 Zhichao Tian1
Zhichao Tian1 Weitao Yao
Weitao Yao- 1Department of Orthopedics, The Affiliated Cancer Hospital of Zhengzhou University, Henan Cancer Hospital, Zhengzhou, China
- 2Department of Orthopedic Oncology, Beijing Jishuitan Hospital, Beijing, China
Recent clinical trials have shown several multi-target tyrosine kinase inhibitors (TKIs) to be effective in the treatment of osteosarcoma. However, these TKIs have a number of targets, and it is yet unclear which of these targets has a key role in osteosarcoma treatment. In this review, we first summarize the TKIs that were studied in clinical trials registered on ClinicalTrials.gov. Further, we compare and discuss the targets of these TKIs. We found that TKIs with promising therapeutic effect for osteosarcoma include apatinib, cabozantinib, lenvatinib, regorafenib, and sorafenib. The key targets for osteosarcoma treatment may include VEGFRs and RET. The receptor tyrosine kinases (RTKs) MET, IGF-1R, AXL, PDGFRs, KIT, and FGFRs might be relevant but unimportant targets for osteosarcoma treatment. Inhibition of one type of RTK for the treatment of osteosarcoma is not effective. It is necessary to inhibit several relevant RTKs simultaneously to achieve a breakthrough in osteosarcoma treatment. This review provides comprehensive information on TKI targets relevant in osteosarcoma treatment, and it will be useful for further research in this field.
Introduction
Osteosarcoma is the most common primary bone malignancy (1). For decades, the treatment model for osteosarcoma has not advanced much. The treatment includes neoadjuvant chemotherapy, surgery, and postoperative chemotherapy, and patients have a 5-years survival rate of about 60% (2). Patients who experience metastatic disease have limited options; they have expected 4-months progression-free survival (PFS) rate of 12% only (3). In recent years, with the great success of tyrosine kinase inhibitor (TKI) use in the treatment of cancers, the treatment for osteosarcoma has entered a new phase.
TKIs are targeted drugs that can specifically inhibit protein tyrosine kinases (PTKs). PTKs are important signaling molecules which having highly regulated activity and are critical components of signaling pathways that control cellular differentiation and proliferation (4). There are 90 PTKs that encoded by human genome (5). These PTKs can be classified into 32 species of non-receptor tyrosine kinases (NRTKs) and 58 species of receptor tyrosine kinases (RTKs) (6). RTKs are involved in the process of extracellular signals into cell, whereas NRTKs regulate intracellular communication (7). RTKs are transmembrane glycoproteins that consist of an extracellular domain, a transmembrane domain, and an intracellular kinase domain (Figure 1) (8). They are activated upon binding to their ligands, and the extracellular signal is transduced to the cytoplasm by phosphorylation of the tyrosine residues on the receptors themselves and on downstream signaling proteins (8). RTKs activate multiple signaling pathways leading to cell migration, differentiation, proliferation, and metabolic changes (9). NRTKs are downstream signaling molecules of RTKs and various other receptors. They do not have transmembrane domains, and are located in nucleus or cytoplasm. NRTKs can be phosphorylated by different RTKs or trans-autophosphorylated upon dimerization (10).
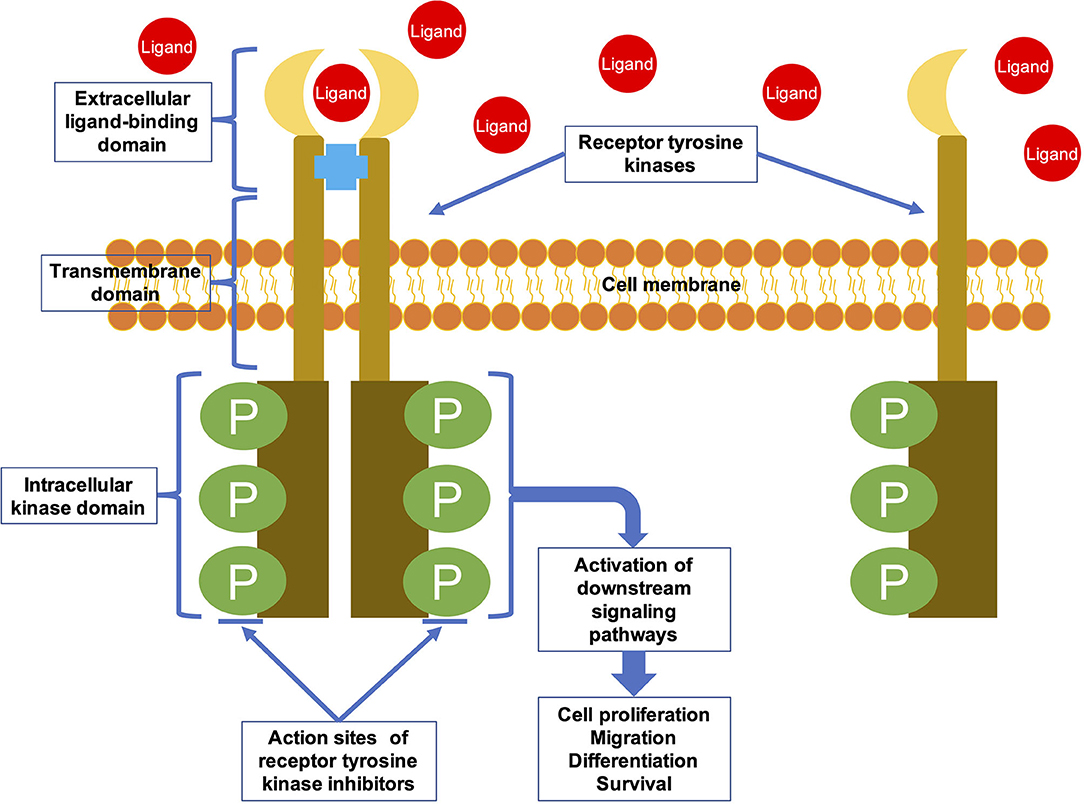
Figure 1. The general structural characteristics and activation mechanism of an RTK. RTKs are transmembrane glycoproteins that consist of an extracellular ligand-binding domain, a transmembrane domain, and an intracellular kinase domain. They are activated by ligand binding and then transduce the extracellular signal to the cytoplasm by phosphorylating tyrosine residues on the receptors themselves (autophosphorylation) and on downstream signaling proteins.
RTK irregularities can lead to a range of diseases. More than half of the oncogene and proto-oncogene products have RTK activities, and their abnormal expression leads to the disorder of cell regulation, which eventually leads to tumorigenesis (11). In addition, RTK irregularities are related to tumor neovascularization, invasion, metastasis, and chemotherapy resistance (4). Therefore, RTKs have become a focus of research on antitumor drugs. Most TKIs are small molecule inhibitors designed to interfere with binding of the RTK intracellular domain with ATP or other substrates, thereby inhibiting the catalytic activity of RTKs (Figure 1) (12, 13). Many of the TKIs inhibit several RTKs as the intracellular domain is relatively conserved among RTKs (14). The targeted drugs that showed efficacy in osteosarcoma treatment are these multi-target TKIs (15).
In this review, we first summarize the TKIs that have shown results in registered clinical trials of osteosarcoma treatment. Further, we compare and discuss the targets of these TKIs. This comprehensive review will be a valuable resource for further research on the use of TKIs in osteosarcoma treatment.
TKIs Reported in Registered Clinicaltrials for Osteosarcoma Treatment
TKIs reported in registered clinical trials on patients with osteosarcoma that have been conducted so far are listed in Table 1. Clinical trials were included herein only if: (1) they were prospective clinical trials, (2) they were registered with ClinicalTrials.gov, (3) their subjects were osteosarcoma patients, (4) the administered pharmacotherapy exclusively included TKIs, with no other drugs, and (5) those with detailed results were retrievable on https://pubmed.ncbi.nlm.nih.gov. There are retrospective studies on other TKIs (pazopanib and sunitinib) in the treatment of osteosarcoma (35, 36). We have not listed them here as retrospective nature is a limitation of such studies.
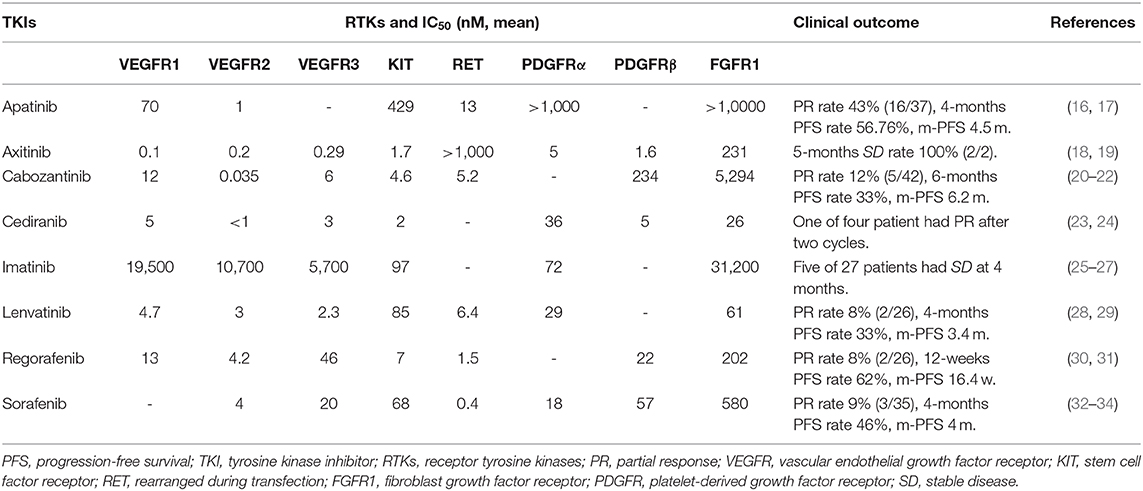
Table 1. Targets and clinical outcomes of TKIs which have shown results in registered clinical trials of osteosarcoma treatment.
