
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 18 September 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01350
This article is part of the Research Topic Revisiting the Metastatic Cascade: Putting Myeloid Cells into Context View all 18 articles
 Serena Lucotti1*
Serena Lucotti1* Ruth J. Muschel2
Ruth J. Muschel2During the process of hematogenous metastasis, tumor cells interact with platelets and their precursors megakaryocytes, providing a selection driver for the metastatic phenotype. Cancer cells have evolved a plethora of mechanisms to engage platelet activation and aggregation. Platelet coating of tumor cells in the blood stream promotes the successful completion of multiple steps of the metastatic cascade. Along the same lines, clinical evidence suggests that anti-coagulant therapy might be associated with reduced risk of metastatic disease and better prognosis in cancer patients. Here, we review experimental and clinical literature concerning the contribution of platelets and megakaryocytes to cancer metastasis and provide insights into the clinical relevance of anti-coagulant therapy in cancer treatment.
Besides their role in hemostasis and wound healing, platelets are a key player during metastatic spread. The idea that platelets support tumor cells during their metastatic endeavors is not new. In 1865, the French doctor Armand Trousseau observed excessive blood clotting in patients with occult carcinoma, including himself, and defined it as the Trousseau syndrome (1, 2). One century later, Gasic showed for the first time that platelets are a prerequisite for experimental metastasis (3, 4). Since then, numerous studies have confirmed the relationship between thrombosis and metastasis. Experimental depletion of platelets by pharmacological or genetic means almost completely abrogates metastasis in a number of mouse models, including but not limited to (3, 5–9). Moreover, inhibition of platelet aggregation through different means, including vitamin K agonists, thrombin inhibitors, non-steroidal anti-inflammatory drugs (NSAIDs), and genetic platelet activation deficiency decrease metastatic colonization to the lungs in experimental models (4, 8–15), supporting the notion that platelet aggregation is a pivotal event during metastatic seeding. In the clinical setting, thrombocytosis (high platelet count, >450,000/mL) and platelet hyperreactivity are associated with cancer progression and a worse prognosis. This correlation has been confirmed across different cancer types, including pancreatic, colorectal, lung, and breast cancer (16–21). Compelling evidence also comes from clinical trials showing reduced risk of metastatic disease in patients under anti-coagulant therapy (22). Nevertheless, anti-coagulant therapy is not yet a component of standard cancer treatment or prophylaxis.
So, what is the role of platelets during the metastatic cascade? Decades of research have uncovered a multifaceted interplay between platelets and tumor cells, whereby platelets directly interact with tumor cells in the bloodstream and support many different aspects of their dissemination. This review first summarizes the biology of platelets and their precursors megakaryocytes and then examines the interaction of tumor cells with platelets and megakaryocytes during the metastatic cascade, including its role in the onset of thromboembolic disease in cancer. Finally, we provide a perspective on the current status of adjuvant anti-coagulant therapy in cancer treatment, highlighting its limitations, and future potential.
Platelets are small membrane bound, blood-borne cell fragments of 2–3 μm in diameter, with a distinctive discoidal shape and lacking a nucleus. They are one of the most abundant type of cells in the blood circulation (150–350 × 106/mL in humans and 1,100 × 106/mL in mice) (23, 24). Thanks to their small size and shape, platelets preferentially marginate toward the outer edge of the blood stream, where lower shear rate and the proximity to vascular endothelium maximizes platelet responsiveness to vascular damage (25–27). Platelets contain numerous cytoplasmatic secretory granules: α-granules and dense granules. The peripheral membrane of α-granules (~50–80 per platelet) has receptors and proteins that are involved in platelet adhesion, angiogenesis and recruitment of immune cells. Dense granules (~3–8 per platelet) contain the low molecular weight agonists of platelet aggregation, involved in the activation, and recruitment of additional platelets to the site of damage (24, 28).
Being anucleate, platelets cannot generate new mRNA and have a short half-life of 8–10 days in humans and 3–5 days in mice. In order to maintain an adequate platelet count, approximately 100 billion platelets are produced every day from their myeloid precursors megakaryocytes (29). Megakaryocytes are giant (50–100 μm in diameter) and very rare cells (~0.01% of bone marrow nucleated cells) that differentiate from common myeloid progenitor cells within specialized osteoblastic and perivascular niches of the bone marrow (30, 31). During development and under pathological conditions megakaryocytes may also be found in the liver and spleen (32–34). Recently, Lefrançais et al. have shown that megakaryocytes of extrapulmonary origin also reside within the lung vasculature in mice, providing evidence that lungs are an active site of platelet biogenesis (35).
The production of platelets is initiated by megakaryocytes through a multistep maturation and developmental process. In response to thrombopoietin (TPO), the amount of cytoskeletal proteins, intracellular granules, and membrane lipids increases in megakaryocytes, leading to a massive enlargement of cytoplasmatic volume accompanied by the formation of an invaginated membrane system (IMS). Concomitantly, numerous DNA replication cycles occur in the absence of cell division (endomitosis), generating a polyploid mature megakaryocyte (35, 36). It is believed that endomitosis serves to increase the amount of lipids, mRNA, and proteins to be transferred to the resulting platelets. Finally, mature megakaryocytes produce membrane and cytoplasmatic protrusion into the blood vessels called preplatelet and proplatelets (35, 36). This process is highly dependent on the reassembly of cytoskeletal filaments that occurs during megakaryocyte maturation and which controls proplatelet elongation and the transportation of granules and organelles from the megakaryocyte cytoplasm into the proplatelet tips. Mature platelets are then produced by spontaneous rounds of fragmentation of proplatelets in the circulation (30, 36). In the bone marrow, thrombopoiesis takes place in the vascular niche and requires the interaction of megakaryocytes with bone marrow endothelial cells and the perivascular extracellular matrix (ECM), as reviewed by (37).
The main functions of platelets are to maintain the integrity of the vascular system by arresting bleeding (hemostasis) and promoting wound healing at sites of vascular injury. Hemostasis involves two parallel and interrelated processes at the site of damage: thrombosis, which is the formation of a platelet aggregate (thrombus), and coagulation, a cascade of cell activation and proteolytic reactions involving different cell types (endothelial cells, platelets, and leukocytes) and soluble proteins (coagulation factors). Coagulation factors are enzymes with serine protease activity present in the circulation as inactive zymogens that undergo activation through proteolytic cleavage (38). These processes culminate with the generation of thrombin, which cleaves soluble fibrinogen into fibrin and leads to the deposition of fibrin fibers and the formation of a (fibrin) clot, a plug of platelets and fibrin mesh (29, 39).
Upon vascular damage or endothelial retraction, components of the endothelial basement membrane and other sub-endothelial extracellular proteins such as von Willebrand Factor (vWF), collagens and fibronectin are exposed to the blood stream. Interaction of integrin receptors expressed on resting platelets with these exposed ligands induces a rapid cascade that leads to the activation of platelets (within seconds) and the formation of a fibrin clot (within minutes). First, the interaction of vWF/collagen with the multimeric complex glycoprotein (GP) Ib-IX-V on the platelet surface causes platelet tethering and rolling on the exposed subendothelium (40–42). This initial and reversible platelet adhesion is followed by intracellular signaling leading to platelet activation, a complex cellular process associated with the activation of Src kinases, increase of cytosolic Ca2+ concentration and the activation of protein kinase C (PKC) and phosphatidylinositol 3 kinase (PI3K) (43, 44). Class I kinases such as PI3Kβ have a pivotal role in Akt phosphorylation and Ca2+ mobilization during thrombus formation (45). Ultimately, these cellular responses lead to the rearrangement of the platelet cytoskeleton, culminating in granule secretion, change of cell shape with production of pseudopodia and platelet spreading, and the “inside-out” conformational change of membrane integrin α2ß1 and αIIbß3 (or GPIIa/IIIb) from a low to a high affinity form (24, 29). The high affinity form of these integrins mediate firm adhesion to collagen (α2ß1), vWF, and fibrin(ogen) (αIIbß3), supporting clot stabilization (46, 47). Thrombus amplification is further sustained by the release of adhesion molecules (e.g., vWF and fibrinogen), coagulation factors (e.g., factors V and IX) and soluble agonists (e.g., ADP, serotonin) and the exposure of additional platelet receptors (GPIb-IX-V, αIIbß3, GPVI, and P-selectin or CD62P) contained in platelet α- and dense granules (46). Activation of platelets also initiates fatty acid oxidation and the de novo synthesis of thromboxane A2 (TXA2), a secondary mediator of thrombus amplification. Together, ADP, thrombin and TXA2 interact with their cognate receptors on other platelets (ADP:P2Y12, thrombin:PAR1/4, TXA2:TP), leading to platelet activation (24, 39, 48). Thus, the initial layer of activated platelets serves as a reactive surface for the tethering, activation, and aggregation of additional platelets.
Other cell types take part in the generation of a fibrin clot, by either serving as an adhesion surface for platelet tethering or by engaging in the coagulation cascade. Activated endothelial cells at the site of vascular injury locally synthesize or expose endothelial cell leukocyte adhesion molecules. Exocytosis of Weibel-Palade bodies in endothelial cells releases vWF, which in turn binds to platelet GP Ib-IX-V and integrin αIIbß3, and exposes P-selectin, which binds to platelet GPIbα and P-selectin ligand (PSGL-1) (42, 49), and E-selectin, which recruit myeloid cells to the site of injury (50, 51). Recruited monocytes, activated endothelial cells and other sub-endothelial stroma cells, such as smooth muscle cells and fibroblasts, express tissue factor (TF, also known as CD142 or thromboplastin), which is the main activator of the coagulation cascade in vivo. When exposed to the blood circulation, TF activates coagulation factor VII and binds to its active form VIIa in a bimolecular complex (TF-VIIa) that initiates the coagulation cascade leading to the activation of thrombin and fibrin deposition (52). Thrombin also cleaves protease activated receptor (PAR)1 and PAR4 on platelets, inducing their activation and aggregation (48). Ultimately, to complete healing, the fibrin clot will be dissolved by fibrinolysis [as reviewed by (53)].
Although platelet have been traditionally studied in the context of blood coagulation, we now know that they are also involved in many other processes such as inflammation, angiogenesis, and innate immunity. These functions have been reviewed by others (54–56) and will not be the focus of the current review.
Tumor cells have adapted to mimic some steps of the hemostatic process and they interact with circulating platelets during their hematogenous transit (Figure 1). This interaction happens within minutes from tumor cell intravasation (7, 57) and relies on an expression pattern triggered by classical oncogenic mutations and microenvironmental cues [as reviewed by (58)].
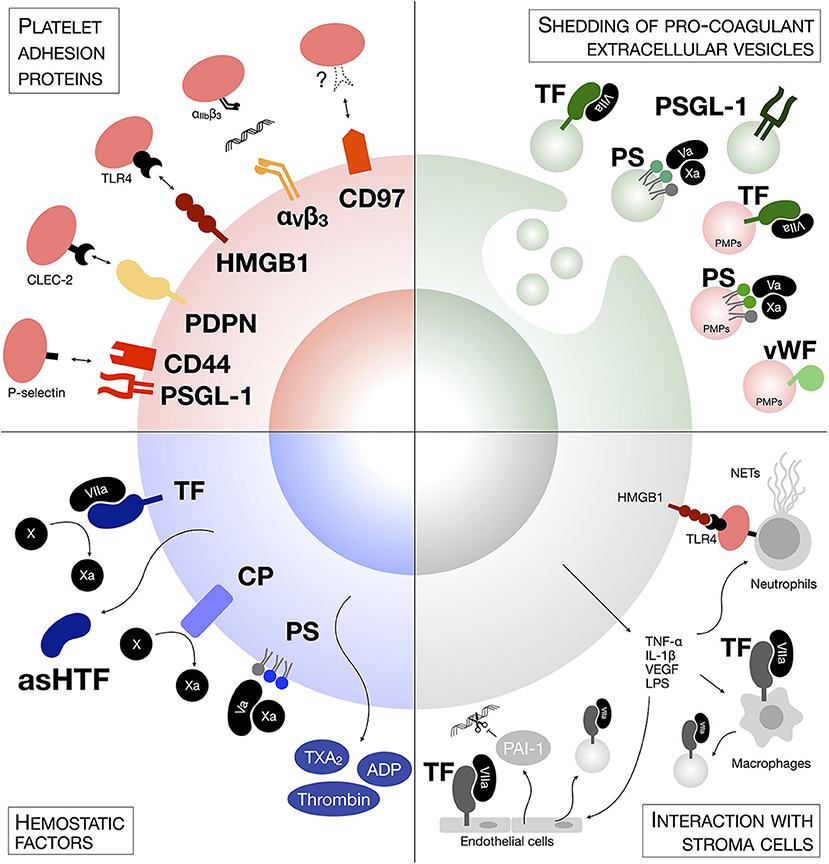
Figure 1. Mechanisms of tumor cells-platelets interaction. Tumor cells generate platelet activation and the formation of microclots on their surface through several mechanisms, including expression of hemostatic factors and adhesion proteins, either on their surface or on the surface of shedded extracellular vesicles, or though the generation of a pro-thrombotic intravascular metastatic niche involving other stroma cells.
Different cancer cells express a variety of hemostatic factors. TF has been found constitutively expressed by most tumor cell lines and metastatic cells express up to 1,000-fold higher levels of TF in comparison to non-metastatic cells (59–61). Epithelial-to-mesenchymal transition (EMT) can also induce TF expression in circulating tumor cells (CTCs) (62). The levels of TF expression on tumor cells correlate with cancer progression and poor prognosis (63). TF-interacting coagulation factor VII has also been found overexpressed in colorectal cancer and hepatocellular carcinoma cells and correlated with hepatic metastasis (64, 65). In addition to engaging the coagulation cascade, both TF and factor VII induce intracellular signaling that supports tumor cell growth, invasion and migration (52, 64, 66). Along the same lines, cancer procoagulant (CP) is a cysteine protease that directly cleaves and activates coagulation factor X, thus inducing the exogenous coagulation cascade and ultimately platelet activation (67, 68). CP is expressed in human malignant tissues, but largely absent in normal tissues (69–71).
Proteins are not the only hemostatic factors in tumor cells; the asymmetric distribution of membrane phospholipids on cancer cells is also responsible for coagulation. The membrane phospholipid phosphatidylserine (PS), which is preferentially localized in the inner leaflets of normal eukaryotic cell membranes (72), has been found highly exposed on the outer leaflet of tumor cells and this exposure is linked to mutations in phospholipid translocases such as flippase (73–75). Intriguingly, metastatic cells have lower flippase activity and thus higher level of PS than their non-invasive counterparts (75). Once on the outer membrane leaflet, the anionic PS creates a negatively charged surface that binds factors Va and Xa, thus initiating the assembly of the prothrombinase complex, and supports the conformational change that activates their proteolytic activity, leading to thrombin deposition, and platelet aggregation (76).
Hemostatic factors are not only expressed on the surface of tumor cells, but they are also released in their soluble form to create a pro-thrombotic niche. Human tumor cell lines secrete a soluble alternatively spliced form of TF (known as asHTF) (77, 78), which can also be found in the plasma of cancer patients (79). Additionally, platelet agonists such as ADP, TXA2, and thrombin have been detected in cell line supernatants and cancer biopsies (80–82) and interact with platelet receptors P2Y12, TP, and PARs, respectively, to initiate platelet aggregation. Hence, tumor cells can activate platelet aggregation also through a paracrine route.
