 Sanjay Premi
Sanjay Premi- Department of Tumor Biology, Moffitt Cancer Center & Research Institute, Tampa, FL, United States
Melanoma is the deadliest type of skin cancer. Human melanomas often show hyperactivity of nitric oxide synthase (NOS) and NADPH oxidase (NOX), which, respectively, generate nitric oxide (NO·) and superoxide (O2·−). The NO· and O2 − react instantly with each other to generate peroxynitrite (ONOO−) which is the driver of melanin chemiexcitation. Melanoma precursors, the melanocytes, are specialized skin cells that synthesize melanin, a potent shield against sunlight's ultraviolet (UV) radiation. However, melanin chemiexcitation paradoxically demonstrates the melanomagenic properties of melanin. In a loop, the NOS activity regulates melanin synthesis, and melanin is utilized by the chemiexcitation pathway to generate carcinogenic melanin-carbonyls in an excited triplet state. These carbonyl compounds induce UV-specific DNA damage without UV. Additionally, the carbonyl compounds are highly reactive and can make melanomagenic adducts with proteins, DNA and other biomolecules. Here we review the role of the melanin chemiexcitation pathway in melanoma initiation, progression, and drug resistance. We conclude by hypothesizing a non-classical, positive loop in melanoma where melanin chemiexcitation generates carcinogenic reactive carbonyl species (RCS) and DNA damage in normal melanocytes. In parallel, NOS and NOX regulate melanin synthesis generating raw material for chemiexcitation, and the resulting RCS and reactive nitrogen species (RNS) regulate cellular proteome and transcriptome in favor of melanoma progression, metastasis, and resistance against targeted therapies.
Introduction
Skin is the largest human organ directly in contact with sunlight's UV rays, a potent carcinogen. Approximately 80% of the mutations in sunlight-induced melanoma are UV signature and cytosine to thymine transitions generated from cytosine-containing cyclobutane pyrimidine dimers (CPDs) (1–3). Due to its unusually wide absorption spectrum, melanin has been considered a potent shield against UV carcinogenicity. Lethal amounts of superoxide and hydrogen peroxide are generated during melanin synthesis (4, 5). Melanocytes survive this stress and remain physiologically normal in skin and in tissue culture. Exogenous factors like UV and chemical pollutants and endogenous factors like spontaneous mutations further elevate the oxidative stress (6–9), which then strongly drives melanocytic transformation into melanoma (10–12). We discovered an additional stress from endogenous activity of nitric oxide synthase (NOS) and NADPH oxidase (NOX) specifically in pigmented melanocytes and not in the isogeneic albino melanocytes (13). Literature also suggests constitutively active NOS and NOX in pigmented melanocytes and melanoma (14–16). The NO· and generated, respectively, by NOS and NOX instantly combine to make peroxynitrite (ONOO−). ONOO− induces melanin chemiexcitation, generating additional CPDs and melanomagenic mutations (13). Additionally, NOS and NOX induce post-translational modifications (PMTs) like nitration and nitrosylation that promote cellular proliferation, invasion, and drug resistance. Melanoma is an aggressive skin cancer where the cells develop resistance against targeted therapies. The role of melanin chemiexcitation, RCS, and RNS remains underrepresented in melanoma. Here we summarize that melanin-synthesis and chemiexcitation are central regulators of melanoma progression and resistance against targeted inhibitors of RAF and RAS.
Role of Melanin Chemiexcitation in Melanomagenesis
Sunlight-induced melanoma exhibits UV signature mutations which arise from CPDs, adducts that are created spontaneously in response to UV exposure. However, we discovered that in response to UVA, the pigmented melanocytes (and not the isogenic albinos) generated CPDs for several hours in the dark, after UV exposure ended (13). Mechanistically, UV-induced reactive nitrogen species (RNS) oxidize melanin to create melanin-carbonyls in a quantum triplet state that has energy equivalent to UV photons. This energy is transferred to DNA to create CPDs in the dark (dark CPD). UV exposure also induces skin pigmentation as a screen against harmful effects of UV exposure. A probable correlation between melanin synthesis, chemiexcitation, and melanomagenesis is discussed below.
Melanin Chemiexcitation and Melanomagenic DNA Damage
Various skin color tones (17, 18) depend upon the type of melanin and its packaging and distribution in melanosomes (19). UV-induced DNA damage and repair triggers melanin synthesis (20). Further, melanin has a wide absorption spectrum, suggesting it to be a potent screen against harmful UV rays (21) and, thus, melanoma. However, pigmented melanocytes are under constitutive oxidative stress owing to their specialized function, melanin synthesis, which generates superoxide and hydrogen peroxide (4, 5, 22–24). How melanocytes survive this oxidative stress is still debatable. Melanin itself can be pro- or antioxidant. Also, the strong intracellular redox in melanocytes is regulated by several autocrine and paracrine factors which are discussed in detail by Denat et al. (25). Adding to this, we described a contradictory, carcinogenic role of melanin in a pathway called melanin chemiexcitation (13) Figure 1. Once activated by UV exposure, the two enzymes nitric oxide synthase (NOS) and NADPH oxidase (NOX) oxidize the melanin into an excited triplet state carbonyl that donates its energy to DNA and generates carcinogenic CPDs in the complete absence of UV. This explains several contradictions in melanoma biology. For example, the most abundant wavelength, UVA in sunlight (320–400 nm), requires melanin for melanoma induction in a hepatocyte growth factor transgenic “humanized skin” mouse model (26). Similarly, spontaneous melanoma initiation was observed in MC1R-truncated, BRAFV600E-mutated mouse models (27). The MC1R truncation leads to higher pheomelanin/eumelanin ratio and golden coat color. Both studies suggested oxidative DNA damage as a leading factor behind melanoma development. However, we suggest a prominent contribution of protein modifications by RNS and RCS, and DNA mutations from chemiexcitation. This is supported by the endogenous NOS and NOX activity in melanocytes and >3-fold higher dark CPD in mice with golden colored (pheomelanin containing) fur (13). In addition to skin melanoma, melanin chemiexcitation might be responsible for UV signature and somatic mutations in other subtypes like acral and uveal melanoma; however, very few investigations have been done to reveal this. In 2017, Rawson et al. described UV signature mutations in ~8% cases of acral melanoma (28). Since the sites of acral melanomas are well-shielded from the sun, we predict that the UV signature in this subtype is probably due to melanin chemiexcitation. In the human eye, the cornea and iris absorb most of the wavelengths below 370 nm, which are most carcinogenic, letting through only ~2% of the longer wavelengths (370–400 nm) (29, 30). These longer wavelengths are comparatively less potent in generating CPD compared to the shorter wavelengths. Accordingly, we predict that the somatic and passenger mutations in the uveal melanoma are a result of melanin chemiexcitation. Alternatively, the UV absorption by iris and retina is not that efficient in children (31, 32), and like skin melanoma, childhood UV exposure is strongly associated with uveal melanoma at a later age. Thus, the actual role of melanin chemiexcitation in inducing melanomagenic mutation in subtypes like acral and uveal melanomas needs detailed investigations and analyses.