Apatinib
Apatinib (Figure 2) was approved for the treatment of metastatic or advanced gastric cancer in China in 2014 (37). Because of its low price, it is widely used off-label in China for treating various malignant tumors, including osteosarcoma (17, 37, 38). Apatinib has fewer confirmed targets compared to other multi-target TKIs; these targets include vascular endothelial growth factor receptor-1 (VEGFR1, IC50 = 70 nM), VEGFR2 (1 nM), rearranged during transfection (RET, 13 nM), stem cell factor receptor (KIT, 429 nM), and v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC, 530 nM) (Table 1) (16).
Figure 2. The chemical structure of protein tyrosine kinases.
In the treatment of osteosarcoma, preclinical studies have shown that apatinib promotes apoptosis and autophagy through VEGFR2/STAT3/BCL-2 signaling pathways in osteosarcoma cells (39), and it inhibits invasion, migration, and PD-L1 expression in osteosarcoma cells (40). Apatinib also attenuates doxorubicin-induced cancer stemness and cancer cell migration of osteosarcoma cells by inhibiting Sox2 (41). Only one phase II trial has been conducted on patients with advanced osteosarcoma (17). In this open-label clinical trial, 37 patients with advanced or metastatic osteosarcoma after failure of standard chemotherapy, received 500 or 750 mg of apatinib once daily in accordance with their body surface area until unacceptable toxicity or disease progression was observed. Consequently, the 4-months PFS was 57% among the apatinib-treated patients, with a median PFS of 4.5 months, and a marked 43% partial response (PR) rate. Doses were reduced or interrupted among 25 of 37 (67%) patients, owing to drug toxicity. The most common grade 3–4 adverse events (AEs) were pneumothorax (16%), wound dehiscence (10%), proteinuria (8%), diarrhea (8%), and palmar-plantar erythrodysesthesia syndrome (8%). No treatment-related deaths were observed. The PR rate in this trial was significantly higher than that reported by retrospective studies (42, 43). It may be due to the high dose of apatinib, which was 750 mg per day in the registered trial, compared with 500 mg per day in retrospective studies. In conclusion, apatinib is an important and effective TKI in osteosarcoma treatment.
Axitinib
Axitinib (Figure 2) is an orally administered TKI of VEGFR1 (0.1 nM), VEGFR2 (0.2 nM), VEGFR3 (0.1–0.3 nM), platelet-derived growth factor receptor-α (PDGFRα, 5.0 nM), PDGFRβ (1.6 nM), Colony stimulating factor 1 receptor (CSF-1R, 73 nM), KIT (1.7 nM), fibroblast growth factor receptor (FGFR1, 231 nM), and RET (>1,000 nM) (Table 1) (18). Axitinib was developed for the treatment of many solid malignancies. It was approved by the Food and Drug Administration (FDA) of the United States to treat advanced renal cell carcinoma (RCC) after failure of other systematic therapies in 2012 (44).
There is no preclinical evidence of axitinib in the treatment of osteosarcoma, and only one clinical trial of axitinib in the treatment of osteosarcoma (19). This phase 1 and pilot consortium trial was about the safety and efficacy of axitinib in adolescents with recurrent or refractory solid tumors. In this trial, 19 patients received axitinib twice daily in continuous 28-days cycles. The most common (>20%) grade 1–2 AEs during treatment included anorexia, anemia, diarrhea, fatigue, hypertension, and nausea. Non-hematological toxicity of grade ≥3 AEs included elevated serum lipase levels and hypertension. Drug exposure and dosage were not associated with hypertension. Five patients presented with stable disease (SD) as the optimal response, including two osteosarcoma patients. One patient with alveolar soft part sarcoma presented a partial response. This result suggests that axitinib has limited efficacy in osteosarcoma compared to other TKIs listed in Table 1. Perhaps that is why there have been few studies on axitinib for osteosarcoma.
Cabozantinib
Cabozantinib (Figure 2) is a fairly multiple target TKI with activity not only for VEGFR2 (0.035 nM) but also for the N-methyl-N0-nitroso-guanidine human osteosarcoma transforming gene RTK (MET, 1.3 nM), KIT (4.6 nM), RET (5.2 nM), VEGFR3 (6 nM), anexelekto RTK (AXL, 7 nM), VEGFR1 (12 nM), and PDGFRβ (234 nM) (21, 22). Registered clinical trials of cabozantinib for prostate (45), lung (46), renal cell (47), and thyroid cancers have been completed (48). It was approved by FDA for the treatment of advanced or metastatic medullary thyroid cancer, RCC and hepatocellular carcinoma (HCC) (49).
In osteosarcoma, preclinical studies have shown that cabozantinib is able to inhibit proliferation and migration of osteosarcoma cells through the ERK and AKT signaling pathways (50). The French sarcoma group had carried out a multicenter, phase II trial involving patients with advanced or metastatic osteosarcoma after failure of other systemic therapy (20). Ninety osteosarcoma patients received cabozantinib orally every day continuous 28-days cycles until unacceptable toxicity or disease progression was observed. The median follow-up duration was 31 months among osteosarcoma patients. Forty-two (93%) osteosarcoma patients were assessable for treatment efficacy through histological and radiological analyses. Five (12%) osteosarcoma patients presented a PR and the median PFS was 6.2 months. The most frequent grade 3 or 4 AEs were hypophosphatemia, increased aspartate aminotransferase levels, palmar-plantar syndrome, pneumothorax, and neutropenia. No treatment-related deaths occurred. The longer median-PFS results represent the best achievement to date in the treatment of osteosarcoma by TKIs.
Cediranib
Cediranib (Figure 2) is an oral multi-target TKI with targets including VEGFR1 (5 nM), VEGFR2 (<1 nM), VEGFR3 (3 nM), KIT (2 nM), PDGFRβ (5 nM), FGFR-1 (26 nM), and PDGFRα (5.0 nM) (Table 1) (23). Cediranib has shown promising activity in a series of malignancies (51), including alveolar soft-part sarcoma, in preclinical models and in clinical trials (52); however, these results are not sufficient to get approval for its routine use until now.
Just like axitinib, there is no preclinical evidence of cediranib being efficient to treat osteosarcoma, and only one phase I trial has been conducted on patients with osteosarcoma (24). In this trial, 16 patients were recruited. Grade 3 or 4 AEs included nausea, vomiting, fatigue, hypertension, and prolonged corrected QT interval. Grade 1 or 2 AEs included palmar-plantar erythrodysesthesia, left ventricular dysfunction, weight loss, elevated thyroid stimulating hormone levels, and headache. One of four patients presented with minor PR as the optimal response. The results were similar to those of axitinib. This may be because the two drugs have similar targets and target sensitivity (Table 1).
Imatinib
Imatinib (Figure 2) is significantly different from the other TKIs presented in Table 1. The main difference is that imatinib does not affect VEGFR1 (19,500 nM), VEGFR2 (10,700 nM), VEGFR3 (5,700 nM), and FGFR1 (31,200 nM). Its sensitive targets include KIT (97 nM), PDGFRα (72 nM), and CSF-1R (291 nM) (25). Imatinib was approved by FDA for the treatment of chronic myeloid leukemia and advanced or metastatic gastrointestinal stroma tumors (GIST) (53, 54).
In preclinical studies, imatinib inhibits proliferation of osteosarcoma cells and inhibits tumor growth in preclinical murine models of osteosarcoma (55). However, another study suggests that imatinib is effective in osteosarcoma treatment only when imatinib is combined with another therapeutic intervention, such as induction of cellular stress, that renders cell survival dependent on PDGF signaling (56). In a phase II multicenter trial on patients with osteosarcoma administered with imatinib (27), five of 27 osteosarcoma patients achieved SD at 4 months after treatment initiation, and various genes in the KIT/PDGFR pathway were either aberrantly expressed or mutated, suggesting that imatinib was not effective against advanced-stage osteosarcoma. This undesirable outcome has led to the exclusion of imatinib from clinical treatment options for osteosarcoma.
Lenvatinib
Lenvatinib (Figure 2) is an oral multi-target TKI with targets including VEGFR1 (4.7 nM), VEGFR2 (3 nM), VEGFR3 (2.3 nM), PDGFRα (29 nM), KIT (85 nM), FGFR-1 (61 nM), and RET (6.4 nM) (Table 1) (28). Phase I trials of lenvatinib were conducted simultaneously worldwide, for the treatment of thyroid cancer, endometrial cancer, RCC, melanoma, colon cancer, and sarcoma (57). Lenvatinib is approved by FDA for the treatment of advanced HCC, radioiodine-refractory differentiated thyroid cancer, and advanced RCC (58).
There are no preclinical studies of lenvatinib in osteosarcoma. An ongoing clinical trial of lenvatinib in relapsed osteosarcoma reported initial results (29). In this trial, patients were aged 2–25 years, and had undergone <2 previous targeted therapies. Twenty-six patients received 14 mg/m2/day of lenvatinib. The most common AEs were diarrhea, proteinuria and hypothyroidism, and the most common grade 3 or 4 AEs were dyspnea and back pain. The 4-months PFS was 33%, the median PFS was 3.4 months, and the PR rate was 8%. This result indicates that lenvatinib has activity against osteosarcoma, and further studies will most probably prove this.
