
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 15 October 2020
Sec. Radiation Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.541281
This article is part of the Research Topic Modeling for Prediction of Radiation-Induced Toxicity to Improve Therapeutic Ratio in the Modern Radiation Therapy Era View all 35 articles
 Michela Carlotta Massi1,2
Michela Carlotta Massi1,2 Francesca Gasperoni3
Francesca Gasperoni3 Francesca Ieva1,2,4*Anna Maria Paganoni1,2,4
Francesca Ieva1,2,4*Anna Maria Paganoni1,2,4 Paolo Zunino1Andrea Manzoni1Nicola Rares Franco1Liv Veldeman5,6Piet Ost5,6Valérie Fonteyne5,6Christopher J. Talbot7Tim Rattay7Adam Webb7
Paolo Zunino1Andrea Manzoni1Nicola Rares Franco1Liv Veldeman5,6Piet Ost5,6Valérie Fonteyne5,6Christopher J. Talbot7Tim Rattay7Adam Webb7 Paul R. Symonds7Kerstie Johnson7
Paul R. Symonds7Kerstie Johnson7 Maarten Lambrecht8
Maarten Lambrecht8 Karin Haustermans8Gert De Meerleer8
Karin Haustermans8Gert De Meerleer8 Dirk de Ruysscher9,10
Dirk de Ruysscher9,10 Ben Vanneste10
Ben Vanneste10 Evert Van Limbergen9,10Ananya Choudhury11Rebecca M. Elliott11Elena Sperk12
Evert Van Limbergen9,10Ananya Choudhury11Rebecca M. Elliott11Elena Sperk12 Carsten Herskind12Marlon R. Veldwijk12
Carsten Herskind12Marlon R. Veldwijk12 Barbara Avuzzi13
Barbara Avuzzi13 Tommaso Giandini14
Tommaso Giandini14 Riccardo Valdagni13,15,16
Riccardo Valdagni13,15,16 Alessandro Cicchetti16
Alessandro Cicchetti16 David Azria17Marie-Pierre Farcy Jacquet18
David Azria17Marie-Pierre Farcy Jacquet18 Barry S. Rosenstein19,20Richard G. Stock19Kayla Collado19
Barry S. Rosenstein19,20Richard G. Stock19Kayla Collado19 Ana Vega21,22,23
Ana Vega21,22,23 Miguel Elías Aguado-Barrera21,22Patricia Calvo22,24Alison M. Dunning25Laura Fachal25,26
Miguel Elías Aguado-Barrera21,22Patricia Calvo22,24Alison M. Dunning25Laura Fachal25,26 Sarah L. Kerns27Debbie Payne28Jenny Chang-Claude29,30Petra Seibold29
Sarah L. Kerns27Debbie Payne28Jenny Chang-Claude29,30Petra Seibold29 Catharine M. L. West11†
Catharine M. L. West11† Tiziana Rancati16† and on behalf of the REQUITE Consortium
Tiziana Rancati16† and on behalf of the REQUITE ConsortiumBackground: REQUITE (validating pREdictive models and biomarkers of radiotherapy toxicity to reduce side effects and improve QUalITy of lifE in cancer survivors) is an international prospective cohort study. The purpose of this project was to analyse a cohort of patients recruited into REQUITE using a deep learning algorithm to identify patient-specific features associated with the development of toxicity, and test the approach by attempting to validate previously published genetic risk factors.
Methods: The study involved REQUITE prostate cancer patients treated with external beam radiotherapy who had complete 2-year follow-up. We used five separate late toxicity endpoints: ≥grade 1 late rectal bleeding, ≥grade 2 urinary frequency, ≥grade 1 haematuria, ≥ grade 2 nocturia, ≥ grade 1 decreased urinary stream. Forty-three single nucleotide polymorphisms (SNPs) already reported in the literature to be associated with the toxicity endpoints were included in the analysis. No SNP had been studied before in the REQUITE cohort. Deep Sparse AutoEncoders (DSAE) were trained to recognize features (SNPs) identifying patients with no toxicity and tested on a different independent mixed population including patients without and with toxicity.
Results: One thousand, four hundred and one patients were included, and toxicity rates were: rectal bleeding 11.7%, urinary frequency 4%, haematuria 5.5%, nocturia 7.8%, decreased urinary stream 17.1%. Twenty-four of the 43 SNPs that were associated with the toxicity endpoints were validated as identifying patients with toxicity. Twenty of the 24 SNPs were associated with the same toxicity endpoint as reported in the literature: 9 SNPs for urinary symptoms and 11 SNPs for overall toxicity. The other 4 SNPs were associated with a different endpoint.
Conclusion: Deep learning algorithms can validate SNPs associated with toxicity after radiotherapy for prostate cancer. The method should be studied further to identify polygenic SNP risk signatures for radiotherapy toxicity. The signatures could then be included in integrated normal tissue complication probability models and tested for their ability to personalize radiotherapy treatment planning.
Radiotherapy represents the most effective non-surgical modality for the potentially curative treatment of prostate cancer. Around a half of survivors underwent radiotherapy as part of their curative care (1), either as single curative treatment or as adjuvant/salvage treatment after radical prostatectomy.
Despite the fact that prognosis is very good in terms of patients' survival rates, it is widely acknowledged that long-term side-effects after radiotherapy can affect a patient's quality-of-life (2–4). A tool able to identify patients likely to develop toxicity could be a crucial step toward personalized radiotherapy with modification of the dose, fractionation, techniques and supportive care. The ultimate goal is to reduce morbidity and improve quality-of-life.
Radiation toxicity is a multifactorial problem, related not only to the cumulative delivered dose, but also to an intrinsic process within tissues responding to cellular injury. Individual genetic background and biological expression pattern, premorbid conditions, concomitant oncological therapies, as well as the cellular microenvironment, could be important factors in the development of side-effects, although their exact contributions are unknown.
With increased interest in this field and relevant data collection on this topic, predictive models have been developed to identify patients likely to develop side effects during radiotherapy (3).
The identification of genetic factors associated with susceptibility to radiation toxicity represents an emerging research area in oncology. A number of different approaches have been explored (5–13), however, the developed models and biomarkers have failed to progress to routine clinical use due to the lack of thorough independent validation.
REQUITE (validating pREdictive models and biomarkers of radiotherapy toxicity to reduce side effects and improve QUalITy of lifE in cancer survivors) was established with the aim of validating models and biomarkers for the prediction of adverse effects following radiotherapy (14–16). In order to address previous limitations in pooling data, in using common toxicity scoring systems and in collecting standardized data, REQUITE carried out an international, multi-center, prospective observational study. A centralized biobank was also established to store blood samples and genome-wide genotyping of single nucleotide polymorphisms (SNPs) was carried out.
The specific purpose of the present study was to attempt to validate genetic risk factors for late toxicity (rectal bleeding and late urinary symptoms) after prostate cancer radiotherapy in the REQUITE population using a deep learning algorithm. This technique aims to identify patient-specific features that define patients with toxicity (“unhealthy”) as outliers with respect to the population of irradiated patients without toxicity (“healthy”).
Deep learning has the potential to overcome the difficulties in replication of results faced by the widespread single-SNP association methods used by genome wide association studies (GWAS). The statistical power of GWAS is limited by a combination of the large number of hypotheses being tested simultaneously and the inherently small effect size of the single SNP (17).
Deep learning approaches, with their intrinsic hierarchical structure (where each layer performs a combination of the outcomes of the previous layers), seem particularly adapt at mimicking complex dependencies within data. The method addresses effectively the following issues: (i) unstable selections of correlated variables and inconsistent selections of linearly dependent genetic variables (18); (ii) strong imbalance between positive and negative outcomes which is usually encountered in studies of radiation toxicity.
REQUITE prostate cancer patients treated with external beam radiotherapy (with/without hormonal therapy, with/without a previous prostatectomy, no brachytherapy) and complete 2-year follow-up were included. Details on the REQUITE population are given in Seibold et al. (14).
Prostate cancer patients were recruited prior to radiotherapy between April 2014 and October 2016. Recruitment was at ten main sites in eight countries (Belgium, France, Germany, Italy, the Netherlands, Spain, UK, US). Conventionally fractionated or hypo-fractionated radiotherapy was prescribed according to local standard-of-care regimens. The patients were followed prospectively for at least 24 months, with longer follow-up encouraged where possible. All patients gave written informed consent. The study was approved by local Ethical Committees and is registered at www.controlled-trials.com (ID ISRCTN98496463).
Demographic, co-morbidity, treatment, physics, longitudinal toxicity (CTCAE v4.0 healthcare professional and patient reported), quality-of-life, and treatment outcome data were collected prospectively using standardized case report forms. CTCAE v4.0 based questionnaires developed to collect patient reported outcomes were adapted from those published elsewhere for the male pelvis (19) and updated to fit with CTCAE v4.0 items.
All patients donated at least two blood samples prior to the start of radiotherapy: an EDTA sample for SNP genotyping plus a PAXgene sample. Genotyping data were generated using the Illumina Infinium OncoArray-500K beadchip. Following standard quality control procedures (20), genotype data were imputed using the 1,000 Genomes Project (version 3) as a reference panel.