Tumor cells express binding proteins that mediate direct activation and adhesion of platelets in the absence of other plasma components. PSGL-1 on tumor cells directly interact with P-selectin exposed on activated platelets (11, 83). Other glycoproteins bearing sialyl-Lewisx structures on the tumor cell surface have also been shown to mediate adhesion to P-selectin-expressing platelets (11). Tumor cell CD44 is also involved in P-selectin binding, either directly or via fibrin (84). Although P-selectin-PSGL-1 engagement requires prior activation of platelets, elegant studies by Furie and Furie using P-selectin- or PSGL-1-null mice suggest their role in fibrin generation together with TF deposition in the growing thrombus (85, 86). Thus, the direct association of tumor cells with platelets through a P-selectin-PSGL-1 interaction is sufficient to mediate the deposition of a fibrin clot on the surface of tumor cells. Podoplanin (PDPN, tumor cells): CLEC-2 (platelet) and HMGB1 (tumor cells): TLR4 (platelets) are other adhesion protein-ligand pairs that support platelet activation and aggregation on tumor cells, and ultimately metastasis (87–91). Additionally, integrin αIIbβ3 on activated platelets has a central role in adhesion to melanoma and breast cancer cells by interacting with tumor cell integrin αVβ3 via fibrinogen (92–94). It is interesting to notice that αVβ3 expression confers a proliferative advantage to breast cancer tumor cells during early stages of brain metastasis, suggesting that platelet promote the survival of cells with higher metastatic potential during their hematogenous transit (95, 96). More recently, Ward et al. have identified the G protein-coupled receptor (GPCR) CD97 on tumor cells as a novel binding protein mediating interaction with platelets. CD97 induces αIIbß3-dependent platelet activation and their aggregation on the surface of tumor cells, although its cognate receptor on platelets still remains to be described (97).
Both hemostatic factors and platelet ligands can be shed by tumor cells in extracellular vesicles, in particular microparticles (MPs, also called microvesicles). MPs are small membrane vesicles of 100–1,000 μm in diameter released by direct membrane budding (98). Already in 1981, Dvorak et al. detected the presence of tumor-derived MPs in vitro and in ascitic fluids of tumor-bearing animals. These MPs could induce fibrin deposition in vivo (99). Tumor cells have been found responsible for the production of these pro-coagulant MPs. MPs expressing TF, PSGL-1, and PS can be detected in the culture medium of tumor cells and in tumor-bearing mice, and mediate thrombin generation and thrombus growth ex vivo and in vivo (77, 100–103). These MPs accumulate in the growing thrombi through a PSGL-1-mediated mechanism and accelerate thrombus growth (86, 100). MPs are found in the blood of cancer patients. Patients with pancreatic, colorectal, brain, prostate, and breast cancer have higher levels of plasma TF/PS-expressing MPs and higher MP-associated pro-thrombotic activity than healthy subjects, especially during advanced stages of disease and after chemotherapy or radiotherapy (104–110). Metastatic cancer patients had particularly high plasma levels of TF+ MPs across a range of cancer types (109).
Certainly not all pro-coagulant MPs in the blood of cancer patients derive from tumor cells. Platelet-derived MPs (PMPs) are the most represented population of MPs in plasma from healthy individuals, accounting for up to 90% of circulating MPs (111–113). Although resting platelets can release MPs (114), most PMPs are produced as a result of platelet activation (111, 115), and are involved in thrombus expansion during hemostasis through the expression of PS, TF, and vWF on their surface (112, 116–118). PMPs are elevated in murine models of cancer and in cancer patients (100, 109). Hence, it is possible that by stimulating platelet aggregation, CTCs induce an increase in platelet-derived MPs, contributing to the pool of circulating pro-coagulant MPs. Interestingly, TF-expressing MPs are also detectable in healthy people, but are not associated with apparent pro-coagulant activity (39). MPs derived from resting platelets lack P-selectin expression, a marker of platelet activation and pro-coagulant protein (111). Hence, cancer-derived MPs and PMPs might express TF in an alternative and readily active conformation, or TF association with negatively charged PS or other adhesion proteins might be required to exert its pro-coagulant function.
Exosomes are a different type of extracellular vesicles of 30–150 nm in diameter that originate in the endocytic pathway and have a pivotal role in mediating short- and long-distance intercellular signaling in both physiological and pathological conditions (119). Although previous evidence suggests that cancer-derived exosomes may initiate thrombosis in vitro and in vivo (120–122), the mechanism and prognostic value of these extracellular vesicles still remains unknown.
As well as directly activating platelets, cancer cells promote a procoagulant niche by altering the thrombotic phenotype in other neighboring stroma cells. Pro-inflammatory cytokines [i.e., TNF-α and interleukin (IL)-1β] and pro-angiogenic factors (i.e., VEGF) released by tumor cells induce the overexpression of TF by endothelial cells and monocytes (123–128) and the release of vWF by endothelial cells (129). Moreover, tumor cell IL-1 induces endothelial secretion of plasminogen activator inhibitor (PAI)-1, an inhibitor of fibrinolysis (130). Tumor cell-derived pro-inflammatory cytokines also induce a peak of stroma-derived MPs contributing to thrombus growth. LPS-stimulated monocytes and endothelial cells release pro-coagulant MPs expressing TF and PSGL-1 (118) and higher number of endothelial-derived MPs can be found in the blood of cancer patients (100, 109). Additionally, tumor cells disseminating to the lung and liver recruit and activate neutrophils to release of extracellular DNA traps (NETs) intravascularly (131, 132). NETs induce platelet aggregation through PS exposure, PMP accumulation and endothelial cell activation, and are associated with increased hypercoagulability and risk of venous thromboembolism in cancer patients (133–135). Interestingly, platelets interacting with tumor cells through the TLR4 axis can also activate NETs formation through a P-selectin-dependent mechanism (136, 137), further amplifying thrombosis.
Per se, metastasis is a highly inefficient process. Experimental evidence suggests that <0.02% of tumor cells entering the circulation end up forming a macroscopic metastases, either in the lungs (0.01% of cells) (138) or liver (0.018% of cells) (139). There are different bottleneck events during the metastatic cascade that reduce the ability of tumor cells to colonize a distant organ. Seminal work by Fidler showed that 125IUDR-labeled B16 melanoma cells arrest in the lungs shortly after intravenous introduction, but only 1% of the injected tumor cells survived in the lungs during the first 24 hours, with most tumor cells dying during the first hour after injection (138). These findings have been confirmed in other models (140, 141) and identified that metastatic inefficiency happens mainly during the intravascular phase of tumor cell dissemination. The circulatory system is in fact a very hostile environment and CTCs are exposed to cell death through immunological, cellular, and physical means. Despite the strong negative selection against tumor cells during the first hour in the blood stream, the intrinsic inefficiency of the metastatic process is not sufficient to abrogate the appearance of distant metastasis. It has been estimated that millions of tumor cells detach from the primary tumor and enter the circulation every day. On such a large scale, 0.02% of surviving cells is no longer a small number, explaining why metastasis is far from rare in cancer patients. A large body of evidence clearly point to platelets-tumor cell interaction as the main reason for tumor cell survival during the intravascular phase of metastasis. Already in 1984, Gorelik et al. had shown that the anti-coagulant heparin dramatically increased the rate of tumor cell elimination during the first day after their injection (142). Platelet depletion during tumor cell presence in the circulation drastically impaired metastatic burden (7, 143). More recently, we have shown that anti-platelet therapy during the intravascular phase of metastasis, but not after tumor cell extravasation, was sufficient to reduce the number of pulmonary metastatic foci subsequently formed (9). Over the years, many mechanisms have been documented for the supporting role of platelets during metastasis, involving a crosstalk between tumor cells and platelets directly or through other stroma cells (Figure 2). Platelets have been found to assist multiple consecutive steps of the metastatic cascade, including tumor cell survival, interaction with the endothelial and immune cells, and transendothelial migration. It is widely accepted that both physical tumor cell-platelet interactions and activation of intracellular signaling pathways in both cell types support these steps of metastasis.
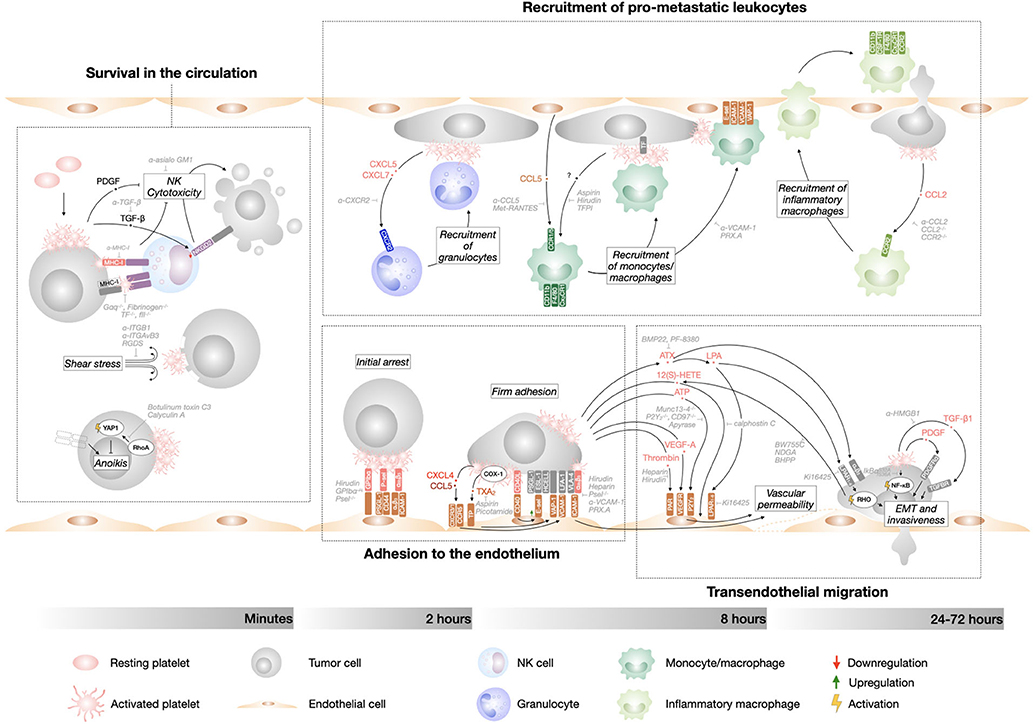
Figure 2. Tumor cell-platelet interplay during the metastatic cascade. Diagram depicting the intravascular steps of the metastatic cascade that are supported by platelet interaction with tumor cells and their timeline. Therapeutic approaches that interfere with the different steps of tumor cells dissemination are indicated in gray.
Patrolling natural killer (NK) cells are the main form of anti-tumor immunosurveillance in the metastatic cascade. NK depletion dramatically increases metastasis to lungs and liver (15, 143–145). Loss of class I major histocompatibility complexes (MHC-I, “missing self”) and up-regulation of surface proteins (“altered-self”) on tumor cells are recognized by NK cells and elicit an anti-tumor response. Cell killing takes place through different mechanisms, including release of cytotoxic granules, engagement of death receptors and secretion of tumor suppressant IFN-γ (146). Platelet cloaking of tumor cells can prevent NK-dependent tumor cell cytolysis (12, 13, 142–144, 147). This concept was firstly elaborated by Nieswandt et al. who showed that the reduction of lung metastasis induced by thrombocytopenia is abrogated by NK cell depletion (143). Interestingly, after tail vein injection of tumor cells, NK depletion supports tumor cell colonization of both lungs, the first vascular bed that they encounter, and liver (142, 143), suggesting that liver colonization in these experimental metastasis models is mainly prevented by NK cell lytic activity. Different mechanisms have been described that explain the NK-suppressive effect of platelets. Platelet express high levels of MHC-I and by coating tumor cells they provide an MHC-I “pseudoexpression” that rescues tumor cell “missing self” and protects them from NK cells recognition (148). TF-dependent platelet and fibrinogen coating have also been found responsible for NK cell evasion (12), potentially via physical shielding of “altered self” and “missing self” expression from immune recognition. Furthermore, activated platelets release soluble factors that induce NK cells quiescence in a paracrine manner. Platelet-derived growth factor (PDGF) inhibit NK cells cytotoxic activity (149, 150) and platelet-derived transforming growth factor (TGF)-ß induces the downregulation of natural killer group 2, member D (NKG2D), a NK cell immunoreceptor that activates anti-tumor reactivity (151).
Additional to NK cells cytotoxicity, hemodynamic shear forces experienced by CTCs in the blood stream induce mechanical damage and death of tumor cells, mainly through increased sensitivity to TNF-related apoptosis-inducing ligand (TRAIL) on NK cells and Smad-dependent cell cycle arrest (152–154). Further, CTCs can undergo cell death due to anoikis, a particular type of apoptosis induced by the disengagement of cell-cell or cell-ECM contacts (155). EMT of CTCs and their clustering in circulating tumor microemboli might provide anoikis resistance (156). The decoration of tumor cells by platelets attenuates tumor cell membrane damage due to shear stress (157). Moreover, platelets bound to tumor cells provide resistance to anoikis by supplying cell-cell contacts and by inducing RhoA-dependent YAP1 activation, which further promotes metastasis (8).
Even when tumor cells survive their first hours in the blood stream, their ability to metastasize is entirely reliant on forming stable interactions with the vascular wall, followed by extravasation. Most tumor cells arrest in capillaries, where they can be trapped due to size restriction, but they can also be found adhered to pre-capillary arteries and portal venules, which are larger in diameter, suggesting the existence of active receptor-ligand interactions (158). The initial and transient interaction of tumor cells with the vessel wall can be mediated by the expression of the selectin family of adhesion molecules, including endothelial P-selectin and E-selectin, which establish and disengage low-affinity bonds that make leukocytes and tumor cells appear to “roll” on the endothelium, at least in vitro (159). Subsequently, firmer adhesion between tumor cells and endothelium is achieved through the expression of a second subset of adhesion molecules, mainly (but not exclusively) integrins and their ligands (159, 160). The principal endothelial integrin ligands are vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1). vWF is also secreted via exocytosis of endothelial cell intracellular granules, the Weibel-Palade bodies (161). Further, basement membrane exposed after endothelial retraction might be another site of anchorage (162). Under physiological conditions, the expression of selectins and integrin ligands on endothelial cells (a process known as endothelial activation) is tightly controlled by transcriptional regulation, exocytosis of intracellular granules and proteolytic cleavage to avoid inappropriate thrombosis or leukocyte recruitment (159).
A growing body of literature describes the role of platelets in supporting the interaction of tumor cells with the vessel wall. Im et al. have shown that tumor cells were less likely to flatten/spread on lung endothelial cells if mice were anticoagulated with hirudin (57). Platelets express adhesion proteins that mediate their rolling (resting platelets) and adhesion (activated platelets) to the vascular wall (163), and thus may form “sticky” bridges between tumor cells and activated endothelial cells or endothelial basement membrane components. For example, Jain et al. have shown that platelets lacking GPIbα, a member of the vWF-binding GPIb-IX-V complex, can still interact with B16 melanoma cells, but dramatically hinder B16 experimental metastasis. This pro-metastatic activity relies solely on the extracellular domain of GPIbα, suggesting that platelet coating of tumor cells and endothelium adhesion, independent of platelet activation, can support metastasis (164). P-selectin on activated platelets interacting with tumor cells mediate their adhesion to endothelial cells and P-selectin−/− mice or platelets support significantly less lung metastasis (83, 145, 165, 166), although infusion of P-selectin+/+ platelets after tumor cell injection partially recovers metastatic nodules, suggesting that platelet P-selectin acts as a pro-metastatic adhesion molecule to both endothelial and tumor cells (167). Integrin αIIbß3, which is found on tumor cells-activated platelet clusters, mediates binding to endothelial cells via ICAM-1 and αVβ3 (163, 165, 168).