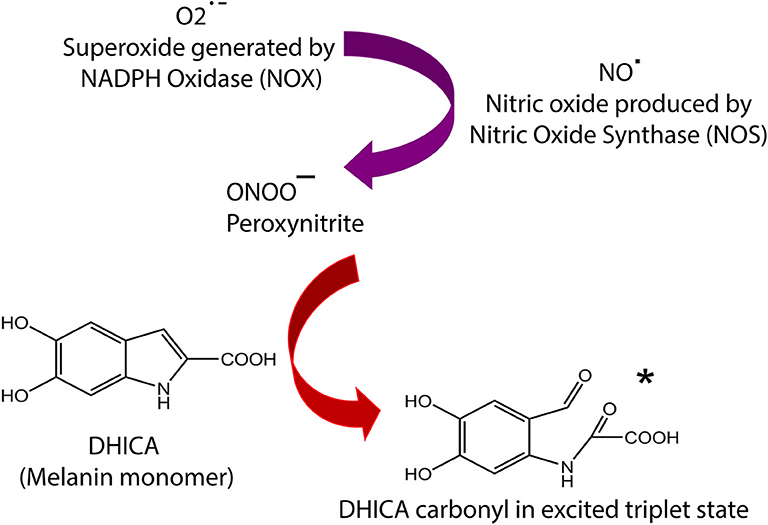
Figure 1. Summary of melanin chemiexcitation pathway. NOS and NOX enzymes are upregulated by UV exposure or endogenous factors, leading to generation of NO·, and O2·−, which instantly combine to produce ONOO−. The peroxynitrite oxidizes melanin an excited triplet state melanin-carbonyl which has energy equivalent to a UV photon. The melanin monomer 5,6-dihydroxyindole-2-carboxylic acid (DHICA) is used as an example. This carbonyl donates its energy to DNA through radiationless transfer, generating pyrimidine dimers in the dark. The remaining carbonyl molecule implements reactive carbonyl species (RNS). Complete details of the chemiexcitation pathway are explained in Premi et al. (13).
Despite distinct proof of the carcinogenicity of melanin, the melanin chemiexcitation pathway is still incomplete. Detailed investigation is required to identify the redox enzymes, location of melanin oxidation, route of melanin fragments to nucleus, mechanism of DNA-melanin interaction, and all the isoforms of NOS and NOX that drive melanin chemiexcitation.
Recently, we investigated if incident CPDs and the ones created by chemiexcitation can be used as genomic dosimeters for early melanoma diagnosis and better prognosis. We identified and quantified rare CPDs at a single base resolution across the genome (33). This revealed CPD hyperspots (ultra-sensitive sites) in pigmented melanocytes that had a precise alignment with the recurrent UV-signature mutations in individual gene promoters of melanomas. We also revealed that several of these are dark CPD hyperspots, created solely by the melanin chemiexcitation. Dark CPDs were confirmed by two methods. First, several sites in the pigmented human melanocytes had CPD accumulated without any UV exposure (the negative control sample) and the amount was almost equal to that of CPD induced immediately after UV exposure ended (Time 0). The same sites had none or negligible CPD in the human skin fibroblasts. Second, we collected CPD data from various time points post UV exposure. Several sites showed increased CPD amount even 7 h after UV exposure ended, compared to the CPDs immediately after UV exposure. Several genomic sites were >200-fold more sensitive for CPD generation in comparison to genome-wide average. Such hypersensitivity can alter melanocytic physiology through transcriptional blockade or epigenetic behavior in addition to the mutations.
Pigment Type, Melanin Chemiexcitation, and Melanoma
The endogenous NOS and NOX activity is specific to pigmented melanocytes and completely absent in syngeneic albino melanocytes. We envision a physiological linkage between melanin synthesis, NOS and NOX activity, and initiation of melanoma from non-cancerous lesions like dysplastic nevi (atypical or benign moles). Nitric oxide produced either by melanocytes themselves (34) or by keratinocytes (35) upregulate the melanin synthesis pathway genes like TYR, TRP1, DCT, and MITF (34, 35). The NO· also promotes pheomelanogenesis (pheomelanin synthesis) by modulating the MC1R gene expression and activity (36). A direct role of NOX in melanin-synthesis has not been demonstrated. However, the mammalian superoxide dismutase is known to oxidize L-3,4-dihydroxyphenylalanine (L-DOPA) (37), one of the initial steps in melanin synthesis. Since melanin synthesis itself produces O2− (22), it is conceivable that melanocytes have chronic expression and activity for superoxide dismutase to eliminate O2·−. Thus, O2·− indirectly promotes melanogenesis through DOPA oxidation by superoxide dismutase. Consequently, in the melanomagenic background, the NOS- and NOX-mediated regulation of melanin synthesis is evidently an important positive-loop that could generate raw materials for the melanin chemiexcitation pathway (13). Moreover, dysplastic nevi (atypical or benign moles) synthesize more pheomelanin compared to eumelanin (38), which is linked with dysregulated and chronic oxidative stress (39). Dysplastic nevi are known risk factors of melanoma development. Compared to eumelanogenesis (eumelanin synthesis), pheomelanogenesis is more hazardous, not only due to higher ROS production but also because it consumes cysteine, an essential component of the cellular antioxidant glutathione (40–42). We believe that the NOS and NOX activity switches eumelanogenesis to pheomelanogenesis and potentially trigger melanoma from dysplastic nevi by upregulating cellular pheomelanin content. The mechanisms behind pheomelanogenesis-mediated promotion of melanoma initiation are completely unknown. We suspect a critical role of the melanin chemiexcitation-mediated generation of CPDs, DNA mutations, microenvironment change, and melanin-carbonyls (discussed below). We believe that NOS- and NOX-mediated promotion of pheomelanogenesis is a pre-requisite for melanoma initiation from dysplastic nevi with BRAF/NRAS mutational background.
Our group is now investigating a NOS and NOX-regulated switch of eumelanogenesis to pheomelanogenesis and characterizes the role of this switch in initiating melanoma from non-cancerous lesions like dysplastic nevi. A direct comparison of MC1R polymorphism, sulfur and pheomelanin content, and melanoma markers among normal melanocytes, dysplastic nevi, and melanoma cells is envisioned to highlight the role of such a switch in the initiation of melanoma. Based on the literature and our own preliminary findings, we also believe that normal melanocytes have a physiological balance of NOS and NOX activity, chemiexcitation, and pigment synthesis. Accordingly, exo- or endogenous carcinogenesis (UV/spontaneous mutations) induces the NOS and NOX hyperactivity and altered pigment synthesis. This leads to chemiexcitation, carcinogenic dark CPDs, and altered redox in normal melanocytes either through RNS or RCS. Combined, these factors initiate melanoma from non-cancerous, RAF/RAS-mutated lesions.
In terms of pigment type and amount, amelanotic or hypomelanotic melanomas are outliers, however, indirectly relatable to melanin chemiexcitation. These are often present on chronically sun-exposed skin in red/blonde hair phenotypes, meaning, the ratio of pheomelanin/eumelanin is very high. This might give a lighter shade to amelanotic melanomas. Also, mortality from amelanotic melanomas is higher than the pigmented ones (43, 44). We propose that the amelanotic melanomas are rich in pheomelanin, and pheomelanin, being more potent compared to eumelanin in melanin chemiexcitation, leads to the poor prognosis of amelanotic melanomas. The misdiagnosis due to lack of color is another clinical factor responsible for poor prognosis in addition to the enhanced DNA damage and passenger mutations from melanin chemiexcitation.