We undertook a comprehensive search of Medline and PubMed databases using the keywords “prostate,” “prostatic,” “radiotherapy,” “radiation,” “irradiation,” “toxicity,” “adverse effects,” “side-effects,” “morbidity,” “injury,” “genetic variation,” “SNP,” “GWAS,” and “polymorphism.” This search identified 60 SNPs published (up to May 31st, 2019) in GWAS patient studies with p < 1.0·10−5 and where findings were adjusted for multiple comparisons OR in studies including a controlled number of SNPs (~102) and using multivariable regularization methods coupled to internal validation to control overfitting.
Forty-three of 60 SNPs were available for the REQUITE population (either directly determined or after imputation) and were included in the analysis. These SNPs were identified in five papers (5, 11, 21–23) and the full list is reported in Table 1.
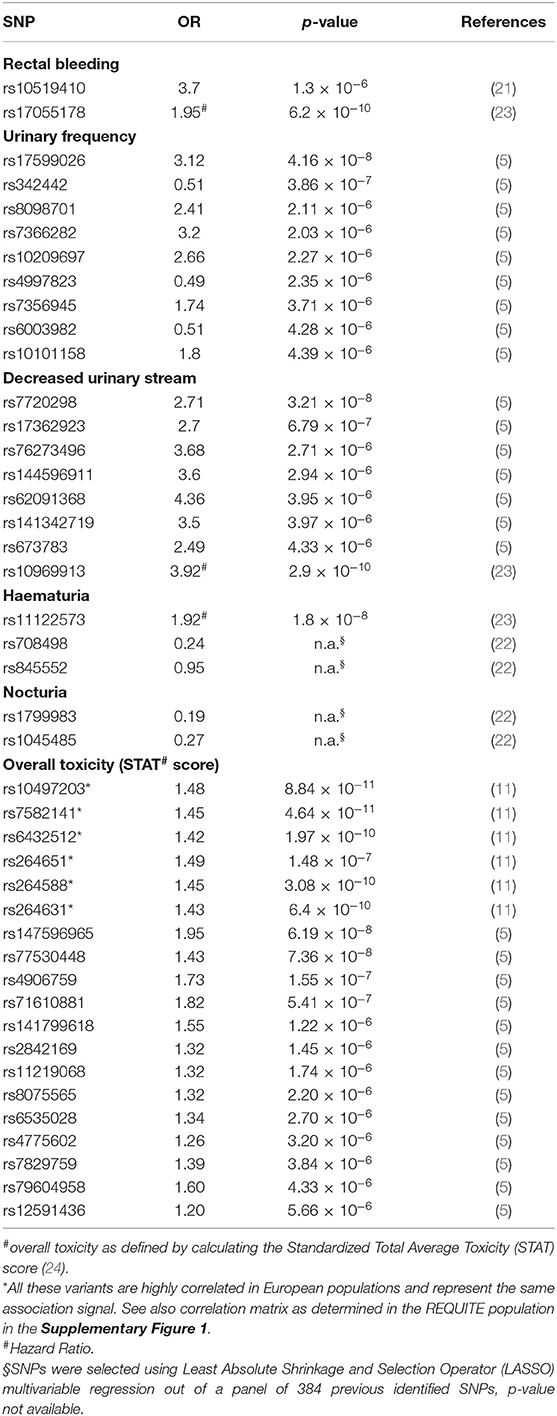
Table 1. Full list of SNPs selected from the literature for validation and associated toxicity endpoint following prostate radiotherapy.
Toxicity endpoints were defined using CTCAE v4.0 scoring reported by health professionals or Patient Reported Outcomes, as detailed for each single endpoint. As the frame of the DSAE is to identify SNPs who would tag a patient as exceptionally “sensitive” to radiation (an “outlier”), patients with other possible known intrinsic higher risk of exhibiting radiation toxicity were always excluded, in particularly patients who had systemic lupus erythematosus, rheumatoid arthritis and other collagen vascular diseases.
The following endpoints were considered:
1. Late rectal bleeding grade≥1 (CTCAE v4.0 scoring): patients exhibiting at least mild bleeding (even requiring no intervention) at 12 or at 24 months. Patients with grade≥1 at baseline and grade ≤ 1 during follow-up were considered as not bleeders; patients with hemorrhoids before radiotherapy treatment were excluded.
2. Late urinary frequency grade≥2 (CTCAE v4.0 scoring): patients with urinary frequency limiting instrumental activities of daily living or if urinary frequency requiting medical management at 12 or at 24 months. Patients with urinary frequency grade≥2 at baseline and grade ≤ 2 during follow-up were considered as not exhibiting this endpoint.
3. Late haematuria grade ≥1 (CTCAE scoring): patients with asymptomatic haematuria (clinical or diagnostic observations only, no intervention indicated) at 12 or 24 months. Patients with haematuria grade≥1 at baseline and grade ≤ 1 during follow-up were considered as not exhibiting the endpoint.
4. Late nocturia grade ≥2 (Patient Reported Outcome): patients declaring need to urinate at least two-three times per night at 12 or 24 months. Patients with nocturia grade≥2 at baseline and grade ≤ 2 during follow-up were considered as not exhibiting the endpoint.
5. Late grade≥1 (Patient Reported Outcome): patients scored with hesitant or dripping stream at 12 or 24 months. Patients with decreased urinary stream grade≥1 at baseline and grade ≤ 1 during follow-up were considered as not exhibiting the endpoint.
Patients who underwent transurethral resection of the bladder and patients on anti-muscarinic drugs (factors which could constitute a confounding factor in the scoring of urinary toxicity) were excluded when considering all urinary endpoints.
The methodology described in Massi et al. (25) was considered. This method proposes a novel feature selection algorithm for the minority class in an imbalanced dataset, i.e., in cases like this dataset, where there is a strong imbalance between the number of patients that are scored as healthy (without side effects) vs. unhealthy (with side effects). The approach uses a representation learning technique, specifically a Deep Sparse AutoEncoder, to obtain the best representation of the majority class (healthy patients in this dataset) and to consequently identify which features (SNPs) distinguish the minority class (unhealthy patients) with respect to the majority class.
An AutoEncoder (AE) is a neural network with an output that reconstructs the input (26). In its simplest version an AE is composed of the input, the output and only a single hidden layer. The input layer in our case is composed of J nodes, one per feature (one per SNP), and we consider a data matrix X, in which each row xi is the vector of SNPs recorded for the patient i, i ∈ {1 ,., N}. The input layer is connected to the hidden layer, hi, through the encoder function, f, such that hi = f (Wxi +b); here W ∈ R H × J denotes the weight matrix and b ∈ RH × 1 the bias vector. Then, the output is the result of the application of a decoder function, g, to the hidden layer hi, such that i = g (W'hi +b'), where W' ∈ R J × H is the weight matrix and b ∈ R J × 1 is the bias vector. Having fixed the functions f and g, the training of the network consists in estimating the corresponding optimal parameters (W, b, W', b'), by minimizing the loss function L(xi, i), which is a function that gives a measure of the similarity between the input and the reconstructed output. In this work, we considered the Euclidean distance as loss function L.
A more sophisticated version of AE (named Deep AE) has multiple hidden layers in which the output of a layer is the input of the next one. Figure 1 depicts a simplified scheme of a Deep AutoEncoder.
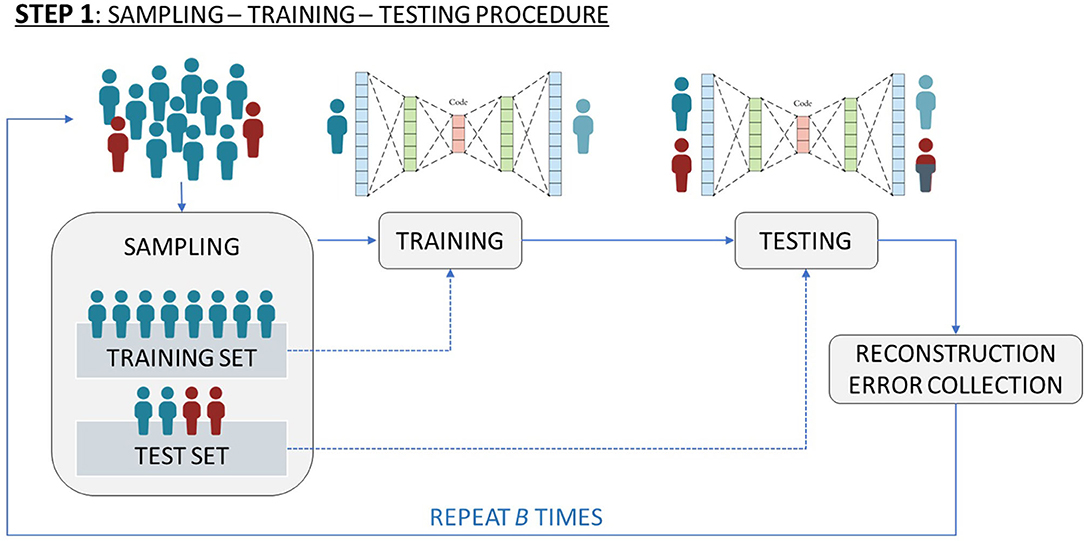
Figure 1. Simplified scheme of a Deep AutoEncoder.
In order to get an effective reconstruction of the input, that allows selection of features that best characterize the input data, we included a penalization term in the loss function. AE algorithms of this type are known as Deep Sparse AEs. Given this framework and with the final goal of validating the SNPs effect on the long-term radiation toxicity, we applied the previously described Deep Sparse AE as follows:
(i) sampling: we sampled S healthy patients (those without toxicity) where S equals the total number of unhealthy patients (those with toxicity). All the unhealthy patients and the S sampled healthy patients form the test set. All the remaining healthy patients constitute the training set.