Regorafenib
Regorafenib (Figure 2) is an orally administered multi-target TKI that targets VEGFR1 (13 nM), VEGFR2 (4.2 nM), VEGFR3 (46 nM), PDGFRβ (22 nM), KIT (7 nM), FGFR-1 (202 nM), and RET (1.5 nM) (Table 1) (30). Regorafenib is the first TKI that showed efficacy in advanced or metastatic colorectal cancer (59), and was approved in 2012 by FDA for this indication. In addition, regorafenib is approved by FDA for the treatment of GIST and HCC (60, 61). It has also been examined in several different tumor types, including relapsed glioblastoma (62), RCC and soft-tissue sarcoma (63, 64). And additional phase II trials in other solid malignant tumors are ongoing (65).
In vitro experiments have shown that regorafenib inhibits osteosarcoma cell growth by inducing apoptosis of cells (66). Two registered clinical trials have been reported on regorafenib in osteosarcoma. The first is a non-comparative, placebo-controlled phase II trial about the efficacy and safety of regorafenib in metastatic osteosarcoma patients (31). In this trial, 38 patients with metastatic osteosarcoma after failure of one or two previous lines of chemotherapy were enrolled. Patients were randomly assigned to receive either regorafenib or a matching placebo. Non-progression was observed in 65% (17 of 26) patients in the regorafenib group. The 12-weeks PFS was 62%, the median PFS was 16.4 weeks, and the PR rate was 8%. The most common grade 3 or 4 AEs included hypertension, hand–foot skin reactions, fatigue, hypophosphatemia, and chest pain. No treatment-related deaths occurred. Another clinical trial was similar to the above (3). This trial enrolled 42 patients with advanced or metastatic osteosarcoma, after failure of at least one prior line of therapy. Study enrolment was terminated early, upon review by the data safety monitoring committee. The median PFS was significantly higher in the regorafenib group than that in the placebo group. And the efficacy and safety were similar to the above trial. These two studies met their primary end point, demonstrating the efficacy and safety of regorafenib in patients with advanced or metastatic osteosarcoma after failure of prior therapy.
Sorafenib
As a multi-target TKI, the targets of sorafenib (Figure 2) include VEGFR2 (4 nM), VEGFR3 (20 nM), PDGFRβ (22 nM), KIT (68 nM), FGFR-1 (580 nM), and RET (0.4 nM) (Table 1) (32, 33). In 2005, sorafenib was approved by FDA for the treatment of advanced RCC (67). Moreover, it was the first TKI approved for patients with radioiodine-refractory differentiated thyroid cancer (68).
Sorafenib is the most studied multi-target TKI in osteosarcoma. Preclinical studies have found that sorafenib blocks tumor growth, tumor angiogenesis, and tumor metastatic potential of osteosarcoma in preclinical murine models (69). Another study has shown that sorafenib targets the PTKs RET and VEGFR2 and suppresses cell proliferation (31). Two registered clinical trials have been reported on sorafenib in osteosarcoma. The first is a phase II trial of sorafenib in patients with advanced or metastatic osteosarcoma after failure of standard treatment (34). In this trial, 35 patients were enrolled, and the 4 months PFS rate was 46%, the median PFS was 4 months, and the PR rate was 9% (3/35). Sorafenib treatment was reduced or briefly interrupted in 46% patients owing to toxicity. Another clinical trial yielded similar results (70). These two clinical trials are milestone achievements for the treatment of advanced osteosarcoma, because they are the first multicenter prospective clinical trials to demonstrate the efficacy of TKIs in osteosarcoma.
Which RTK is the Key Target?
The targets of the aforementioned multi-target TKIs, namely RTKs, are listed in Table 1 and Figure 3. It can be seen that only KIT is the common target of all multi-target TKIs mentioned above. Other RTKs that are targets of majority TKIs include VEGFRs, RET, PDGFRs, and FGFR1. To determine which RTK is the key target for osteosarcoma, we continue to review the relevant literature.
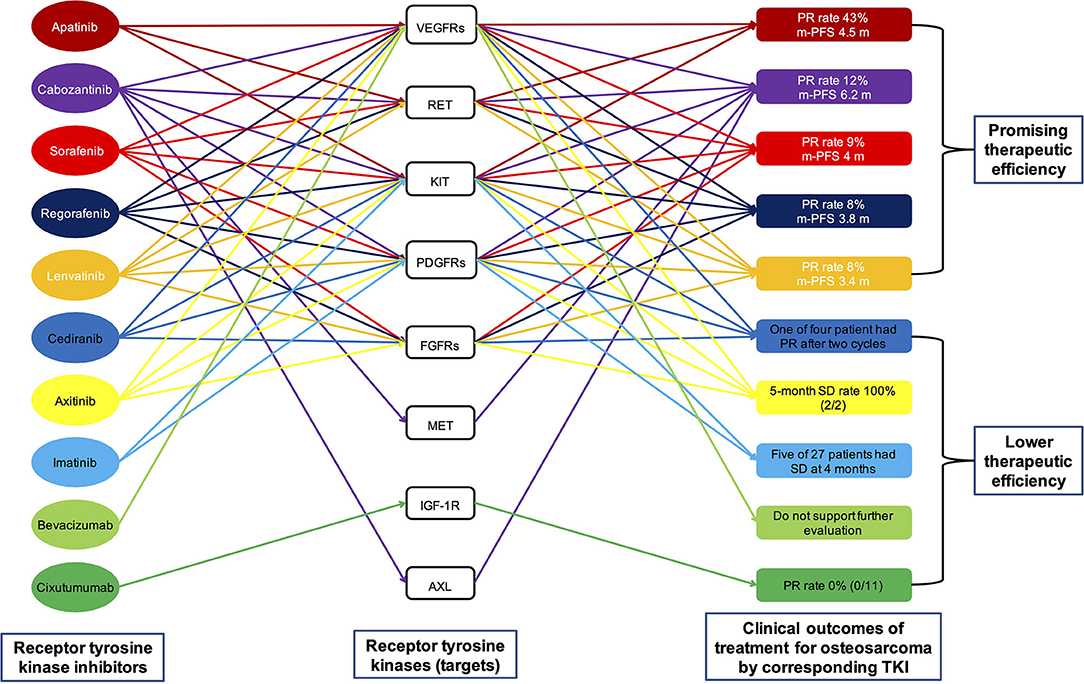
Figure 3. A visual interaction map of the different targets of the different drugs. Preclinical data indicate that all eight targets shown in the figure play an important role in the progression of osteosarcoma. However, clinical trials in osteosarcoma have demonstrated the low efficacy of single-target therapy by inhibiting VEGFs (by bevacizumab), KIT and PDGFRs (by imatinib), and IGF-1R (by cixutumumab). The results of these clinical trials suggest that the inhibition of one type of targets in the treatment of osteosarcoma is not feasible. PFS, progression-free survival; TKI, tyrosine kinase inhibitor; PR, partial response; VEGFR, vascular endothelial growth factor receptor; KIT, stem cell factor receptor; RET, rearranged during transfection; FGFR1, fibroblast growth factor receptor; PDGFR, platelet-derived growth factor receptor; SD, stable disease.
VEGFRs
VEGF is a highly specific vascular endothelial growth factor, which can promote vascular permeability, extracellular matrix degeneration, endothelial cell migration, proliferation, and angiogenesis (71). VEGF signaling pathways have three RTKs, namely VEGFR1, VEGFR2, and VEGFR3 (72). VEGFR1 has the strongest binding affinity to VEGF and more thorough phosphorylation, but it has less effect on the activation of intracellular signaling intermediates (73). VEGFR2 is the dominant receptor that mediates the biological effect and pro-angiogenic functions of VEGF, which is closely related to cell division and chemotaxis (74). And this pathway has been under research focus on priority for the development of antiangiogenic therapies (75). VEGFR3 exists only in lymphoid endothelial cells in adults. In a variety of cancer types, VEGFR3 is associated with tumor lymphangiogenesis, lymph node invasion and metastasis (76). Currently, anti-angiogenic therapies targeting VEGF/VEGFRs signaling pathway include anti-VEGF antibodies (Bevacizumab) (77), anti-VEGFR2 antibodies (ramucirumab) (78), and TKIs (14). Both bevacizumab and ramucirumab have been approved by FDA for use in therapy of a series of malignancies. At present, more than a dozen types of TKIs that target VEGFRs have been approved by FDA for the treatment of cancer therapy.
Regarding osteosarcoma, a systematic review showed that high VEGF expression in osteosarcoma patients were associated with lower PFS rate. In addition, osteosarcoma patients with high VEGF expression were associated with shorter overall survival (OS) (79). These findings suggested that targeting the VEGF signaling pathway may be an effective treatment for osteosarcoma. However, this is not the case. The clinical outcomes of targeting VEGF/VEGFR signaling pathways in osteosarcoma are not always favorable. The clinical efficacy of bevacizumab in osteosarcoma was limited (Figure 3) (80), and there is not even a single study on the clinical treatment of osteosarcoma with ramucirumab until now. As shown in Table 1 and Figure 3, TKIs—including apatinib, cabozantinib, lenvatinib, regorafenib, and sorafenib—having better therapeutic effect on osteosarcoma include VEGFRs as targets, whereas TKI (imatinib) whose targets did not include VEGFRs had poor therapeutic effect on osteosarcoma. This seems to prove that VEGFRs are essential for TKIs to be effective in osteosarcoma treatment. However, not all multi-target TKIs that target VEGFRs are effective in treating osteosarcoma; for example, axitinib and cediranib (Table 1 and Figure 3). In conclusion, therapy that targets VEGF/VEGFR is an important and effective anticancer method. However, the efficiency of inhibiting just VEGF/VEGFR in osteosarcoma treatment is limited.