The rolling and adhesion of platelet-tumor cell emboli requires activation of endothelial cells, as shown by evidence that genetic depletion or pharmacological inhibition of P-selectin and vascular adhesion protein-1 (VAP-1) expression in endothelial cells dramatically hinders metastasis in different platelet-proficient in vivo models (83, 145, 165, 169, 170). Platelet microthrombi on tumor cells induce endothelial cell expression of E-selectin, VCAM-1, and VAP-1 in lung endothelial cells and anti-coagulant therapy dramatically reduces endothelial activation to basal levels (9, 170, 171). Both direct and paracrine signaling have been found responsible for platelet-dependent initiation of endothelial cell activation. Direct interaction of CD40L on activated platelets with endothelial CD40 triggers expression of E-selectin, VCAM-1, and ICAM-1 on endothelial cells in vitro and in vivo (172). Interestingly, CD40L on T cells mediates their interaction with antigen presenting cells and B-cells, suggesting that activated platelets might also have a role in linking innate and adaptive immune responses. Additional to direct contact, pro-inflammatory cytokines released by activated platelets are known activators of endothelial cells, for example CXCL4 and CCL5 (173). Our lab has shown that TXA2 derived from platelets interacting with tumor cells intravascularly induces the expression of E-selectin and VCAM-1 by endothelial cells in the proximity of tumor cells-platelets emboli (9). Tumor cells can then establish firm adhesion with activated endothelial cells via expressed selectin ligands [e.g., PSGL-1, CD44v, CD24, HCELL, E-selectin ligand 1 (ESL-1); (174–178)] and integrins [e.g., α4ß1/VLA-4, αLß2/LFA-1, and α3β1; (162, 179–182)].
It is important to mention that the initial arrest of tumor cells, in particular in pulmonary circulation, seems to be largely independent of platelet interaction with tumor cells. Although the association of tumor cells αVβ3 with platelets αIIbβ3 mediate their adhesion to collagen I in vitro and increase lung metastasis in vivo (94, 183), its role in the adhesion to endothelial cells is not understood. Heparin treatment (142) or platelet depletion (7) does not affect the number of tumor cells arrested in the lungs at 10 minutes after injection, suggesting that the physical entrapment of tumor cells in the lung capillaries remains a main mechanism of initial arrest.
The interaction of tumor cells with leukocytes, including myeloid cells and lymphocytes, has been observed for a long time. More recently, evidence has emerged indicating that platelets play a pivotal role in coordinating the formation of transient tumor cell-immune cell intravascular niches. Labelle et al. have shown that early after intravasation, tumor cells lodging in the lung vasculature are found decorated with granulocytes (CD11b+ MMP9+ Ly6G+) that support metastatic seeding and subsequent metastasis (7). Importantly, tumor cell interaction with platelets leading to release of soluble chemoattractants is necessary for the recruitment of these granulocytes, including the granulocyte CXCR2 ligands CXCL5/7. Neutrophils might be part of these early microemboli, as they are recruited by platelet-derived chemoattractants and adhere to activated platelets and endothelium (184). Platelet-neutrophil interaction through TLR4 and P-selectin and platelet-derived TXA2, CXCL4, and vWF induce intravascular NETosis, which greatly supports seeding and progression of pulmonary metastasis (132). Tumor cells actively adhere to intravascular NETs through β1 integrin in hepatic sinusoids (185), suggesting that NETs-dependent trapping could support tumor cell arrest in much larger vessels than capillaries. Tumor cell-platelet-granulocyte emboli start to dissolve after 4 hours and are followed by a second wave of immune cells recruitment driven by platelets. Gil-Bernabe et al. have described the recruitment of a subset of undifferentiated monocytes/macrophages (CD11b+ F4/80+ CX3CR1+ CD11c− Ly6C−) to disseminating tumor cells (15). This recruitment depends on TF expression by tumor cells, leading to the deposition of microclots on tumor cells that establish direct interaction with the monocytes. Microemboli containing monocytes/macrophages, tumor cells, and platelets form within the vasculature from 2 hours after tumor cell introduction and reach their maximum volume at 8 hours, but are dissolved by 24 hours. The recruitment of these myeloid cells is promoted by the release of CCL5 by activated endothelial cells and their expression of the adhesion molecules VCAM-1, VAP-1, and E-selectin, all induced by clots on tumor cells (50, 170, 171). Selective depletion or functional impairment of monocytes/macrophages significantly reduced tumor cell number during the first day post-injection, suggesting that patrolling monocytes/macrophages support tumor cell survival in the circulation during early phases of metastasis (15). Finally, a third wave of inflammatory monocytes are recruited to tumor cells through CCL2-CCR2 signaling (186). Platelets are a main source of CCL2, which is stored in their α-granules (96). After diapedesis, these monocytes differentiate into metastasis-associated macrophages (MAMs), characterized by an inflammatory phenotype (F4/80+ CSF1-R+ CD11b+ Ly6C− CX3CR1high CCR2high) (187). These MAMs are localized in the extravascular space and are recruited to the proximity of extravasating tumor cells within 24–72 hours after injection, and support tumor cell extravasation and initial growth (186, 187). Although tumor cell-platelet interaction still takes place at the moment of initial MAM recruitment, it is still unknown if platelets are involved in their chemotaxis and differentiation.
Interestingly, the recruitment of pro-metastatic immune cells by platelets in metastatic organs can happen before tumor cell colonization, at the level of pre-metastatic niche. Our lab has shown that the population of Cx3CR1+ monocytes/macrophages in the lungs increases in pre-metastatic tumor bearing mice, but not in mice injected with TF-deficient tumor cells or in mice that are anticoagulated with aspirin or hirudin during lung preconditioning (9, 15). Hence, circulating platelets activated at the primary tumor can carry signals to distant organs, supporting tumor cell homing.
Once adhered to the vascular wall, tumor cells cross the endothelial barrier in a process called transendothelial migration (TEM), which happens during the first 3 days after entering the circulation, although the timing is highly dependent on the cell type and the secondary organ (188). Similar to leukocyte diapedesis, tumor cell TEM preferentially takes place by a paracellular route and the acquisition of an invasive phenotype and is accompanied by an increase in vascular permeability (159). Several mechanisms have been proposed to account for the increase in vascular permeability during metastatic seeding, including transient disruption of endothelial cell junctions (189), cytoskeletal rearrangements leading to endothelial retraction (159), and apoptosis/necroptosis of endothelial cells, resulting in the irreversible opening of the endothelial barrier (190, 191). Vascular permeability is induced locally in the proximity of tumor cell-platelet microemboli and might be potentiated by further exposure of subendothelial vWF, TF, and collagen. Thrombin interaction with PAR receptors on endothelial cells induces endothelial cell retraction (192). Adenine triphosphate (ATP) is released by platelet dense granules in response to interaction with tumor cells and can bind to P2Y2 receptors on endothelial cells, resulting in the opening of vascular junctions and TEM (97, 193). Similarly, autotaxin (ATX) derived from tumor cell-stimulated platelets and its product lysophosphatidic acid (LPA) interact with tumor cells αVβ3 and LPA receptors (LPAR), respectively, and promote TEM and bone metastasis of breast cancer (194). LPA also increases the permeability of cerebral microvascular endothelial cells directly (195). Furthermore, 12(S)-HETE, a product of arachidonic acid metabolism derived from tumor cells and interacting platelets, induces cytoskeletal rearrangement in endothelial cells, leading to their retraction (196–198). VEGF-A from activated platelets might also induce endothelial permeability and TEM, as does VEGF-A delivered by inflammatory MAMs in lungs (186). Direct contact between tumor cell-platelets clusters and endothelial cells also induces endothelial permeability. Interaction of endothelial VCAM-1 with tumor cell VLA-4 and/or endothelial ICAM-1 with platelet αIIbß3 triggers an “outside-in” signaling cascade in endothelial cells that induces the digestion of tight junctions, cytoskeletal rearrangement, and endothelial retraction (199, 200). Caspase-dependent apoptosis of endothelial cells has also been observed in response to bacteria-activated platelets (201). Although endothelial necroptosis has been characterized as a novel mechanism of TEM during metastasis, platelet do not seem to contribute (191).
Concomitant with inducing vascular permeability, tumor cells need to undergo dynamic changes of cell shape to move through the vascular wall. Although it is not a prerequisite for intravasation, EMT of CTCs is associated with the acquisition of mesenchymal markers, allowing greater motility (202), as does the subsequent formation of proteolytically active protrusions called invadopodia through RHO- and ROCK-dependent polymerization of actin fibers at their leading edge (203–205). The acquisition of this invasive phenotype by tumor cells is supported by their association with platelets, as shown by the fact that cancer cells exposed to activated platelets have a higher capacity for ECM degradation and tissue infiltration (206). This phenotype is sustained far longer than the transient interaction between tumor cell-platelet, whose emboli are dissolved within 24 hours (15, 207). Evidence suggests that this process takes place in two-wave kinetics. Initially (within 16 hours), CD97-mediated interaction with platelets induces release of platelet LPA and activates RHO in tumor cells, promoting tumor cell invasiveness (97). Later on (from 40 hours), platelets induce a gene expression signature in tumor cells that initiates EMT and metastatic seeding, including pro-metastatic mmp9, ccl2, and serpine1 (14). Labelle et al. elegantly showed that both the physical interaction with platelets, leading to NF-kB activation in tumor cells, and the paracrine signaling of TGF-β1 released by platelet cooperate to induce this EMT program (14). TGF-β1 is stored in platelet granules (208) and released upon tumor cell dependent platelet activation and direct receptor-ligand interactions, including HMGB1:TLR4 (91). Although the effect of platelet on tumor cell protrusion has not been described, the upregulation of PDGF receptor α (PDGFRα) in tumor cells undergoing EMT has been associated with invadopodia formation and stabilization (205). PDGF is readily released by the α-granules of activated platelets, suggesting that they might initiate invadopodia formation by tumor cells.
Despite extensive evidence for the facilitation of metastasis by platelets prior to completion of extravasation, the evidence for platelet involvement in later stages of metastasis as a general rule is tenuous. Although platelet depletion decreased the proliferation and viability of tumor cells in mice (209), our lab and others observed that anticoagulant therapy, blockade of platelet activation and of tumor cell-platelet adhesion molecules failed to affect tumor growth both at the primary and at the secondary site (9, 12, 91, 209, 210). Extravascular platelets can be detected in primary tumors, where they support tumor cell proliferation and local invasion (33, 211, 212). It is not known whether they are similarly present in metastases. Although this would seem likely, such evidence has not been confirmed in metastasis models.
A potential role of platelets in angiogenesis has been reported. They support vessel maturation by promoting endothelial junction formation/endothelial VE-cadherin expression (211) and pericyte coverage of vessels (33). Additionally, platelets store or take up anti-angiogenic (e.g., endostatin, thrombospondin-1, and CXCL4) and pro-angiogenic factors (e.g., such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and PDGF), which are segregated in different types of α-granules and selectively released (96, 211, 213–217). The release of pro-angiogenic molecules has been observed both at the primary and at the metastatic site and is associated with better blood perfusion and vessel stability (209, 211). MPs from activated platelets can also support angiogenesis (218). Platelet could potentially affect tumor growth. Growth factors contained in platelet granules and PMPs might promote tumor cell proliferation (96), noting that anti-proliferative effects of PMPs were also reported (219). These data raise questions of whether platelet pro-angiogenic and pro-mitogenic effects might impact tumor progression, highlighting the need of further research in this field.
In addition Magnus et al. have shown that TF expression by tumor cells is associated with exit from tumor cell dormancy (220), but its possible role in metastatic dormancy is not clear. Effects of TF on metastatic dormancy might be due to intracellular signaling downstream of the TF receptor but could be completely independent of tumor cell-platelet interactions. Nevertheless, Albrengues et al. have elegantly shown that NETs support the awakening of dormant disseminated tumor cells in the lungs by cleaving subendothelial laminin, which in turns interacts with α3β1 integrin on dormant cancer cells and induces cell cycle progression (221). Taking into consideration that activated platelets induce NETosis through soluble mediators (184), sustained and systemic platelet activation in cancer patients might contribute to cancer recurrence through activation of dormant cells. Further research is needed to determine whether there is a contribution of platelets to metastatic dormancy.
Altogether, platelets interacting with a tumor cell are active biosuppliers of many assets needed to survive in the blood stream, adhere to the endothelium and extravasate. Hence, cancer cells that can induce the deposition of microclots on their surface have the potential to initiate metastasis, irrespective of cancer type, site of metastasis, and oncogenic mutations. This might explain the widespread pro-metastatic effect of platelet activation across many different cancer types.
Whereas, the contribution of platelets to metastasis has been extensively characterized, the role of their precursor megakaryocytes is less well-defined. In general, cancer is associated with increased megakaryopoiesis. Mice with ovarian tumors have higher numbers of bone marrow and spleen megakaryocytes, which correlated with their increased platelet counts. A similar correlation between bone marrow megakaryocytes and platelet count is found in women with ovarian cancer (33). Higher counts of bone marrow megakaryocytes, pro-platelets and platelets were found in pediatric chronic myeloid leukemia (222) and in metastatic breast cancer (223). Higher numbers of pulmonary megakaryocytes were also observed in patients with lung metastases (224). The increase of megakaryocytes during neoplasia might be partially traced back to TPO, whose plasma concentration is significantly higher in cancer patients and is predictive of poorer response and survival (225, 226). Tumor cells might directly secrete TPO (227) or alternatively induce its production by stroma cells. In ovarian cancer, interleukin-6 (IL-6) produced by the primary tumor stimulates the production of TPO by hepatic cells, the physiological source of TPO, which in turn induces megakaryocyte maturation and platelet production in the bone marrow (33), as a systemic effect and possibly prior to metastatic dissemination. Similarly, highly metastatic mammary adenocarcinomas are associated with increased numbers of bone marrow megakaryocytes in rats, yet in the absence of bony metastases (228). Other pre-clinical evidence suggests that bone marrow megakaryopoiesis takes place after tumor cell colonization. Jackson et al. have shown that the number of megakaryocytes in the bone marrow increased after intracardial injection of breast cancer cells in mice (metastatic cancer to bones), but not after orthotopic injection in the mammary fat pad (localized cancer) (223). They further showed that tumor cell interaction with osteoblasts led to the release of soluble factors that induced proliferation of megakaryocytes (223).