Reactive Carbonyl Species (RCS) and Melanoma
Several reactive carbonyl species have been described in vivo (45). We added melanin-carbonyls to this list, which are generated by NOS- and NOX-mediated chemiexcitation in pigmented melanocytes (13). After energy transfer, the melanin-carbonyls remain chemically active and induce RCS in pigmented melanocytes. Carbonyl compounds are known to deplete glutathione (46, 47). In response to a physiological UV dose, the non-melanocytic (non-pigmented) cells recover their GSH content quickly. On the contrary, pigmented melanocytes maintain a very low GSH/GSSG ratio for >4 h (Premi et al., 13). This can be attributed to either the melanin-carbonyl-mediated trapping of glutathione or the upregulation of pheomelanogenesis, which consumes cysteine. RCS cause tissue disintegration (48–50) and promote proliferative cell-signaling in several human malignancies including melanoma (50–52). Notably, the carbonyl scavengers were shown to induce pronounced apoptosis in human melanoma cell lines via the loss of mitochondrial membrane potential (53). This suggests the addiction of melanoma to the carbonyl compounds. We believe that the NOS and NOX hyperactivity, pigmentation, and resulting melanin chemiexcitation is a chronic source of melanin-carbonyls, aiding melanoma survival through unknown mechanisms. Accordingly, we classify melanin-carbonyls as potent, “non-classical” melanoma carcinogens.
Carbonyl stress can induce protein dysfunction and DNA damage. Due to their high reactivity, the well-known carbonyl sources like 4-hydroxynonenal, glyoxal, and methylglyoxal are known to interact with the free amine or sulfhydryl groups of the proteins (54–58). Accordingly, the RCS prefers arginine, cysteine, and lysine residues (59). This interaction is generally covalent and alters normal protein function (60, 61). Some studies also suggest DNA damage through glyoxal- or methylglyoxal-mediated histone glycoxidation followed by DNA strand breaks (62).
Oxidation of cellular proteins to a protein carbonyl (protein carbonylation) is frequently detected in plasma and cellular proteins in colorectal cancer patients (61, 63, 64), Hodgkin's lymphoma (65), bladder cancer (66), prostate cancer (67), and breast cancer (68). However, protein carbonylation and adduction of carbonyls to proteins are fundamentally different. The type of interaction between melanin-carbonyls and proteins/DNA is still undetermined since the chemical nature, structure, and reactivity of melanin-carbonyls is unknown. We are investigating the interaction of melanin-carbonyls with short, double-stranded DNA fragments and simple proteins like BSA in an in vitro chemical reaction between synthetic melanin and synthetic peroxynitrite (13). The aim is to identify DNA vs. melanin-carbonyl and protein vs. melanin-carbonyl adducts using HPLC and mass spectrometry.
Reactive Carbonyl Species and Tumor Immunology
Melanin chemiexcitation produces an α,β-unsaturated melanin-carbonyl (13). Naturally occurring α,β-unsaturated carbonyls are immunosuppressive. Curcumin is one example that regulates JAK-STAT, AP-1, and κB in immune cells, thereby affecting the inflammatory cytokines (69, 70). Curcumin is antiproliferative for rat lymphocytes where it promotes apoptosis (71). The biology of curcumin is explained by Michael addition where its α,β-unsaturated, β-diketo moiety interacts with -SH groups on proteins through irreversible 1,4 addiction reaction (72). This also explains the antioxidant properties of curcumin where the β-diketo moiety neutralizes transition metal toxicity through chelation. Michael addition might have toxic side effects; however, it can block carcinogenesis by inducing enzymes like glutathione transferase and quinone reductase, which actively inhibit carcinogenesis.
Two other naturally occurring carbonyl compounds, chalcones and zerumbone, also induce intrinsic apoptosis in T lymphocytes by promoting the activation of caspase 3 and 9 (73–77). Chalcones are petal pigments that have α-,β-unsaturated carbonyl groups linking two aryl rings (78), and zerumbone is derived from the zingiberaceae family. A prominently anti-inflammatory chalcone that suppresses T cell proliferation is xanthohumol, which is extracted from hops (79). Several other examples of the immunosuppressive effects of α,β-unsaturated carbonyls on innate and adaptive response have been discussed in detail by Arshad et al. (80). Accordingly, the natural α,β-unsaturated carbonyls need further investigation for their therapeutic uses in autoimmune disorders. Moreover, adducts of proteins with carbonyls like malondialdehyde (MDA) and 4-hydroxynonenal(4-HNE) interfere with antibody production in pulmonary diseases (81) and several immune disorders (82–84). Adducts of formaldehyde carbonyls and proteins are known to bias the immune system toward a hypersensitive Th2 response (85). Owing to NOS–NOX hyperactivity and pigmentation, melanoma is envisioned to have a chronic melanin-carbonyl stress. No interaction of the melanin-carbonyls with proteins or other cellular molecules has been explored so far. We believe that melanin-carbonyls might suppress immune response against melanoma by modulating the tumor microenvironment. To explore this, our group is employing mass spectrometry–based strategies to first identify a “melanin-carbonyl modified” global proteome in the melanoma microenvironment and analyze the downstream effectors for their role in modulating immune response.
The participants of chemiexcitation are also known for immunomodulation. One of the prime combinatorial functions of NOS and NOX is production of ONOO−leading to posttranslational modifications (PTMs) like nitrosylation and nitration. Just like the chalcones and zerumbone, peroxynitrite is known to induce apoptosis in human thymocytes (86). Nitration/nitrosylation reduces the immunogenicity of peptides, antigen presentation by MHC, or TCR functions (87, 88). The RNS exposure down-regulates CD4, CD8, and chemokine receptors, thereby impairing T cell stimulation and migration (89). Nitration is known to block cytotoxic T cells from infiltrating non-melanoma tumors through sparsely known mechanisms (64, 90). In this regard, we believe that in the melanoma microenvironment, elevated NOS and NOX activity suppresses immune response through PTMs which alter TCR signaling, T cell activation, and chemotaxis. Matched detection of chemotaxis and activation markers such as CCL2, CCL5, CXCL12, CD3, CD4, CD8, CTLA-4, PD-1, and CXCR4 and nitration in melanoma tissue will establish the role of NOS and NOX in inhibiting the T cell infiltration/activation in melanoma.
Chemiexcitation Pathway and Melanoma Drug Resistance
In addition to inducing melanin chemiexcitation and RCS in normal melanocytes, NOS and NOX enzymes are also known to be hyperactive in melanoma. Cellular pathways regulated by these enzymes are still debatable. However, scattered reports suggest their role in regulating proliferative cell signaling, apoptosis, and melanoma invasion.