KIT
The KIT is a kind of RTK, and its ligand is stem cell factor (81). KIT also contains an extracellular domain, a transmembrane domain, and an intracellular domain (82). After binding with a ligand, KIT induces multiple signaling cascades, which are essential for biological processes including cell differentiation, proliferation, and migration (83). KIT plays important roles in the functioning of nervous system, kidney, gastrointestinal tract, bones, lungs, and pancreas (81). Abnormal expression of KIT has been documented in various neoplasms (82, 84). These results have encouraged researchers to work on the use of KIT as a potential target in the treatment of malignant tumors.
Although KIT has been widely studied in neoplastic and non-oncological diseases, there are few preclinical and clinical studies on KIT in osteosarcoma. In 2007, a study showed that tumor specimens from 20 out of 100 patients with osteosarcoma were KIT-positive and that OS were not different between patients with and without KIT expression (85). In 2011, another study found that KIT was expressed in 46.15% patients with osteosarcoma and that KIT-positive tumors had worse response to chemotherapy (86). The results of both studies point to the possibility that KIT is not a key player in proliferation and metastasis of osteosarcoma. Although all the TKIs effective in osteosarcoma listed in Table 1 and Figure 3 include KIT as a target, there are still three TKIs (axitinib, cediranib, and imatinib) that include KIT as a target but are not effective in osteosarcoma. Based on the limited evidence described above, we surmise that KIT is not a key RTK in osteosarcoma treatment and that targeting KIT alone in osteosarcoma treatment is inefficient.
RET
The RTK RET is coded by the gene RET (87). As an RTK, RET also has characteristic composition of extracellular domain, transmembrane domain, and intracellular kinase domain (88). RET binds with the ligand–coreceptor complex of glial cell line-derived neurotrophic factor family ligands and glial cell line-derived neurotrophic factor family receptor alpha (89). The RET-bound complex then leads to phosphorylation of intracellular kinase domain, which activates downstream signaling pathways. RET signals are associated with many RET-mediated functions (90). RET mutations have been documented in a variety of organ systems (91–93). Abnormal expression of RET has been observed in many malignancies (94, 95). These results have led to the use of RET as a capable target in cancer therapy. Several multi-target TKIs targeting RET are approved by the FDA for cancer therapy and non-oncologic disease treatment (94). Selective RET inhibitors selpercatinib and pralsetinib are undergoing clinical trials respectively, with preliminary results demonstrating partial response and low incidence of serious AEs (96, 97).
The role of RET in osteosarcoma has been far less studied than in other malignancies. Using phosphoproteomic screening, RET was first identified in vitro as a receptor that can promote behavior of metastatic osteosarcoma cells and is thought to be a potential therapeutic target for osteosarcoma (98). It has also been confirmed that the overexpression of RET is associated with chemotherapeutic resistance induced by cisplatin and bortezomib, and with the increase of stem cell-like properties of osteosarcoma (99–101). There are many clinical trials on the role of RET in osteosarcoma. As shown in Table 1 and Figure 3, all multi-target TKIs (apatinib, cabozantinib, lenvatinib, regorafenib, and sorafenib) with high efficiency in osteosarcoma are inhibitors of RET. Further, the three multi-target TKIs (axitinib, cediranib, and imatinib) with low efficiency in osteosarcoma do not inhibit RET. In conclusion, RET is an important and potentially critical target for osteosarcoma treatment, and it may be equally important as VEGFRs. The role of RET in osteosarcoma has not been studied in detail and warrants further study.
PDGFRs
PDGF is named for its origin in platelets. It exists in normal physiological state in platelets. When blood clots, PDGF is released by disintegrated platelets and activated (102). PDGF signaling pathway consists of four ligands and two receptors (PDGFR-α and PDGFR-β) (103). Ligand-induced receptor dimerization of PDGF receptors leads to autophosphorylation of the receptors (103, 104). A series of downstream signaling molecules bind to specific phosphotyrosines in the intracellular domain of PDGFRs and mediate intracellular signal transduction (105, 106). Activation of these pathways leads to cell proliferation and migration (103, 107). PDGFs/PDGFRs are expressed in many normal human cells and are involved in the normal development and physiological process of important organs (105). The PDGFs/PDGFRs pathway play an important role in the development and metastasis of cancers (104). Given the association between PDGF over-activity and malignant tumors, different kinds of PDGF inhibitors have been developed. These inhibitors include the fully human IgG1 monoclonal antibody against PDGFRα named olaratumab (108), and multi-target TKIs such as cediranib, imatinib, and sorafenib (Table 1).
Many studies have confirmed that PDGFs/PDGFRs signaling pathway play an important role in the proliferation and migration of osteosarcoma (109, 110). However, the expression of PDGFs/PDGFRs in osteosarcoma has a significant heterogeneity (111), and the association of PDGFs expression with osteosarcoma prognosis has not yet been established. Some studies have suggested that high expression of PDGFs is associated with poor prognosis of osteosarcoma patients (111, 112). On the contrary, other studies have shown that OS and PFS analysis was not different between osteosarcoma patients on the basis of PDGFs/PDGFRs expression level (113, 114). The use of PDGFR inhibitor alone in clinical trials is not efficacy, whereas its combination with other drugs seems more promising (27, 115–117). As shown in Table 1 and Figure 3, axitinib and cediranib are effective inhibitors of PDGFRs, yet their efficacy in osteosarcoma is as poor as imatinib. Apatinib, which is more effective in osteosarcoma, does not target PDGFRs. The PDGFs/PDGFRs signaling pathway may not to be the driver of osteosarcoma, especially in the presence of other signaling pathways such as VEGFs. Therefore, the application of PDGFs/PDGFRs inhibitor would be expected to be effective only if used in combination with another drug.
FGFRs
FGFRs are a subfamily of RTKs, with four subtypes including FGFR1, FGFR2, FGFR3, and FGFR4 (118). As RTKs, FGFRs also has characteristic composition of extracellular domain, transmembrane domain, and intracellular kinase domain that transduces downstream signaling (119). The ligands of FGFRs comprise 22 growth factors, which are classified into seven subgroups (120). FGFs/FGFRs signaling pathway are activated through FGFs binding to FGFRs, which depends on heparan sulfate, a glycosaminoglycan (121). Activation of FGFRs results in receptor dimerization, transphosphorylation of receptor kinase domains, and activation of downstream signaling pathways (122, 123). FGFs/FGFRs signaling pathway play an important role in many physiological processes such as embryogenesis, tissue repair, wound healing, and inflammation (119). Aberrant activation of the pathway due to FGFRs amplification, mutations, or gene fusions has been observed in many malignancies (124). Currently, several multi-target or selective TKIs that target FGFRs have been approved by FDA for the treatment of malignancies (125, 126).
Regarding osteosarcoma, a study revealed low expression of FGFR-2 and FGFR-3 across standard and patient-derived osteosarcoma cells (127). Another study, which included 352 osteosarcoma samples, detected FGFR1 amplification in 18% of the chemotherapy resistant osteosarcoma patients (128). This suggests that FGFR1 amplification is associated with poor response of osteosarcoma to chemotherapy. Another study showed that deregulated FGFRs signaling plays an important role in osteosarcoma formation and the development of lung metastases (129). There are no clinical trials of selective FGFR inhibitors in osteosarcoma. There are many clinical trials of multi-target FGFR TKIs in osteosarcoma. As shown in Table 1 and Figure 3, cediranib, a multi-target FGFR1 TKI, is not effective in the osteosarcoma treatment. However, apatinib and cabozantinib, which do not target FGFR1, can effectively treat osteosarcoma. To sum up, it is clear that FGFR signaling pathways might be relevant but is unimportant in osteosarcoma treatment.
Other RTKs in Osteosarcoma
By summarizing the clinical trials of various multi-target TKIs in osteosarcoma and their targets, we have identified the above five RTKs that may be associated in the treatment of osteosarcoma. However, the key RTKs for osteosarcoma seem to be more than that.
MET
MET kinase is a heterodimer protein (130). As an RTK, MET contains a characteristically extracellular domain, transmembrane domain, and intracellular kinase domain (131). The binding of its ligand, hepatocyte growth factor, results in MET dimerization, transphosphorylation of receptor kinase domains, and activation of downstream signaling pathways (132). MET signaling pathway play an important role in many physiological processes (132, 133). Aberrant activation of the pathway due to MET amplification, mutations, or gene fusions has been observed in many malignancies and degenerative diseases (133, 134). Several small-molecule inhibitors and monoclonal antibodies of MET are in the development (134).
Regarding osteosarcoma, a study has shown that overexpression of the MET caused primary osteoblasts to transform into osteosarcoma cells (135). Another study suggested that MET receptor is aberrantly expressed in almost all the human osteosarcoma cells (136). Other studies also found that inhibiting the MET signaling pathway can increase apoptotic rate and suppress the migration, proliferation, and invasion of osteosarcoma cells (137, 138). There is no report about the treatment of osteosarcoma with a selective MET inhibitor. However, the multi-target MET inhibitor cabozantinib has a high efficiency in osteosarcoma (Table 1 and Figure 3) (20). All this evidence suggests that MET may be one of the drivers of osteosarcoma.
IGF-1R
IGF-1R is a hetero-tetrameric transmembrane glycoprotein with RTK activity, which is ubiquitously expressed in various human cell types and tissues (139). As an RTK, IGF-1R has a typical extracellular domain, transmembrane domain, and intracellular tyrosine kinase domain (136, 140). It plays an important role in growth and various physiological functions, including differentiation, development, apoptosis, and metabolism by binding to its ligand IGF-1 (141). Aberrant activation of IGF-1 has been observed in many malignancies (142). There are many new drugs being developed to target IGF/IGF-1R signaling pathway (139).