Megakaryocytes are found in the main sites of blood-borne metastasis such as bone marrow, lungs and liver, as well as the circulation (229). Evidence is pointing to a role for megakaryocytes in cancer metastasis, although both pro- and anti-metastatic effects have been observed. Shirai et al. have shown that reduction of TPO synthesis in hepatic cells through silencing of the THPO gene results in slower metastatic progression of PyMT mammary tumors in mice and in lower numbers of metastatic lung nodules (230). In contrast, Tpo−/− mice lacking more than 90% of bone marrow megakaryocytes develop more widespread and aggressive metastasis than their wild type counterpart (223), suggesting an anti-metastatic role of megakaryocytes. On the same line, TPO-driven megakaryopoiesis in the bone marrow decreases the incidence and size of tumor bone lesions (231). Interestingly, the number of circulating megakaryocytes has a trend to be associated with better survival and lower risk of metastatic disease in prostate cancer patients (229). There are different possible explanations for these contrasting roles of megakaryocytes during metastasis. On one hand, megakaryocytes can support metastasis through thrombopoiesis, and thrombocytosis is associated with cancer progression (16–20). Stone et al. have shown that paraneoplastic thrombocytosis in cancer patients can result from increased megakaryopoiesis through the IL-6/TPO axis (33). Importantly, high concentrations of plasma IL-6 (>10 pg/mL) are associated with lower overall patient survival, and inhibition of IL-6 synthesis by tumor cells restores normal platelet numbers and improves disease control (33). Similarly, an anti-IL-6 monoclonal antibody (sarilumab) used for treatment of rheumatoid arthritis is associated with a decreased platelet count (232). On the other hand, megakaryocytes can support the formation of (pre-)metastatic niches that affect tumor skeletal growth. In the bone marrow vascular niche, megakaryocyte crosstalk with osteoblasts, osteoclasts, and endothelial cells controls bone homeostasis [as reviewed by (37)]. Megakaryocytes release a plethora of osteoblast growth factors, such as fibroblast growth factor (FGF)-2 and TGF-β (37), that can support metastatic growth. Concomitantly, megakaryocytes suppress bone resorption by inhibiting osteoclast activity, with possible anti-metastatic consequences due to reduced release of bone matrix-bound growth factors and suppression of the well-described vicious cycle of osteolytic bone metastasis (233). Additionally, megakaryocytes store in their granules and secrete an array of pro- and anti-angiogenic factors (37, 213, 214, 216), which could control the vascularity of bone niches during skeletal metastasis. During bone metastasis, tumor cells hijack this crosstalk. In tumor bearing-mice, mature bone marrow megakaryocytes, and platelets express and release higher levels of the anti-angiogenic thrombospondin (TSP)-1 (234). Furthermore, the direct contact of megakaryocytes with prostate cancer cells induces cell cycle arrest and apoptosis (231). Hence, the direct interface of megakaryocytes with tumor cells can result in both pro- and anti-metastatic consequences.
In conclusion, the role of megakaryocytes during metastasis has only started to be appreciated. Although separating the direct effect of megakaryocytes on disseminated tumor cells from their thrombopoietic function will be challenging, further research is needed to reconcile the observed pro- and anti-metastatic effects of megakaryocytes and to understand the role of megakaryocytes on metastasis to secondary thrombopoiesis sites, such as lungs and liver.
A common complication of cancer is the onset of a hypercoagulable state that promotes thromboembolism (TE), which is the second leading cause of cancer-related morbidity and mortality (235, 236). Cancer patients have a 4- to 7-fold higher risk of TE than the general population (237), in particular if undergoing chemotherapy (238). Emboli in the circulation can lead to arterial thromboembolism (ATE), manifesting as myocardial infarction and ischemic stroke, and venous thromboembolism (VTE), which leads to events such as deep venous thrombosis and pulmonary embolism. Although ATE affects an average of 4.7% of all cancer patients (236), VTE events are far more common in cancer patients. The incidence of VTE varies between different types of cancer, with the highest incidence in pancreatic cancer (30–57% of patients) (239), followed by brain tumors (up to 31.7%) (240), and lung cancer (up to 21.5%) (241). Cancer-associated VTE causes a 3 to 10-fold higher risk of death and is consistently associated with worse prognosis, including lower overall survival and higher mortality (242). Advanced-stage tumors are largely associated with a greater risk of TE and cancer patients with TE have higher risk of tumor progression (236, 242). TE can be the first manifestation of an undiagnosed cancer and 90% patients with VTE have underlying metastatic cancer at the time of the event (108, 243). These associations highlight the link between thrombosis and the metastatic cascade and point to the pro-coagulant properties of metastatic cells as the underlying mechanism of VTE in cancer patients.
The expression of hemostatic factors and adhesion proteins in CTCs and tumor biopsies has been associated with poor prognosis. TF expression in malignant cells is an independent prognostic factor of tumor progression and VTE risk across a range of cancer types, including pancreatic (244, 245), glioma (246), colorectal cancer (247), breast cancer (248), and non-small cell lung cancer (249, 250). CD97 was highly expressed on CTCs from blood of metastatic prostate cancer and in their bone metastases (97). Similarly, expression of selectin ligands by tumor cells leads to a poorer prognosis (251).
Pro-thrombotic MPs have been proposed to play a major role in the pathogenesis of disseminated VTE in cancer patients. Not only do cancer patients have higher levels of circulating pro-coagulant MPs than healthy people, but cancer patients with VTE also have significantly higher plasma numbers of TF-expressing MPs and increased pro-coagulant MP activity in comparison to cancer patients without VTE (101, 108, 109, 252, 253). The concentration of TF-bearing MPs was associated with a higher risk of TE and higher mortality rate in some studies (101, 106, 108, 252), but not in others (110, 253). Also, the detection of TF+ MPs failed to predict the occurrence of TE events (252). Thus, the prognostic value of tumor-derived TF+ MPs remains controversial, suggesting the existence of TF-independent pathways of thrombosis induced by extracellular vesicles.
In contrast, a growing body of literature on PMPs supports their potential contribution to VTE in cancer patients. Previous reports have shown increased plasma levels of PMPs in cancer patients vs. healthy controls, with increased levels correlated with tumor grade (254–256). Importantly, high plasma PMP levels are correlated with poor overall survival from prostate cancer and higher risk of metastasis in both prostate and gastric cancer, where PMPs can be used as predictors of metastatic disease with high sensitivity and specificity (255, 257). Cancer patients with a history of VTE have higher plasma levels of PMPs than cancer patients with no prior VTE events (113, 256). These vesicles are derived from both resting and activated platelets and only a minority expressed TF (113, 256). Importantly, Bucciarelli et al. observed that PMPs could be an independent predictor of overall VTE risk, supporting the use of PMPs as a biomarker to identify patients at high risk of VTE, with or without cancer. Although further research is needed to understand the casual relationship between PMPs, VTE, and cancer metastasis, these reports suggest that plasma PMPs could be used as prognostic factors for cancer diagnosis, progression and VTE occurrence, and might be employed to guide the implementation of preventive thromboprophylaxis in high risk patients, irrespective of cancer status.
Considering the central role of platelets in the onset of metastasis and VTE, it follows that anti-coagulant drugs might be used to prevent VTE or metastasis altogether. Several families of anti-coagulant drugs that can target different aspects of the coagulation cascade, including thrombosis (aspirin and antagonists of P2Y12 and PARs) and coagulation factors (heparins, factor X inhibitors), have been evaluated in clinical trials as possible adjuvant therapies for cancer patients (Table 1). By affecting the normal hemostatic process, these drugs are often associated with side effects such as hemorrhage (275, 276) and therapies with higher efficacy, safety, and ease of administration have been evaluated over time.
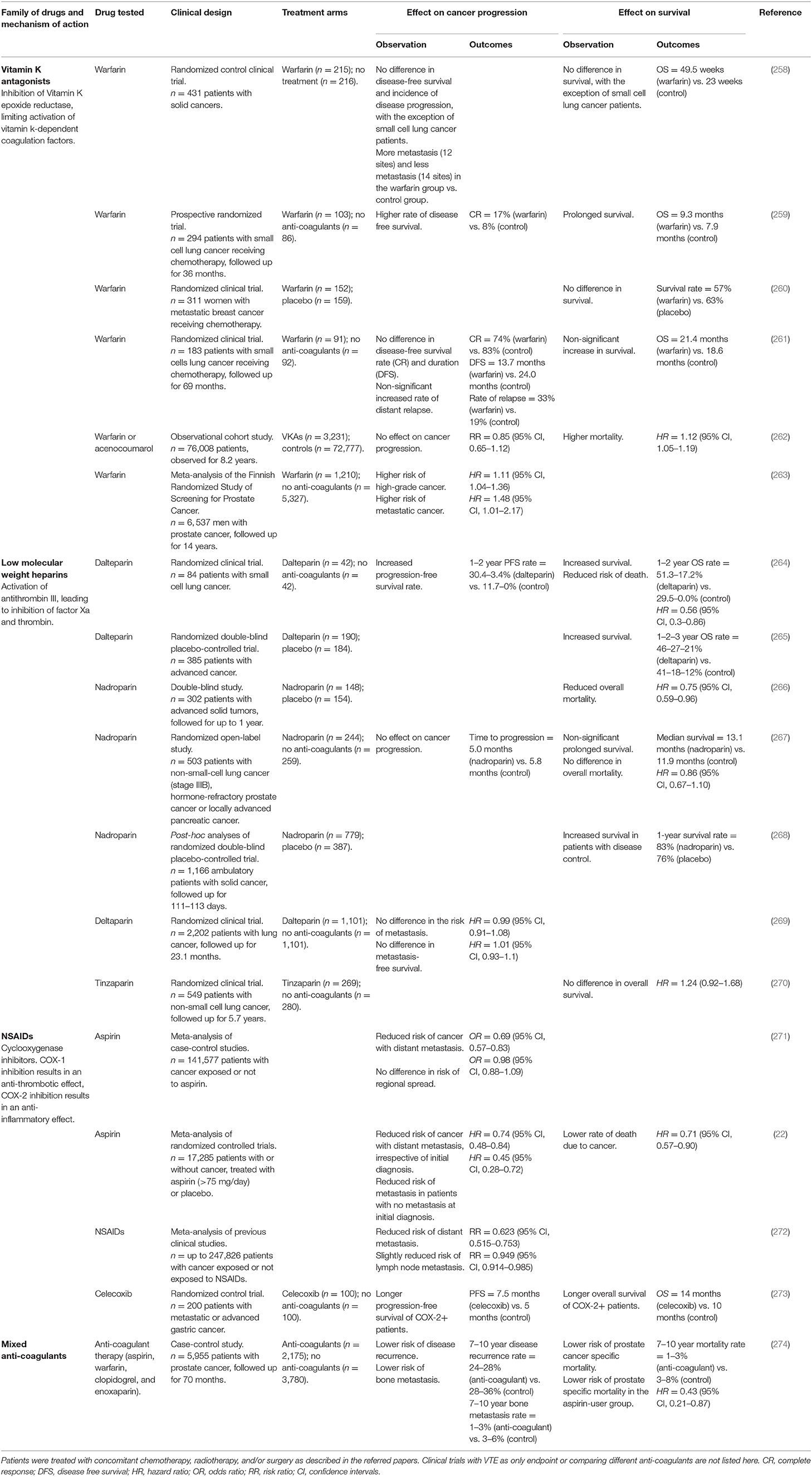
Table 1. List of clinical trial comparing the effect of adjuvant anti-coagulant drugs to control (no-treatment or placebo treatment) in terms of cancer progression, metastatic disease, and survival.
Historically, vitamin K antagonists (VKAs) such as warfarin were employed as the standard of care in cancer patients when they were the only anticoagulant pharmaceutical available. Despite promising pre-clinical data, long-term use of VKAs failed to show any effect on the risk of metastatic disease in most studies (258–263), and was generally associated with a higher risk of bleeding and higher mortality rate than other anticoagulants (277, 278). Low molecular weight heparins (LMWHs) were introduced later on. They showed a higher efficacy than unfractionated heparins and could be self-administered subcutaneously at home instead of intravenously in hospitalized patients (279). LMWHs were found to reduce cancer-related mortality and prolong survival of cancer patients with better prognosis at the start of randomization [e.g., disease control; (265, 266, 268)], consistent with its prevention of metastatic disease. However, increased overall survival and reduced VTE recurrence due to LMWHs has only been identified in some studies (264, 266, 267), and not others (269, 270), questioning further clinical evaluation of LMWHs as possible metastasis/VTE-preventive agents. These results could be partially due to the advanced cancer stage of enrolled patients, where pre-existing occult metastasis could not be affected by anti-coagulant therapy. Additionally, the anti-metastatic effect of LMWHs might be equally due to their inhibition of coagulation factors (e.g., thrombin and factor Xa) and their binding to selectins and integrins, affecting interactions between tumor cells, immune cells, and the vascular wall directly (280). The pleiotropic effects of LMWHs make it difficult to draw definitive conclusions from these trials.
More recently, other anticoagulants have also shown discouraging results in clinical trials. Despite promising pre-clinical data, the ADP P2Y12 receptor antagonist prasugrel (TRITON-TIMI 38 trial) (281, 282) and the P2Y12 inhibitors thienopyridines (clopidogrel and prasugrel) (283) increased the incidence of newly diagnosed solid cancers and the risk of cancer-related death. Similar effects were seen upon treatment with the PAR1 antagonist vorapaxar (TRACER trial) (284) and the factor Xa inhibitor apixaban (APPRAISE-2 trial) (285). The reason of this effect is still not clear, and additional data are needed to address the safety of these and other anticoagulant drugs.
Less controversial results come from clinical trials evaluating NSAIDs as an adjuvant therapy for cancer patients, which have been overall associated with longer progression-free survival and reduced risk of distant metastasis (271–274, 286). The strongest case is presented by the cyclooxygenase (COX)-1 and−2 inhibitor aspirin. Long-term aspirin treatment (≥5–10 years) has proven to reduce cancer incidence and mortality, in particular due to reduction in colorectal adenomas and cancer (22, 287–291). This is possibly related to the inhibitory effect of aspirin on COX-2, which is involved in the early carcinogenesis of colorectal adenocarcinoma (292), or to other COX-independent targets that are affected by higher doses of aspirin. Aspirin treatment is also associated with an increased T cell infiltration in ovarian cancer, potentially paving the way to use aspirin in combination with checkpoint inhibitors for tumor control (293). In addition to its effect on primary cancers, the meta-analysis of case-control studies and randomized control trials by Rothwell et al. has shown that regular use of aspirin reduced the risk of metastatic cancer, particularly pulmonary metastasis (271, 286). Results were particularly impressive for patients with no evidence of metastasis at randomization, for whom the risk of developing in-trial metastasis was more than halved (HR = 0.45, 95% CI 0.28–0.71, p = 0.0009). Further this reduction occurred in many cancer types for which COX-2 is not known to be important in its carcinogenesis. The risk of cancer-related deaths was significantly reduced by low-dose aspirin (<300 mg/day), which is mainly anti-thrombotic, but not high-dose aspirin (≥300 mg/day), which is both anti-thrombotic and anti-inflammatory (286), reinforcing the hypothesis that aspirin prevents metastasis through its effect on platelet aggregation. More recently, Yang et al. have found a reduced number of blood CTCs in colorectal cancer patients treated with aspirin, consistent with its effect on tumor cell survival in the circulation and metastatic seeding (294). Our group has conclusively shown that this metastasis-preventive effect of aspirin relies on the inhibition of COX-1 and its downstream product TXA2 in platelets, which is responsible for the generation of a favorable intravascular niche promoting the survival and metastatic seeding of disseminating tumor cells (9). Importantly, we showed that untreated mice infused with aspirin-treated platelets harbor significantly fewer lung metastasis, supporting the notion that the anti-metastatic effect of aspirin does not rely on extra-platelet targets. In light of these results, we propose that more selective inhibitors of TXA2, such as the dual TXAS inhibitor and TP antagonist picotamide, would achieve the same anti-metastatic result while sparing other gastroprotective COX-1 products, resulting in a better anti-thrombotic profile and less side effects. Future clinical trials might address the efficacy and safety of picotamide as adjuvant therapeutic intervention in cancer patients.
Other families of anti-platelet drugs, such as inhibitors of αIIbβ3 (e.g., tirofiban and eptifibatide) and selectins (e.g., crizanlizumab and rivipansel), have FDA indications for the treatment of cardiovascular events, but their effect on cancer progression has not been evaluated yet. Additionally, reduction of paraneoplastic thrombopoiesis through IL-6 neutralization might represent a valuable approach to reduce metastatic spreading (33).