(ii) training: we trained the network only on the previously specified training set. The idea here was to learn how to best represent healthy patients. The result of this step is the estimate of the neural network characteristics (weight and bias vectors, encoder and decoder functions).
(iii) testing: we tested the estimated network on the previously specified test set. The result of this step is a matrix of Reconstruction Errors, R ∈ R(2S) × J. Considering the previous step and the fact that unhealthy patients are the minority class, the rows of R which are related to unhealthy patients should contain higher values with respect to those rows of R associated to healthy patients.
(iv) SNP identification: we identified which SNPs are associated with the highest Reconstruction Error. Further details on this step are given at the end of this section.
The steps (i)-(iii) are repeated 50 times in order to reduce a possible selection bias induced by the sampling step (i), thus obtaining 50 R matrices.
In order to identify which features should be selected for characterizing the minority class with respect to the majority class, in step (iv) the average Reconstruction Error per feature per class is computed according to that proposed in Massi et al. (25), which means computing two vectors (one for the unhealthy patients and one for the healthy patients), both made by J elements. Then, we investigated the distribution of the difference, Δ, between the average Reconstruction Errors related to unhealthy patients and the average Reconstruction Errors related to healthy patients. See Figure 2 for a schematic representation of the above described workflow.
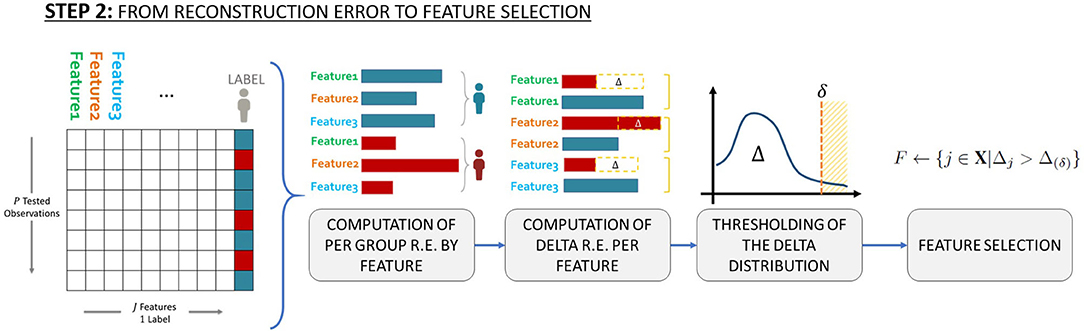
Figure 2. Schematic representation of the workflow used to identify which features to select to characterize the minority class (i.e., patients with toxicity) with respect to the majority class (patients without toxicity).
Finally, to define which SNPs are associated with late toxicity endpoints, we set possible thresholds equal to the 70-th, 80-th, the 90-th and the 95-th percentiles of the distribution of the Reconstruction Error differences, Δ. This means that we investigated the SNPs associated with the top 30%, the top 20%, the top 10% and the top 5% differences. These thresholds identity the effect size of identified SNPs, a large effect size (Odds Ratio>2) for SNPs in the 90-th/95-th percentiles, a moderate (Odds Ratio~2) and small (Odds Ratio <2) effect size for SNPs in the 80-th and 70-th percentiles, respectively.
For the interested reader, in this section we provide some more specific details regarding the development and specific implementation of the DSAE for the applications described in this paper. For more details on the methodology, its strenghts and all model's hyperparameters mentioned below, refer to the description in Massi et al. (25).
The experiments were implemented and carried out using Python Keras framework for Deep Learning with Tensorflow as backend.
For better comparability of results in the experiments we structured the DSAEs included in the sampling-training-testing procedure with the same architecture and hyperparameters for all five endpoints. In particular, all the encoders of the DSAEs were composed of an input layer with J = 43 nodes (one per SNP), followed by a sequence of hidden layers of 40, 30 (with hyperbolic tangent activation function) and 20 nodes, respectively. To the 20 nodes of the innermost hidden layer we applied a sigmoidal activation function to foster the sparsity induced by the penalization term (weighted with λ=10e-5). The decoder architecture of all DSAEs was specular to the encoder, with a sequence of layers with 30 and 40 nodes, followed by an output layer of J = 43 nodes. The training of the DSAE for each of the B = 50 iterations was performed for 400 epochs, exploiting the Adam optimization algorithm with its default parameters (learning rate equal to 0.001).
REQUITE enrolled 1,681 prostate cancer patients who were treated with external beam radiotherapy without brachytherapy. One thousand four hundred and fifty patients with complete 2-year follow-up were available for analysis. Forty-nine patients were excluded because of an intrinsic higher risk of exhibiting radiation toxicity, due to their co-morbidities (patients with a diagnosis of systemic lupus erythematosus, rheumatoid arthritis and other collagen vascular diseases). Details on the clinical characteristics of the cohorts selected for each toxicity endpoint are given in Supplementary Tables 1,2.
One hundred and sixty of 1,366 available patients (11.7%) had late rectal bleeding grade≥1. Figure 3 shows the differences between averaged Reconstruction Errors between the two classes (i.e., differences between red and blue columns). The largest part of the differences is close to zero (red line in the bottom panel of Figure 3). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) select SNPs associated with the toxicity outcome with different effect size. Table 2 lists results for the SNPs previously reported to be associated with late rectal bleeding and overall toxicity in comparison with SNPs selected by the DSAE in the REQUITE cohort. For late rectal bleeding eight SNPs were identified, two SNPs previously associated with overall toxicity (red stars in Figure 3) and six SNPs previously found to be associated with urinary toxicity.
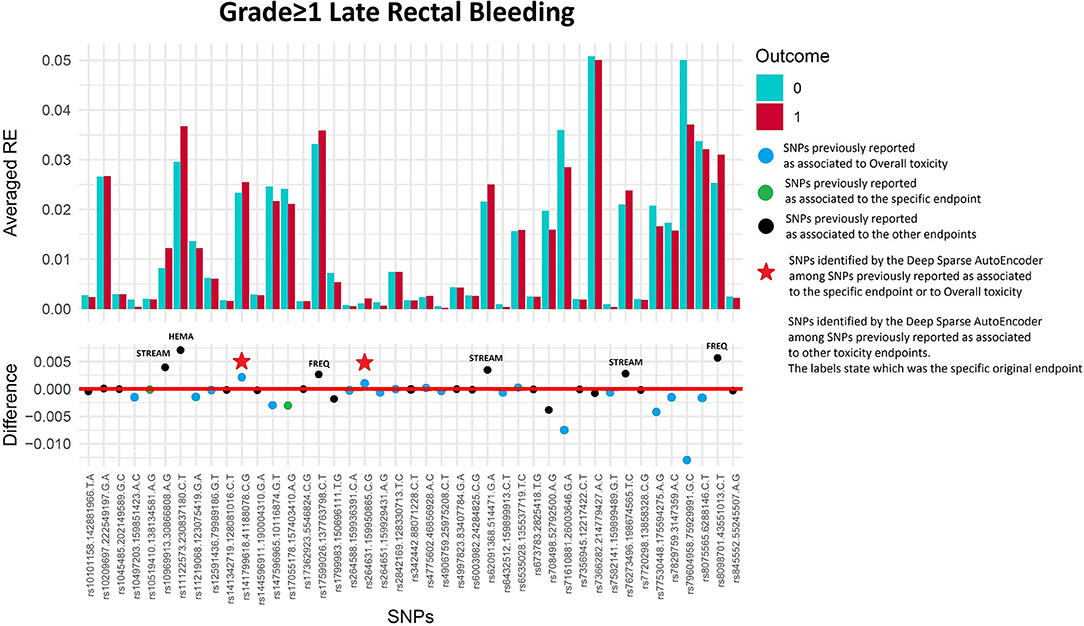
Figure 3. Results for late rectal bleeding grade≥1 from the Deep Sparse AutoEncoder. The 43 considered SNPs are reported in the x-axis and the averaged Reconstruction Errors (RE) are reported in the y-axis (top panel), red columns refer to patients with toxicity, while blue columns refer to patients without toxicity. In the lower panel the difference between averaged Reconstruction Errors between the two classes are represented for each SNP (i.e., differences between red and blue columns). For most SNPs, the difference is close to zero (red line in the bottom panel of the figure). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) are selecting SNPs associated to the toxicity outcome. Green circles refer to SNPs that were previously identified as associated with late rectal bleeding, while blue circles refer to SNPs that were previously associated with overall toxicity as defined by calculation of the Standardized Total Average Toxicity (STAT) score (24). Red stars indicate SNPs (either specific for this endpoint or related to overall toxicity) defining patients with toxicity as outliers with respect to the characteristics of patients without toxicity. Labels show SNPs that not directly associated with late rectal bleeding/overall toxicity, but contributing to their identification. The label states for which toxicity endpoint the SNPs were originally associated with in the literature: FREQ=urinary frequency, HEMA=haematuria, NOCT=nocturia, STREAM=decreased urinary stream.
