 Rafael Rosell
Rafael Rosell Martyna Filipska
Martyna Filipska Imane Chaib
Imane Chaib Fernando Laguia
Fernando Laguia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
GENERAL COMMENTARY article
Front. Oncol., 10 September 2020
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01726
A Commentary on
Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer
by Lu, Y., Xue, J., Deng, T., Zhou, X., Yu, K., Deng, L., et al. (2020). Nat. Med. 26, 732–740. doi: 10.1038/s41591-020-0840-5
CRISPR/Cas9 ribonucleoprotein-mediated editing has been used to disrupt the PDCD1 gene encoding programmed cell death-1 (PD-1) in human T cells, resulting in a significantly reduced PD-1 expression without affecting T cell viability during prolonged in vitro culture. Modified T-cells show up-regulation of IFN-gamma production and enhanced cytotoxicity (1) and also augmented chimeric antigen receptor (CAR) T cell cytokine production and cytotoxicity toward PD-L1-expressing cancer cells in vitro (2). Moreover, genome-wide CRISPR-Cas9 screening has served to identify novel therapeutic targets. For example, the mRNA decapping enzyme scavenger (DCPS) gene, which is essential for acute myeloid leukemia (AML) survival, thus allowing treatment of AML with RG3039, a DCPS inhibitor used to treat spinal muscular atrophy (3). Similarly, CRISPR-Cas9 genome-wide screening in multiple myeloma cells identifies mechanisms whereby CSN9 signalosome subunits modulate cereblon expression levels and sensitivity to immunomodulatory drugs, i.e., lenalidomine and pomalidomide (4). A CRISPR-assisted telomerase-activating gene expression system, using Cas9 as an effector gene, killed various cancer cell lines, including A549, PANC-1, and others, without affecting normal cells (5). Other genome-wide CRISPR screening in CD8 T cells in the context of immunotherapy have identified a RNA Helicase Dhx37 that modulates NF-kB function and suppresses T cell production in breast cancer. Targeting DHX37 is suggested could be of significant clinical relevance (6)
A phase I clinical trial of CRISPR-Cas9 PD-1-edited T cells in non-small-cell lung cancer demonstrates that the disruption of PD-1 on T cells generally induced grade 1 or 2 adverse events, and the most frequent events were lymphopenia, fatigue, leukopenia, fever, arthralgia, and skin rash. Flow cytometric analysis demonstrated decreased PD-1 expression, with median disruption of 46.3% in edited T cells. NGS targeted sequencing of the PD-1 gene indicated that the median editing efficiency of all 12 patients was 5.8%. A significant increase in the proportion of CD8+IFN-gamma+ cells was found in edited T cells (7).
PD-1 receptor is mainly expressed on mature cytotoxic T lymphocytes in peripheral tissues and the tumor microenvironment. PD-1 expression is inducible upon the activation of T cells, and PD-1 acts as a coinhibitory receptor that functions as immune checkpoint to maintain the peripheral immune tolerance and prevents autoimmunity (8). PD-1 ligation by PD-L1 expressed on tumors cells transduces signaling via the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the immunoreceptor tyrosine-based inhibitor switch motif (ITISM) of the PD-1 cytoplasmic tail, which further inhibits the PI3K/AKT, MAPK/ERK1/2, and/or mTOR, thus suppressing tumor growth (9). A new discovery shows that PDCD1 knockdown in non-small-cell lung cancer cell lines, NCi-H1299 and Calu-1, reduced the mRNA and protein levels in PD-1 depleted cells compared to control cells. Moreover, PDCD1 silencing resulted in increased cell proliferation and colony formation. After PDCD1 knockdown, phospho(p)-AKT and pERK1/2 levels were increased in both cell lines. Over-expressing PDCD1 increased mRNA and protein levels, inhibited cell proliferation and colony formation in NCI-H1299 and Calu-1 cells with decreased