In conclusion, clinical trials so far have shown that there is no linear relationship between anticoagulation and metastasis prevention, but the effect largely depends on the drug used, the cancer type and the risk-benefit balance. These considerations have delayed the introduction of anticoagulant drugs as adjuvant therapy in cancer patients. A relatively weak and low-cost anticoagulant like aspirin has been by far the most successful drug in reducing the risk of metastatic cancer, in addition to reducing cancer incidence. It is possible that the inhibition of platelet COX-1/TXA2 signaling achieved by aspirin and TXA2 inhibitors, but not other anti-platelet drugs, is essential to disrupt the formation of intravascular pro-metastatic niches, which are required for early metastatic seeding. Although other anti-coagulant might affect later stages of metastasis, this effect might not be sufficient to achieve metastasis prevention (9). Although a more complete benefit-harm evaluation will have to be made in the future, the risk of major bleeding associated with long-term aspirin treatment, especially for exposure shorter than 5 years, might be counterbalanced by a reduced risk of vascular events and a significant reduction of cancer-related deaths (22). In 2015, the international Phase III clinical trial Add-Aspirin has started to address the effect of low (100 mg/day) or medium (300 mg/day) dose of aspirin or placebo on patients with non-metastatic solid tumors (breast, colorectal, gastro-esophageal, and prostate cancer) over a period of 5 years of more. Following up on excellent results on tolerability and toxicity, this trial will help understand the rate of survival, cancer recurrence, metastasis appearance, and the overall benefit-harm profile associated with aspirin use (295, 296).
This review highlights the multifaceted interplay between tumor cells, megakaryocytes and platelets during hematogenous metastasis. Undoubtedly, the ability of tumor cells to interact with platelets confers a strong positive drive toward metastatic dissemination. The activation of platelets by tumor cells in the blood stream converts the physiologically “resting” intravascular niche into an active tumor-permissive environment, characterized by repressed immunosurveillance, and activated endothelial and myeloid cells. The advantage provided by platelets to the metastatic process can explain why cancers evolve to promote paraneoplastic megakaryopoiesis and thrombopoiesis to support their progression. The current knowledge on the contribution of platelets and megakaryocytes to metastasis opens promising therapeutic avenues. In particular, targeting platelets rather than the more genetically instable tumor cells might be a promising strategy for metastasis prevention. In support of this, experimental studies clearly indicate that abrogation of platelet interaction with tumor cells ultimately reduces metastasis across a wide range of cancer types and metastatic sites. We would expect thromboembolic complications to be reduced as well. How to target platelets, however, in cancer patient care is still a work-in-progress. For some anticoagulants, the extremely deleterious side effects due to the impairment of hemostasis is overshadowing any survival advantages associated with decreased cancer progression. Lessons from previous clinical trials suggest that the best strategy would be to target tumor cell-platelet interactions while leaving physiological platelet functions and stroma-platelet crosstalk unaffected. Further research is required to expand our understanding of the molecular mechanisms underlying these processes and to develop safer adjuvant or neoadjuvant therapies for cancer patients.
This manuscript was written by SL and RM. All authors contributed to the article and approved the submitted version.
SL is the recipient of Cancer Research UK Oxford Centre DPhil Prize Studentship BBRTJW00. RM is funded by Cancer Research UK, grant C5255/A23755.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Trousseau A, Bazire PV, Cormack JR. Lectures on Clinical Medicine, Delivered at the Hotel-Dieu. Paris: New Sydenham Society (1872).
3. Gasic GJ, Gasic TB, Stewart CC. Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci USA. (1968) 61:46–52. doi: 10.1073/pnas.61.1.46
4. Gasic GJ, Gasic TB, Galanti N, Johnson T, Murphy S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int J Cancer. (1973) 11:704–18. doi: 10.1002/ijc.2910110322
5. Bakewell SJ, Nestor P, Prasad S, Tomasson MH, Dowland N, Mehrotra M, et al. Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proc Natl Acad Sci USA. (2003) 100:14205–10. doi: 10.1073/pnas.2234372100
6. Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. (2004) 104:397–401. doi: 10.1182/blood-2004-02-0434
7. Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci USA. (2014) 111:E3053–61. doi: 10.1073/pnas.1411082111
8. Haemmerle M, Taylor ML, Gutschner T, Pradeep S, Cho MS, Sheng J, et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. (2017) 8:310. doi: 10.1038/s41467-017-00411-z
9. Lucotti S, Cerutti C, Soyer M, Gil-Bernabe AM, Gomes AL, Allen PD, et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J Clin Invest. (2019) 129:1845–62. doi: 10.1172/JCI121985
10. Agostino D, Cliffton EE, Girolami A. Effect of prolonged coumadin treatment on the production of pulmonary metastases in the rat. Cancer. (1966) 19:284–8. doi: 10.1002/1097-0142(196602)19:2<284::AID-CNCR2820190223>3.0.CO;2-0
11. Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA. (2001) 98:3352–7. doi: 10.1073/pnas.061615598
12. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. (2005) 105:178–85. doi: 10.1182/blood-2004-06-2272
13. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood. (2007) 110:133–41. doi: 10.1182/blood-2007-01-065995
14. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. (2011) 20:576–90. doi: 10.1016/j.ccr.2011.09.009
15. Gil-Bernabe AM, Ferjancic S, Tlalka M, Zhao L, Allen PD, Im JH, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. (2012) 119:3164–75. doi: 10.1182/blood-2011-08-376426
16. Costantini V, Zacharski LR, Moritz TE, Edwards RL. The platelet count in carcinoma of the lung and colon. Thromb Haemost. (1990) 64:501–5. doi: 10.1055/s-0038-1647347
17. Inoue K, Kohashikawa K, Suzuki S, Shimada M, Yoshida H. Prognostic significance of thrombocytosis in renal cell carcinoma patients. Int J Urol. (2004) 11:364–7. doi: 10.1111/j.1442-2042.2004.00808.x
18. Brown KM, Domin C, Aranha GV, Yong S, Shoup M. Increased preoperative platelet count is associated with decreased survival after resection for adenocarcinoma of the pancreas. Am J Surg. (2005) 189:278–82. doi: 10.1016/j.amjsurg.2004.11.014
19. Ayhan A, Bozdag G, Taskiran C, Gultekin M, Yuce K, Kucukali T. The value of preoperative platelet count in the prediction of cervical involvement and poor prognostic variables in patients with endometrial carcinoma. Gynecol Oncol. (2006) 103:902–5. doi: 10.1016/j.ygyno.2006.05.034
20. Brockmann MA, Giese A, Mueller K, Kaba FJ, Lohr F, Weiss C, et al. Preoperative thrombocytosis predicts poor survival in patients with glioblastoma. Neuro Oncol. (2007) 9:335–42. doi: 10.1215/15228517-2007-013
21. Cooke NM, Egan K, Mcfadden S, Grogan L, Breathnach OS, O'leary J, et al. Increased platelet reactivity in patients with late-stage metastatic cancer. Cancer Med. (2013) 2:564–70. doi: 10.1002/cam4.86
22. Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. (2012) 379:1602–12. doi: 10.1016/S0140-6736(11)61720-0
23. Jirouskova M, Shet AS, Johnson GJ. A guide to murine platelet structure, function, assays, and genetic alterations. J Thromb Haemost. (2007) 5:661–9. doi: 10.1111/j.1538-7836.2007.02407.x
24. Gremmel T, Frelinger AL III, Michelson AD. Platelet Physiology. Semin Thromb Hemost. (2016) 42:191–204. doi: 10.1055/s-0035-1564835
25. Nesbitt WS, Westein E, Tovar-Lopez FJ, Tolouei E, Mitchell A, Fu J, et al. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. (2009) 15:665–73. doi: 10.1038/nm.1955
26. Tokarev AA, Butylin AA, Ataullakhanov FI. Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophys J. (2011) 100:799–808. doi: 10.1016/j.bpj.2010.12.3740
27. Tokarev AA, Butylin AA, Ermakova EA, Shnol EE, Panasenko GP, Ataullakhanov FI. Finite platelet size could be responsible for platelet margination effect. Biophys J. (2011) 101:1835–43. doi: 10.1016/j.bpj.2011.08.031
28. Thon JN, Italiano JE. Platelets: production, morphology and ultrastructure. Handb Exp Pharmacol. (2012) 210:3–22. doi: 10.1007/978-3-642-29423-5_1
29. Michelson A, Cattaneo M, Frelinger A, Newman P. Platelets 4th ed. Elsevier/Academic Press (2019). Available online at: https://www.sciencedirect.com/book/9780128134566/platelets?via=ihub=
30. Guo T, Wang X, Qu Y, Yin Y, Jing T, Zhang Q. Megakaryopoiesis and platelet production: insight into hematopoietic stem cell proliferation and differentiation. Stem Cell Investig. (2015) 2:3. doi: 10.3978/j.issn.2306-9759.2015.02.01
31. Noetzli LJ, French SL, Machlus KR. New insights into the differentiation of megakaryocytes from hematopoietic progenitors. Arterioscler Thromb Vasc Biol. (2019) 39:1288–300. doi: 10.1161/ATVBAHA.119.312129
32. Thachil J. The contribution of megakaryocytes to liver fibrosis. Gut. (2009) 58:1565. doi: 10.1136/gut.2008.176503
33. Stone RL, Nick AM, Mcneish IA, Balkwill F, Han HD, Bottsford-Miller J, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. (2012) 366:610–8. doi: 10.1056/NEJMoa1110352
34. Brouard N, Jost C, Matthias N, Albrecht C, Egard S, Gandhi P, et al. A unique microenvironment in the developing liver supports the expansion of megakaryocyte progenitors. Blood Adv. (2017) 1:1854–66. doi: 10.1182/bloodadvances.2016003541
35. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. (2017) 544:105–9. doi: 10.1038/nature21706
36. Machlus KR, Italiano JEJr. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. (2013) 201:785–96. doi: 10.1083/jcb.201304054
37. Psaila B, Lyden D, Roberts I. Megakaryocytes, malignancy and bone marrow vascular niches. J Thromb Haemost. (2012) 10:177–88. doi: 10.1111/j.1538-7836.2011.04571.x
38. Schenone M, Furie BC, Furie B. The blood coagulation cascade. Curr Opin Hematol. (2004) 11:272–7. doi: 10.1097/01.moh.0000130308.37353.d4
39. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. (2008) 359:938–49. doi: 10.1056/NEJMra0801082
40. Fredrickson BJ, Dong JF, Mcintire LV, Lopez JA. Shear-dependent rolling on von Willebrand factor of mammalian cells expressing the platelet glycoprotein Ib-IX-V complex. Blood. (1998) 92:3684–93. doi: 10.1182/blood.V92.10.3684
41. Jackson SP, Mistry N, Yuan Y. Platelets and the injured vessel wall– “rolling into action”: focus on glycoprotein Ib/V/IX and the platelet cytoskeleton. Trends Cardiovasc Med. (2000) 10:192–7. doi: 10.1016/S1050-1738(00)00062-1
42. Kulkarni S, Dopheide SM, Yap CL, Ravanat C, Freund M, Mangin P, et al. A revised model of platelet aggregation. J Clin Invest. (2000) 105:783–91. doi: 10.1172/JCI7569
43. Jackson SP, Nesbitt WS, Kulkarni S. Signaling events underlying thrombus formation. J Thromb Haemost. (2003) 1:1602–12. doi: 10.1046/j.1538-7836.2003.00267.x
44. Kasirer-Friede A, Cozzi MR, Mazzucato M, De Marco L, Ruggeri ZM, Shattil SJ. Signaling through GP Ib-IX-V activates alpha IIb beta 3 independently of other receptors. Blood. (2004) 103:3403–11. doi: 10.1182/blood-2003-10-3664
45. Valet C, Severin S, Chicanne G, Laurent PA, Gaits-Iacovoni F, Gratacap MP, et al. The role of class I, II and III PI 3-kinases in platelet production and activation and their implication in thrombosis. Adv Biol Regul. (2016) 61:33–41. doi: 10.1016/j.jbior.2015.11.008
46. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, Mcredmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. (2004) 103:2096–104. doi: 10.1182/blood-2003-08-2804
47. Szanto T, Joutsi-Korhonen L, Deckmyn H, Lassila R. New insights into von Willebrand disease and platelet function. Semin Thromb Hemost. (2012) 38:55–63. doi: 10.1055/s-0031-1300952
48. Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost. (2016) 14:918–30. doi: 10.1111/jth.13302
49. Laubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol. (2010) 20:169–77. doi: 10.1016/j.semcancer.2010.04.005
50. Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. (2007) 101:234–47. doi: 10.1161/CIRCRESAHA.107.151860b
51. Cheung LS, Raman PS, Balzer EM, Wirtz D, Konstantopoulos K. Biophysics of selectin-ligand interactions in inflammation and cancer. Phys Biol. (2011) 8:015013. doi: 10.1088/1478-3975/8/1/015013
52. Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F. Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost. (2011) 9(Suppl. 1):306–15. doi: 10.1111/j.1538-7836.2011.04318.x
53. Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. (2015) 29:17–24. doi: 10.1016/j.blre.2014.09.003
54. Sabrkhany S, Griffioen AW, Oude Egbrink MG. The role of blood platelets in tumor angiogenesis. Biochim Biophys Acta. (2011) 1815:189–96. doi: 10.1016/j.bbcan.2010.12.001
55. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
56. Habets KL, Huizinga TW, Toes RE. Platelets and autoimmunity. Eur J Clin Invest. (2013) 43:746–57. doi: 10.1111/eci.12101
57. Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA, et al. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. (2004) 64:8613–9. doi: 10.1158/0008-5472.CAN-04-2078
58. Garnier D, Magnus N, D'asti E, Hashemi M, Meehan B, Milsom C, et al. Genetic pathways linking hemostasis and cancer. Thromb Res. (2012) 129(Suppl. 1):S22–9. doi: 10.1016/S0049-3848(12)70012-9
59. Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci USA. (1992) 89:11832–6. doi: 10.1073/pnas.89.24.11832
60. Kataoka H, Uchino H, Asada Y, Hatakeyama K, Nabeshima K, Sumiyoshi A, et al. Analysis of tissue factor and tissue factor pathway inhibitor expression in human colorectal carcinoma cell lines and metastatic sublines to the liver. Int J Cancer. (1997) 72:878–84. doi: 10.1002/(SICI)1097-0215(19970904)72:5<878::AID-IJC27>3.0.CO;2-3
61. Ettelaie C, Collier ME, Featherby S, Benelhaj NE, Greenman J, Maraveyas A. Analysis of the potential of cancer cell lines to release tissue factor-containing microvesicles: correlation with tissue factor and PAR2 expression. Thromb J. (2016) 14:2. doi: 10.1186/s12959-016-0075-3
62. Bourcy M, Suarez-Carmona M, Lambert J, Francart ME, Schroeder H, Delierneux C, et al. Tissue factor induced by epithelial-mesenchymal transition triggers a procoagulant state that drives metastasis of circulating tumor cells. Cancer Res. (2016) 76:4270–82. doi: 10.1158/0008-5472.CAN-15-2263
63. Van Den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood. (2012) 119:924–32. doi: 10.1182/blood-2011-06-317685
64. Koizume S, Jin MS, Miyagi E, Hirahara F, Nakamura Y, Piao JH, et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. (2006) 66:9453–60. doi: 10.1158/0008-5472.CAN-06-1803
65. Tang JQ, Fan Q, Wu WH, Jia ZC, Li H, Yang YM, et al. Extrahepatic synthesis of coagulation factor VII by colorectal cancer cells promotes tumor invasion and metastasis. Chin Med J. (2010) 123:3559–65. doi: 10.3760/cma.j.issn.0366-6999.2010.24.011
66. Tsai MC, Chen KD, Wang CC, Huang KT, Wu CH, Kuo IY, et al. Factor VII promotes hepatocellular carcinoma progression through ERK-TSC signaling. Cell Death Discov. (2015) 1:15051. doi: 10.1038/cddiscovery.2015.51
67. Olas B, Mielicki WP, Wachowicz B, Krajewski T. Cancer procoagulant stimulates platelet adhesion. Thromb Res. (1999) 94:199–203. doi: 10.1016/S0049-3848(98)00214-X
68. Olas B, Wachowicz B, Mielicki WP, Buczynski A. Free radicals are involved in cancer procoagulant-induced platelet activation. Thromb Res. (2000) 97:169–75. doi: 10.1016/S0049-3848(99)00129-2
69. Donati MB, Gambacorti-Passerini C, Casali B, Falanga A, Vannotti P, Fossati G, et al. Cancer procoagulant in human tumor cells: evidence from melanoma patients. Cancer Res. (1986) 46:6471–4.
