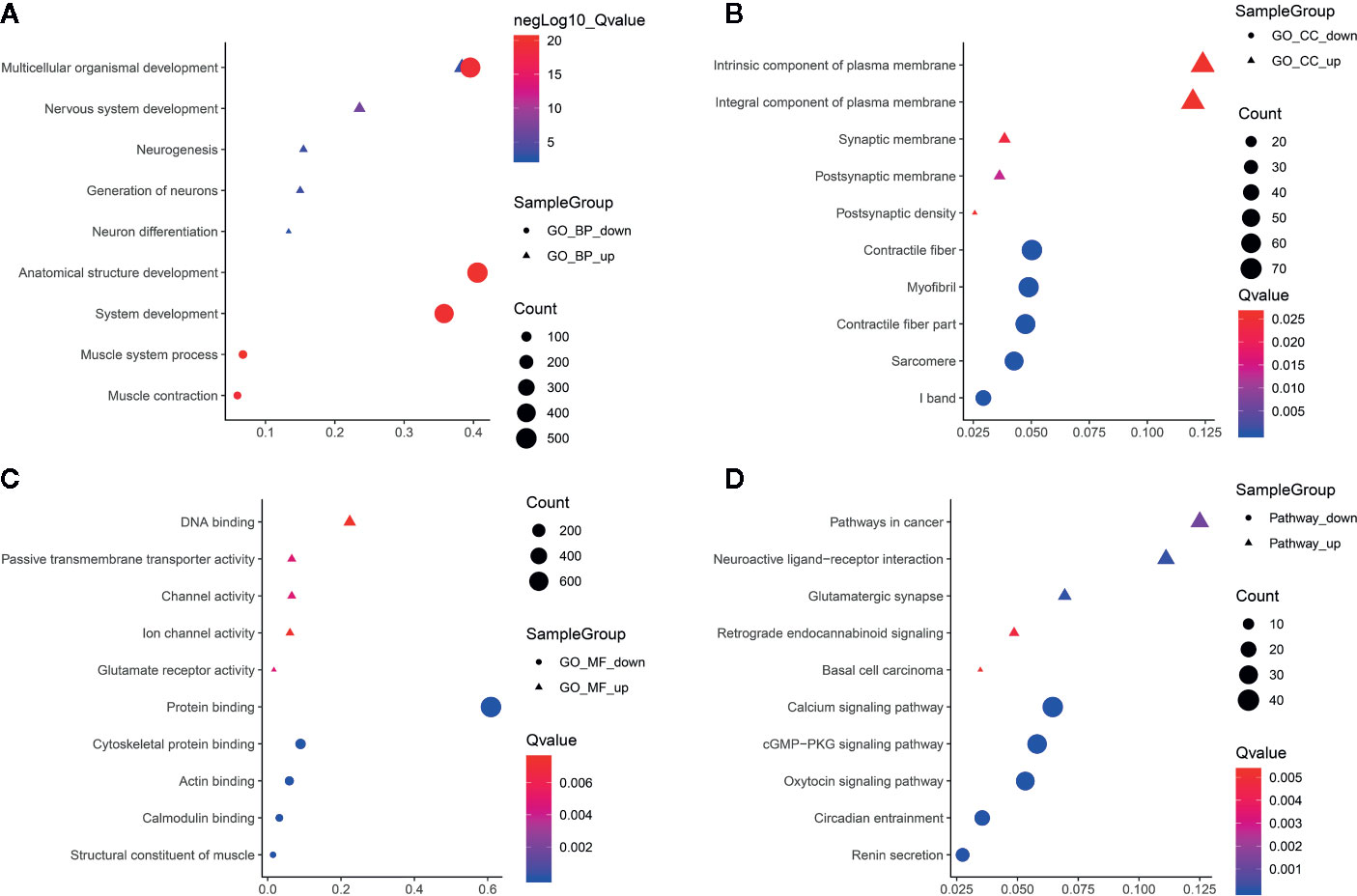
Figure 5 Functional enrichment analysis results of protein coding genes harboring differentially methylated m6A sites. The top ten enrichment results of (A) biological process (BP); (B) cellular component (CC); (C) molecular function (MF) and (D) KEGG.
Functional enrichment analyses of differentially methylated lncRNA-associated genes were performed. For the BP category, hypermethylated or hypomethylated lncRNAs-associated genes were in a significant correlation with system development (Figure 6A). For the CC category, hypermethylated lncRNAs-associated genes were significantly associated with neuron part, neuron projection and early endosome membrane (Figure 6B), while hypomethylated lncRNAs-associated genes were mainly enriched in contractile fiber part, contractile fiber and sarcomere (Figure 6B). For the MF category, upmethylated lncRNAs-associated genes were mainly enriched in cation binding, ion binding and metal ion binding (Figure 6C), while downmethylated lncRNAs-associated genes were involved in structural constituent of muscle, protein binding and actin filament binding (Figure 6C). KEGG pathway analysis results showed that hypermethylated or hypomethylated lncRNAs-associated genes were significantly correlated with transcriptional misregulation in cancer (Figure 6D).