NOS and NOX Activity in Melanoma
Melanomas often show higher NOS activity (15, 16, 91, 92), which is strongly correlated with the poor survival of patients (91, 92). The well-known BRAF and NRAS mutations in melanoma (93, 94) lead to the constitutive activation of the p44/p42 MAPK pathway, which upregulates iNOS expression (95, 96). The resulting NO· release prolongs the survival of melanoma cells by inhibiting apoptosis (16, 97, 98). The proposed mechanism for this inhibition is NOS-mediated nitration/nitrosation of Caspase 3, Hif-1α, and Phd2, conferring resistance to cisplatin (99). Additionally, NOS inhibition upregulates apoptotic proteins like Bax, Caspase-1, Caspase-3, Caspase-6, and Mdm2. The fact that this apoptosis upregulation can be prevented by inducing NOS or by exogenous chemical sources of NO· (100) demonstrates the potential role of NOS in employing melanoma drug-resistance. Conversely, NOS inhibition downregulates Bcl-2, decreases intratumoral microvessel density, and increases intratumoral apoptosis in vivo (97, 101). Inhibiting NO· production leads to cell cycle arrest at G2-M stage followed by apoptosis (100). NOX family members upregulate the redox-sensitive signaling pathways involved in tumorigenesis. NOX1 is overexpressed in melanoma cells and controls melanoma invasion by regulating matrix metaloproteinase-2 (MMP-2) transcription (102, 103). Similarly, NOX4 knockdown arrests the cell cycle at G2-M stage, thus abolishing melanoma proliferation and tumor formation (103). NOX4 also induces the phosphorylation of focal adhesion kinase, which positively modulates cell motility through cellular-Src kinase (104). In addition, Akt induction of NOX4 leads to ROS production which, along with Akt, leads to NF-κB activation, vertical growth of melanoma, and resistance to apoptosis (105, 106).
NOS and NOX Activity and Melanoma Drug Resistance
Melanoma drug resistance is a huge clinical challenge. Several signaling pathways and secondary and tertiary mutations have been explored; however, none explain it perfectly. We believe that the NOS and NOX activity is at the center of drug resistance. There is a strong correlation between upregulated NOS and NOX activity and melanoma progression through the inhibition of apoptosis and/or promotion of cell viability. NOS- and NOX-mediated RNS induces posttranslational modifications (PTMs) like nitrosylation and nitration. We believe this regulates phosphorylation/dephosphorylation, DNA mutations, and other cellular events leading to apoptosis inhibition and drug resistance. Accordingly, inhibiting NOS–NOX will reactivate the intrinsic apoptotic pathway proteins like SMACS, Bcl-2 family, caspases, and cytochrome c. Caspases and cytochrome c are of special interest because PTMs like nitration/nitrosylation are known to inhibit their apoptotic function (107, 108). The MAPK pathway, which is constitutively activated by BRAF and NRAS mutations in melanoma, is positively associated with iNOS expression (95). Also, NF-κB is the mediator between MAPK and NOS expression (109), and NOX4 is known to induce NF-κB production (105, 106). This suggests a direct interaction and functional correlation between NOS and NOX, BRAF/NRAS mutations, melanoma progression, and drug-resistance. Irrespective of PTMs, lung cancer cells are known to survive apoptosis by triggering p53 expression in response to NO·/O2−-mediated DNA damage (110). Also, NO·-mediated upregulation of DNA-PKcs protects cells from DNA-damaging antitumor agents (111), and p53 is a major substrate for DNA-PKcs. However, this will promote clonal growth p53 mutated melanoma cells, which is not yet known. Thus, NOS- and NOX-mediated PTMs and downstream cellular events appear to be the best plausible strategy employed by melanoma cells to evade drug-mediated apoptosis.
We previously reported endogenous nuclear nitration, specifically in pigmented melanocytes, which is further elevated several-fold by UV (13). We are now investigating the physiological relevance of endogenous NOS and NOX activity in normal melanocytes to identify a baseline activity. We believe the NOS and NOX activity help melanocytes in evading apoptosis induction from the lethal oxidative stress generated during melanin synthesis. Secondly, we are identifying NOS- and NOX-induced, posttranslationally modified melanoma proteome (PMMP) and characterizing its role in cell survival, proliferation, and resistance against targeted therapies.
Chemiexcitation Pathway and Gene Expression
The NOS- and NOX-mediated RNS, especially NO· and ONOO− are envisaged to regulate gene expression by modulating transcription factors and histone methylases/acetylases through either metal-nitrosyl complexes or direct nitration/nitrosylation (112). This is poorly defined in eukaryotic systems. In yeast, the NO·-mediated nitrosylation of cysteines on the transcription factor Ace1 inhibits the transcription of its target gene CUP1 (113). NF-κB nitrosylation inhibits its DNA binding (114). NO· also facilitates the nuclear transport of the IκBα, which displaces NF-κB from DNA and inhibits cytokine expression in endothelial cells (115). Contrary to this, NO· stimulates IKKα activity that degrades Iκb, thereby allowing NF-κB to enter the nucleus and regulate transcription (116). NO· ejects Zn from the zinc finger transcription factors through metal nitrosyl complexes, leading to loss of function of several transcription factors like Egr-1 and Sp1 (117, 118). In addition to affecting transcription factors, NO· also modulates the epigenetic regulation of gene expression. Using unnaturally high doses of chemical sources of NO· and ONOO−, several experimental approaches suggest an active role of NOS and NOX in regulating histone PTMs (119). Histone-modifying enzymes, particularly lysine demethylases (LSD) are amine oxidases, majority of which are Fe(II)-dependent dioxygenases that use ketoglutarate and oxygen. Nitric oxide attacks the iron core in their catalytic pocket, inhibiting demethylation and upregulating histone methylation (120). NO·-nitrosated GAPDH aids in degrading lysine methylases, thus making lysine available for acetylation. Further, NO· activates a phosphatase which reduces global histone acetylation by activating a histone de-acetylase (HDAC) (121). On the contrary, NO· is also known to nitrosylate cysteine residues on HDACs that blocks their binding to chromatin. These inconsistencies in the actual role of NOS and NOX in modulating gene expression has not been addressed so far. Why NO· upregulates certain genes but suppresses others is probably explainable by either methylation or acetylation alterations. A bigger concern is chemical sources and physiologically irrelevant concentrations of NO· being used in most of these studies. Also, most of the studies have been conducted on non-melanocytic cells. Melanomas and melanocytes have an endogenous NOS and NOX hyperactivity, which provides a unique opportunity to investigate the role of RNS and RCS in regulating gene expression in a physiological setup.
Clinical Significance and Concluding Remarks
Based on our previous research, preliminary experiments, and the literature, it is evident that melanin chemiexcitation is a non-classical and novel pathogenesis pathway in melanoma. We believe that melanin, NOS and NOX activity, and melanin chemiexcitation are at the center of melanomagenesis and drug resistance and regulate a unique, non-classical and unidirectional positive feedback loop Figure 2. In normal melanocytes, melanin-synthesis produces lethal oxidative stress which can kill any non-melanocytic cell. However, the endogenous/physiological/baseline NOS and NOX activity protect melanocytes through PTMs and RCS. In response to an exogenous stress like UV or endogenous carcinogenic event like spontaneous mutations, NOS and NOX activity and melanin chemiexcitation are enhanced several-fold. In melanoma, the MAPK mutations like BRAFV600E are known to further upregulate NOS and NOX activity and expression. In parallel, the RNS upregulate melanin synthesis as a raw material for chemiexcitation that generates RCS.