70. Falanga A, Consonni R, Marchetti M, Locatelli G, Garattini E, Passerini CG, et al. Cancer procoagulant and tissue factor are differently modulated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood. (1998) 92:143–51. doi: 10.1182/blood.V92.1.143.413k18_143_151
71. Molnar S, Guglielmone H, Lavarda M, Rizzi ML, Jarchum G. Procoagulant factors in patients with cancer. Hematology. (2007) 12:555–9. doi: 10.1080/10245330701521416
72. Nagata S, Suzuki J, Segawa K, Fujii T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. (2016) 23:952–61. doi: 10.1038/cdd.2016.7
73. Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. (1991) 51:3062–6.
74. Riedl S, Rinner B, Asslaber M, Schaider H, Walzer S, Novak A, et al. In search of a novel target - phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim Biophys Acta. (2011) 1808:2638–45. doi: 10.1016/j.bbamem.2011.07.026
75. Vallabhapurapu SD, Blanco VM, Sulaiman MK, Vallabhapurapu SL, Chu Z, Franco RS, et al. Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium. Oncotarget. (2015) 6:34375–88. doi: 10.18632/oncotarget.6045
76. Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. (2003) 42:423–38. doi: 10.1016/S0163-7827(03)00025-0
77. Yu JL, Rak JW. Shedding of tissue factor (TF)-containing microparticles rather than alternatively spliced TF is the main source of TF activity released from human cancer cells. J Thromb Haemost. (2004) 2:2065–7. doi: 10.1111/j.1538-7836.2004.00972.x
78. Hobbs JE, Zakarija A, Cundiff DL, Doll JA, Hymen E, Cornwell M, et al. Alternatively spliced human tissue factor promotes tumor growth and angiogenesis in a pancreatic cancer tumor model. Thromb Res. (2007) 120(Suppl. 2):S13–21. doi: 10.1016/S0049-3848(07)70126-3
79. Goldin-Lang P, Tran QV, Fichtner I, Eisenreich A, Antoniak S, Schulze K, et al. Tissue factor expression pattern in human non-small cell lung cancer tissues indicate increased blood thrombogenicity and tumor metastasis. Oncol Rep. (2008) 20:123–8. doi: 10.3892/or.20.1.123
80. Boukerche H, Berthier-Vergnes O, Penin F, Tabone E, Lizard G, Bailly M, et al. Human melanoma cell lines differ in their capacity to release ADP and aggregate platelets. Br J Haematol. (1994) 87:763–72. doi: 10.1111/j.1365-2141.1994.tb06736.x
81. Heinmoller E, Weinel RJ, Heidtmann HH, Salge U, Seitz R, Schmitz I, et al. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol. (1996) 122:735–44. doi: 10.1007/BF01209121
82. Ekambaram P, Lambiv W, Cazzolli R, Ashton AW, Honn KV. The thromboxane synthase and receptor signaling pathway in cancer: an emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. (2011) 30:397–408. doi: 10.1007/s10555-011-9297-9
83. Kim YJ, Borsig L, Varki NM, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci USA. (1998) 95:9325–30. doi: 10.1073/pnas.95.16.9325
84. Alves CS, Burdick MM, Thomas SN, Pawar P, Konstantopoulos K. The dual role of CD44 as a functional P-selectin ligand and fibrin receptor in colon carcinoma cell adhesion. Am J Physiol Cell Physiol. (2008) 294:C907–16. doi: 10.1152/ajpcell.00463.2007
85. Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature. (1992) 359:848–51. doi: 10.1038/359848a0
86. Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. (2003) 197:1585–98. doi: 10.1084/jem.20021868
87. Kato Y, Fujita N, Kunita A, Sato S, Kaneko M, Osawa M, et al. Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J Biol Chem. (2003) 278:51599–605. doi: 10.1074/jbc.M309935200
88. Suzuki-Inoue K, Kato Y, Inoue O, Kaneko MK, Mishima K, Yatomi Y, et al. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. (2007) 282:25993–6001. doi: 10.1074/jbc.M702327200
89. Kato Y, Kaneko MK, Kunita A, Ito H, Kameyama A, Ogasawara S, et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. (2008) 99:54–61. doi: 10.1111/j.1349-7006.2007.00634.x
90. Takagi S, Sato S, Oh-Hara T, Takami M, Koike S, Mishima Y, et al. Platelets promote tumor growth and metastasis via direct interaction between Aggrus/podoplanin and CLEC-2. PLoS ONE. (2013) 8:e73609. doi: 10.1371/journal.pone.0073609
91. Yu LX, Yan L, Yang W, Wu FQ, Ling Y, Chen SZ, et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat Commun. (2014) 5:5256. doi: 10.1038/ncomms6256
92. Boukerche H, Berthier-Vergnes O, Tabone E, Dore JF, Leung LL, Mcgregor JL. Platelet-melanoma cell interaction is mediated by the glycoprotein IIb-IIIa complex. Blood. (1989) 74:658–63. doi: 10.1182/blood.V74.2.658.658
93. Nierodzik ML, Plotkin A, Kajumo F, Karpatkin S. Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. J Clin Invest. (1991) 87:229–36. doi: 10.1172/JCI114976
94. Felding-Habermann B, Habermann R, Saldivar E, Ruggeri ZM. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J Biol Chem. (1996) 271:5892–900. doi: 10.1074/jbc.271.10.5892
95. Lorger M, Krueger JS, O'neal M, Staflin K, Felding-Habermann B. (2009). Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc Natl Acad Sci USA 106, 10666–10671. doi: 10.1073/pnas.0903035106
96. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. (2011) 11:123–34. doi: 10.1038/nrc3004
97. Ward Y, Lake R, Faraji F, Sperger J, Martin P, Gilliard C, et al. Platelets promote metastasis via binding tumor CD97 leading to bidirectional signaling that coordinates transendothelial migration. Cell Rep. (2018) 23:808–22. doi: 10.1016/j.celrep.2018.03.092
98. Van Niel G, D'angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. (2018) 19:213–28. doi: 10.1038/nrm.2017.125
99. Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, Bitzer AM, et al. Tumor shedding and coagulation. Science. (1981) 212:923–4. doi: 10.1126/science.7195067
100. Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med. (2009) 206:1913–27. doi: 10.1084/jem.20082297
101. Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, et al. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. (2009) 15:6830–40. doi: 10.1158/1078-0432.CCR-09-0371
102. Wang JG, Geddings JE, Aleman MM, Cardenas JC, Chantrathammachart P, Williams JC, et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood. (2012) 119:5543–52. doi: 10.1182/blood-2012-01-402156
103. Rousseau A, Van Dreden P, Khaterchi A, Larsen AK, Elalamy I, Gerotziafas GT. Procoagulant microparticles derived from cancer cells have determinant role in the hypercoagulable state associated with cancer. Int J Oncol. (2017) 51:1793–800. doi: 10.3892/ijo.2017.4170
104. Zwicker JI, Furie BC, Kos CA, Larocca T, Bauer KA, Furie B. Trousseau's syndrome revisited: tissue factor-bearing microparticles in pancreatic cancer. Blood. (2005) 106:259. doi: 10.1182/blood.V106.11.259.259
105. Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, et al. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. (2007) 97:119–23. doi: 10.1160/TH06-03-0141
106. Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost. (2007) 5:520–7. doi: 10.1111/j.1538-7836.2007.02369.x
107. Haubold K, Rink M, Spath B, Friedrich M, Chun FK, Marx G, et al. Tissue factor procoagulant activity of plasma microparticles is increased in patients with early-stage prostate cancer. Thromb Haemost. (2009) 101:1147–55. doi: 10.1160/TH08-10-0654
108. Tesselaar ME, Romijn FP, Van Der Linden IK, Bertina RM, Osanto S. Microparticle-associated tissue factor activity in cancer patients with and without thrombosis. J Thromb Haemost. (2009) 7:1421–3. doi: 10.1111/j.1538-7836.2009.03504.x
109. Campello E, Spiezia L, Radu CM, Bulato C, Castelli M, Gavasso S, et al. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb Res. (2011) 127:473–7. doi: 10.1016/j.thromres.2011.01.002
110. Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, Marosi C, et al. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J Thromb Haemost. (2012) 10:1363–70. doi: 10.1111/j.1538-7836.2012.04754.x
111. Flaumenhaft R, Dilks JR, Richardson J, Alden E, Patel-Hett SR, Battinelli E, et al. Megakaryocyte-derived microparticles: direct visualization and distinction from platelet-derived microparticles. Blood. (2009) 113:1112–21. doi: 10.1182/blood-2008-06-163832
112. Zahra S, Anderson JA, Stirling D, Ludlam CA. Microparticles, malignancy and thrombosis. Br J Haematol. (2011) 152:688–700. doi: 10.1111/j.1365-2141.2010.08452.x
113. Bucciarelli P, Martinelli I, Artoni A, Passamonti SM, Previtali E, Merati G, et al. Circulating microparticles and risk of venous thromboembolism. Thromb Res. (2012) 129:591–7. doi: 10.1016/j.thromres.2011.08.020
114. Cauwenberghs S, Feijge MA, Harper AG, Sage SO, Curvers J, Heemskerk JW. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. (2006) 580:5313–20. doi: 10.1016/j.febslet.2006.08.082
115. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. (1999) 94:3791–9. doi: 10.1182/blood.V94.11.3791
116. Biro E, Sturk-Maquelin KN, Vogel GM, Meuleman DG, Smit MJ, Hack CE, et al. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J Thromb Haemost. (2003) 1:2561–8. doi: 10.1046/j.1538-7836.2003.00456.x
117. Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, et al. Procoagulant microparticles: disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol. (2006) 26:2594–604. doi: 10.1161/01.ATV.0000246775.14471.26
118. Owens AP III, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. (2011) 108:1284–97. doi: 10.1161/CIRCRESAHA.110.233056
119. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-mediated metastasis: communication from a distance. Dev Cell. (2019) 49:347–60. doi: 10.1016/j.devcel.2019.04.011
120. Lima LG, Leal AC, Vargas G, Porto-Carreiro I, Monteiro RQ. Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb Res. (2013) 132:450–6. doi: 10.1016/j.thromres.2013.07.026
121. Gomes FG, Sandim V, Almeida VH, Rondon AMR, Succar BB, Hottz ED, et al. Breast-cancer extracellular vesicles induce platelet activation and aggregation by tissue factor-independent and -dependent mechanisms. Thromb Res. (2017) 159:24–32. doi: 10.1016/j.thromres.2017.09.019
122. Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. (2017) 7:6438. doi: 10.1038/s41598-017-06893-7
123. Colucci M, Balconi G, Lorenzet R, Pietra A, Locati D, Donati MB, et al. Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest. (1983) 71:1893–6. doi: 10.1172/JCI110945
124. Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MAJr. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci USA. (1986) 83:4533–7. doi: 10.1073/pnas.83.12.4533
125. Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, et al. Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med. (1990) 172:1535–45. doi: 10.1084/jem.172.6.1535
126. Kirchhofer D, Tschopp TB, Hadvary P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Invest. (1994) 93:2073–83. doi: 10.1172/JCI117202
127. Vrana JA, Stang MT, Grande JP, Getz MJ. Expression of tissue factor in tumor stroma correlates with progression to invasive human breast cancer: paracrine regulation by carcinoma cell-derived members of the transforming growth factor beta family. Cancer Res. (1996) 56:5063–70.
128. Falanga A, Panova-Noeva M, Russo L. Procoagulant mechanisms in tumour cells. Best Pract Res Clin Haematol. (2009) 22:49–60. doi: 10.1016/j.beha.2008.12.009
129. Brock TA, Dvorak HF, Senger DR. Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am J Pathol. (1991) 138:213–21.
130. Nachman RL, Hajjar KA, Silverstein RL, Dinarello CA. Interleukin 1 induces endothelial cell synthesis of plasminogen activator inhibitor. J Exp Med. (1986) 163:1595–600. doi: 10.1084/jem.163.6.1595
131. Cools-Lartigue J, Spicer J, Mcdonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. (2013) 123:3446–58. doi: 10.1172/JCI67484
132. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. (2016) 8:361ra138. doi: 10.1126/scitranslmed.aag1711
133. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
134. Mauracher LM, Posch F, Martinod K, Grilz E, Daullary T, Hell L, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. (2018) 16:508–18. doi: 10.1111/jth.13951
135. Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q, et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine. (2020) 53:102671. doi: 10.1016/j.ebiom.2020.102671
136. Clark SR, Ma AC, Tavener SA, Mcdonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
137. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
138. Fidler IJ. Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-Iodo-2′ -deoxyuridine23. J Natl Cancer Inst. (1970) 45:773–82.
139. Luzzi KJ, Macdonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. (1998) 153:865–73. doi: 10.1016/S0002-9440(10)65628-3
140. Glaves D, Weiss L. Initial tumor cell arrest in animals of defined coagulative status. Int J Cancer. (1978) 21:741–6. doi: 10.1002/ijc.2910210611
141. Liotta LA, Vembu D, Saini RK, Boone C. In vivo monitoring of the death rate of artificial murine pulmonary micrometastases. Cancer Res. (1978) 38:1231–6.
142. Gorelik E, Bere WW, Herberman RB. Role of NK cells in the antimetastatic effect of anticoagulant drugs. Int J Cancer. (1984) 33:87–94. doi: 10.1002/ijc.2910330115
143. Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. (1999) 59:1295–300.
144. Wiltrout RH, Herberman RB, Zhang SR, Chirigos MA, Ortaldo JR, Green KMJr, et al. Role of organ-associated NK cells in decreased formation of experimental metastases in lung and liver. J Immunol. (1985) 134:4267–75.
145. Coupland LA, Chong BH, Parish CR. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. (2012) 72:4662–71. doi: 10.1158/0008-5472.CAN-11-4010
146. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
147. Hanna N. Role of natural killer cells in control of cancer metastasis. Cancer Metastasis Rev. (1982) 1:45–64. doi: 10.1007/BF00049480
148. Placke T, Orgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. (2012) 72:440–8. doi: 10.1158/0008-5472.CAN-11-1872
149. Gersuk GM, Chang WC, Pattengale PK. Inhibition of human natural killer cell activity by platelet-derived growth factor. II. Membrane binding studies, effects of recombinant IFN-alpha and IL-2, and lack of effect on T cell and antibody-dependent cellular cytotoxicity. J Immunol. (1988) 141, 4031–8.