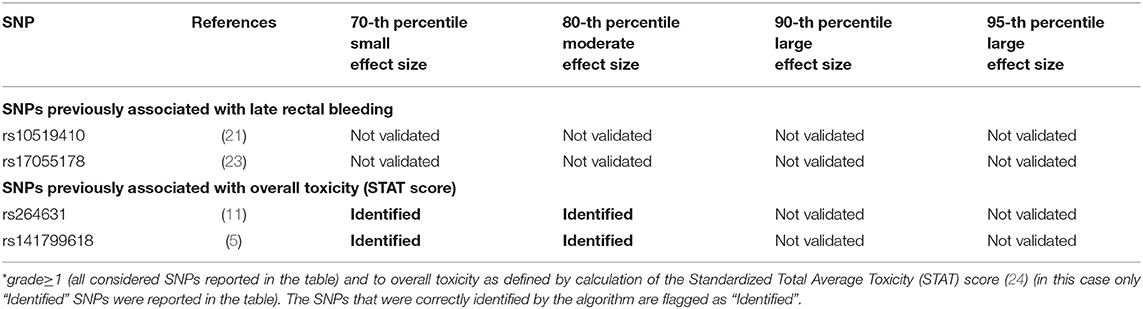
Table 2. Deep Sparse AutoEncoder testing of SNPs associated with Late Rectal Bleeding*.
Fifty-six of 1,334 available patients (4.2%) experienced late urinary frequency grade≥2. Patients were excluded from the analysis if they had urinary frequency grade≥2 at baseline (n = 26), they underwent transurethral resection of the bladder (n = 31) or were using anti-muscarinic drugs (n = 10). Figure 4 and Table 3 show that the DSAE analysis identified 14 SNPs: four already reported as associated with urinary frequency (rs17599026, rs8098701, rs7366282, rs10209697), four associated with overall toxicity, one previously associated with bleeding and five with other urinary symptoms.
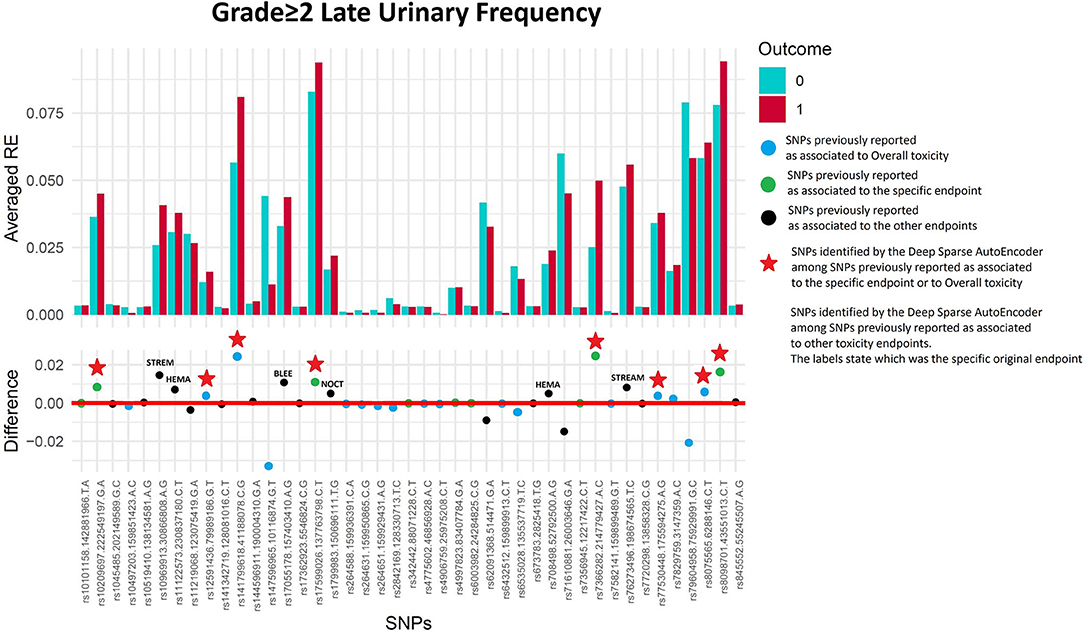
Figure 4. Results for late urinary frequency grade≥2 from the Deep Sparse AutoEncoder. The 43 considered SNPs are reported in the x-axis and the averaged Reconstruction Errors (RE) are reported in the y-axis (top panel), red columns refer to patients with toxicity, while blue columns refer to patients without toxicity. In the lower panel the difference between averaged Reconstruction Errors between the two classes are represented for each SNP (i.e., differences between red and blue columns). For most SNPs, the difference is close to zero (red line in the bottom panel of the figure). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) are selecting SNPs associated to the toxicity outcome. Green circles refer to SNPs that were previously identified as associated with late urinary frequency, while blue circles refer to SNPs that were previously associated with overall toxicity as defined by calculation of the Standardized Total Average Toxicity (STAT) score (24). Red stars indicate SNPs (either specific for this endpoint or related to overall toxicity) defining patients with toxicity as outliers with respect to the characteristics of patients without toxicity. Labels show SNPs that not directly associated with late urinary frequency/overall toxicity, but contributing to their identification. The label states for which toxicity endpoint the SNPs were originally associated with in the literature: BLEE=rectal bleeding, HEMA=haematuria, NOCT=nocturia, STREAM=decreased urinary stream.
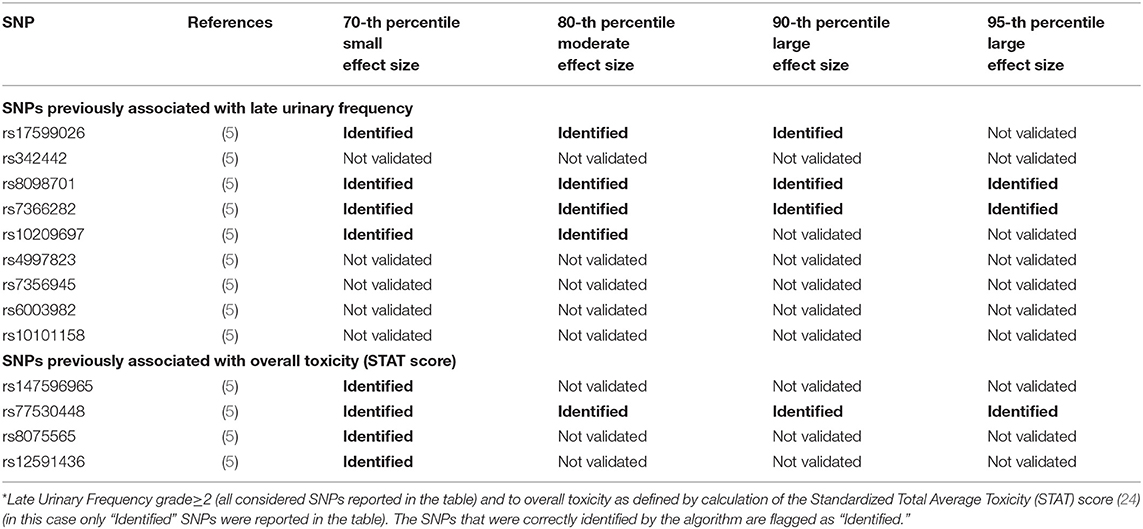
Table 3. Results from Deep Sparse AutoEncoder testing of SNPs associated with Urinary Frequency*.
Seventy-four of 1,343 available patients (5.5%) experienced late haematuria grade≥1. Seventeen patients were excluded from the analysis because they had haematuria at baseline grade≥1, while 41 were excluded because underwent transurethral resection of the bladder or were using anti-muscarinic drugs. Figure 5 and Table 4 report DSAE results for this endpoint: 10 SNPs were identified. Two SNPs already associated with haematuria (rs708498 and rs845552), five SNPs associated with overall toxicity, and three SNPs with other urinary symptoms.
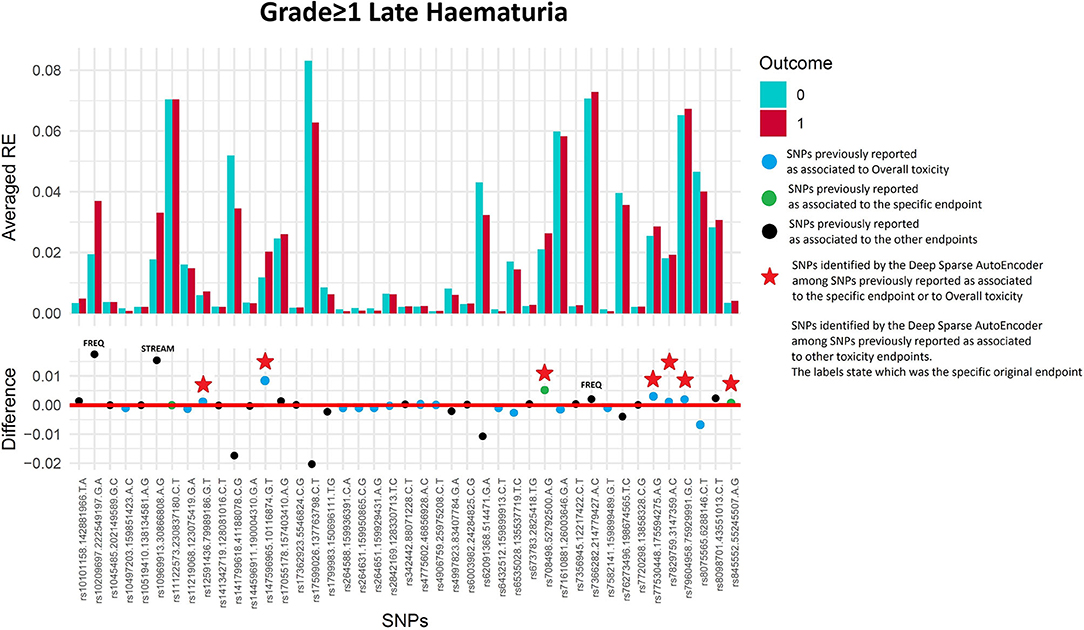
Figure 5. Results for late haematuria grade≥1 from the Deep Sparse AutoEncoder. The 43 considered SNPs are reported in the x-axis and the averaged Reconstruction Errors (RE) are reported in the y-axis (top panel), red columns refer to patients with toxicity, while blue columns refer to patients without toxicity. In the lower panel the difference between averaged Reconstruction Errors between the two classes are represented for each SNP (i.e., differences between red and blue columns). For most SNPs, the difference is close to zero (red line in the bottom panel of the figure). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) are selecting SNPs associated to the toxicity outcome. Green circles refer to SNPs that were previously identified as associated with late haematuria, while blue circles refer to SNPs that were previously associated with overall toxicity as defined by calculation of the Standardized Total Average Toxicity (STAT) score (24). Red stars indicate SNPs (either specific for this endpoint or related to overall toxicity) defining patients with toxicity as outliers with respect to the characteristics of patients without toxicity. Labels show SNPs that not directly associated with late haematuria/overall toxicity, but contributing to their identification. The label states for which toxicity endpoint the SNPs were originally associated with in the literature: BLEE=rectal bleeding, FREQ=urinary frequency, NOCT=nocturia, STREAM=decreased urinary stream.