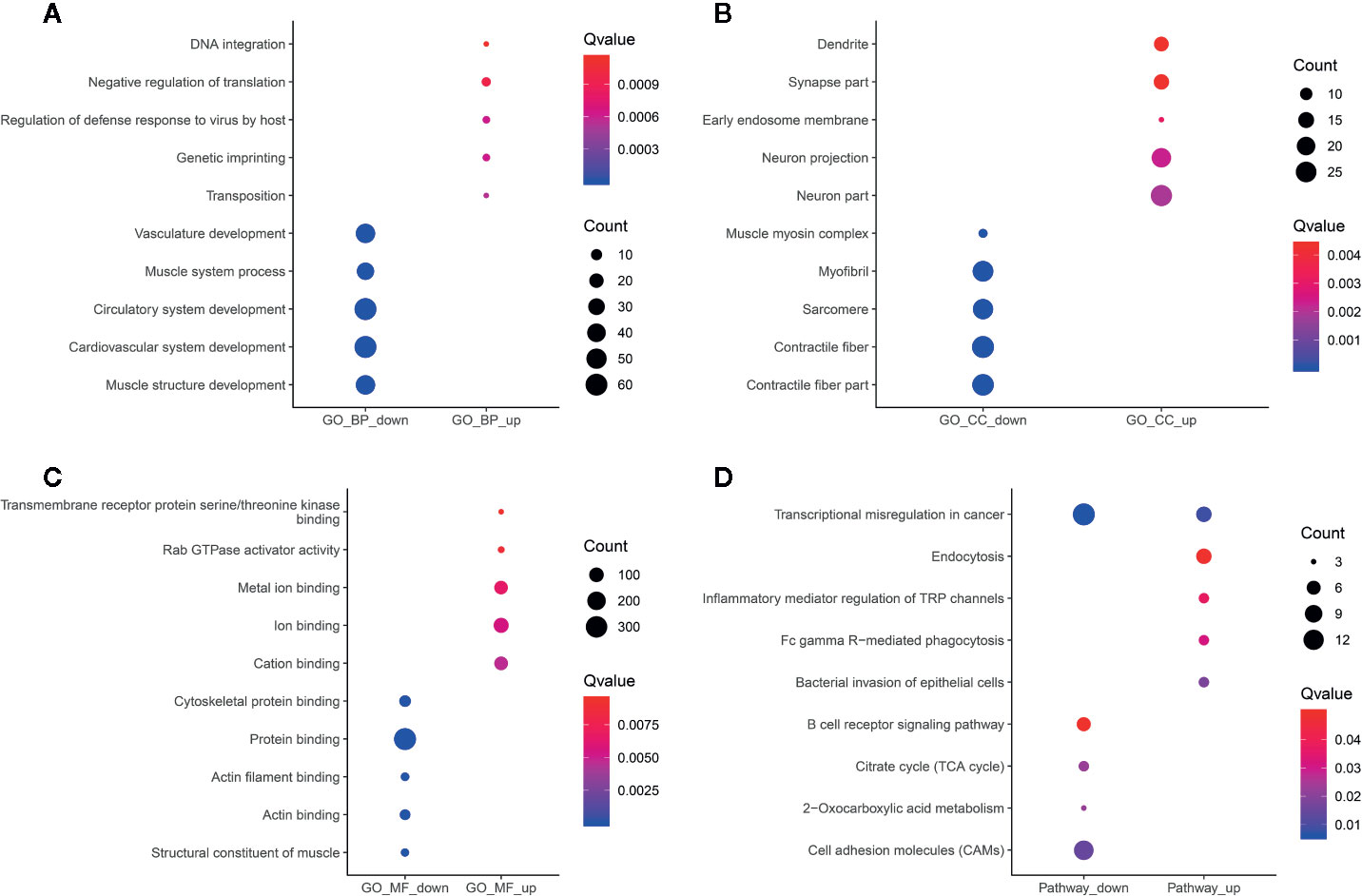
Figure 6 Functional enrichment analysis results of differentially methylated m6A sites-contained lncRNAs-associated genes. The top ten enrichment results of (A) biological process (BP); (B) cellular component (CC); (C) molecular function (MF) and (D) KEGG.
In Figure 7A, hypermethylated circRNAs-associated genes were mainly enriched in epithelial or epithelium cell migration processes, while hypomethylated circRNAs-associated genes were significantly associated with organismal processes. For the CC category, these circRNAs-associated genes were involved in postsynaptic density or membrane, synapse myofibril and contractile fiber (Figure 7B). For the MF category, upmethylated or downmethylated circRNAs-associated genes were mainly enriched in anion binding (Figure 7C). KEGG pathway analysis results showed that these circRNAs-associated genes were involved in adherens junction (Figure 7D).