p-AKT and p-ERK1/2 levels (10). A non-small-cell lung cancer patient having high PD-1 expression in the tumor biopsy had rapid progression to anti-PD-1 therapy. The finding was validated with the detection of PD-1 transcript in lung cancer cells in resected lung cancer tissues. Knockout or antibody blockade of PD-1 enhanced M109 (mouse lung cancer cell line) viability in vitro. Also, PD-1 blockade accelerated growth of M109-xenograft tumors (11). Immunoblot analysis disclosed that PD-1 is expressed by NSCLC cell lines and that the expressed PD-1 was 55 KDa in size, similar to the size of T-cell expressed PD-1. Furthermore, flow cytometry showed that PD-1 is expressed in a sub-population of all examined cancer cells (10). Simultaneous overexpression of both PDCD1 and PDCD1LG1, which encodes PD-L1, significantly decreased cell proliferation, P-AKT, and p-ERK levels compared to cells transfected with PDCD1, PDCD1LG1, or the control (10). The treatment of Calu-1, SW480, HT-29 and other cell lines with PD-1-targeted nivolumab, or pembrolizumab, increased cell proliferation and higher p-AKT and p-ERK levels than the cells treated with an isotype control antibody. The same observations were seen in xenografted cells in mice (10). The aforementioned results raise doubts about the adequacy of CRISPR-Cas9-mediated-PD-1 disruption in T cells (1, 7). The clinical consequences of Wang et al. (10) are unknown at this moment, however it offers a new view that the tumor cell intrinsic PD-1/PD-L1 axis suppresses tumor growth and inhibits AKT and ERK1/2 signaling pathways and could prevent the interaction with PD-1 expressing cells. As in cancer cells, PD-1 also regulates PI3K/AKT, MAPK/ERK1/2 and mammalian target of rapamycin (mTOR) pathways in T cells (9). Further understanding of the tumor-suppressor function of cancer cell PD-1 receptor expression can provide a new perspective in cancer management. Treatment of lung cancer is currently based on immunotherapy with antibodies against PD-1 or PD-L1, alone or in combination with chemotherapy. The use of pembrolizumab alone, or in combination with chemotherapy, as well as atezolizumab plus chemotherapy, is recommended for stage IV non-small-cell lung cancer patients without driver alterations (EGFR or ALK) according to the American Society of Clinical Oncology and Ontario Health joint guideline (12). Tumor PD-L1 expression by immunohistochemistry is considered the standard practice. Response rate is low, with median progression free survival, that is generally short-lived (12). It is possible, as above described, that if tumor PD-1 is in equilibrium with tumor PD-L1, the disruption of intratumoral PD1-PD-L1 induced by monoclonal antibodies that block PD-1 (nivolumab or pembrolizumab) or PD-L1 (atezolizumab, durvalumab and avelumab) can provoke tumor hyper-progression which has been reported in several studies (13) (Figure 1). The figure summarizes the previous reports on the conflicting role that PD-1 or PD-L1 monoclonal antibodies could have in causing tumor progression. A phase 1 clinical trial of multiplex CRISPR-Cas9 editing to engineer T cells in three cancer patients was reported. Two genes encoding the endogenous T cell receptor (TCR) chains, TRAC and TRBC, were deleted in T cells to reduce TCR mispairing and to enhance the expression of a synthetic, cancer specific TCR transgene (NY-ESO-1). PDCD1 was also removed. Modified T cells persisted for up to 9 months, suggesting the feasibility of CRISPR gene editing for cancer immunotherapy (14). The preliminary results are enticing and shed light on the plausibility that multiplex CRISPR-Cas9 editing to engineer T cells could be more effective in cancer patients.
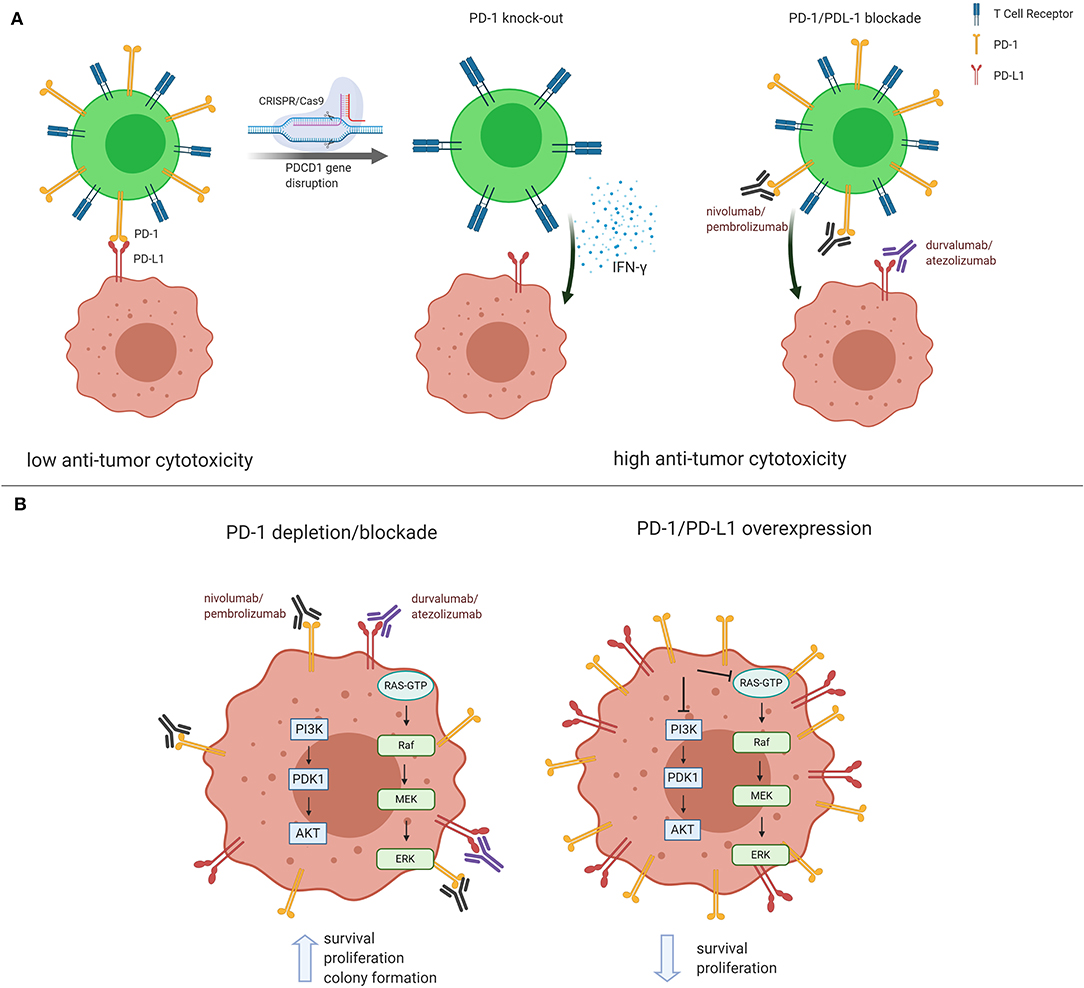