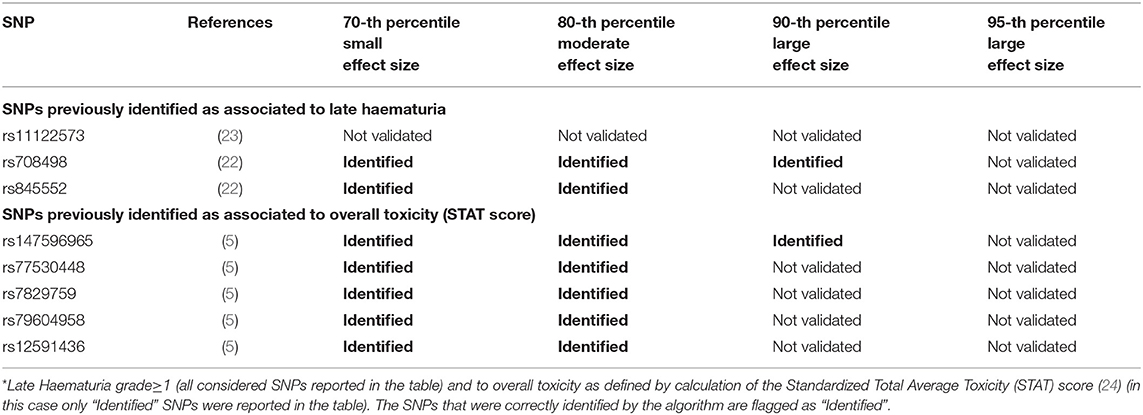
Table 4. Results from Deep Sparse AutoEncoder testing of SNPs associated with Late Haematuria*.
Two hundred and twenty-three patients out of 1,250 available patients (17.8%) experienced late nocturia grade≥2. One hundred and ten patients were excluded from analysis because they had nocturia grade≥2 at baseline, while 41 were excluded because underwent transurethral resection of the bladder or were using anti-muscarinic drugs. Figure 6 and Table 5 report results for the validation through DSAE in the REQUITE population. Eleven SNPs were identified: one SNP already found to be associated with nocturia, four with overall toxicity, one with bleeding and five with other urinary symptoms.
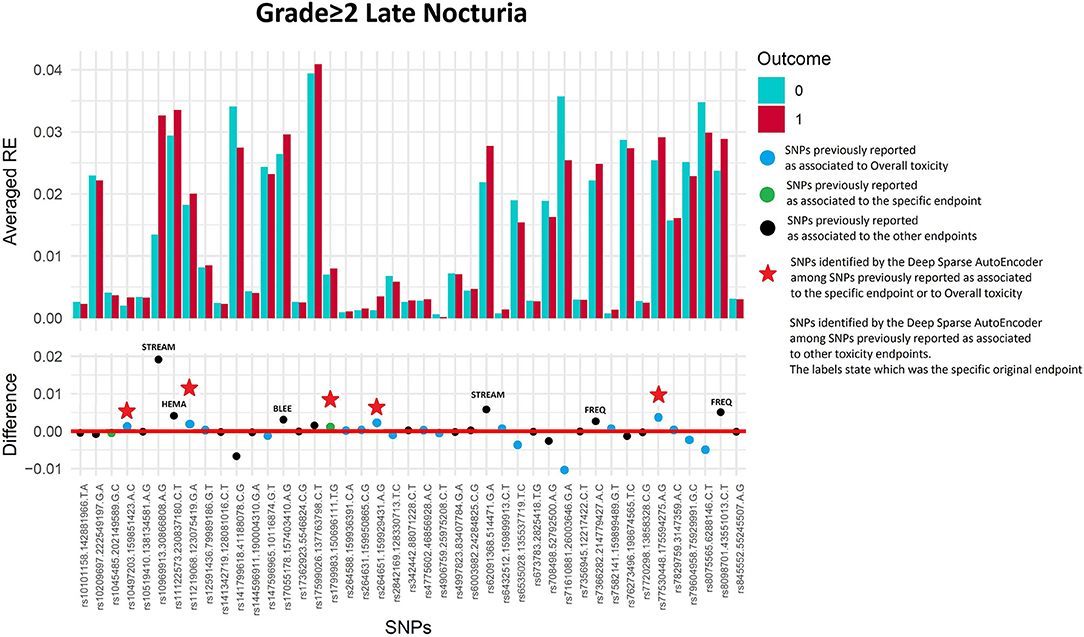
Figure 6. Results for late nocturia grade≥2 from the Deep Sparse AutoEncoder. The 43 considered SNPs are reported in the x-axis and the averaged Reconstruction Errors (RE) are reported in the y-axis (top panel), red columns refer to patients with toxicity, while blue columns refer to patients without toxicity. In the lower panel the difference between averaged Reconstruction Errors between the two classes are represented for each SNP (i.e., differences between red and blue columns). For most SNPs, the difference is close to zero (red line in the bottom panel of the figure). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) are selecting SNPs associated to the toxicity outcome. Green circles refer to SNPs that were previously identified as associated with late nocturia, while blue circles refer to SNPs that were previously associated with overall toxicity as defined by calculation of the Standardized Total Average Toxicity (STAT) score (24). Red stars indicate SNPs (either specific for this endpoint or related to overall toxicity) defining patients with toxicity as outliers with respect to the characteristics of patients without toxicity. Labels show SNPs that not directly associated with late nocturia/overall toxicity, but contributing to their identification. The label states for which toxicity endpoint the SNPs were originally associated with in the literature: BLEE=rectal bleeding, FREQ=urinary frequency, HEMA=haematuria, STREAM=decreased urinary stream.
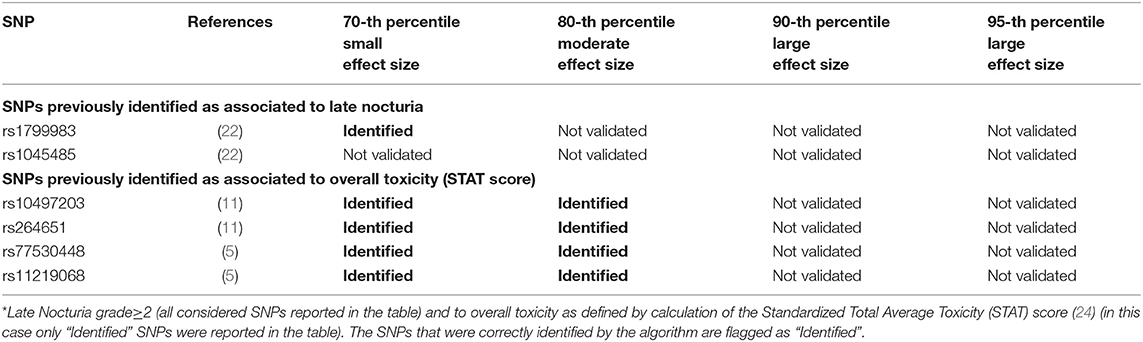
Table 5. Results from Deep Sparse AutoEncoder testing of SNPs associated with Late Nocturia*.
Two hundred and eleven out of 1,234 available patients (17.1%) experienced late decreased stream grade≥1. One hundred and twenty-six patients were excluded from analysis because they had decreased stream grade≥1 at baseline, while 41 were excluded because underwent transurethral resection of the bladder or were using anti-muscarinic drugs. Eleven SNPs were selected: two SNPs previously identified for decreased urinary stream (rs76273496 and rs673783), two for overall toxicity, six for other urinary symptoms and one for bleeding (Figure 7 and Table 6).