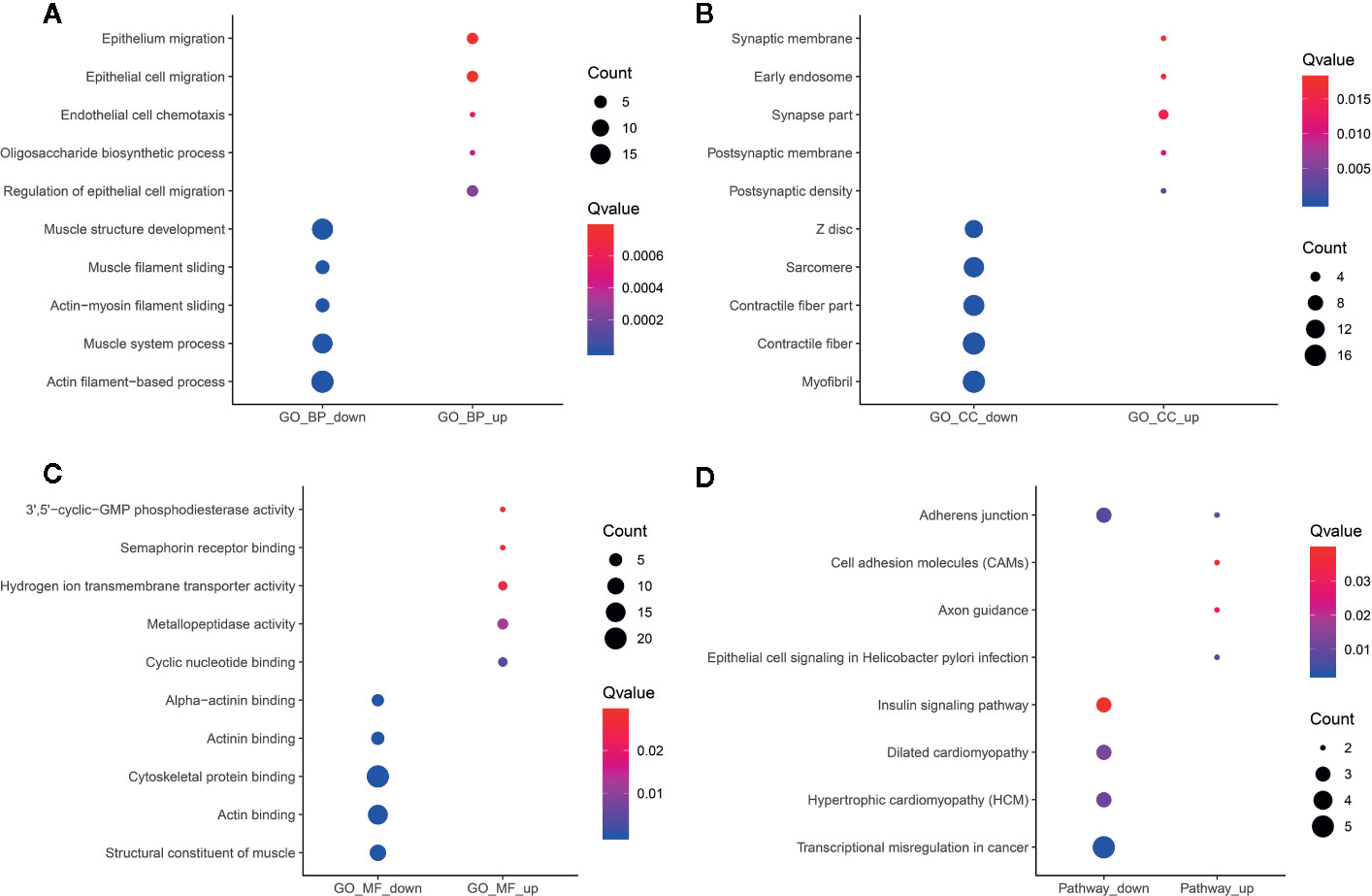
Figure 7 Functional enrichment analysis results of differentially methylated m6A circRNAs-associated genes. The top ten enrichment results of (A) biological process (BP); (B) cellular component (CC); (C) molecular function (MF) and (D) KEGG.
Potential RBPs within differentially methylated m6A sites were explored using RMBase database (http://rna.sysu.edu.cn/rmbase/; version: 2.0) (26). In total, 158 RBPs within differentially methylated m6A sites were identified, which were depicted as heat maps (Figure 8A). GO and KEGG enrichment analyses were then performed based on these RBPs. As shown in Figure 8B, for the BP term, these RBPs were mainly enriched in mRNA metabolic process, mRNA processing, RNA processing and RNA splicing. As for the CC term, these proteins were significantly associated with nuclear, nucleoplasm and ribonucleoprotein (Figure 8C). For the MF term, RNA binding was mainly enriched (Figure 8D). KEGG pathway results showed that these RBPs were significantly associated with spliceosome, mRNA surveillance pathway, RNA transport and ribosome biogenesis in eukaryotes (Figure 8E). These results indicated the key roles of these RBPs in regulation of gene expression, which could be involved in m6A modifications in ameloblastoma.