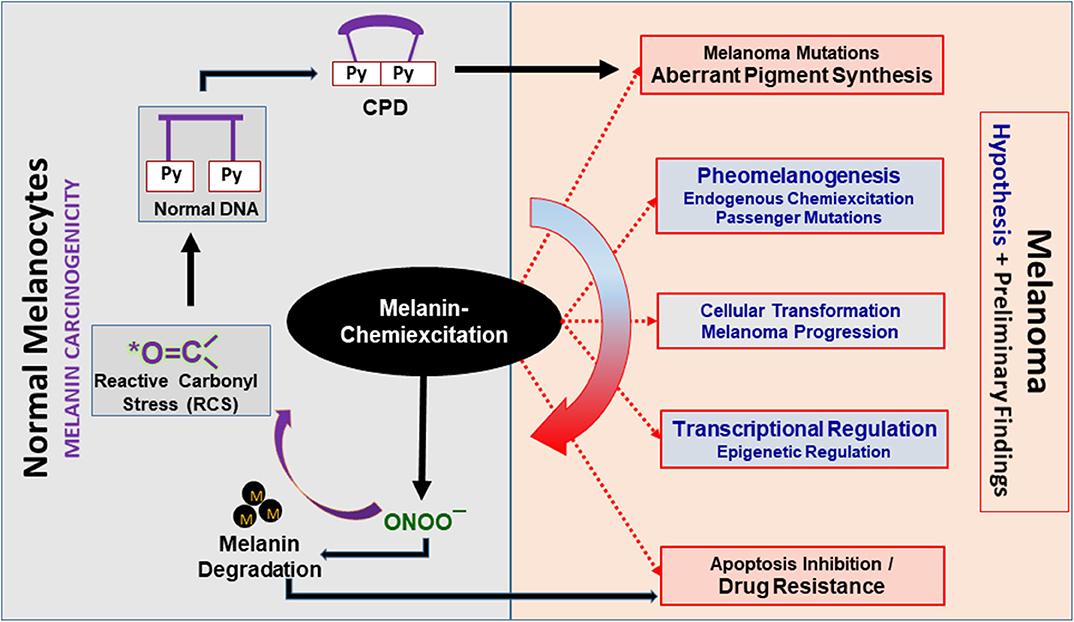
Figure 2. Melanin chemiexcitation as a central regulator of melanoma biology. Melanin chemiexcitation generates cyclobutane pyrimidine dimers (CPD) in the dark that induce melanomagenic mutations. In parallel, the reactive nitrogen and carbonyl species (RNS and RCS) generated during chemiexcitation regulate pigment synthesis, cellular transformations, posttranslational modifications, and gene expression. Coordinatively, this promotes melanoma progression and drug resistance through apoptosis inhibition. Black outlines have experimental proof whereas, the red outlines with gray filling and blue fonts are partially supported by experimental data. Black “M” dots represent melanin polymers, oligomers, or granules.
In conclusion, NOS–NOX activity induce melanomagenic mutation through chemiexcitation mediated CPDs that might initiate melanoma. Secondly, the NOS–NOX activity-induced PTMs inhibit apoptogenic signaling leading to resistance to the targeted therapy. Additionally, since carbonyl scavengers induce apoptosis in melanoma, we propose that the carbonyl compounds inhibit apoptosis through unknown mechanisms. Detailed investigation of NOS and NOX activity is envisaged to either identify novel targets for new drugs or enhance the efficacy of current standard-of-care antimelanoma strategies.
Author Contributions
SP conceived the idea, wrote the manuscript, and also made the figures.
Funding
This work was supported by institutional startup fund from Moffitt Cancer Center & Research Institute to SP.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. (2012) 44:1006–14. doi: 10.1038/ng.2359
2. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell. (2012) 150:251–63. doi: 10.1016/j.cell.2012.06.024
4. Tomita Y, Hariu A, Kato C, Seiji M. Radical production during tyrosinase reaction, dopa-melanin formation, and photoirradiation of dopa-melanin. J Invest Dermatol. (1984) 82:573–6. doi: 10.1111/1523-1747.ep12261311
5. Jenkins NC, Grossman D. Role of melanin in melanocyte dysregulation of reactive oxygen species. Biomed Res Int. (2013) 2013:908797. doi: 10.1155/2013/908797
6. Scharffetter-Kochanek K, Wlaschek M, Brenneisen P, Schauen M, Blaudschun R, Wenk J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol Chem. (1997) 378:1247–57.
7. Hanson KM, Clegg RM. Observation and quantification of ultraviolet-induced reactive oxygen species in ex vivo human skin. Photochem Photobiol. (2002) 76:57–63. doi: 10.1562/0031-8655(2002)0760057OAQOUI2.0.CO2
8. Zhang X, Rosenstein BS, Wang Y, Lebwohl M, Wei H. Identification of possible reactive oxygen species involved in ultraviolet radiation-induced oxidative DNA damage. Free Radic Biol Med. (1997) 23:980–5. doi: 10.1016/S0891-5849(97)00126-3
9. Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. (2004) 43:326–35. doi: 10.1111/j.1365-4632.2004.02222.x
10. Fried L, Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. (2008) 21:117–22. doi: 10.1111/j.1755-148X.2008.00451.x
11. Meyskens FL Jr, Farmer PJ, Yang S, Anton-Culver H. New perspectives on melanoma pathogenesis and chemoprevention. Recent Res Cancer Res. (2007) 174:191–5. doi: 10.1007/978-3-540-37696-5_16
12. Liu-Smith F, Dellinger R, Meyskens FL Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch Biochem Biophys. (2014) 563:51–5. doi: 10.1016/j.abb.2014.04.007
13. Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, et al. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science. (2015) 347:842–7. doi: 10.1126/science.1256022
14. Fecker LF, Eberle J, Orfanos CE, Geilen CC. Inducible nitric oxide synthase is expressed in normal human melanocytes but not in melanoma cells in response to tumor necrosis factor-alpha, interferon-gamma, and lipopolysaccharide. J Invest Dermatol. (2002) 118:1019–25. doi: 10.1046/j.1523-1747.2002.01744.x
15. Massi D, Franchi A, Sardi I, Magnelli L, Paglierani M, Borgognoni L, et al. Inducible nitric oxide synthase expression in benign and malignant cutaneous melanocytic lesions. J Pathol. (2001) 194:194–200. doi: 10.1002/1096-9896(200106)194:2<194::AID-PATH851>3.0.CO;2-S
16. Grimm EA, Ellerhorst J, Tang CH, Ekmekcioglu S. Constitutive intracellular production of iNOS and NO in human melanoma: possible role in regulation of growth and resistance to apoptosis. Nitric Oxide. (2008) 19:133–7. doi: 10.1016/j.niox.2008.04.009
17. Barsh GS. What controls variation in human skin color? PLoS Biol. (2003) 1:E27. doi: 10.1371/journal.pbio.0000027
18. Pichon LC, Landrine H, Corral I, Hao Y, Mayer JA, Hoerster KD. Measuring skin cancer risk in African Americans: is the fitzpatrick skin type classification scale culturally sensitive? Ethn Dis. (2010) 20:174–9.