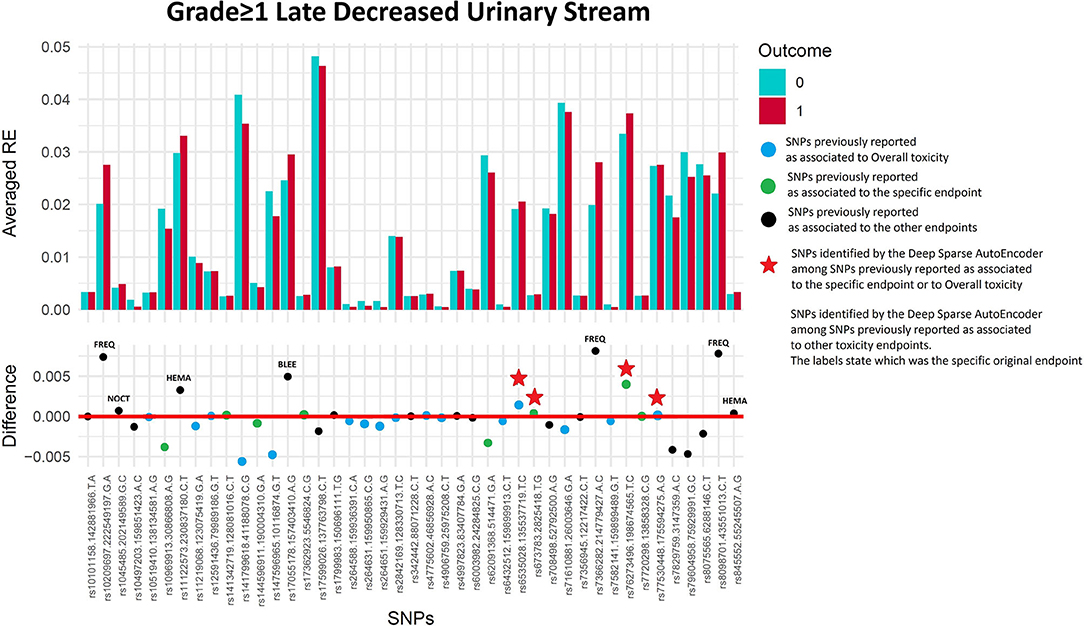
Figure 7. Results for late decreased urinary stream grade≥1 from the Deep Sparse AutoEncoder. The 43 considered SNPs are reported in the x-axis and the averaged Reconstruction Errors (RE) are reported in the y-axis (top panel), red columns refer to patients with toxicity, while blue columns refer to patients without toxicity. In the lower panel the difference between averaged Reconstruction Errors between the two classes are represented for each SNP (i.e., differences between red and blue columns). For most SNPs, the difference is close to zero (red line in the bottom panel of the figure). The chosen thresholds in this difference (i.e., highest 30, 20, 10, and 5% differences) are selecting SNPs associated to the toxicity outcome. Green circles refer to SNPs that were previously identified as associated with late decreased urinary stream, while blue circles refer to SNPs that were previously associated with overall toxicity as defined by calculation of the Standardized Total Average Toxicity (STAT) score (24). Red stars indicate SNPs (either specific for this endpoint or related to overall toxicity) defining patients with toxicity as outliers with respect to the characteristics of patients without toxicity. Labels show SNPs that not directly associated with late decreased urinary stream /overall toxicity, but contributing to their identification. The label states for which toxicity endpoint the SNPs were originally associated with in the literature: BLEE=rectal bleeding FREQ=urinary frequency, HEMA=haematuria, NOCT=nocturia.
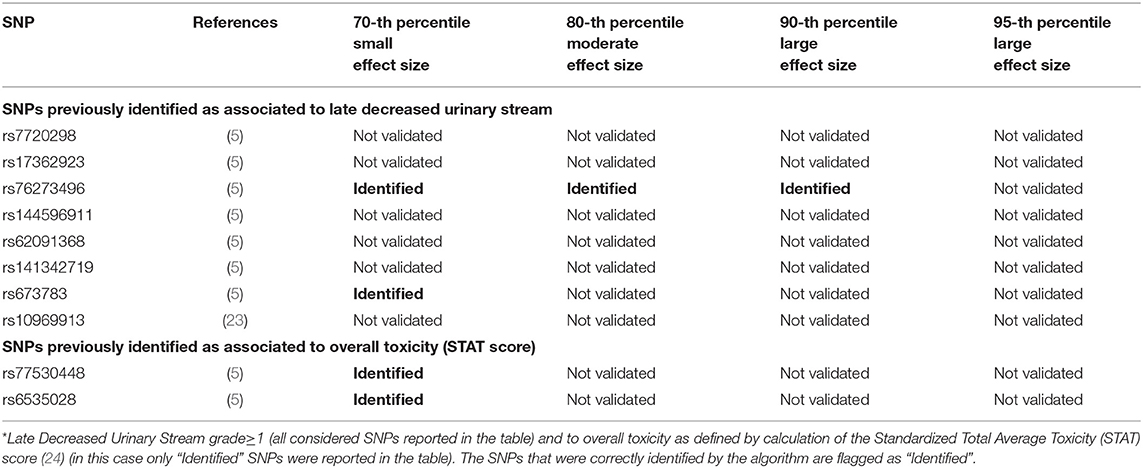
Table 6. Results from Deep Sparse AutoEncoder testing of SNPs associated with Late Decreased Urinary Stream*.
A simple validation approach, using univariate logistic analysis, identified eight SNPs with p < 0.05 (range 0.01–0.05), none of them is validated when considering the Bonferroni correction for multiple testing, which would require p < 0.0011 in this case. Detailed results are presented in Supplementary Table 4.
In recent years Normal Tissue Complication Probability (NTCP) models have been developed to attempt to predict before the start of treatment patients at risk of long-term radiation toxicity. These recent developments were also characterized by the shift from NTCP dose-based modeling to the wider field of more “comprehensive” predictive models. In the speculative case that two patients receive exactly the “same dose distribution,” the risk of toxicity is always modulated by the single individual profile.
The fact that “dose is not enough” was clear from the early days of radiobiology but is receiving constantly growing attention in the current “omics” epoch (Bentzen, 2006): the availability of individual information characterizing patients and potentially influencing their reactions to radiation is increasingly important, especially in the era of image-guided radiotherapy that can spare the organs at risk in most patients.
The purpose of any predictive model in oncology is to provide valid outcome predictions for new patients. Essentially, the main interest of a dataset used to develop a model is to learn for the future. Systematic validation in multi-center collaborative settings hence is a crucial aspect in the process of predictive modeling. REQUITE is the largest multi-center observational study in this field to date, collecting standardized data longitudinally. The study was specifically designed to enable validation of models and biomarkers that predict a patient's risk of developing long-term side-effects following radiotherapy.
The present work focused on the validation of findings from previous GWAS of radiation toxicity after radiotherapy for prostate cancer. To the best of our knowledge, few validation studies in this frame have been conducted so far. Barnett et al. (13) performed an independent validation study of 92 SNPs in 46 genes in a large cohort of breast (976 patients) and prostate (637 patients) cancer patients who received radiotherapy. They focused on five rectal (bleeding, proctitis, sphincter control, stool frequency, tenesmus) and four urinary endpoints (frequency, nocturia, incontinence, and decreased stream) reported by patients 2 years after radiotherapy. An additional endpoint of overall toxicity as measured by the STAT score was also considered. None of the investigated associations was confirmed after adjustment for multiple comparisons.
Genome-wide radiogenomic studies are identifying and validating SNPs. However, to date these studies have relied on the classical single marker association test (both in the discovery and validation setting), which is hampered by the need for multiple-testing corrections. For typical study sizes, this method can detect only relatively large effect size and has limited power to identify reliably modest effects from the many SNPs that are likely to contribute to a polygenic risk profile associated with radiation toxicity. Genome-wide studies miss SNPs that make small but real contributions to risk.
Machine learning has already been proposed as a promising alternative approach to estimate overall genetic risk (27). The approach can identify multiple SNPs with small effects that together but not individually reach genome-wide significance. Two studies have already proposed machine learning methods to identify SNP-based signatures associated with late toxicity after radiotherapy for prostate cancer (27, 28).
Here, we extended the use of machine learning methods by using a method that addresses an important limitation of studies on radiation toxicity: the imbalance of classes, with a lower frequency of patients with vs. without late toxicity. This imbalance is important because it can lead to sub-optimal solutions (29), even when datasets are used for validation. As a first step in testing our approach, we attempted to and were successful in validating previously reported associations identified in studies based on classical single marker association tests. The next step will be a de novo analysis to identify SNPs with smaller individual effects.
Dealing with imbalance requires non-classical statistical solutions. Here, we explore novel methods for feature selection that come from the Deep Learning research field (25). Indeed, deep learning approaches, with their intrinsic hierarchical structure (where each layer realizing a combination of the previous layer), seem particularly adept at mimicking complex dependencies within data. Deep learning has already been applied and shown to have potential in similar bioinformatics research areas, such as for modeling the competition between splice sites (30) and in predicting RNA- and DNA-binding specificity (31).
We used DSAE to obtain the best possible representation of the majority class (without toxicity) and so to identify which features (SNPs) distinguish the minority class (with toxicity). The encoder and decoder functions are usually non-linear (i.e., sigmoid, hyperbolic tangent, rectified linear unit etc.), which enables a better reconstruction of the input by the capture of complex non-linear relationships among SNPs. Training on healthy patients allows the overall SNP pattern of normal radio-sensitivity to be established. Testing measures the “distance” between each new patient and the pattern of normal radio-sensitivity to identify SNPs associated with the highest reconstruction errors (i.e., highest distances) between the pattern of normality and the SNP profile of patients scored with toxicity (i.e., radio-sensitive patients). The distribution of the reconstructed errors allows identification and classification of SNPs with very large/large effect (SNPs associated with the top 95th percentile and 90th percentile of the distribution of reconstructed errors) and with moderate/small effects (SNPs associated with the top 80th percentile and 70th percentile of the distribution of reconstructed errors).