Figure 1. Conflicting role of PD-1 and PD-L1 in cancer management. (A) In the classical understanding of PD-1/PD-L1 regulation, PD-1 expressed on mature cytotoxic T lymphocytes reduces anti-tumor cytotoxicity via binding to tumor PD-L1. Both PD-1/PD-L1 CRISPR/Cas9-mediated PD-1 knockdown and blockade in T cells result in higher anti-tumor cytotoxicity. (B) Recent reports show that PD-1/PD-L1 blockade and/or silencing lead to PI3K/AKT and MEK/ERK1/2 pathway activation, thus inducing tumor growth and survival. The opposite effect is observed when PD-1/PD-L1 are found overexpressed in cancer cells. Figure created using BioRender.
In our opinion, in addition to assessing patients for PD-L1 expression in the tumor, the expression of PD-1 in the tumor could serve as a potential biomarker to define efficacy or tumor progression following immunotherapy with either anti-PD-1 o anti-PD-L1 monoclonal antibodies (13).
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Work in the RR's group is partially supported by a grant from La Caixa Foundation, a Spanish Association Against Cancer (AECC) grant (PROYE18012ROSE) and funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 765492.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. (2016) 6:20070. doi: 10.1038/srep20070
2. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
3. Yamauchi T, Masuda T, Canver MC, Seiler M, Semba Y, Shboul M, et al. Genome-wide CRISPR-Cas9 screen identifies leukemia-specific dependence on a pre-mRNA metabolic pathway regulated by DCPS. Cancer Cell. (2018) 33:386–400 e5. doi: 10.1016/j.ccell.2018.01.012
4. Liu J, Song T, Zhou W, Xing L, Wang S, Ho M, et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia. (2019) 33:171–80. doi: 10.1038/s41375-018-0205-y
5. Dai W, Xu X, Wang D, Wu J, Wang J. Cancer therapy with a CRISPR-assisted telomerase-activating gene expression system. Oncogene. (2019) 38:4110–24. doi: 10.1038/s41388-019-0707-8
6. Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. (2019) 178:1189–204 e23. doi: 10.1016/j.cell.2019.07.044
7. Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. (2020) 26:732–40. doi: 10.1038/s41591-020-0840-5
8. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. (2016) 375:1767–78. doi: 10.1056/NEJMra1514296
9. Xia Y, Jeffrey Medeiros L, Young KH. Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies. Biochim Biophys Acta. (2016) 1865:58–71. doi: 10.1016/j.bbcan.2015.09.002
10. Wang X, Yang X, Zhang C, Wang Y, Cheng T, Duan L, et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc Natl Acad Sci USA. (2020) 117:6640–50. doi: 10.1073/pnas.1921445117
11. Du S, McCall N, Park K, Guan Q, Fontina P, Ertel A, et al. Blockade of tumor-expressed PD-1 promotes lung cancer growth. Oncoimmunology. (2018) 7:e1408747. doi: 10.1080/2162402X.2017.1408747
12. Hanna NH, Schneider BJ, Temin S, Baker S Jr, Brahmer J, Ellis PM, et al. Therapy for stage IV non-small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. J Clin Oncol. (2020) 38:1608–32. doi: 10.1200/JCO.19.03022
13. Santarpia M, Aguilar A, Chaib I, Cardona AF, Fancelli S, Laguia F, et al. Non-small-cell lung cancer signaling pathways, metabolism, and PD-1/PD-L1 antibodies. Cancers. (2020) 12:1475. doi: 10.3390/cancers12061475
Keywords: PD-1, PD-L1, T Cells, CRISPR, PI3K, Akt, MAPK, NSCLC
Citation: Rosell R, Filipska M, Chaib I, Lligé D and Laguia F (2020) Commentary: Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Front. Oncol. 10:1726. doi: 10.3389/fonc.2020.01726
Received: 04 June 2020; Accepted: 03 August 2020;
Published: 10 September 2020.
Edited by:
John Maher, King's College London, United KingdomReviewed by:
Dimitrios Laurin Wagner, Charité – Universitätsmedizin Berlin, GermanyCopyright © 2020 Rosell, Filipska, Chaib, Lligé and Laguia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Rosell, cnJvc2VsbEBpY29uY29sb2dpYS5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.