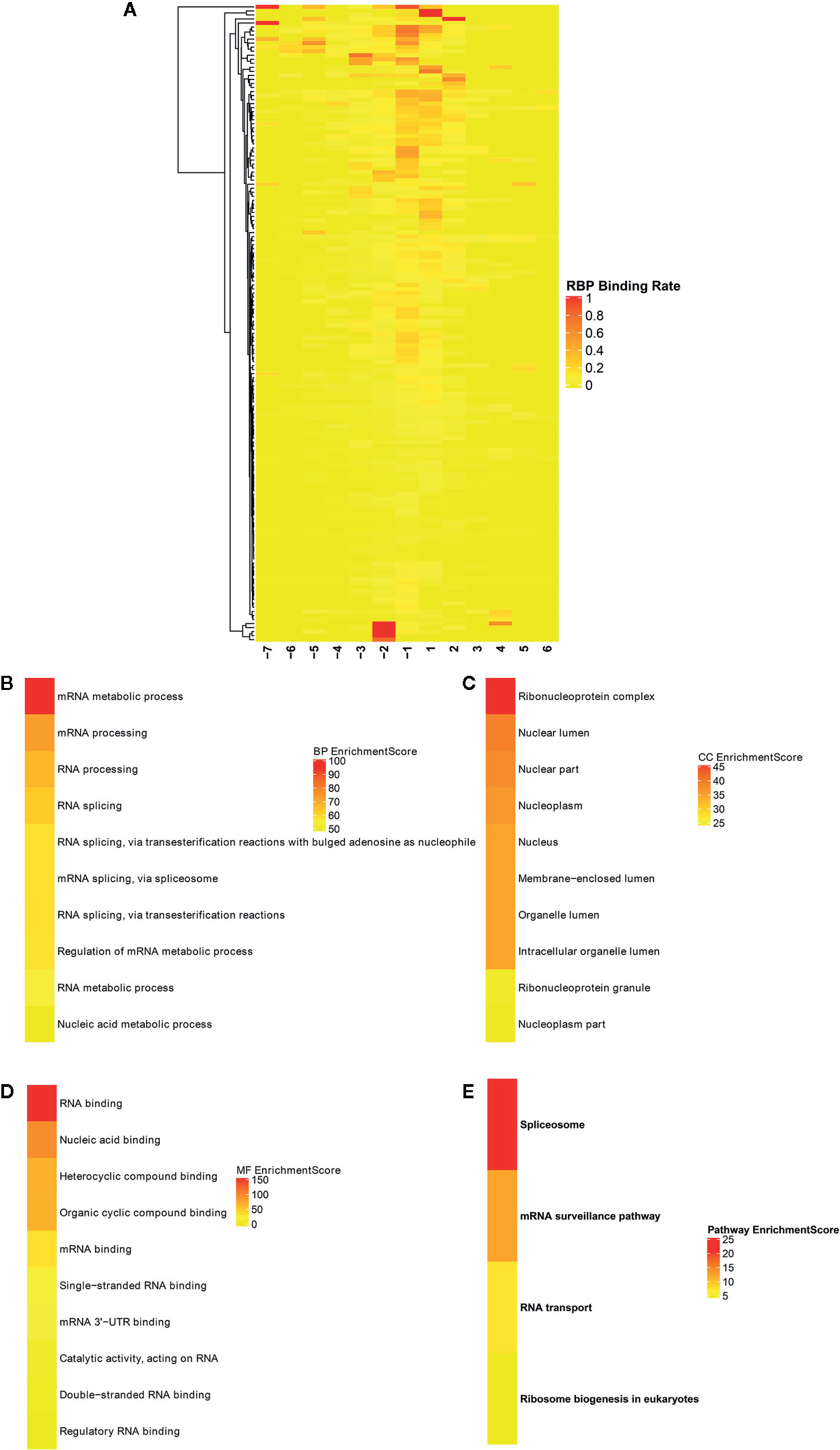
Figure 8 Identification of RNA binding proteins (RBPs) within differentially methylated m6A sites. (A) Heatmaps showing all 158 RBPs in ameloblastoma tissues and adjacent normal oral tissues. The top ten functional enrichment analysis results including (B) biological process (BP); (C) cellular component (CC); (D) membrane function (MF) and (E) KEGG based on these RBPs, respectively.
We comprehensively analyzed MeRIP-seq and RNA-seq data in ameloblastoma and normal tissues. The results showed that, the expression levels of 689 hypermethylated genes tended to be up-regulated in ameloblastoma compared to normal oral tissues (Figure 9A). Furthermore, 482 hypermethylated genes were up-regulated in ameloblastoma, while 337 hypomethylated genes tended to be up-regulated. One thousand two hundred thirty-one hypomethylated and down-regulated genes were found, indicating that the expression levels of downmethylated genes tended to be down-regulated in ameloblastoma compared to normal oral tissues. We further analyzed the expression levels of genes containing differentially methylated m6A sites in five regions between ameloblastoma and normal oral tissues. As depicted in Figure 9B, the expression levels of these mRNAs in five regions were all higher in ameloblastoma tissues than normal oral tissues. Moreover, the CDS region had the highest fractions of mRNAs among five different regions (Figure 9C). In Figures 10A–C, we displayed the general locations of upmethylated m6A sites in ameloblastoma- or other oral diseases-related mRNA [HOXC13 (27)], lncRNA [HOXC13-AS (27)] and circRNA [hsa_circ_0086414 (28)] in ameloblastoma compared to adjacent normal oral tissues.