150. Gersuk GM, Westermark B, Mohabeer AJ, Challita PM, Pattamakom S, Pattengale PK. Inhibition of human natural killer cell activity by platelet-derived growth factor (PDGF). III. Membrane binding studies and differential biological effect of recombinant PDGF isoforms. Scand J Immunol. (1991) 33:521–32. doi: 10.1111/j.1365-3083.1991.tb02522.x
151. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. (2009) 69:7775–83. doi: 10.1158/0008-5472.CAN-09-2123
152. Chang SF, Chang CA, Lee DY, Lee PL, Yeh YM, Yeh CR, et al. Tumor cell cycle arrest induced by shear stress: Roles of integrins and Smad. Proc Natl Acad Sci USA. (2008) 105:3927–32. doi: 10.1073/pnas.0712353105
153. Barnes JM, Nauseef JT, Henry MD. Resistance to fluid shear stress is a conserved biophysical property of malignant cells. PLoS ONE. (2012) 7:e50973. doi: 10.1371/journal.pone.0050973
154. Mitchell MJ, King MR. Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via trimeric death receptors. New J Phys. (2013) 15:015008. doi: 10.1088/1367-2630/15/1/015008
155. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
156. Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. (2012) 2012:306879. doi: 10.1155/2012/306879
157. Egan K, Cooke N, Kenny D. Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis. (2014) 31:697–704. doi: 10.1007/s10585-014-9660-7
158. Chambers AF, Groom AC, Macdonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. (2002) 2:563–72. doi: 10.1038/nrc865
159. Reymond N, D'agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. (2013) 13:858–70. doi: 10.1038/nrc3628
160. Strell C, Entschladen F. Extravasation of leukocytes in comparison to tumor cells. Cell Commun Signal. (2008) 6:10. doi: 10.1186/1478-811X-6-10
161. Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. (2013) 11(Suppl. 1):192–201. doi: 10.1111/jth.12225
162. Wang H, Fu W, Im JH, Zhou Z, Santoro SA, Iyer V, et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol. (2004) 164:935–41. doi: 10.1083/jcb.200309112
163. Coenen DM, Mastenbroek TG, Cosemans J. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. (2017) 130:2819–28. doi: 10.1182/blood-2017-04-780825
164. Jain S, Zuka M, Liu J, Russell S, Dent J, Guerrero JA, et al. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc Natl Acad Sci USA. (2007) 104:9024–8. doi: 10.1073/pnas.0700625104
165. Dardik R, Savion N, Kaufmann Y, Varon D. Thrombin promotes platelet-mediated melanoma cell adhesion to endothelial cells under flow conditions: role of platelet glycoproteins P-selectin and GPIIb-IIIA. Br J Cancer. (1998) 77:2069–75. doi: 10.1038/bjc.1998.349
166. Kim YJ, Borsig L, Han HL, Varki NM, Varki A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am J Pathol. (1999) 155:461–72. doi: 10.1016/S0002-9440(10)65142-5
167. Qi CL, Wei B, Ye J, Yang Y, Li B, Zhang QQ, et al. P-selectin-mediated platelet adhesion promotes the metastasis of murine melanoma cells. PLoS ONE. (2014) 9:e91320. doi: 10.1371/journal.pone.0091320
168. Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med. (1998) 187:329–39. doi: 10.1084/jem.187.3.329
169. Ludwig RJ, Boehme B, Podda M, Henschler R, Jager E, Tandi C, et al. Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res. (2004) 64:2743–50. doi: 10.1158/0008-5472.CAN-03-1054
170. Ferjancic S, Gil-Bernabe AM, Hill SA, Allen PD, Richardson P, Sparey T, et al. VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood. (2013) 121:3289–97. doi: 10.1182/blood-2012-08-449819
171. Laubli H, Spanaus KS, Borsig L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood. (2009) 114:4583–91. doi: 10.1182/blood-2008-10-186585
172. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. doi: 10.1038/35393
173. Gleissner CA, Von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. (2008) 28:1920–7. doi: 10.1161/ATVBAHA.108.169417
174. Aigner S, Ramos CL, Hafezi-Moghadam A, Lawrence MB, Friederichs J, Altevogt P, et al. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J. (1998) 12:1241–51. doi: 10.1096/fasebj.12.12.1241
175. Napier SL, Healy ZR, Schnaar RL, Konstantopoulos K. Selectin ligand expression regulates the initial vascular interactions of colon carcinoma cells: the roles of CD44v and alternative sialofucosylated selectin ligands. J Biol Chem. (2007) 282:3433–41. doi: 10.1074/jbc.M607219200
176. Kappelmayer J, Nagy BJr. The interaction of selectins and PSGL-1 as a key component in thrombus formation and cancer progression. Biomed Res Int. (2017) 2017:6138145. doi: 10.1155/2017/6138145
177. Dimitroff CJ, Descheny L, Trujillo N, Kim R, Nguyen V, Huang W, et al. Identification of leukocyte E-selectin ligands, P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res. (2005) 65:5750–60. doi: 10.1158/0008-5472.CAN-04-4653
178. Burdick MM, Chu JT, Godar S, Sackstein R. HCELL is the major E- and L-selectin ligand expressed on LS174T colon carcinoma cells. J Biol Chem. (2006) 281:13899–905. doi: 10.1074/jbc.M513617200
179. Wawryk SO, Novotny JR, Wicks IP, Wilkinson D, Maher D, Salvaris E, et al. The role of the LFA-1/ICAM-1 interaction in human leukocyte homing and adhesion. Immunol Rev. (1989) 108:135–61. doi: 10.1111/j.1600-065X.1989.tb00016.x
180. Okahara H, Yagita H, Miyake K, Okumura K. Involvement of very late activation antigen 4 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) in tumor necrosis factor alpha enhancement of experimental metastasis. Cancer Res. (1994) 54:3233–6.
181. Garofalo A, Chirivi RG, Foglieni C, Pigott R, Mortarini R, Martin-Padura I, et al. Involvement of the very late antigen 4 integrin on melanoma in interleukin 1-augmented experimental metastases. Cancer Res. (1995) 55:414–9.
182. Sokeland G, Schumacher U. The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol Cancer. (2019) 18:12. doi: 10.1186/s12943-018-0937-3
183. Felding-Habermann B, O'toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA. (2001) 98:1853–8. doi: 10.1073/pnas.98.4.1853
184. Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet Interaction with Innate Immune Cells. Transfus Med Hemother. (2016) 43:78–88. doi: 10.1159/000444807
185. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer. (2017) 140:2321–30. doi: 10.1002/ijc.30635
186. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
187. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE. (2009) 4:e6562. doi: 10.1371/journal.pone.0006562
188. Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. (2012) 2:1091–9. doi: 10.1158/2159-8290.CD-12-0329
189. Garcia-Roman J, Zentella-Dehesa A. Vascular permeability changes involved in tumor metastasis. Cancer Lett. (2013) 335:259–69. doi: 10.1016/j.canlet.2013.03.005
190. Kebers F, Lewalle JM, Desreux J, Munaut C, Devy L, Foidart JM, et al. Induction of endothelial cell apoptosis by solid tumor cells. Exp Cell Res. (1998) 240:197–205. doi: 10.1006/excr.1998.3935
191. Strilic B, Yang L, Albarran-Juarez J, Wachsmuth L, Han K, Muller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. (2016) 536:215–8. doi: 10.1038/nature19076
192. Bogatcheva NV, Garcia JG, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry. (2002) 67:75–84. doi: 10.1023/A:1013904231324
193. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. (2013) 24:130–7. doi: 10.1016/j.ccr.2013.05.008
194. Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood. (2014) 124:3141–50. doi: 10.1182/blood-2014-04-568683
195. Sarker MH, Hu DE, Fraser PA. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation. (2010) 17:39–46. doi: 10.1111/j.1549-8719.2010.00001.x
196. Tang DG, Diglio CA, Honn KV. 12(S)-HETE-induced microvascular endothelial cell retraction results from PKC-dependent rearrangement of cytoskeletal elements and alpha V beta 3 integrins. Prostaglandins. (1993) 45:249–67. doi: 10.1016/0090-6980(93)90051-8
197. Honn KV, Tang DG, Grossi I, Duniec ZM, Timar J, Renaud C, et al. Tumor cell-derived 12. (S)-hydroxyeicosatetraenoic acid induces microvascular endothelial cell retraction. Cancer Res. (1994) 54:565–74.
198. Honn KV, Tang DG, Grossi IM, Renaud C, Duniec ZM, Johnson CR, et al. Enhanced endothelial cell retraction mediated by 12(S)-HETE: a proposed mechanism for the role of platelets in tumor cell metastasis. Exp Cell Res. (1994) 210:1–9. doi: 10.1006/excr.1994.1001
199. Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. (2009) 61:22–32. doi: 10.1016/S1734-1140(09)70004-0
200. Schlesinger M, Bendas G. Vascular cell adhesion molecule-1 (VCAM-1)–an increasing insight into its role in tumorigenicity and metastasis. Int J Cancer. (2015) 136:2504–14. doi: 10.1002/ijc.28927
201. Kuckleburg CJ, Tiwari R, Czuprynski CJ. Endothelial cell apoptosis induced by bacteria-activated platelets requires caspase-8 and−9 and generation of reactive oxygen species. Thromb Haemost. (2008) 99:363–72. doi: 10.1160/TH07-07-0474
202. Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. (2019) 49:361–74. doi: 10.1016/j.devcel.2019.04.010
203. Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. (2009) 122:3015–24. doi: 10.1242/jcs.034561
205. Eckert MA, Yang J. Targeting invadopodia to block breast cancer metastasis. Oncotarget. (2011) 2:562–8. doi: 10.18632/oncotarget.301
206. Pang JH, Coupland LA, Freeman C, Chong BH, Parish CR. Activation of tumour cell ECM degradation by thrombin-activated platelet membranes: potentially a P-selectin and GPIIb/IIIa-dependent process. Clin Exp Metastasis. (2015) 32:495–505. doi: 10.1007/s10585-015-9722-5
207. Laubli H, Stevenson JL, Varki A, Varki NM, Borsig L. L-selectin facilitation of metastasis involves temporal induction of Fut7-dependent ligands at sites of tumor cell arrest. Cancer Res. (2006) 66:1536–42. doi: 10.1158/0008-5472.CAN-05-3121
208. Assoian RK, Sporn MB. Type beta transforming growth factor in human platelets: release during platelet degranulation and action on vascular smooth muscle cells. J Cell Biol. (1986) 102:1217–23. doi: 10.1083/jcb.102.4.1217
209. Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. (2008) 68:6851–8. doi: 10.1158/0008-5472.CAN-08-0718
210. Jain S, Russell S, Ware J. Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. J Thromb Haemost. (2009) 7:1713–7. doi: 10.1111/j.1538-7836.2009.03559.x
211. Li R, Ren M, Chen N, Luo M, Deng X, Xia J, et al. Presence of intratumoral platelets is associated with tumor vessel structure and metastasis. BMC Cancer. (2014) 14:167. doi: 10.1186/1471-2407-14-167
212. Haemmerle M, Bottsford-Miller J, Pradeep S, Taylor ML, Choi HJ, Hansen JM, et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J Clin Invest. (2016) 126:1885–96. doi: 10.1172/JCI85086
213. Ben-Ezra J, Sheibani K, Hwang DL, Lev-Ran A. Megakaryocyte synthesis is the source of epidermal growth factor in human platelets. Am J Pathol. (1990) 137:755–9.
214. Mohle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci USA. (1997) 94:663–8. doi: 10.1073/pnas.94.2.663
215. Wartiovaara U, Salven P, Mikkola H, Lassila R, Kaukonen J, Joukov V, et al. Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb Haemost. (1998) 80:171–5. doi: 10.1055/s-0037-1615158
216. Italiano JEJr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. (2008) 111:1227–33. doi: 10.1182/blood-2007-09-113837
217. Klement GL, Yip TT, Cassiola F, Kikuchi L, Cervi D, Podust V, et al. Platelets actively sequester angiogenesis regulators. Blood. (2009) 113:2835–42. doi: 10.1182/blood-2008-06-159541
218. Kim HK, Song KS, Chung JH, Lee KR, Lee SN. Platelet microparticles induce angiogenesis in vitro. Br J Haematol. (2004) 124:376–84. doi: 10.1046/j.1365-2141.2003.04773.x
219. Michael JV, Wurtzel JGT, Mao GF, Rao AK, Kolpakov MA, Sabri A, et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood. (2017) 130:567–80. doi: 10.1182/blood-2016-11-751099
220. Magnus N, Garnier D, Meehan B, Mcgraw S, Lee TH, Caron M, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci USA. (2014) 111:3544–9. doi: 10.1073/pnas.1314118111
221. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
222. Hussein K, Stucki-Koch A, Gohring G, Kreipe H, Suttorp M. Increased megakaryocytic proliferation, pro-platelet deposition and expression of fibrosis-associated factors in children with chronic myeloid leukaemia with bone marrow fibrosis. Leukemia. (2017) 31:1540–6. doi: 10.1038/leu.2017.73
223. Jackson W III, Sosnoski DM, Ohanessian SE, Chandler P, Mobley A, Meisel KD, Mastro AM. Role of megakaryocytes in breast cancer metastasis to bone. Cancer Res. (2017) 77:1942–54. doi: 10.1158/0008-5472.CAN-16-1084
224. Soares FA. Increased numbers of pulmonary megakaryocytes in patients with arterial pulmonary tumour embolism and with lung metastases seen at necropsy. J Clin Pathol. (1992) 45:140–2. doi: 10.1136/jcp.45.2.140
225. Werynska B, Ramlau R, Podolak-Dawidziak M, Jankowska R, Prajs I, Usnarska-Zubkiewicz L, et al. Serum thrombopoietin levels in patients with reactive thrombocytosis due to lung cancer and in patients with essential thrombocythemia. Neoplasma. (2003) 50:447–51.
226. Elmahdi S, Muramatsu H, Narita A, Ismael O, Hama A, Nishio N, et al. Markedly high plasma thrombopoietin (TPO) level is a predictor of poor response to immunosuppressive therapy in children with acquired severe aplastic anemia. Pediatr Blood Cancer. (2016) 63:659–64. doi: 10.1002/pbc.25820
227. Besbes S, Shah S, Al-Dybiat I, Mirshahi S, Helfer H, Najah H, et al. Thrombopoietin secretion by human ovarian cancer cells. Int J Cell Biol. (2017) 2017:1873834. doi: 10.1155/2017/1873834
228. McGary CT, Miele ME, Welch DR. Highly metastatic 13762NF rat mammary adenocarcinoma cell clones stimulate bone marrow by secretion of granulocyte-macrophage colony-stimulating factor/interleukin-3 activity. Am J Pathol. (1995) 147:1668–81.