Regarding osteosarcoma, a study showed that the relative expression of IGF-1R in osteosarcoma was significantly higher than that in corresponding non-cancerous bones. The expression of IGF-1R is closely related to distant metastasis and prognosis of osteosarcoma. Higher IGF-1R expression in osteosarcoma patients is associated with poorer survival (143). Extensive screening and validation strategies identified IGF-1R as one of the specific RTKs that can activated and promote the phenotype of osteosarcoma cells in vitro (98). However, despite encouraging preclinical data, clinical trials of IGF-1R inhibitors for osteosarcoma have not yielded satisfactory results. In a phase II trial of the IGF-1R monoclonal antibody R1507 in patients with osteosarcoma, only two PRs were observed in 38 patients, with median PFS of 5.7 weeks (144). In another phase II trial of the IGF-1R antibody cixutumumab in children with osteosarcoma, no PRs were observed in 11 patients (Figure 3) (145). In general, IGF-1R might be one of the drivers of osteosarcoma, but inhibiting it alone is clearly not an option.
AXL
AXL kinase contains a characteristically extracellular domain, transmembrane domain, and intracellular domain (146). Upon high-affinity binding to its ligand, growth arrest-specific protein 6 (GAS6), undergoes homodimerization, and subsequent transautophosphorylation within the intracellular kinase domain, thus activating downstream signaling pathways (147). The Gas6/AXL signaling pathway play an important role in many physiological processes (148). The aberrant expression of Gas6/AXL has been observed in many malignancies (147). Different small-molecule inhibitors and monoclonal antibodies of AXL have been developed (148).
There have been identified AXL as one of the specific RTKs that can activated and promote the phenotype of osteosarcoma cells in vitro (149). Higher AXL expression in osteosarcoma patients is associated with poorer survival (150). Knockdown of AXL in osteosarcoma cells leads to decreased proliferation and increased apoptosis (151). In addition, miR-199a-3p and lncRNA DANCR regulate the progression of osteosarcoma through targeting AXL (152). A retrospective study showed that one in five patients with osteosarcoma treated with an AXL inhibitor, sunitinib, showed PR (36). In an phase II trial of the AXL inhibitor cabozantinib in osteosarcoma, the 6-months PFS rate was 33%, the median PFS was 6.2 months, and the PR rate was 12% (Figure 3) (20). It should be noted that both sunitinib and cabozantinib are multi-target TKIs. At present, there is no report about the efficacy of single-target drugs against AXL in osteosarcoma. From the above evidence, we suspect that AXL is also a driver of osteosarcoma.
Discussion
In this review, we first summarized the multitarget TKIs that were reported in registered clinical trials in patients with osteosarcoma. We compared the targets of these TKIs and found that VEGFRs and RET may be the key RTKs for the treatment of osteosarcoma. We further showed that KIT, PDGFRs, and FGFRs might be relevant but unimportant RTKs for osteosarcoma. In addition, we reviewed the literature and found that MET, IGF-1R, and AXL may also be relevant targets for the treatment of osteosarcoma.
Preclinical studies indicate that all eight RTKs mentioned above play an important role in the progression of osteosarcoma. However, clinical trials in osteosarcoma have demonstrated the low efficacy of single-target therapy by inhibiting VEGFs (by bevacizumab) (80), KIT and PDGFRα (by imatinib) (115), and IGF-1R (by cixutumumab or R1507) (Figure 3) (144, 145). The results of these clinical trials indicate that the inhibition of one type of RTKs in the treatment of osteosarcoma is not feasible. The molecular mechanism indicates that there is a large amount of crossover and overlap among the downstream signaling pathways of RTKs. For example, SRC kinase, which plays a key role in the development of osteosarcoma (153), can be activated, respectively by VEGFRs (154), KIT (81), RET (90), PDGFRs (103), IGF-1R (155), and AXL (148). Therefore, it can be determined that inhibition of one type of RTKs is not effective in the treatment of osteosarcoma. It is necessary to inhibit several key RTKs simultaneously in order to achieve a breakthrough in the treatment of osteosarcoma. For example, cabozantinib, which can simultaneously inhibit VEGFRs, KIT, RET, PDGFRβ, MET, and AXL (21, 22), has the best effect in the treatment of osteosarcoma, with a median PFS of 6.2 months, compared to other TKIs listed in Table 1 and Figure 3 (20).
This review serves as a good reference in the field of RTK-targeted osteosarcoma treatment. First, this study can provide reference for new drug research and development. For example, researchers have discontinued studies on several kinds of IGF-1R antibodies because of poor clinical trial results. However, these single-target TKIs are ideal for combination therapy. Researchers should try to use these single-target drugs in combination with other drugs. Secondly, it provides reference for drug screening. Researchers can roughly determine whether a drug is effective for osteosarcoma based on the literature review of the eight RTKs mentioned above. Finally, according to this review, we can treat osteosarcoma precisely to reduce the side effects and improve the curative effect. For example, the combination of the single-target drugs of abovementioned eight RTKs can not only maximize the efficacy but also avoid the side effects associated with ineffective targets.
Obviously, there is still a lot of important works need to be done. Firstly, there are errors in IC50 values of some targets of each drug (21, 22, 32). These errors will mislead researchers while selecting drugs and cause confusion during follow-up research. Therefore, it is necessary to correct the IC50 values of each target of each TKI. Secondly, controlled clinical trials are needed to compare the effectiveness of various TKIs in the treatment of osteosarcoma. There is little work in this area. The current clinical trials of TKIs in osteosarcoma are all about one drug. Only controlled clinical trials can accurately compare the efficiency differences of each TKIs in osteosarcoma. Finally, this study is only a preliminary summary of these RTKs. Relevant signaling pathways and mechanisms still need to be studied further.
We can say that the current use of multi-target TKIs in the treatment of osteosarcoma is only a stopgap. The main drawback of this stopgap approach is the side effects associated with ineffective targets. With the emergence of various selective TKIs, the combination therapy using different selective (or single-target) TKIs in osteosarcoma will be the future trend.
Conclusion
Currently, TKIs with promising therapeutic effect for osteosarcoma include apatinib, cabozantinib, lenvatinib, regorafenib, and sorafenib. Key RTKs for osteosarcoma treatment may include VEGFRs and RET. The receptors MET, IGF-1R, AXL, PDGFRs, KIT, and FGFRs might be relevant but unimportant RTKs for osteosarcoma. Inhibition of one type of RTK is not effective in the treatment of osteosarcoma. It is necessary to inhibit several relevant RTKs simultaneously in order to achieve a breakthrough in osteosarcoma treatment.