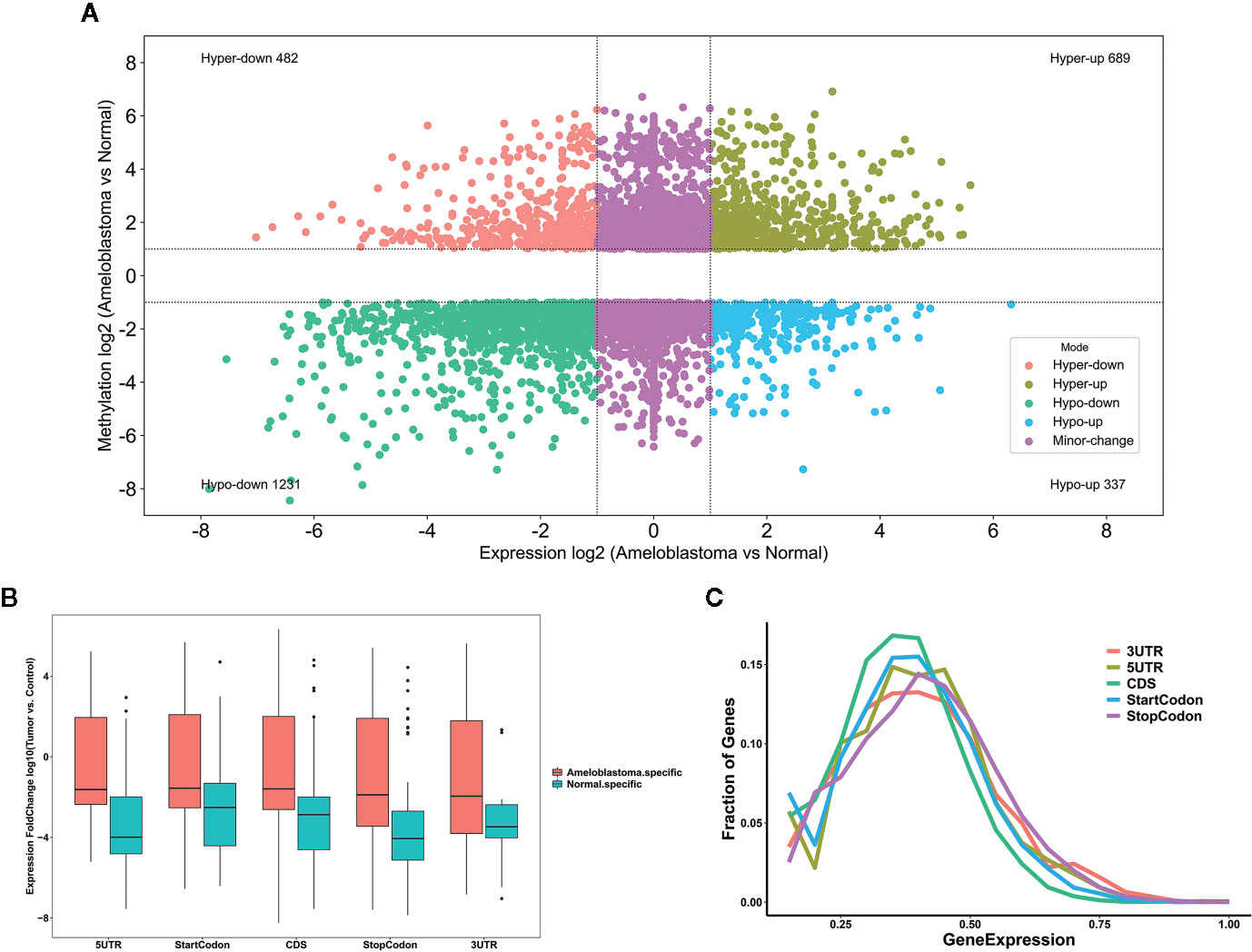
Figure 9 Comprehensive analysis of MeRIP-seq and RNA-seq data in ameloblastoma and normal tissues. (A) Scatter plot showing the distribution of mRNAs with a significant change both in m6A and mRNA levels between ameloblastoma tissues and normal oral tissues. (B) Box plot depicting the expression levels of genes in different five regions between ameloblastoma tissues and normal oral tissues. (C) Relative expression levels of genes in different five regions.
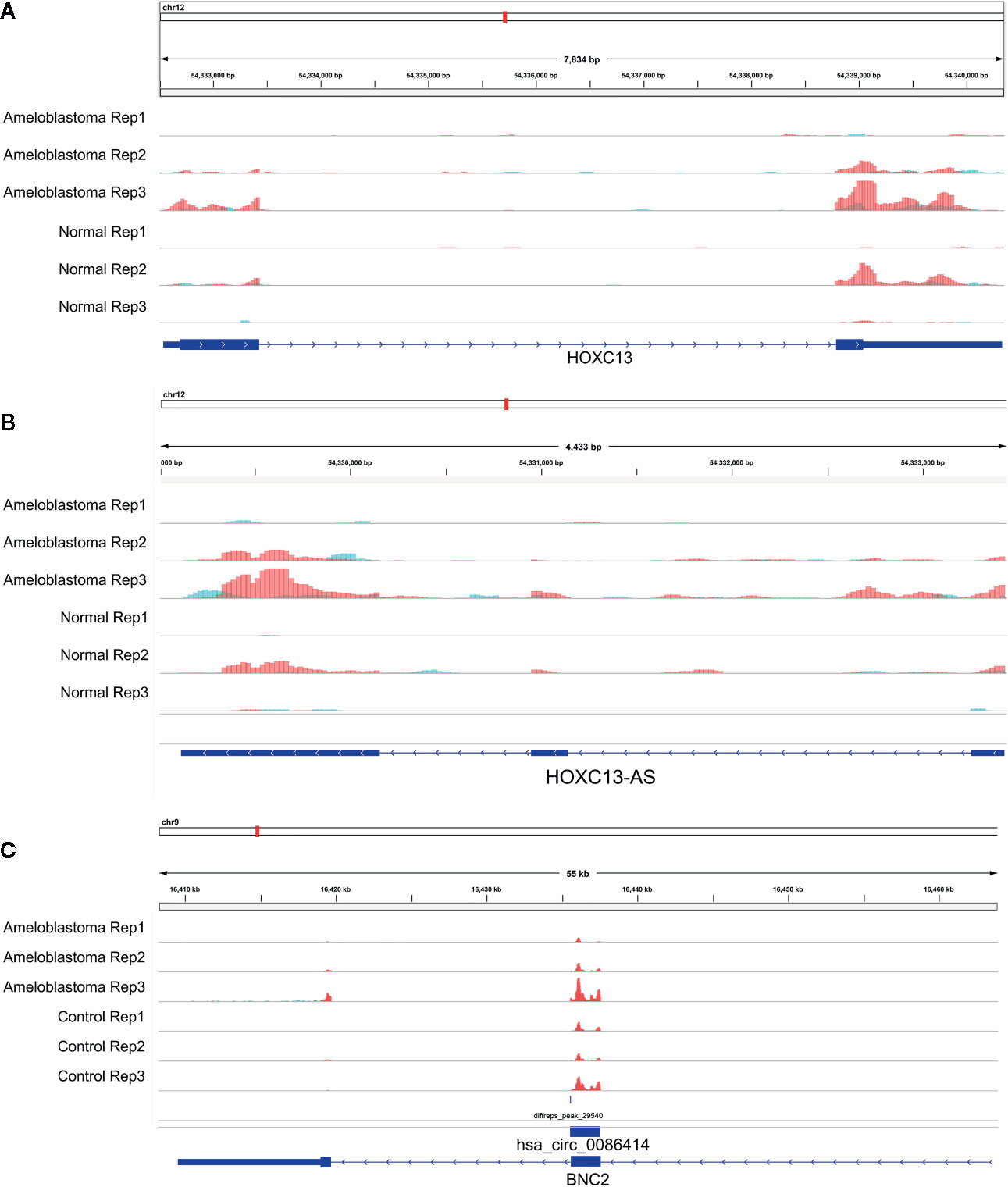
Figure 10 Representative differentially methylated mRNA, lncRNA and circRNA in ameloblastoma and adjacent normal oral tissues. (A) HOXC13; (B) HOXC13-AS; (C) hsa_circ_0086414.
m6A is the most abundant mRNA modification in mammals. It has been well recognized that m6A is involved in a variety of aspects of mRNA metabolism such as mRNA translation and mRNA decay (29). Growing evidence emphasizes its important RNA biology as a hallmark of cancer (30–33). However, RNA modification in ameloblastoma is rarely reviewed. In this study, we comprehensively analyzed the differences in m6A modification between human ameloblastoma and normal oral tissues. We identified differentially methylated mRNAs and many important biological pathways. Furthermore, lncRNAs and circRNAs harboring m6A peaks were identified and these lncRNA- and circRNA-associated genes were involved in many key biological processes.
Our MeRIP-seq analysis results revealed that there were highly overlapping and non-overlapping m6A peaks within mRNAs, lncRNAs and circRNAs between ameloblastoma tissues and adjacent normal oral tissues. Consistent with previous studies, most of m6A-motified mRNAs, lncRNAs and circRNAs contained only one m6A peak (23). The m6A modification mainly occurs in the RRACH sequence (where R = A or G, H = A, C, or U). Herein, we found that the m6A peaks within mRNAs, lncRNAs and circRNAs were respectively enriched in the AAACU, GAACU and AAACC sequences for ameloblastoma tissues compared to normal tissues. Consistent with other tumors, m6A was most often enriched in the CDS and stop codon regions for ameloblastoma (34).