229. Xu L, Mao X, Guo T, Chan PY, Shaw G, Hines J, et al. The novel association of circulating tumor cells and circulating megakaryocytes with prostate cancer prognosis. Clin Cancer Res. (2017) 23:5112–22. doi: 10.1158/1078-0432.CCR-16-3081
230. Shirai T, Revenko AS, Tibbitts J, Ngo ATP, Mitrugno A, Healy LD, et al. Hepatic thrombopoietin gene silencing reduces platelet count and breast cancer progression in transgenic MMTV-PyMT mice. Blood Adv. (2019) 3:3080–91. doi: 10.1182/bloodadvances.2019000250
231. Li X, Koh AJ, Wang Z, Soki FN, Park SI, Pienta KJ, et al. Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. J Bone Miner Res. (2011) 26:125–34. doi: 10.1002/jbmr.204
232. Wells AF, Parrino J, Mangan EK, Paccaly A, Lin Y, Xu C, et al. Immunogenicity of sarilumab monotherapy in patients with rheumatoid arthritis who were inadequate responders or intolerant to disease-modifying antirheumatic drugs. Rheumatol Ther. (2019) 6:339–52. doi: 10.1007/s40744-019-0157-3
233. Esposito M, Guise T, Kang Y. The biology of bone metastasis. Cold Spring Harb Perspect Med. (2018) 8:a031252. doi: 10.1101/cshperspect.a031252
234. Zaslavsky A, Baek KH, Lynch RC, Short S, Grillo J, Folkman J, et al. Platelet-derived thrombospondin-1 is a critical negative regulator and potential biomarker of angiogenesis. Blood. (2010) 115:4605–13. doi: 10.1182/blood-2009-09-242065
235. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol. (2005) 6:401–10. doi: 10.1016/S1470-2045(05)70207-2
236. Tuzovic M, Herrmann J, Iliescu C, Marmagkiolis K, Ziaeian B, Yang EH. Arterial thrombosis in patients with cancer. Curr Treat Options Cardiovasc Med. (2018) 20:40. doi: 10.1007/s11936-018-0635-x
237. Blom JW, Vanderschoot JP, Oostindier MJ, Osanto S, Van Der Meer FJ, Rosendaal FR. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost. (2006) 4:529–35. doi: 10.1111/j.1538-7836.2006.01804.x
238. Khorana AA, Connolly GC. Assessing risk of venous thromboembolism in the patient with cancer. J Clin Oncol. (2009) 27:4839–47. doi: 10.1200/JCO.2009.22.3271
239. Menapace LA, Peterson DR, Berry A, Sousou T, Khorana AA. Symptomatic and incidental thromboembolism are both associated with mortality in pancreatic cancer. Thromb Haemost. (2011) 106:371–8. doi: 10.1160/TH10-12-0789
240. Brandes AA, Scelzi E, Salmistraro G, Ermani M, Carollo C, Berti F, et al. Incidence of risk of thromboembolism during treatment high-grade gliomas: a prospective study. Eur J Cancer. (1997) 33:1592–6. doi: 10.1016/S0959-8049(97)00167-6
241. Delmonte A, Mariotti M, Scarpi E, Ulivi P, Gavelli G, Rossi A, et al. Venous thromboembolic events in advanced adenocarcinoma of the lung: impact on prognosis according to platinum therapies and presence of driver mutations. Ann Oncol. (2015) 26:vi83. doi: 10.1093/annonc/mdv343.33
242. Khorana AA. Venous thromboembolism and prognosis in cancer. Thromb Res. (2010) 125:490–3. doi: 10.1016/j.thromres.2009.12.023
243. Rickles FR, Patierno S, Fernandez PM. Tissue factor, thrombin, and cancer. Chest. (2003) 124:58–68S. doi: 10.1378/chest.124.3_suppl.58S
244. Kakkar AK, Lemoine NR, Scully MF, Tebbutt S, Williamson RC. Tissue factor expression correlates with histological grade in human pancreatic cancer. Br J Surg. (1995) 82:1101–4. doi: 10.1002/bjs.1800820831
245. Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Hu YC, et al. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin Cancer Res. (2007) 13:2870–5. doi: 10.1158/1078-0432.CCR-06-2351
246. Guan M, Jin J, Su B, Liu WW, Lu Y. Tissue factor expression and angiogenesis in human glioma. Clin Biochem. (2002) 35:321–5. doi: 10.1016/S0009-9120(02)00312-0
247. Seto S, Onodera H, Kaido T, Yoshikawa A, Ishigami S, Arii S, et al. Tissue factor expression in human colorectal carcinoma: correlation with hepatic metastasis and impact on prognosis. Cancer. (2000) 88:295–301. doi: 10.1002/(SICI)1097-0142(20000115)88:2<295::AID-CNCR8>3.0.CO;2-U
248. Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissue factor expression in breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer. (2000) 83:164–70. doi: 10.1054/bjoc.2000.1272
249. Koomagi R, Volm M. Tissue-factor expression in human non-small-cell lung carcinoma measured by immunohistochemistry: correlation between tissue factor and angiogenesis. Int J Cancer. (1998) 79:19–22. doi: 10.1002/(SICI)1097-0215(19980220)79:1<19::AID-IJC4>3.0.CO;2-Z
250. Sawada M, Miyake S, Ohdama S, Matsubara O, Masuda S, Yakumaru K, et al. Expression of tissue factor in non-small-cell lung cancers and its relationship to metastasis. Br J Cancer. (1999) 79:472–7. doi: 10.1038/sj.bjc.6690073
251. Laubli H, Borsig L. Altered cell adhesion and glycosylation promote cancer immune suppression and metastasis. Front Immunol. (2019) 10:2120. doi: 10.3389/fimmu.2019.02120
252. Manly DA, Wang J, Glover SL, Kasthuri R, Liebman HA, Key NS, et al. Increased microparticle tissue factor activity in cancer patients with venous thromboembolism. Thromb Res. (2010) 125:511–2. doi: 10.1016/j.thromres.2009.09.019
253. Van Doormaal F, Kleinjan A, Berckmans RJ, Mackman N, Manly D, Kamphuisen PW, et al. Coagulation activation and microparticle-associated coagulant activity in cancer patients. An exploratory prospective study. Thromb Haemost. (2012) 108:160–5. doi: 10.1160/TH12-02-0099
254. Kim HK, Song KS, Park YS, Kim CM, Lee KR. Method comparison of flow cytometric assay of platelet microparticles and changes of platelet microparticles during cancer chemotherapy. Thromb Haemost. (2002) 87:547–8. doi: 10.1055/s-0037-1613043
255. Kim HK, Song KS, Park YS, Kang YH, Lee YJ, Lee KR, et al. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur J Cancer. (2003) 39:184–91. doi: 10.1016/S0959-8049(02)00596-8
256. Fricke A, Ullrich PV, Cimniak AFV, Becherer C, Follo M, Heinz J, et al. Levels of activated platelet-derived microvesicles in patients with soft tissue sarcoma correlate with an increased risk of venous thromboembolism. BMC Cancer. (2017) 17:527. doi: 10.1186/s12885-017-3515-y
257. Helley D, Banu E, Bouziane A, Banu A, Scotte F, Fischer AM, et al. Platelet microparticles: a potential predictive factor of survival in hormone-refractory prostate cancer patients treated with docetaxel-based chemotherapy. Eur Urol. (2009) 56:479–84. doi: 10.1016/j.eururo.2008.06.038
258. Zacharski LR, Henderson WG, Rickles FR, Forman WB, Cornell CJ Jr, Forcier RJ, et al. Effect of warfarin anticoagulation on survival in carcinoma of the lung, colon, head and neck, and prostate. Final report of VA cooperative study #75. Cancer. (1984) 53, 2046–52. doi: 10.1002/1097-0142(19840515)53:10<2046::AID-CNCR2820531007>3.0.CO;2-F
259. Chahinian AP, Propert KJ, Ware JH, Zimmer B, Perry MC, Hirsh V, et al. A randomized trial of anticoagulation with warfarin and of alternating chemotherapy in extensive small-cell lung cancer by the Cancer and Leukemia Group B. J Clin Oncol. (1989) 7:993–1002. doi: 10.1200/JCO.1989.7.8.993
260. Levine M, Hirsh J, Gent M, Arnold A, Warr D, Falanga A, et al. Double-blind randomised trial of a very-low-dose warfarin for prevention of thromboembolism in stage IV breast cancer. Lancet. (1994) 343:886–9. doi: 10.1016/S0140-6736.9490008-6
261. Maurer LH, Herndon J. E. 2nd, Hollis DR, Aisner J, Carey RW, Skarin AT, Perry MC, et al. Randomized trial of chemotherapy and radiation therapy with or without warfarin for limited-stage small-cell lung cancer: a Cancer and Leukemia Group B study. J Clin Oncol. (1997) 15:3378–87. doi: 10.1200/JCO.1997.15.11.3378
262. Pengo V, Noventa F, Denas G, Pengo MF, Gallo U, Grion AM, et al. Long-term use of vitamin K antagonists and incidence of cancer: a population-based study. Blood. (2011) 117:1707–9. doi: 10.1182/blood-2010-08-304758
263. Kinnunen PT, Murtola TJ, Talala K, Taari K, Tammela TL, Auvinen A. Warfarin use and prostate cancer risk in the Finnish Randomized Study of screening for prostate cancer. Scand J Urol. (2016) 50:413–9. doi: 10.1080/21681805.2016.1228085
264. Altinbas M, Coskun HS, Er O, Ozkan M, Eser B, Unal A, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost. (2004) 2:1266–71. doi: 10.1111/j.1538-7836.2004.00871.x
265. Kakkar AK, Levine MN, Kadziola Z, Lemoine NR, Low V, Patel HK, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the fragmin advanced malignancy outcome study (FAMOUS). J Clin Oncol. (2004) 22:1944–8. doi: 10.1200/JCO.2004.10.002
266. Klerk CP, Smorenburg SM, Otten HM, Lensing AW, Prins MH, Piovella F, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. (2005) 23:2130–5. doi: 10.1200/JCO.2005.03.134
267. Van Doormaal FF, Di Nisio M, Otten HM, Richel DJ, Prins M, Buller HR. Randomized trial of the effect of the low molecular weight heparin nadroparin on survival in patients with cancer. J Clin Oncol. (2011) 29:2071–6. doi: 10.1200/JCO.2010.31.9293
268. Barni S, Bonizzoni E, Verso M, Gussoni G, Petrelli F, Perrone T, et al. The effect of low-molecular-weight heparin in cancer patients: the mirror image of survival? Blood. (2014) 124:155–6. doi: 10.1182/blood-2014-03-561761
269. Macbeth F, Noble S, Evans J, Ahmed S, Cohen D, Hood K, et al. Randomized phase III trial of standard therapy plus low molecular weight heparin in patients with lung cancer: FRAGMATIC Trial. J Clin Oncol. (2016) 34:488–94. doi: 10.1200/JCO.2015.64.0268
270. Meyer G, Besse B, Doubre H, Charles-Nelson A, Aquilanti S, Izadifar A, et al. Anti-tumour effect of low molecular weight heparin in localised lung cancer: a phase III clinical trial. Eur Respir J. (2018) 52:1801220. doi: 10.1183/13993003.01220-2018
271. Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. (2012) 13:518–27. doi: 10.1016/S1470-2045(12)70112-2
272. Zhao X, Xu Z, Li H. NSAIDs use and reduced metastasis in cancer patients: results from a meta-analysis. Sci Rep. (2017) 7:1875. doi: 10.1038/s41598-017-01644-0
273. Guo Q, Li Q, Wang J, Liu M, Wang Y, Chen Z, et al. A comprehensive evaluation of clinical efficacy and safety of celecoxib in combination with chemotherapy in metastatic or postoperative recurrent gastric cancer patients: a preliminary, three-center, clinical trial study. Medicine. (2019) 98:e16234. doi: 10.1097/MD.0000000000016234
274. Choe KS, Cowan JE, Chan JM, Carroll PR, D'amico AV, Liauw SL. Aspirin use and the risk of prostate cancer mortality in men treated with prostatectomy or radiotherapy. J Clin Oncol. (2012) 30:3540–4. doi: 10.1200/JCO.2011.41.0308
275. Harter K, Levine M, Henderson SO. Anticoagulation drug therapy: a review. West J Emerg Med. (2015) 16:11–7. doi: 10.5811/westjem.2014.12.22933
276. Lloyd AJ, Dewilde S, Noble S, Reimer E, Lee AYY. What impact does venous thromboembolism and bleeding have on cancer patients' quality of life? Value Health. (2018) 21:449–55. doi: 10.1016/j.jval.2017.09.015
277. Meyer G, Marjanovic Z, Valcke J, Lorcerie B, Gruel Y, Solal-Celigny P, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med. (2002) 162:1729–35. doi: 10.1001/archinte.162.15.1729
278. Hull RD, Pineo GF, Brant RF, Mah AF, Burke N, Dear R, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med. (2006) 119:1062–72. doi: 10.1016/j.amjmed.2006.02.022
279. Koopman MM, Prandoni P, Piovella F, Ockelford PA, Brandjes DP, Van Der Meer J, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group N Engl J Med. (1996) 334:682–7. doi: 10.1056/NEJM199603143341102
280. Bendas G, Borsig L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol. (2012) 2012:676731. doi: 10.1155/2012/676731
281. Wiviott SD, Braunwald E, Mccabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. (2007) 357:2001–15. doi: 10.1056/NEJMoa0706482
282. Floyd JS, Serebruany VL. Prasugrel as a potential cancer promoter: review of the unpublished data. Arch Intern Med. (2010) 170:1078–80. doi: 10.1001/archinternmed.2010.154
283. Mauri L, Kereiakes DJ, Yeh RW, Driscoll-Shempp P, Cutlip DE, Steg PG, et al. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med. (2014) 371:2155–66. doi: 10.1056/NEJMoa1409312
284. FDA (Center for Drug Evaluation and Research). Cross-Discipline Team Leader Review on Vorapaxar. (2014). Available online at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204886Orig1s000SumR.pdf NDA 294-886. (accessed April 18, 2014).
285. FDA (Center for Drug Evaluation and Research). Apixaban Clinical Review. (2012). Available online at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202155Orig1s000MedR.pdf (accessed December 17, 2012).
286. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. (2012) 379:1591–601. doi: 10.1016/S0140-6736(12)60209-8
287. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. (1991) 325:1593–6. doi: 10.1056/NEJM199112053252301
288. Flossmann E, Rothwell PM, British Doctors Aspirin T The UK-TIA Aspirin Trial. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. (2007) 369:1603–13. doi: 10.1016/S0140-6736(07)60747-8
289. Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. (2010) 376:1741–50. doi: 10.1016/S0140-6736(10)61543-7
290. Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. (2011) 378:2081–7. doi: 10.1016/S0140-6736(11)61049-0
291. Elwood PC, Morgan G, Pickering JE, Galante J, Weightman AL, Morris D, et al. Aspirin in the treatment of cancer: reductions in metastatic spread and in mortality: a systematic review and meta-analyses of published studies. PLoS ONE. (2016) 11:e0152402. doi: 10.1371/journal.pone.0152402
292. Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. (2010) 29:781–8. doi: 10.1038/onc.2009.421
293. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
294. Yang L, Lv Z, Xia W, Zhang W, Xin Y, Yuan H, et al. The effect of aspirin on circulating tumor cells in metastatic colorectal and breast cancer patients: a phase II trial study. Clin Transl Oncol. (2018) 20:912–21. doi: 10.1007/s12094-017-1806-z
295. Coyle C, Cafferty FH, Rowley S, Mackenzie M, Berkman L, Gupta S, et al. ADD-ASPIRIN: a phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp Clin Trials. (2016) 51:56–64. doi: 10.1016/j.cct.2016.10.004
Keywords: platelets, megakaryocytes, cancer metastasis, coagulation, anti-coagulant therapy, thromboembolism
Citation: Lucotti S and Muschel RJ (2020) Platelets and Metastasis: New Implications of an Old Interplay. Front. Oncol. 10:1350. doi: 10.3389/fonc.2020.01350
Received: 16 April 2020; Accepted: 26 June 2020;
Published: 18 September 2020.
Edited by:
George S. Karagiannis, Albert Einstein College of Medicine, United StatesReviewed by:
Robert Campbell, The University of Utah, United StatesCopyright © 2020 Lucotti and Muschel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serena Lucotti, c2VsNDAwMkBtZWQuY29ybmVsbC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.