The DSAE successfully validated multiple SNPs contributing to an increased risk of toxicity. Some SNPs were already associated with the specific considered endpoint, others were previously associated with overall toxicity, and some were previously associated with other toxicities.
As common in GWAS, many significant SNPs lie in non-coding regions, and it is premature to speculate on their functional significance. We refer readers to the original publications which discuss possible gene functions (5, 11, 23), but give an example to illustrate likely clinical relevance. DSAE validated two SNPs previously associated with haematuria, rs708498 and rs845552, which are located in the PTGER2 and EGFR genes, respectively. PTGER2 (widely distributed in humans) encodes Prostaglandin E2 receptor 2. Irradiation causes hypermethylation of this antifibrotic gene (32). EGFR has been shown to play a critical role in TGF-β1 dependent fibroblast to myofibroblast differentiation (33). These two SNPs were also identified for urinary stream (rs845552) and urinary frequency (rs708498).
The main strength of our study is use of a large international prospective multi-center cohort of patients treated with modern radiotherapy techniques and fractionation schemes. The patients were specifically enrolled to validate models and biomarkers for predicting radiation toxicity, and the study design involved a standardized data collection scheme for collecting healthcare professional and patient-reported outcomes. The extensive role of data management also allowed for quality assurance of data collected, and we used “real world” data coming from “data-farming” (34).
A possible limitation of our study was use of 2-year follow-up toxicity data. The REQUITE study is still maturing, normal tissue reactions in the intestinal and urinary tract develop gradually from 6 months after radiotherapy till to around 3 years for the intestinal syndrome and to 5 years for the urinary syndrome. Recent additional funding is allowing extension of the REQUITE study with the aim of reaching standardized collection of follow-up data till year 5.
The use of grade 1 and grade 2 events is another possible limitation of this study. As the application of deep learning techniques requires a suitable number of events, the choice of mild or moderate (when possible) toxicity was forced by the number of morbidity events registered in the REQUITE population. The low number of severe toxicity is for sure a reflection of modern radiotherapy techniques which allow a substantial sparing of normal tissues, at least for the case of prostate cancer irradiation. Yet, some grade 1 and grade 2 toxicity can assume a chronic behavior, with substantial impact on the quality of life of long term survivors, for example, this could happen, for grade 2 urinary frequency and nocturia which are impairing daily activities and the quality of sleep for many years (35). A further point, more associated to research rather to clinical activity, is related to the possibility that the same genes/variants predispose to severe toxicity that predispose to low-grade toxicity. A realistic hypothesis is that some genes/variants will be common and others will be unique to severe toxicities. For example, ATM seems to be important for both mild and severe toxicity, though the particular variants differ with common SNPs associated with any toxicity, but rare mutations associated with severe toxicity. We think we can make a good case that genes identified via GWAS of mild toxicity represent good candidates for subsequent sequencing studies to identify rare mutations that may be associated with severe toxicities. Probably there are at least some biologic mechanisms common to both mild and severe toxicity, though the optimal genomic signature for each may differ. Our work still adds value by pointing to the candidate genes or loci that are likely important for both.
We have shown our approach is worth studying further and the next step would be to use it to identify patterns of SNPs to define polygenic risk scores that can be included into integrated normal tissue complication probability models, together with validated dosimetric and clinical risk factors.
The DSAE methodology underlines that, within the current RT, experiencing no toxicity could be considered as the “normal” situation, with patients with mild/moderate toxicity being outliers. The possible knowledge of the single patient intrinsic radiosensitivity and the identification of these outlier subjects could help in tailoring decision making. This should not entail changing the probability of tumor control to avoid mild/moderate side-effects, yet it should be focused on maximizing uncomplicated tumor control, even considering the patient inclination toward the different side-effects. The availability of such models would be relevant for the clinic, allowing the single patient optimization, thus constituting an important step toward the implementation of predictive modeling in the clinic. This approach would allow tailoring of therapeutic approach (i.e., active surveillance vs. prostatectomy vs. brachytherapy vs. external beam radiotherapy) and of doses (both to tumor and organs at risk) to the specific patient anatomy, clinical situation and individual biology. Combining biological stratification with toxicity reducing techniques (such as imaging fusion, image guidance, fractionation and reduced margins for Planning Target Volume) could further decrease treatment related toxicity rates and allow for dose escalation to enhance tumor control. Integrated predictive models will also be an essential tool in the design of interventional trials to modify the radiotherapy strategies. A detailed discussion of the potential ways in which biomarker/SNP assays might be implemented in routine clinical practice can be found in Azria et al. (7).
Other future work could study the possibility of “scaling” the use of DSEAs to the discovery of new genetic signatures using the whole GWAS information available in the REQUITE population, thus achieving the possibility of considering millions of features to detect outliers.
A deep learning approach can validate SNPs associated with toxicity after radiotherapy. The method can identify complex SNP signatures for multiple toxicity endpoints and should be studied further to extract polygenic risk scores to include in integrated normal tissue complication probability models that could be used to personalize radiotherapy planning.
Funding for the five year REQUITE project ended on 30th September 2018. REQUITE does not benefit financially from supplying data and/or samples to researchers, but does make a charge to cover its costs and support continued maintenance of the database and biobank beyond the ending of the funding period. To facilitate this continued access to researchers, the REQUITE Steering Committee approved a tiered cost recovery model for access to data and/ or samples. Contact REQUITE (cmVxdWl0ZUBtYW5jaGVzdGVyLmFjLnVr) for more information on pricing.
The REQUITE study was reviewed and approved by North West - Great Manchester East Ethics Committee (UK, reference 14 NW 0035) and by the local Ethics Committees of all participating centers. The patients provided their written informed consent to participate in this study and for the publication of the data included in this article.
MM, AP, FG, TRan, and CW: study design. MM, FG, and TRan: study development. AP, FI, AM, PZ, RE, and JC-C: coordination/supervision of the study. LV, PO, VF, TRat, PRS, KJ, ML, KH, GdM, DdR, BV, EvL, ACh, ES, CH, MV, BA, RV, DA, M-PJ, RS, KC, and PC: patient enrolment and follow-up. CT, TG, ACi, BR, AV, and MA-B: collection of the data. LF, AD, SK, and DP: SNP assay. JC-C, PS, AW, and RE: trial and data management. MM, FG, NF, FI, AP, AM, and TRan: statistical analysis. MM, FG, NF, TRan, and CW: draft of the paper. All authors: critical revision of the manuscript/final approval.