Author Contributions
ZT and WY: conceptualization. XN: methodology. ZT: software, writing, and original draft preparation. WY: resources. XN and WY: writing, review, and editing. All authors have read and agreed to the published version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Misaghi A, Goldin A, Awad M, Kulidjian AA. Osteosarcoma: a comprehensive review. Sicot J. (2018) 4:12. doi: 10.1051/sicotj/2017028
2. Marina NM, Smeland S, Bielack SS, Bernstein M, Jovic G, Krailo MD, et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol. (2016) 17:1396–408. doi: 10.1016/s1470-2045(16)30214-5
3. Davis LE, Bolejack V, Ryan CW, Ganjoo KN, Loggers ET, Chawla S, et al. Randomized double-blind phase II study of regorafenib in patients with metastatic osteosarcoma. J Clin Oncol. (2019) 37:1424–31. doi: 10.1200/jco.18.02374
4. Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang YS. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. (2018) 17:36. doi: 10.1186/s12943-018-0801-5
5. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. (2002) 298:1912–34. doi: 10.1126/science.1075762
6. Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. (2002) 27:514–20. doi: 10.1016/s0968-0004(02)02179-5
7. Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. (2010) 13:1–14. doi: 10.1007/s10456-009-9160-6
8. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
9. Gschwind A, Fische OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. (2004) 4:361–70. doi: 10.1038/nrc1360
10. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. (2000) 69:373–98. doi: 10.1146/annurev.biochem.69.1.373
11. Drake JM, Lee JK, Witte ON. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol. (2014) 34:1722–32. doi: 10.1128/mcb.01592-13
12. Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. (2005) 353:172–87. doi: 10.1056/NEJMra044389
13. Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. (2004) 303:1800–5. doi: 10.1126/science.1095920
14. Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. (2019) 12:5. doi: 10.1186/s13045-019-0718-5
15. Wilding CP, Elms ML, Judson I, Tan AC, Jones RL, Huang PH. The landscape of tyrosine kinase inhibitors in sarcomas: looking beyond pazopanib. Expert Rev Anticancer Ther. (2019) 19:971–91. doi: 10.1080/14737140.2019.1686979
16. Tian S, Quan H, Xie C, Guo H, Lu F, Xu Y, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. (2011) 102:1374–80. doi: 10.1111/j.1349-7006.2011.01939.x
17. Xie L, Xu J, Sun X, Tang X, Yan T, Yang R, et al. Apatinib for advanced osteosarcoma after failure of standard multimodal therapy: an open label phase ii clinical trial. Oncologist. (2019) 24:e542–50. doi: 10.1634/theoncologist.2018-0542
18. Choueiri TK. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr Opin Investig Drugs. (2008) 9:658–71. doi: 10.2174/157015908784533851
19. Geller JI, Fox E, Turpin BK, Goldstein SL, Liu X, Minard CG, et al. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: a Children's Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer. (2018) 124:4548–55. doi: 10.1002/cncr.31725
20. Italiano A, Mir O, Mathoulin-Pelissier S, Penel N, Piperno-Neumann S, Bompas E, et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2020) 21:446–55. doi: 10.1016/S1470-2045(19)30825-3
21. Roy S, Narang BK, Rastogi SK, Rawal RK. A novel multiple tyrosine-kinase targeted agent to explore the future perspectives of anti-angiogenic therapy for the treatment of multiple solid tumors: cabozantinib. Anticancer Agents Med Chem. (2015) 15:37–47. doi: 10.2174/1871520614666140902153840
22. Bowles DW, Kessler ER, Jimeno A. Multi-targeted tyrosine kinase inhibitors in clinical development: focus on XL-184 (cabozantinib). Drugs Today. (2011) 47:857–68. doi: 10.1358/dot.2011.47.11.1688487
23. Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave R, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. (2005) 65:4389–400. doi: 10.1158/0008-5472.Can-04-4409
24. Fox E, Aplenc R, Bagatell R, Chuk MK, Dombi E, Goodspeed W, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. (2010) 28:5174–81. doi: 10.1200/JCO.2010.30.9674
25. Waller CF. Imatinib mesylate. Recent Results Cancer Res. (2018) 212:1–27. doi: 10.1007/978-3-319-91439-8_1
26. Manley PW, Stiefl N, Cowan-Jacob SW, Kaufman S, Mestan J, Wartmann M, et al. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem. (2010) 18:6977–86. doi: 10.1016/j.bmc.2010.08.026
27. Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Meyers PA, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol. (2009) 27:3148–53. doi: 10.1200/JCO.2008.20.5054
28. Hussein Z, Mizuo H, Hayato S, Namiki M, Shumaker R. Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small-molecule, multitargeted tyrosine kinase inhibitor. Eur J Drug Metab Pharmacokinet. (2017) 42:903–14. doi: 10.1007/s13318-017-0403-4
29. Gaspar N, Casanova M, Sirvent F. Single-agent expansion cohort of lenvatinib (LEN) and combination dose-finding cohort of LEN + etoposide (ETP) + ifosfamide (IFM) in patients (pts) aged 2 to # 25 years with relapsed/refractory osteosarcoma (OS). J Clin Oncol. (2018) 36:abstr11527. doi: 10.1093/annonc/mdz283.009
30. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. (2011) 129:245–55. doi: 10.1002/ijc.25864
31. Duffaud F, Mir O, Boudou-Rouquette P, Piperno-Neumann S, Penel N, Bompas E, et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. (2019) 20:120–33. doi: 10.1016/s1470-2045(18)30742-3
32. Mao WF, Shao MH, Gao PT, Ma J, Li HJ, Li GL, et al. The important roles of RET, VEGFR2 and the RAF/MEK/ERK pathway in cancer treatment with sorafenib. Acta Pharmacol Sin. (2012) 33:1311–8. doi: 10.1038/aps.2012.76
33. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. (2004) 64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443
34. Grignani G, Palmerini E, Dileo P, Asaftei SD, D'Ambrosio L, Pignochino Y, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol. (2012) 23:508–16. doi: 10.1093/annonc/mdr151
35. Longhi A, Paioli A, Palmerini E, Cesari M, Abate ME, Setola E, et al. Pazopanib in relapsed osteosarcoma patients: report on 15 cases. Acta Oncol. (2019) 58:124–8. doi: 10.1080/0284186X.2018.1503714
36. Penel-Page M, Ray-Coquard I, Larcade J, Girodet M, Bouclier L, Rogasik M, et al. Off-label use of targeted therapies in osteosarcomas: data from the French registry OUTC'S (Observatoire de l'Utilisation des Therapies Ciblees dans les Sarcomes). BMC Cancer. (2015) 15:854. doi: 10.1186/s12885-015-1894-5
37. Scott LJ. Apatinib: a review in advanced gastric cancer and other advanced cancers. Drugs. (2018) 78:747–58. doi: 10.1007/s40265-018-0903-9
38. Liu G, Wang C, He Y, E M. Application effect of apatinib in patients with failure of standard treatment for advanced malignant tumours. BMC Pharmacol Toxicol. (2019) 20:61. doi: 10.1186/s40360-019-0362-2
39. Liu K, Ren T, Huang Y, Sun K, Bao X, Wang S, et al. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis. (2017) 8:e3015. doi: 10.1038/cddis.2017.422
40. Zheng B, Ren T, Huang Y, Guo W. Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem Biophys Res Commun. (2018) 495:1695–701. doi: 10.1016/j.bbrc.2017.12.032
41. Tian ZC, Wang JQ, Ge H. Apatinib ameliorates doxorubicin-induced migration and cancer stemness of osteosarcoma cells by inhibiting Sox2 via STAT3 signalling. J Orthop Translat. (2020) 22:132–41. doi: 10.1016/j.jot.2019.07.003
42. Tian Z, Gu Z, Wang X, Liu Z, Yao W, Wang J, et al. Efficacy and safety of apatinib in treatment of osteosarcoma after failed standard multimodal therapy: an observational study. Medicine. (2019) 98:e15650. doi: 10.1097/MD.0000000000015650
43. Zhu B, Li J, Xie Q, Diao L, Gai L, Yang W. Efficacy and safety of apatinib monotherapy in advanced bone and soft tissue sarcoma: an observational study. Cancer Biol Ther. (2018) 19:198–204. doi: 10.1080/15384047.2017.1416275
44. Keating GM. Axitinib: a review in advanced renal cell carcinoma. Drugs. (2015) 75:1903–13. doi: 10.1007/s40265-015-0483-x
45. Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. (2016) 34:3005–13. doi: 10.1200/jco.2015.65.5597
46. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. (2016) 17:1653–60. doi: 10.1016/s1470-2045(16)30562-9
47. Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. (2016) 17:917–27. doi: 10.1016/s1470-2045(16)30107-3
48. Cabanillas ME, de Souza JA, Geyer S, Wirth LJ, Menefee ME, Liu SV, et al. Cabozantinib as salvage therapy for patients with tyrosine kinase inhibitor-refractory differentiated thyroid cancer: results of a multicenter phase II international thyroid oncology group trial. J Clin Oncol. (2017) 35:3315–21. doi: 10.1200/jco.2017.73.0226
49. Leavitt J, Copur MS. FDA approved uses of cabozantinib. Oncology. (2019) 33:685004. Available online at: https://www.cancernetwork.com/view/fda-approveduses-cabozantinib
50. Fioramonti M, Fausti V, Pantano F, Iuliani M, Ribelli G, Lotti F, et al. Cabozantinib affects osteosarcoma growth through a direct effect on tumor cells and modifications in bone microenvironment. Sci Rep. (2018) 8:4177. doi: 10.1038/s41598-018-22469-5
51. Sahade M, Caparelli F, Hoff PM. Cediranib: a VEGF receptor tyrosine kinase inhibitor. Future Oncol. (2012) 8:775–81. doi: 10.2217/fon.12.73
52. Judson I, Morden JP, Kilburn L, Leahy M, Benson C, Bhadri V, et al. Cediranib in patients with alveolar soft-part sarcoma (CASPS): a double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol. (2019) 20:1023–34. doi: 10.1016/s1470-2045(19)30215-3
53. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. (2017) 376:917–27. doi: 10.1056/NEJMoa1609324
54. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. (2009) 373:1097–104. doi: 10.1016/s0140-6736(09)60500-6
55. Gobin B, Moriceau G, Ory B, Charrier C, Brion R, Blanchard F, et al. Imatinib mesylate exerts anti-proliferative effects on osteosarcoma cells and inhibits the tumour growth in immunocompetent murine models. PLoS ONE. (2014) 9:e90795. doi: 10.1371/journal.pone.0090795
56. Yamaguchi SI, Ueki A, Sugihara E, Onishi N, Yaguchi T, Kawakami Y, et al. Synergistic antiproliferative effect of imatinib and adriamycin in platelet-derived growth factor receptor-expressing osteosarcoma cells. Cancer Sci. (2015) 106:875–82. doi: 10.1111/cas.12686
57. Suyama K, Iwase H. Lenvatinib: a promising molecular targeted agent for multiple cancers. Cancer Control. (2018) 25:1073274818789361. doi: 10.1177/1073274818789361
58. Zschabitz S, Grullich C. Lenvantinib: a tyrosine kinase inhibitor of VEGFR 1-3, FGFR 1-4, PDGFRalpha, KIT and RET. Recent Results Cancer Res. (2018) 211:187–98. doi: 10.1007/978-3-319-91442-8_13
59. Andre T, Raymond E, de Gramont A. Regorafenib for metastatic colorectal cancer. Lancet. (2013) 381:1536–7. doi: 10.1016/s0140-6736(13)60975-7
60. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblomm H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. (2013) 381:295–302. doi: 10.1016/s0140-6736(12)61857-1
61. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 389:56–66. doi: 10.1016/s0140-6736(16)32453-9
62. Lombardi G, De Salvo GL, Brandes AA, Eoli M, Ruda R, Faedi M, et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. (2019) 20:110–9. doi: 10.1016/s1470-2045(18)30675-2
63. Gyawali B, Prasad V. Me too-drugs with limited benefits—the tale of regorafenib for HCC. Nat Rev Clin Oncol. (2018) 15:62. doi: 10.1038/nrclinonc.2017.190
64. Mir O, Brodowicz T, Italiano A, Wallet J, Blay JY, Bertucci F, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2016) 17:1732–42. doi: 10.1016/s1470-2045(16)30507-1
65. Ettrich TJ, Seufferlein T. Regorafenib. Recent Results Cancer Res. (2018) 211:45–56. doi: 10.1007/978-3-319-91442-8_3
66. Pan PJ, Liu YC, Hsu FT. Protein kinase B and extracellular signal-regulated kinase inactivation is associated with regorafenib-induced inhibition of osteosarcoma progression in vitro and in vivo. J Clin Med. (2019) 8:900. doi: 10.3390/jcm8060900
67. Motzer RJ, Nosov D, Eisen T, Bondarenko I, Lesovoy V, Lipatov O, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a phase III trial. J Clin Oncol. (2013) 31:3791–9. doi: 10.1200/jco.2012.47.4940
68. Escudier B, Worden F, Kudo M. Sorafenib: key lessons from over 10 years of experience. Expert Rev Anticancer Ther. (2019) 19:177–89. doi: 10.1080/14737140.2019.1559058
69. Pignochino Y, Grignani G, Cavalloni G, Motta M, Tapparo M, Bruno S, et al. Sorafenib blocks tumour growth, angiogenesis and metastatic potential in preclinical models of osteosarcoma through a mechanism potentially involving the inhibition of ERK1/2, MCL-1 and ezrin pathways. Mol Cancer. (2009) 8:118. doi: 10.1186/1476-4598-8-118
70. Grignani G, Palmerini E, Ferraresi V, D'Ambrosio L, Bertulli R, Asaftei SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol. (2015) 16:98–107. doi: 10.1016/s1470-2045(14)71136-2
71. Quan GM, Choong PF. Anti-angiogenic therapy for osteosarcoma. Cancer Metastasis Rev. (2006) 25:707–13. doi: 10.1007/s10555-006-9031-1
72. Steeghs N, Nortier JW, Gelderblom H. Small molecule tyrosine kinase inhibitors in the treatment of solid tumors: an update of recent developments. Ann Surg Oncol. (2007) 14:942–53. doi: 10.1245/s10434-006-9227-1
73. Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. (1994) 269:26988–95.
74. Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. (2015) 29:1298–315. doi: 10.1101/gad.263202.115
75. Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. (2010) 11:1000–17. doi: 10.2174/138945010791591395
76. Deng Y, Zhang X, Simons M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler Thromb Vasc Biol. (2015) 35:421–9. doi: 10.1161/atvbaha.114.304881
77. Raouf S, Bertelli G, Ograbek A, Field P, Tran I. Real-world use of bevacizumab in metastatic colorectal, metastatic breast, advanced ovarian and cervical cancer: a systematic literature review. Future Oncol. (2019) 15:543–61. doi: 10.2217/fon-2018-0480
78. Sanchez-Gastaldo A, Gonzalez-Exposito R, Garcia-Carbonero R. Ramucirumab clinical development: an emerging role in gastrointestinal tumors. Target Oncol. (2016) 11:479–87. doi: 10.1007/s11523-016-0419-8
79. Chen D, Zhang YJ, Zhu KW, Wang WC. A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumour Biol. (2013) 34:1895–9. doi: 10.1007/s13277-013-0733-z
80. Navid F, Santana VM, Neel M, McCarville MB, Shulkin BL, Wu J, et al. A phase II trial evaluating the feasibility of adding bevacizumab to standard osteosarcoma therapy. Int J Cancer. (2017) 141:1469–77. doi: 10.1002/ijc.30841
81. Martinez-Anton A, Gras D, Bourdin A, Dubreuil P, Chanez P. KIT as a therapeutic target for non-oncological diseases. Pharmacol Ther. (2019) 197:11–37. doi: 10.1016/j.pharmthera.2018.12.008
82. Roskoski R. The role of small molecule Kit protein-tyrosine kinase inhibitors in the treatment of neoplastic disorders. Pharmacol Res. (2018) 133:35–52. doi: 10.1016/j.phrs.2018.04.020
83. Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. (2012) 92:1619–49. doi: 10.1152/physrev.00046.2011
84. Abbaspour Babaei M, Kamalidehghan B, Saleem M, Huri HZ, Ahmadipour F. Receptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cells. Drug Des Devel Ther. (2016) 10:2443–59. doi: 10.2147/DDDT.S89114
85. Sulzbacher I, Birner P, Toma C, Wick N, Mazalm PR. Expression of c-kit in human osteosarcoma and its relevance as a prognostic marker. J Clin Pathol. (2007) 60:804–7. doi: 10.1136/jcp.2005.032839
86. Miiji LN, Petrilli AS, Di Cesare S, Odashiro AN, Burnier MN, de Toledom SR, et al. C-kit expression in human osteosarcoma and in vitro assays. Int J Clin Exp Pathol. (2011) 4:775–81. doi: 10.1002/pbc.22223
87. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. (1985) 42:581–8. doi: 10.1016/0092-8674(85)90115-1
88. Borrello MG, Ardini E, Locati LD, Greco A, Licitra L, Pierotti MA. RET inhibition: implications in cancer therapy. Expert Opin Ther Targets. (2013) 17:403–19. doi: 10.1517/14728222.2013.758715
89. Kim M, Kim DJ. GFRA1: a novel molecular target for the prevention of osteosarcoma chemoresistance. Int J Mol Sci. (2018) 19:41078. doi: 10.3390/ijms19041078
90. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. (2014) 14:173–86. doi: 10.1038/nrc3680
91. Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. (2008) 45:1–14. doi: 10.1136/jmg.2007.053959
92. Santoro M, Carlomagno F. Central role of RET in thyroid cancer. Cold Spring Harb Perspect Biol. (2013) 5:a009233. doi: 10.1101/cshperspect.a009233
93. Rosell R, Karachaliou N. RET inhibitors for patients with RET fusion-positive and RET wild-type non-small-cell lung cancer. Lancet Oncol. (2016) 17:1623–5. doi: 10.1016/s1470-2045(16)30557-5
94. Li AY, McCusker MG, Russo A, Scilla KA, Gittens A, Arensmeyer K, et al. RET fusions in solid tumors. Cancer Treat Rev. (2019) 81:101911. doi: 10.1016/j.ctrv.2019.101911
95. Plaza-Menacho I, Mologni L, McDonald NQ. Mechanisms of RET signaling in cancer: current and future implications for targeted therapy. Cell Signal. (2014) 26:1743–52. doi: 10.1016/j.cellsig.2014.03.032
96. Dolgin E. LOXO-292 reins in RET-driven tumors. Cancer Discov. (2018) 8:904–5. doi: 10.1158/2159-8290.Cd-nb2018-070
97. Poh A. BLU-667 controls RET-altered thyroid cancers. Cancer Discov. (2019) 9:OF5. doi: 10.1158/2159-8290.Cd-nb2019-084
98. Rettew AN, Young ED, Lev DC, Kleinerman ES, Abdul-Karim FW, Getty PJ, et al. Multiple receptor tyrosine kinases promote the in vitro phenotype of metastatic human osteosarcoma cell lines. Oncogenesis. (2012) 1:e34. doi: 10.1038/oncsis.2012.34
99. Luo J, Xia Y, Yin Y, Luo J, Liu M, Zhang H, et al. ATF4 destabilizes RET through non-classical GRP78 inhibition to enhance chemosensitivity to bortezomib in human osteosarcoma. Theranostics. (2019) 9:6334–53. doi: 10.7150/thno.36818
100. Kim M, Jung JY, Choi S, Lee H, Morales LD, Koh JT, et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy. (2017) 13:149–68. doi: 10.1080/15548627.2016.1239676
101. Chen Y, Huang W, Sun W, Zheng B, Wang C, Luo Z, et al. LncRNA MALAT1 promotes cancer metastasis in osteosarcoma via activation of the PI3K-Akt signaling pathway. Cell Physiol Biochem. (2018) 51:1313–26. doi: 10.1159/000495550
102. Roskoski R. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol Res. (2018) 129:65–83. doi: 10.1016/j.phrs.2018.01.021
103. Heldin CH. Targeting the PDGF signaling pathway in the treatment of non-malignant diseases. J Neuroimmune Pharmacol. (2014) 9:69–79. doi: 10.1007/s11481-013-9484-2
104. Heldin CH, Lennartsson J, Westermark B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J Intern Med. (2018) 283:16–44. doi: 10.1111/joim.12690
105. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. (2008) 22:1276–312. doi: 10.1101/gad.1653708
106. Xu J, Xie L, Guo W. PDGF/PDGFR effects in osteosarcoma and the “add-on” strategy. Clin Sarcoma Res. (2018) 8:15. doi: 10.1186/s13569-018-0102-1
107. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. (1999) 79:1283–316. doi: 10.1152/physrev.1999.79.4.1283
108. Andrick BJ, Gandhi A. Olaratumab: a novel platelet-derived growth factor receptor α-inhibitor for advanced soft tissue sarcoma. Ann Pharmacother. (2017) 51:1090–8. doi: 10.1177/1060028017723935
109. Takagi S, Takemoto A, Takami M, Oh-Hara T, Fujita N. Platelets promote osteosarcoma cell growth through activation of the platelet-derived growth factor receptor-Akt signaling axis. Cancer Sci. (2014) 105:983–8. doi: 10.1111/cas.12464
110. Zhang D, Cui G, Sun C, Lei L, Lei L, Williamson RA, et al. Hypoxia promotes osteosarcoma cell proliferation and migration through enhancing platelet-derived growth factor-BB/platelet-derived growth factor receptor-beta axis. Biochem Biophys Res Commun. (2019) 512:360–6. doi: 10.1016/j.bbrc.2019.03.040
111. Oda Y, Wehrmann B, Radig K, Walter H, Rose I, Neumann W, et al. Expression of growth factors and their receptors in human osteosarcomas. Immunohistochemical detection of epidermal growth factor, platelet-derived growth factor and their receptors: its correlation with proliferating activities and p53 expression. Gen Diagn Pathol. (1995) 141:97–103. doi: 10.1016/j.palaeo.2004.11.019
112. Sulzbacher I, Birner P, Trieb K, Traxler M, Lang S, Chott A. Expression of platelet-derived growth factor-AA is associated with tumor progression in osteosarcoma. Mod Pathol. (2003) 16:66–71. doi: 10.1097/01.Mp.0000043522.76788.0a
113. Sulzbacher I, Birner P, Dominkus M, Pichlhofer B, Mazal PR. Expression of platelet-derived growth factor-alpha receptor in human osteosarcoma is not a predictor of outcome. Pathology. (2010) 42:664–8. doi: 10.3109/00313025.2010.520310
114. Abdeen A, Chou AJ, Healey JH, Khanna C, Osborne TS, Hewitt SM, et al. Correlation between clinical outcome and growth factor pathway expression in osteogenic sarcoma. Cancer. (2009) 115:5243–50. doi: 10.1002/cncr.24562