We identified differentially m6A modified mRNAs between ameloblastoma and normal oral tissues. To uncover the functions of m6A in ameloblastoma, we performed functional enrichment analysis of differentially methylated mRNAs. Our results revealed that mRNAs with abnormal m6A modification were significantly enriched in organism developmental processes, indicating that m6A could be involved in the development of ameloblastoma. It has been confirmed that lncRNAs play critical roles in mediating the regulation of transcription and post-transcription (35). LncRNAs are involved in chromatin organization, transcriptional, and posttranscriptional regulation (36, 37). It has been reported that lncRNA X-inactive-specific transcript (XIST) may induce the transcriptional silencing of genes on the X chromosome. As an example, RBM15 and RBM15B recruited METTL3 to methylate XIST. Furthermore, silencing RBM15, RBM15B, or METTL3 may impair XIST-mediated transcriptional repression both in vitro and in vivo (38). It has been well recognized that lncRNAs with m6A modification are common in human cancers (39). Up to date, only a few lncRNAs have been functionally characterized. As an example, a previous study has found that demethylated lncRNA inhibits pancreatic cancer cell motility (40). Herein, we observed the differences in m6A modification between human ameloblastoma and normal oral tissues, revealing a potential role for m6A-modified lncRNAs in the development of ameloblastoma. However, further experiments should be required to confirm these results. Although m6A is recognized as an abundant co-transcriptional modification in mRNAs and ncRNAs (41, 42) including circRNAs (43), it is involved in many aspects of post-transcriptional mRNA metabolism (44–46). CircRNAs exhibit patterns of m6A modifications that are distinct from those of mRNAs. However, little is known about the influences of m6A modification on circRNA biology in cells. Typically, circRNA is thought to be a co-transcript produced by canonical linear mRNA splicing that occurs in the nucleus. Herein, we identified differentially m6A-modified circRNAs in human ameloblastoma tissues than oral normal tissues. Recent findings have demonstrated that the export of circNSUN2 from the nucleus to the cytoplasm is dependent on m6A modification (20). Thus, m6A-modified circRNA plays a functional role in the progression of tumors, which may serve as a potential molecular marker.