19. Stamatas GN, Zmudzka BZ, Kollias N, Beer JZ. Non-invasive measurements of skin pigmentation in situ. Pigment Cell Res. (2004) 17:618–26. doi: 10.1111/j.1600-0749.2004.00204.x
20. Gilchrest BA, Eller MS. DNA photodamage stimulates melanogenesis and other photoprotective responses. J Investig Dermatol Symp Proc. (1999) 4:35–40. doi: 10.1038/sj.jidsp.5640178
21. Brenner M, Hearing VJ. The protective role of melanin against UV damage in human skin. Photochem Photobiol. (2008) 84:539–49. doi: 10.1111/j.1751-1097.2007.00226.x
22. Koga S, Nakano M, Tero-Kubota S. Generation of superoxide during the enzymatic action of tyrosinase. Arch Biochem Biophys. (1992) 292:570–5. doi: 10.1016/0003-9861(92)90032-R
23. Mastore M, Kohler L, Nappi AJ. Production and utilization of hydrogen peroxide associated with melanogenesis and tyrosinase-mediated oxidations of DOPA and dopamine. FEBS J. (2005) 272:2407–15. doi: 10.1111/j.1742-4658.2005.04661.x
24. Munoz-Munoz JL, Garcia-Molina F, Varon R, Tudela J, Garcia-Canovas F, Rodriguez-Lopez JN. Generation of hydrogen peroxide in the melanin biosynthesis pathway. Biochim Biophys Acta. (2009) 1794:1017–29. doi: 10.1016/j.bbapap.2009.04.002
25. Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel-Malek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. (2014) 134:1512–8. doi: 10.1038/jid.2014.65
26. Noonan FP, Zaidi MR, Wolnicka-Glubisz A, Anver MR, Bahn J, Wielgus A, et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat Commun. (2012) 3:884. doi: 10.1038/ncomms1893
27. Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. (2012) 491:449–53. doi: 10.1038/nature11624
28. Rawson RV, Johansson PA, Hayward NK, Waddell N, Patch AM, Lo S, et al. Unexpected UVR and non-UVR mutation burden in some acral and cutaneous melanomas. Lab Invest. (2017) 97:130–45. doi: 10.1038/labinvest.2016.143
29. Norren DV, Vos JJ. Spectral transmission of the human ocular media. Vision Res. (1974) 14:1237–44. doi: 10.1016/0042-6989(74)90222-3
30. Mallet JD, Gendron SP, Drigeard Desgarnier MC, Rochette PJ. Implication of ultraviolet light in the etiology of uveal melanoma: a review. Photochem Photobiol. (2014) 90:15–21. doi: 10.1111/php.12161
31. Dillon J, Wang RH, Atherton SJ. Photochemical and photophysical studies on human lens constituents. Photochem Photobiol. (1990) 52:849–54. doi: 10.1111/j.1751-1097.1990.tb08692.x
32. Mallet JD, Rochette PJ. Wavelength-dependent ultraviolet induction of cyclobutane pyrimidine dimers in the human cornea. Photochem Photobiol Sci. (2013) 12:1310–8. doi: 10.1039/c3pp25408a
33. Premi S, Han L, Mehta S, Knight J, Zhao DJ, Palmatier MA, et al. Genomic sites hypersensitive to ultraviolet radiation. Proc Natl Acad Sci USA. (2019) 116:24196–205. doi: 10.1073/pnas.1907860116
34. Romero-Graillet C, Aberdam E, Biagoli N, Massabni W, Ortonne JP, Ballotti R. Ultraviolet B radiation acts through the nitric oxide and cGMP signal transduction pathway to stimulate melanogenesis in human melanocytes. J Biol Chem. (1996) 271:28052–6. doi: 10.1074/jbc.271.45.28052
35. Romero-Graillet C, Aberdam E, Clement M, Ortonne JP, Ballotti R. Nitric oxide produced by ultraviolet-irradiated keratinocytes stimulates melanogenesis. J Clin Invest. (1997) 99:635–42. doi: 10.1172/JCI119206
36. Dong Y, Cao J, Wang H, Zhang J, Zhu Z, Bai R, et al. Nitric oxide enhances the sensitivity of alpaca melanocytes to respond to alpha-melanocyte-stimulating hormone by up-regulating melanocortin-1 receptor. Biochem Biophys Res Commun. (2010) 396:849–53. doi: 10.1016/j.bbrc.2010.05.001
37. Brennan T, Shapiro H, Edelstein L, Snyder LM. Demonstration of dopa oxidase activity by purified mammalian superoxide dismutase using polyacrylamide gel electrophoretic separation. AGE. (1978) 1:56–9. doi: 10.1007/BF02439875
38. Salopek TG, Yamada K, Ito S, Jimbow K. Dysplastic melanocytic nevi contain high levels of pheomelanin: quantitative comparison of pheomelanin/eumelanin levels between normal skin, common nevi, and dysplastic nevi. Pigment Cell Res. (1991) 4:172–9. doi: 10.1111/j.1600-0749.1991.tb00435.x
39. Pavel S, van Nieuwpoort F, van der Meulen H, Out C, Pizinger K, Cetkovska P, et al. Disturbed melanin synthesis and chronic oxidative stress in dysplastic naevi. Eur J Cancer. (2004) 40:1423–30. doi: 10.1016/j.ejca.2003.11.035
40. Ezzahir A. The influence of melanins on the photoperoxidation of lipids. J Photochem Photobiol B. (1989) 3:341–9. doi: 10.1016/1011-1344(89)80038-7
41. Chedekel MR, Smith SK, Post PW, Pokora A, Vessell DL. Photodestruction of pheomelanin: role of oxygen. Proc Natl Acad Sci USA. (1978) 75:5395–9. doi: 10.1073/pnas.75.11.5395
42. Koch WH, Chedekel MR. Photochemistry and photobiology of melanogenic metabolites: formation of free radicals. Photochem Photobiol. (1987) 46:229–38. doi: 10.1111/j.1751-1097.1987.tb04761.x
43. Thomas NE, Kricker A, Waxweiler WT, Dillon PM, Busman KJ, From L. Comparison of clinicopathologic features and survival of histopathologically amelanotic and pigmented melanomas: a population-based study. JAMA Dermatol. (2014) 150:1306–14. doi: 10.1001/jamadermatol.2014.1348
44. Strazzulla LC, Li X, Zhu K, Okhovat JP, Lee SJ, Kim CC. Clinicopathologic, misdiagnosis, and survival differences between clinically amelanotic melanomas and pigmented melanomas. J Am Acad Dermatol. (2019) 80:1292–8. doi: 10.1016/j.jaad.2019.01.012
45. Niki E. Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med. (2009) 47:469–84. doi: 10.1016/j.freeradbiomed.2009.05.032
46. Li W, Maloney RE, Circu ML, Alexander JS, Aw TY. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: role for glutathione-dependent metabolism of methylglyoxal. Free Radic Biol Med. (2013) 54:51–61. doi: 10.1016/j.freeradbiomed.2012.10.552
47. Okouchi M, Okayama N, Aw TY. Preservation of cellular glutathione status and mitochondrial membrane potential by N-acetylcysteine and insulin sensitizers prevent carbonyl stress-induced human brain endothelial cell apoptosis. Curr Neurovasc Res. (2009) 6:267–78. doi: 10.2174/156720209789630348