115. Bond M, Bernstein ML, Pappo A, Schultz KR, Krailo M, Blaney SM, et al. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children's Oncology Group study. Pediatr Blood Cancer. (2008) 50:254–8. doi: 10.1002/pbc.21132
116. Chiorean EG, Sweeney C, Youssoufian H, Qin A, Dontabhaktuni A, Loizos N, et al. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor alpha (PDGFRalpha) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2014) 73:595–604. doi: 10.1007/s00280-014-2389-9
117. Lowery CD, Blosser W, Dowless M, Knoche S, Stephens J, Li H, et al. Olaratumab exerts antitumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor alpha. Clin Cancer Res. (2018) 24:847–57. doi: 10.1158/1078-0432.CCR-17-1258
118. Porta R, Borea R, Coelho A, Khan S, Araujo A, Reclusa P, et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. (2017) 113:256–67. doi: 10.1016/j.critrevonc.2017.02.018
119. Ghedini GC, Ronca R, Presta M, Giacomini A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev Anticancer Ther. (2018) 18:861–72. doi: 10.1080/14737140.2018.1491795
120. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. (2011) 149:121–30. doi: 10.1093/jb/mvq121
121. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. (2009) 8:235–53. doi: 10.1038/nrd2792
122. Naing A. A new screening tool for FGFR inhibitor treatment? Lancet Oncol. (2019) 20:1340–2. doi: 10.1016/s1470-2045(19)30490-5
123. Zhou WY, Zheng H, Du XL, Yang JL. Characterization of FGFR signaling pathway as therapeutic targets for sarcoma patients. Cancer Biol Med. (2016) 13:260–8. doi: 10.20892/j.issn.2095-3941.2015.0102
124. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. (2016) 22:259–67. doi: 10.1158/1078-0432.Ccr-14-3212
125. Liu FT, Li NG, Zhang YM, Xie WC, Yang SP, Lu T, et al. Recent advance in the development of novel, selective and potent FGFR inhibitors. Eur J Med Chem. (2020) 186:111884. doi: 10.1016/j.ejmech.2019.111884
126. Facchinetti F, Hollebecque A, Bahleda R, Loriot Y, Olaussen KA, Massard C, et al. Facts and new hopes on selective FGFR inhibitors in solid tumors. Clin Cancer Res. (2020) 26:764–74. doi: 10.1158/1078-0432.CCR-19-2035
127. Hassan SE, Bekarev M, Kim MY, Lin J, Piperdi S, Gorlick R, et al. Cell surface receptor expression patterns in osteosarcoma. Cancer. (2012) 118:740–9. doi: 10.1002/cncr.26339
128. Fernanda Amary M, Ye H, Berisha F, Khatri B, Forbes G, Lehovsky K, et al. Fibroblastic growth factor receptor 1 amplification in osteosarcoma is associated with poor response to neo-adjuvant chemotherapy. Cancer Med. (2014) 3:980–7. doi: 10.1002/cam4.268
129. Weekes D, Kashima TG, Zandueta C, Perurena N, Thomas DP, Sunters A, et al. Regulation of osteosarcoma cell lung metastasis by the c-Fos/AP-1 target FGFR1. Oncogene. (2016) 35:2852–61. doi: 10.1038/onc.2015.344
130. Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. (2011) 3:S21–35. doi: 10.1177/1758834011422557
131. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. Eur J Cancer. (2010) 46:1260–70. doi: 10.1016/j.ejca.2010.02.028
132. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. (2003) 22:309–25. doi: 10.1023/a:1023768811842
133. Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. (2013) 39:793–801. doi: 10.1016/j.ctrv.2013.02.001
134. Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. (2017) 3:148–53. doi: 10.1016/j.cdtm.2017.06.002
135. Patane S, Avnet S, Coltella N, Costa B, Sponza S, Olivero M, et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res. (2006) 66:4750–7. doi: 10.1158/0008-5472.CAN-05-4422
136. Coltella N, Manara MC, Cerisano V, Trusolino L, Di Renzo MF, Scotlandi K, et al. Role of the MET/HGF receptor in proliferation and invasive behavior of osteosarcoma. FASEB J. (2003) 17:1162–4. doi: 10.1096/fj.02-0576fje
137. Zhou Q, Hu T, Xu Y. Anticancer potential of TUG1 knockdown in cisplatin-resistant osteosarcoma through inhibition of MET/Akt signalling. J Drug Target. (2020) 28:204–11. doi: 10.1080/1061186X.2019.1644651
138. Xie W, Xiao J, Wang T, Zhang D, Li Z. MicroRNA-876-5p inhibits cell proliferation, migration and invasion by targeting c-Met in osteosarcoma. J Cell Mol Med. (2019) 23:3293–301. doi: 10.1111/jcmm.14217
139. Osher E, Macaulay VM. Therapeutic targeting of the IGF axis. Cells. (2019) 8:80895. doi: 10.3390/cells8080895
140. Li H, Batth IS, Qu X, Xu L, Song N, Wang R. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer. (2017) 16:6. doi: 10.1186/s12943-016-0576-5
141. Pillai RN, Ramalingam SS. Inhibition of insulin-like growth factor receptor: end of a targeted therapy? Transl Lung Cancer Res. (2013) 2:14–22. doi: 10.3978/j.issn.2218-6751.2012.11.05
142. Chmielowski B. Insulin-like growth factor 1 receptor inhibitors: where do we come from? What are we? Where are we going? Cancer. (2014) 120:2384–7. doi: 10.1002/cncr.28727
143. Wang YH, Han XD, Qiu Y, Xiong J, Yu Y, Wang B, et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J Surg Oncol. (2012) 105:235–43. doi: 10.1002/jso.22077
144. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. (2014) 120:2448–56. doi: 10.1002/cncr.28728
145. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2014) 61:452–6. doi: 10.1002/pbc.24605
146. Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. (2014) 14:769–85. doi: 10.1038/nrc3847
147. Shen Y, Chen X, He J, Liao D, Zu X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. (2018) 198:99–111. doi: 10.1016/j.lfs.2018.02.033
148. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. (2019) 18:153. doi: 10.1186/s12943-019-1090-3
149. Rettew AN, Getty PJ, Greenfield EM. Receptor tyrosine kinases in osteosarcoma: not just the usual suspects. Adv Exp Med Biol. (2014) 804:47–66. doi: 10.1007/978-3-319-04843-7_3
150. Han J, Tian R, Yong B, Luo C, Tan P, Shen J, et al. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem Biophys Res Commun. (2013) 435:493–500. doi: 10.1016/j.bbrc.2013.05.019
151. Zhang Y, Tang YJ, Man Y, Pan F, Li ZH, Jia LS. Knockdown of AXL receptor tyrosine kinase in osteosarcoma cells leads to decreased proliferation and increased apoptosis. Int J Immunopathol Pharmacol. (2013) 26:179–88. doi: 10.1177/039463201302600117
152. Jiang N, Wang X, Xie X, Liao Y, Liu N, Liu J, et al. lncRNA DANCR promotes tumor progression and cancer stemness features in osteosarcoma by upregulating AXL via miR-33a-5p inhibition. Cancer Lett. (2017) 405:46–55. doi: 10.1016/j.canlet.2017.06.009
153. Chen Q, Zhou Z, Shan L, Zeng H, Hua Y, Cai Z. The importance of Src signaling in sarcoma. Oncol Lett. (2015) 10:17–22. doi: 10.3892/ol.2015.3184
154. Lee M, Choy WC, Abid MR. Direct sensing of endothelial oxidants by vascular endothelial growth factor receptor-2 and c-Src. PLoS ONE. (2011) 6:e28454. doi: 10.1371/journal.pone.0028454
Keywords: TKIs, RTKs, target therapy, VEGFRs, RET, osteosarcoma
Citation: Tian Z, Niu X and Yao W (2020) Receptor Tyrosine Kinases in Osteosarcoma Treatment: Which Is the Key Target? Front. Oncol. 10:1642. doi: 10.3389/fonc.2020.01642
Received: 20 February 2020; Accepted: 27 July 2020;
Published: 28 August 2020.
Edited by:
Kwong Tsang, Precision Biologics, Inc., United StatesReviewed by:
Leonardo Scapozza, Université de Genève, SwitzerlandHanqing Liu, Jiangsu University, China
Copyright © 2020 Tian, Niu and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weitao Yao, eXd0MDAwMDEmI3gwMDA0MDsxNjMuY29t