RBPs act as m6A readers or functional factors in m6A modification (47). Our data identified 158 RBPs within differentially methylated m6A sites between ameloblastoma and normal oral tissues. As shown in functional enrichment analysis, these RBPs were mainly enriched in various processes of RNA metabolism. M6A modification changes the expression of target genes, thereby affecting cellular processes and physiological functions. In this study, we comprehensively analyzed MeRIP-seq and RNA-seq data in ameloblastoma and normal tissues. Our data showed that 689 hypermethylated and 482 hypermethylated genes was highly expressed in ameloblastoma compared to normal oral tissues. Furthermore, among hypomethylated genes, 337 was up-regulated and 1,231 was down-regulated in ameloblastoma tissues in comparison to normal oral tissues, indicating that m6A could be involved in regulation of gene expression. For example, our previous study has confirmed that HOXC13 and HOXC13-AS are both highly expressed in ameloblastoma tissues (27). Their hypermethylation was found in ameloblastoma than normal oral tissues. hsa_circ_0086414 has been detected in oral tissues (28). In this study, we found that hsa_circ_0086414 was differentially m6A-modified in ameloblastoma tissues in comparison to normal oral tissues. Our data indicates that m6A could participate in tumor progression through the modification of tumor-related genes.
m6A modification is involved in almost every step in mRNA metabolism. Furthermore, it also affects the processing of lncRNAs and circRNAs. Our findings provide the first m6A modification landscape in ameloblastoma. Differentially expressed mRNAs with hyper-methylated or hypo-methylated m6A modifications are identified, which may help observe the mechanisms of m6A-mediated gene expression regulation. In further studies, we will evaluate the biological relevance and clinical value of m6A in human ameloblastoma.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The study was reviewed and approved by the Ethics Committee of School and Hospital of Stomatology, China Medical University (2019012). The patients/participants provided their written informed consent to participate in this study.
MZ conceived and designed the study. XN and JX conducted most of the experiments and data analysis, and wrote the manuscript. JL, LC, and XQ participated in collecting data and helped to draft the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the National Natural Science Foundation of China (81072197 and 81470758).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
m6A, N6-methyladenosine; mRNA, messenger RNA; lncRNA, long no-coding RNA; circRNA, circular RNA; snRNA, small nuclear RNA; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; RBPs, RNA binding proteins; FC, fold change.
1. Zhang J, Wang Y, Fan C, Xiao X, Zhang Q, Xu T, et al. Interleukin-8/beta-catenin mediates epithelial-mesenchymal transition in ameloblastoma. Oral Dis (2019) 25:1964–71. doi: 10.1111/odi.13173
2. Yoshimoto S, Tanaka F, Morita H, Hiraki A, Hashimoto S. Hypoxia-induced HIF-1alpha and ZEB1 are critical for the malignant transformation of ameloblastoma via TGF-beta-dependent EMT. Cancer Med (2019) 8:7822–32. doi: 10.1002/cam4.2667
3. Siar CH, Ng KH. Epithelial-to-mesenchymal transition in ameloblastoma: focus on morphologically evident mesenchymal phenotypic transition. Pathology (2019) 51:494–501. doi: 10.1002/cam4.2667
4. Bian Z, Zhang J, Li M, Feng Y, Wang X, Zhang J, et al. LncRNA-FEZF1-AS1 Promotes Tumor Proliferation and Metastasis in Colorectal Cancer by Regulating PKM2 Signaling. Clin Cancer Res (2018) 24:4808–19. doi: 10.1158/1078-0432.CCR-17-2967
5. Gao X, Wang G, Zhang YK, Zhong M. Expression and mechanism of regulation of PP2A/Pr65 in ameloblastoma. Surgeon (2014) 12:129–33. doi: 10.1016/j.surge.2013.07.003
6. Zhong M, Wang J, Gong YB, Li JC, Zhang B, Hou L. Expression of HOXC13 in ameloblastoma. Zhonghua Kou Qiang Yi Xue Za Zhi (2007) 42:43–6.
7. Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer (2019) 18:87. doi: 10.1186/s12943-019-1014-2
8. Tatomer DC, Wilusz JE. An Unchartered Journey for Ribosomes: Circumnavigating Circular RNAs to Produce Proteins. Mol Cell (2017) 66:1–2. doi: 10.1016/j.molcel.2017.03.011
9. Meyer KD, Jaffrey SR. Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
10. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell (2017) 169:1187–200. doi: 10.1016/j.cell.2017.05.045
11. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol (2018) 20:1074–83. doi: 10.1038/s41556-018-0174-4
12. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol (2019) 15:88–94. doi: 10.1038/s41589-018-0184-3
13. Atkinson SR, Marguerat S, Bahler J. Exploring long non-coding RNAs through sequencing. Semin Cell Dev Biol (2012) 23:200–5. doi: 10.1016/j.semcdb.2011.12.003
14. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet (2016) 17:47–62. doi: 10.1038/nrg.2015.10
15. Diniz MG, Franca JA, Vilas-Boas FAS, de Souza FTA, Calin GA, Gomez RS, et al. The long noncoding RNA KIAA0125 is upregulated in ameloblastomas. Pathol Res Pract (2019) 215:466–9. doi: 10.1016/j.prp.2018.12.030
16. Liu H, Xu Y, Yao B, Sui T, Lai L, Li Z. A novel N6-methyladenosine (m6A)-dependent fate decision for the lncRNA THOR. Cell Death Dis (2020) 11:613. doi: 10.1038/s41419-020-02833-y
17. Das A, Gorospe M, Panda AC. The coding potential of circRNAs. Aging (2018) 10:2228–9. doi: 10.18632/aging.101554
18. Panda AC, Grammatikakis I, Munk R, Gorospe M, Abdelmohsen K. Emerging roles and context of circular RNAs. Wiley interdisciplinary reviews RNA. Wiley Interdiscip Rev RNA (2017) 8. doi: 10.1002/wrna.1386
19. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res (2017) 27:626–41. doi: 10.1038/cr.2017.31
20. Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun (2019) 10:4695. doi: 10.1038/s41467-019-12651-2
21. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
22. Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant Review with the Integrative Genomics Viewer. Cancer Res (2017) 77:e31–e4. doi: 10.1158/0008-5472.CAN-17-0337
23. Chen Y, Zhou C, Sun Y, He X, Xue D. m(6)A RNA modification modulates gene expression and cancer-related pathways in clear cell renal cell carcinoma. Epigenomics (2020) 12:87–99. doi: 10.2217/epi-2019-0182
24. Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics (2011) 27:1653–9. doi: 10.1093/bioinformatics/btr261
25. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature (2012) 485:201–6. doi: 10.1038/nature11112
26. Xuan JJ, Sun WJ, Lin PH, Zhou KR, Liu S, Zheng LL, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res (2018) 46:D327–d34. doi: 10.1093/nar/gkx934
27. Sun Y, Niu X, Wang G, Qiao X, Chen L, Zhong M. A Novel lncRNA ENST00000512916 Facilitates Cell Proliferation, Migration and Cell Cycle Progression in Ameloblastoma. Onco Targets Ther (2020) 13:1519–31. doi: 10.2147/OTT.S236158
28. Li L, Zhang ZT. Hsa_circ_0086414 Might Be a Diagnostic Biomarker of Oral Squamous Cell Carcinoma. Med Sci Monit (2020) 26:e919383. doi: 10.12659/MSM.919383
29. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol (2017) 18:31–42. doi: 10.1038/nrm.2016.132
30. Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol (2014) 15:R14. doi: 10.1186/gb-2014-15-1-r14
31. Marcel V, Catez F, Diaz JJ. Ribosome heterogeneity in tumorigenesis: the rRNA point of view. Mol Cell Oncol (2015) 2:e983755. doi: 10.4161/23723556.2014.983755
32. Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer? Nat Rev Cancer (2012) 12:84–8. doi: 10.1038/nrc3195
33. Kirchner S, Ignatova Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet (2015) 16:98–112. doi: 10.1038/nrg3861
34. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer (2019) 18:176. doi: 10.1186/s12943-019-1109-9
35. Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis (2018) 9:124. doi: 10.1038/s41419-017-0129-x
36. Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol (2016) 18:431–42. doi: 10.1038/ncb3328
37. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell (2013) 152:1298–307. doi: 10.1016/j.cell.2013.02.012
38. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature (2016) 537:369–73. doi: 10.1038/nature19342
39. Jacob R, Zander S, Gutschner T. The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs. Int J Mol Sci (2017) 18:2387. doi: 10.3390/ijms18112387
40. He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, et al. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 48:838–46. doi: 10.1159/000491915
41. Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet (2014) 15:127–50. doi: 10.1146/annurev-genom-090413-025405
42. Gilbert WV, Bell TA, Schaening C. Messenger RNA modifications: Form, distribution, and function. Sci (New York NY) (2016) 352:1408–12. doi: 10.1126/science.aad8711
43. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep (2017) 20:2262–76. doi: 10.1016/j.celrep.2017.08.027
44. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell (2016) 61:507–19. doi: 10.1016/j.molcel.2016.03.004
45. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505:117–20. doi: 10.1038/nature12730
46. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
Keywords: m6A modification, ameloblastoma, messenger RNA, long noncoding RNA, circular RNA
Citation: Niu X, Xu J, Liu J, Chen L, Qiao X and Zhong M (2020) Landscape of N6-Methyladenosine Modification Patterns in Human Ameloblastoma. Front. Oncol. 10:556497. doi: 10.3389/fonc.2020.556497
Received: 28 April 2020; Accepted: 21 September 2020;
Published: 14 October 2020.
Edited by:
Wantao Chen, Shanghai Jiao Tong University, ChinaReviewed by:
Gaurisankar Sa, Bose Institute, IndiaCopyright © 2020 Niu, Xu, Liu, Chen, Qiao and Zhong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Zhong, emhvbmdtaW5nX29yYWxAYWxpeXVuLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.