REQUITE received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 601826. FG was supported by MRC unit programme MC_UU_00002/5. ACh, RE, and CW were supported by the NIHR Manchester Biomedical Research Center. LF was supported by the European Union's Horizon 2020 Research and Innovation Programme under Marie Sklodowska-Curie grant agreement number 656144. TRan was supported by Fondazione Italo Monzino. ACi was supported by AIRC IG 21479. AV was supported by Spanish Instituto de Salud Carlos III (ISCIII) funding, an initiative of the Spanish Ministry of Economy and Innovation partially supported by European Regional Development FEDER Funds (INT15/00070; INT16/00154; INT17/00133; PI19/01424; PI16/00046; PI13/02030; PI10/00164), and through the Autonomous Government of Galicia (Consolidation and structuring program: IN607B). TRat is currently an NIHR Clinical Lecturer. He was previously funded by a National Institute of Health Research (NIHR) Doctoral Research Fellowship (DRF 2014-07-079). This publication represents independent research. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. SK was supported by grant K07CA187546 from the National Cancer Institute (NCI).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.541281/full#supplementary-material
1. Cooperberg MR, Carroll PR. Trends in management for patients with localized prostate cancer, 1990-2013. JAMA. (2015) 314:80–2. doi: 10.1001/jama.2015.6036
2. Zelefsky MJ, Poon BY, Eastham J, Vickers A, Pei X, Scardino PT. Longitudinal assessment of quality of life after surgery, conformal brachytherapy, and intensity-modulated radiation therapy for prostate cancer. Radiother Oncol. (2016) 118:85–91. doi: 10.1016/j.radonc.2015.11.035
3. Landoni V, Fiorino C, Cozzarini C, Sanguineti G, Valdagni R, Rancati T. Predicting toxicity in radiotherapy for prostate cancer. Phys Med. (2016) 32:521–32. doi: 10.1016/j.ejmp.2016.03.003
4. Rancati T, Palorini F, Cozzarini C, Fiorino C, Valdagni R. Understanding urinary toxicity after radiotherapy for prostate cancer: first steps forward. Tumori. (2017) 103:395–404. doi: 10.5301/tj.5000681
5. Kerns SL, Dorling L, Fachal L, Bentzen S, Pharoah PD, Barnes DR, et al. Meta-analysis of genome wide association studies identifies genetic markers of late toxicity following radiotherapy for prostate cancer. EBioMedicine. (2016) 10:150–63. doi: 10.1016/j.ebiom.2016.07.022
6. El Naqa I, Kerns SL, Coates J, Luo Y, Speers C, West CML, et al. Radiogenomics and radiotherapy response modeling. Phys Med Biol. (2017) 62:R179–206. doi: 10.1088/1361-6560/aa7c55
7. Azria D, Lapierre A, Gourgou S, De Ruysscher D, Colinge J, Lambin P, et al. Data-based radiation oncology: design of clinical trials in the toxicity biomarkers era. Front Oncol. (2017) 7:83. doi: 10.3389/fonc.2017.00083
8. Herskind C, Talbot CJ, Kerns SL, Veldwijk MR, Rosenstein BS, West CM. Radiogenomics: a systems biology approach to understanding genetic risk factors for radiotherapy toxicity? Cancer Lett. (2016) 382:95–109. doi: 10.1016/j.canlet.2016.02.035
9. Andreassen CN, Rosenstein BS, Kerns SL, Ostrer H, De Ruysscher D, Cesaretti JA, et al. Individual patient data meta-analysis shows a significant association between the ATM rs1801516 SNP and toxicity after radiotherapy in 5456 breast and prostate cancer patients. Radiother Oncol. (2016) 121:431–9. doi: 10.1016/j.radonc.2016.06.017
10. Kerns SL, West CM, Andreassen CN, Barnett GC, Bentzen SM, Burnet NG, et al. Radiogenomics: the search for genetic predictors of radiotherapy response. Future Oncol. (2014) 10:2391–406. doi: 10.2217/fon.14.173
11. Fachal L, Gómez-Caamaño A, Barnett GC, Peleteiro P, Carballo AM, Calvo-Crespo P, et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat Genet. (2014) 46:891–84. doi: 10.1038/ng.3020
12. Barnett GC, Thompson D, Fachal L, Kerns S, Talbot C, Elliott RM, et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother Oncol. (2014) 111:178–85. doi: 10.1016/j.radonc.2014.02.012
13. Barnett GC, Coles CE, Elliott RM, Baynes C, Luccarini C, Conroy D, et al. Independent validation of genes and polymorphisms reported to be associated with radiation toxicity: a prospective analysis study. Lancet Oncol. (2012) 13:65–77. doi: 10.1016/S1470-2045(11)70302-3
14. Seibold P, Webb A, Aguado-Barrera ME, Azria D, Bourgier C, Brengues M, et al. REQUITE: a prospective multicentre cohort study of patients undergoing radiotherapy for breast, lung or prostate cancer. Radiother Oncol. (2019) 138:59–67. doi: 10.1016/j.radonc.2019.04.034
15. De Ruysscher D, Defraene G, Ramaekers BLT, Lambin P, Briers E, Stobart H, et al. Optimal design and patient selection for interventional trials using radiogenomic biomarkers: a REQUITE and radiogenomics consortium statement. Radiother Oncol. (2016) 121:440–6. doi: 10.1016/j.radonc.2016.11.003
16. West C, Azria D, Chang-Claude J, Davidson S, Lambin P, Rosenstein B, et al. The REQUITE project: validating predictive models and biomarkers of radiotherapy toxicity to reduce side-effects and improve quality of life in cancer survivors. Clin Oncol. (2014) 26:739–42. doi: 10.1016/j.clon.2014.09.008
17. Ioannidis JP, Trikalinos TA, Khoury MJ. Implications of small effect sizes of individual genetic variants on the design and interpretation of genetic association studies of complex diseases. Am J Epidemiol. (2006) 164:609–14. doi: 10.1093/aje/kwj259
18. Michalopoulos I, Pavlopoulos GA, Malatras A, Karelas A, Kostadima MA, et al. Human gene correlation analysis (HGCA): a tool for the identification of transcriptionally co-expressed genes. BMC Res Notes. (2012) 5:265. doi: 10.1186/1756-0500-5-265
19. Farnell DJ, Mandall P, Anandadas C, Routledge J, Burns MP, Logue JP, et al. Development of a patient-reported questionnaire for collecting toxicity data following prostate brachytherapy. Radiother Oncol. (2010) 97:136–42. doi: 10.1016/j.radonc.2010.05.011
20. Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The oncoarray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev. (2017) 26:126–35. doi: 10.1158/1055-9965.EPI-16-0106
21. Kerns SL, Stock RG, Stone NN, Blacksburg SR, Rath L, Vega A, et al. Genome-wide association study identifies a region on chromosome 11q14.3 associated with late rectal bleeding following radiation therapy for prostate cancer. Radiother Oncol. (2013) 107:372–6. doi: 10.1016/j.ijrobp.2013.06.343
22. De Langhe S, De Meerleer G, De Ruyck K, Ost P, Fonteyne V, De Neve W, et al. Integrated models for the prediction of late genitourinary complaints after high-dose intensity modulated radiotherapy for prostate cancer: making informed decisions. Radiother Oncol. (2014) 112:95–9. doi: 10.1016/j.radonc.2014.04.005
23. Kerns SL, Fachal L, Dorling L, Barnett GC, Baran A, Peterson DR, et al. Radiogenomics consortium genome-wide association study meta-analysis of late toxicity after prostate cancer radiotherapy. J Natl Cancer Inst. (2020) 112:179–90. doi: 10.1093/jnci/djz075
24. Barnett GC, West CM, Coles CE, Pharoah PD, Talbot CJ, Elliott RM, et al. Standardized total average toxicity score: a scale- and grade-independent measure of late radiotherapy toxicity to facilitate pooling of data from different studies. Int J Radiat Oncol Biol Phys. (2012) 82:1065–74. doi: 10.1016/j.ijrobp.2011.03.015
25. Massi M, Ieva F, Gasperoni F, Paganoni AM. Minority class feature selection through semi-supervised deep sparse autoencoders. Milano: Mox Report - Politecnico di Milano (2019)
26. Hinton GE, Salakhutdinov RR. Reducing the dimensionality of data with neural networks. Science. (2006) 313:504–7. doi: 10.1126/science.1127647
27. Oh JH, Kerns S, Ostrer H, Powell SN, Rosenstein B, Deasy JO. Computational methods using genome-wide association studies to predict radiotherapy complications and to identify correlative molecular processes. Sci Rep. (2017) 7:43381. doi: 10.1038/srep43381
28. Lee S, Kerns S, Ostrer H, Rosenstein B, Deasy JO, Oh JH. Machine learning on a genome-wide association study to predict late genitourinary toxicity after prostate radiation therapy. Int J Radiat Oncol Biol Phys. (2018) 101:128–35. doi: 10.1016/j.ijrobp.2018.01.054
29. Yin L, Ge Y, Xiao K, Wang X, Quan X. Feature selection for high-dimensional imbalanced data. Neurocomputing. (2013) 105:3–11. doi: 10.1016/j.neucom.2012.04.039
30. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. (2015) 347:1254806. doi: 10.1126/science.1254806
31. Alipanahi B, Delong A, Weirauch MT, Frey BDNA- J and RNA-binding proteins by deep learning. Nat Biotechnol. (2015) 33:831–8. doi: 10.1038/nbt.3300
32. Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, et al. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol. (2010) 177:2245–55. doi: 10.2353/ajpath.2010.100446
33. Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, et al. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem. (2013) 288:14824–38. doi: 10.1074/jbc.M113.451336
34. Mayo CS, Kessler ML, Eisbruch A, Weyburne G, Feng M, Hayman JA, et al. The big data effort in radiation oncology: data mining or data farming? Adv Radiat Oncol. (2016) 1:260–71. doi: 10.1016/j.adro.2016.10.001
Keywords: prostate cancer, late toxicity, snps, deep learning, autoencoder, validation
Citation: Massi MC, Gasperoni F, Ieva F, Paganoni AM, Zunino P, Manzoni A, Franco NR, Veldeman L, Ost P, Fonteyne V, Talbot CJ, Rattay T, Webb A, Symonds PR, Johnson K, Lambrecht M, Haustermans K, De Meerleer G, de Ruysscher D, Vanneste B, Van Limbergen E, Choudhury A, Elliott RM, Sperk E, Herskind C, Veldwijk MR, Avuzzi B, Giandini T, Valdagni R, Cicchetti A, Azria D, Jacquet M-P, Rosenstein BS, Stock RG, Collado K, Vega A, Aguado-Barrera ME, Calvo P, Dunning AM, Fachal L, Kerns SL, Payne D, Chang-Claude J, Seibold P, West CML and Rancati T (2020) A Deep Learning Approach Validates Genetic Risk Factors for Late Toxicity After Prostate Cancer Radiotherapy in a REQUITE Multi-National Cohort. Front. Oncol. 10:541281. doi: 10.3389/fonc.2020.541281
Received: 08 March 2020; Accepted: 02 September 2020;
Published: 15 October 2020.
Edited by:
Timothy James Kinsella, Warren Alpert Medical School of Brown University, United StatesReviewed by:
Wei Zhao, Stanford University, United StatesCopyright © 2020 Massi, Gasperoni, Ieva, Paganoni, Zunino, Manzoni, Franco, Veldeman, Ost, Fonteyne, Talbot, Rattay, Webb, Symonds, Johnson, Lambrecht, Haustermans, De Meerleer, de Ruysscher, Vanneste, Van Limbergen, Choudhury, Elliott, Sperk, Herskind, Veldwijk, Avuzzi, Giandini, Valdagni, Cicchetti, Azria, Jacquet, Rosenstein, Stock, Collado, Vega, Aguado-Barrera, Calvo, Dunning, Fachal, Kerns, Payne, Chang-Claude, Seibold, West and Rancati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Ieva, ZnJhbmNlc2NhLmlldmFAcG9saW1pLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.