48. Baynes JW, Thorpe SR. Glycoxidation and lipoxidation in atherogenesis. Free Radic Biol Med. (2000) 28:1708–16. doi: 10.1016/S0891-5849(00)00228-8
49. Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res. (2001) 56:1–21. doi: 10.1210/rp.56.1.1
50. Wondrak GT, Cervantes-Laurean D, Roberts MJ, Qasem JG, Kim M, Jacobson EL, et al. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem Pharmacol. (2002) 63:361–73. doi: 10.1016/S0006-2952(01)00915-7
51. Wondrak GT, Cervantes-Laurean D, Jacobson EL, Jacobson MK. Histone carbonylation in vivo and in vitro. Biochem J. (2000) 351:769–77. doi: 10.1042/bj3510769
52. Iersel ML, Ploemen JP, Struik I, van Amersfoort C, Keyzer AE, Schefferlie JG, et al. Inhibition of glutathione S-transferase activity in human melanoma cells by alpha,beta-unsaturated carbonyl derivatives. Effects of acrolein, cinnamaldehyde, citral, crotonaldehyde, curcumin, ethacrynic acid, and trans-2-hexenal. Chem Biol Interact. (1996) 102:117–32. doi: 10.1016/S0009-2797(96)03739-8
53. Wondrak GT, Jacobson MK, Jacobson EL. Antimelanoma activity of apoptogenic carbonyl scavengers. J Pharmacol Exp Ther. (2006) 316:805–14. doi: 10.1124/jpet.105.094953
54. Davydov VV, Dobaeva NM, Bozhkov AI. Possible role of alteration of aldehyde's scavenger enzymes during aging. Exp Gerontol. (2004) 39:11–6. doi: 10.1016/j.exger.2003.08.009
55. Li H, Wang J, Kaphalia B, Ansari GA, Khan MF. Quantitation of acrolein-protein adducts: potential biomarker of acrolein exposure. J Toxicol Environ Health A. (2004) 67:513–24. doi: 10.1080/15287390490276539
56. Hashimoto M, Sibata T, Wasada H, Toyokuni S, Uchida K. Structural basis of protein-bound endogenous aldehydes. Chemical and immunochemical characterizations of configurational isomers of a 4-hydroxy-2-nonenal-histidine adduct. J Biol Chem. (2003) 278:5044–51. doi: 10.1074/jbc.M210129200
57. Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci USA. (1992) 89:4544–8. doi: 10.1073/pnas.89.10.4544
58. Hauptlorenz S, Esterbauer H, Moll W, Pumpel R, Schauenstein E, Puschendorf B. Effects of the lipidperoxidation product 4-hydroxynonenal and related aldehydes on proliferation and viability of cultured Ehrlich ascites tumor cells. Biochem Pharmacol. (1985) 34:3803–9. doi: 10.1016/0006-2952(85)90428-9
59. Rabbani N, Thornalley PJ. Glycation research in amino acids: a place to call home. Amino Acids. (2012) 42:1087–96. doi: 10.1007/s00726-010-0782-1
60. Coussens LM, Werb Z. Inflammation and cancer. Nature. (2002) 420:860–7. doi: 10.1038/nature01322
61. Shen Y, Ma J, Yan R, Ling H, Li X, Yang W, et al. Impaired self-renewal and increased colitis and dysplastic lesions in colonic mucosa of AKR1B8-deficient mice. Clin Cancer Res. (2015) 21:1466–76. doi: 10.1158/1078-0432.CCR-14-2072
62. Roberts MJ, Wondrak GT, Laurean DC, Jacobson MK, Jacobson EL. DNA damage by carbonyl stress in human skin cells. Mutat Res. (2003) 522:45–56. doi: 10.1016/S0027-5107(02)00232-4
63. Chang D, Wang F, Zhao YS, Pan HZ. Evaluation of oxidative stress in colorectal cancer patients. Biomed Environ Sci. (2008) 21:286–9. doi: 10.1016/S0895-3988(08)60043-4
64. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
65. Morabito F, Cristani M, Saija A, Stelitano C, Callea V, Tomaino A, et al. Lipid peroxidation and protein oxidation in patients affected by Hodgkin's lymphoma. Media Inflamm. (2004) 13:381–3. doi: 10.1080/09629350400008760
66. Yilmaz IA, Akcay T, Cakatay U, Telci A, Ataus S, Yalcin V. Relation between bladder cancer and protein oxidation. Int Urol Nephrol. (2003) 35:345–50. doi: 10.1023/B:UROL.0000022920.93994.ba
67. Goswami K, Nandeesha H, Koner BC, Nandakumar DN. A comparative study of serum protein-bound sialic acid in benign and malignant prostatic growth: possible role of oxidative stress in sialic acid homeostasis. Prostate Cancer Prostatic Dis. (2007) 10:356–9. doi: 10.1038/sj.pcan.4500965
68. Rossner P Jr, Terry MB, Gammon MD, Agrawal M, Zhang FF, Ferris JS, et al. Plasma protein carbonyl levels and breast cancer risk. J Cell Mol Med. (2007) 11:1138–48. doi: 10.1111/j.1582-4934.2007.00097.x
69. Bright JJ. Curcumin and autoimmune disease. Adv Exp Med Biol. (2007) 595:425–51. doi: 10.1007/978-0-387-46401-5_19
70. Jackson JK, Higo T, Hunter WL, Burt HM. The antioxidants curcumin and quercetin inhibit inflammatory processes associated with arthritis. Inflamm Res. (2006) 55:168–75. doi: 10.1007/s00011-006-0067-z
71. Sikora E, Bielak-Zmijewska A, Piwocka K, Skierski J, Radziszewska E. Inhibition of proliferation and apoptosis of human and rat T lymphocytes by curcumin, a curry pigment. Biochem Pharmacol. (1997) 54:899–907. doi: 10.1016/S0006-2952(97)00251-7
72. Priyadarsini KI. The chemistry of curcumin: from extraction to therapeutic agent. Molecules. (2014) 19:20091–112. doi: 10.3390/molecules191220091
73. Schwartz A, Middleton E Jr. Comparison of the effects of quercetin with those of other flavonoids on the generation and effector function of cytotoxic T lymphocytes. Immunopharmacology. (1984) 7:115–26. doi: 10.1016/0162-3109(84)90061-4
74. Middleton E Jr, Drzewiecki G. Flavonoid inhibition of human basophil histamine release stimulated by various agents. Biochem Pharmacol. (1984) 33:3333–8. doi: 10.1016/0006-2952(84)90102-3
75. Rahman HS, Rasedee A, How CW, Abdul AB, Zeenathul NA, Othman HH, et al. Zerumbone-loaded nanostructured lipid carriers: preparation, characterization, and antileukemic effect. Int J Nanomed. (2013) 8:2769–81. doi: 10.2147/IJN.S45313
76. Rahman HS, Rasedee A, Chartrand MS, Othman HH, Yeap SK, Namvar F. Zerumbone induces G2/M cell cycle arrest and apoptosis via mitochondrial pathway in Jurkat cell line. Nat Prod Commun. (2014) 9:1237–42. doi: 10.1177/1934578X1400900904
77. Rahman HS, Rasedee A, Abdul AB, Zeenathul NA, Othman HH, Yeap SK, et al. Zerumbone-loaded nanostructured lipid carrier induces G2/M cell cycle arrest and apoptosis via mitochondrial pathway in a human lymphoblastic leukemia cell line. Int J Nanomed. (2014) 9:527–38. doi: 10.2147/IJN.S54346
78. Goodarzi M, Saeys W, de Araujo MC, Galvao RK, Vander Heyden Y. Binary classification of chalcone derivatives with LDA or KNN based on their antileishmanial activity and molecular descriptors selected using the successive projections algorithm feature-selection technique. Eur J Pharm Sci. (2014) 51:189–95. doi: 10.1016/j.ejps.2013.09.019
79. Gao X, Deeb D, Liu Y, Gautam S, Dulchavsky SA, Gautam SC. Immunomodulatory activity of xanthohumol: inhibition of T cell proliferation, cell-mediated cytotoxicity and Th1 cytokine production through suppression of NF-kappaB. Immunopharmacol Immunotoxicol. (2009) 31:477–84. doi: 10.1080/08923970902798132
80. Arshad L, Jantan I, Bukhari SN, Haque MA. Immunosuppressive effects of natural alpha,beta-unsaturated carbonyl-based compounds, and their analogs and derivatives, on immune cells: a review. Front Pharmacol. (2017) 8:22. doi: 10.3389/fphar.2017.00022
81. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2011) 184:796–802. doi: 10.1164/rccm.201010-1605OC
82. Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, et al. Pathological aspects of lipid peroxidation. Free Radic Res. (2010) 44:1125–71. doi: 10.3109/10715762.2010.498478
83. Lucas A, Morales J, Velando A. Differential effects of specific carotenoids on oxidative damage and immune response of gull chicks. J Exp Biol. (2014) 217:1253–62. doi: 10.1242/jeb.098004
84. Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. (2008) 153:6–20. doi: 10.1038/sj.bjp.0707395
85. Moghaddam A, Olszewska W, Wang B, Tregoning JS, Helson R, Sattentau QJ, et al. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines. Nat Med. (2006) 12:905–7. doi: 10.1038/nm1456
86. Moulian N, Truffault F, Gaudry-Talarmain YM, Serraf A, Berrih-Aknin S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood. (2001) 97:3521–30. doi: 10.1182/blood.V97.11.3521
87. Birnboim HC, Lemay AM, Lam DK, Goldstein R, Webb JR. Cutting edge: MHC class II-restricted peptides containing the inflammation-associated marker 3-nitrotyrosine evade central tolerance and elicit a robust cell-mediated immune response. J Immunol. (2003) 171:528–32. doi: 10.4049/jimmunol.171.2.528
88. Hardy LL, Wick DA, Webb JR. Conversion of tyrosine to the inflammation-associated analog 3'-nitrotyrosine at either TCR- or MHC-contact positions can profoundly affect recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells. J Immunol. (2008) 180:5956–62. doi: 10.4049/jimmunol.180.9.5956
89. Kasic T, Colombo P, Soldani C, Wang CM, Miranda E, Roncalli M, et al. Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol. (2011) 41:1843–9. doi: 10.1002/eji.201040868
90. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci USA. (2018) 115:10094–9. doi: 10.1073/pnas.1800695115
91. Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC, et al. Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin Cancer Res. (2000) 6:4768–75.
92. Ekmekcioglu S, Ellerhorst JA, Prieto VG, Johnson MM, Broemeling LD, Grimm EA. Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer. (2006) 119:861–6. doi: 10.1002/ijc.21767
93. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
94. Gorden A, Osman I, Gai W, He D, Huang W, Davidson A, et al. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. (2003) 63:3955–7.
95. Ellerhorst JA, Ekmekcioglu S, Johnson MK, Cooke CP, Johnson MM, Grimm EA. Regulation of iNOS by the p44/42 mitogen-activated protein kinase pathway in human melanoma. Oncogene. (2006) 25:3956–62. doi: 10.1038/sj.onc.1209419
96. Kuo CL, Ho FM, Chang MY, Prakash E, Lin WW. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem. (2008) 103:931–40. doi: 10.1002/jcb.21466
97. Salvucci O, Carsana M, Bersani I, Tragni G, Anichini A. Antiapoptotic role of endogenous nitric oxide in human melanoma cells. Cancer Res. (2001) 61:318–26.
98. Tang CH, Grimm EA. Depletion of endogenous nitric oxide enhances cisplatin-induced apoptosis in a p53-dependent manner in melanoma cell lines. J Biol Chem. (2004) 279:288–98. doi: 10.1074/jbc.M310821200
99. Godoy LC, Anderson CT, Chowdhury R, Trudel LJ, Wogan GN. Endogenously produced nitric oxide mitigates sensitivity of melanoma cells to cisplatin. Proc Natl Acad Sci USA. (2012) 109:20373–8. doi: 10.1073/pnas.1218938109
100. Zheng M, Ekmekcioglu S, Walch ET, Tang CH, Grimm EA. Inhibition of nuclear factor-kappaB and nitric oxide by curcumin induces G2/M cell cycle arrest and apoptosis in human melanoma cells. Melanoma Res. (2004) 14:165–71. doi: 10.1097/01.cmr.0000129374.76399.19
101. Sikora AG, Gelbard A, Davies MA, Sano D, Ekmekcioglu S, Kwon J, et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin Cancer Res. (2010) 16:1834–44. doi: 10.1158/1078-0432.CCR-09-3123
102. Liu F, Gomez Garcia AM, Meyskens FL Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J Invest Dermatol. (2012) 132:2033–41. doi: 10.1038/jid.2012.119
103. Yamaura M, Mitsushita J, Furuta S, Kiniwa Y, Ashida A, Goto Y, et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. (2009) 69:2647–54. doi: 10.1158/0008-5472.CAN-08-3745
104. Ribeiro-Pereira C, Moraes JA, Souza Mde J, Laurindo FR, Arruda MA, Barja-Fidalgo C. Redox modulation of FAK controls melanoma survival–role of NOX4. PLoS ONE. (2014) 9:e99481. doi: 10.1371/journal.pone.0099481
105. Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, et al. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol Med. (2002) 8:1–8. doi: 10.1007/BF03401997
106. Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. (2007) 117:719–29. doi: 10.1172/JCI30102
107. Garcia-Heredia JM, Diaz-Moreno I, Diaz-Quintana A, Orzaez M, Navarro JA, Hervas M, et al. Specific nitration of tyrosines 46 and 48 makes cytochrome c assemble a non-functional apoptosome. FEBS Lett. (2012) 586:154–8. doi: 10.1016/j.febslet.2011.12.007
108. Torok NJ, Higuchi H, Bronk S, Gores GJ. Nitric oxide inhibits apoptosis downstream of cytochrome C release by nitrosylating caspase 9. Cancer Res. (2002) 62:1648–53.
109. Uffort DG, Grimm EA, Ellerhorst JA. NF-kappaB mediates mitogen-activated protein kinase pathway-dependent iNOS expression in human melanoma. J Invest Dermatol. (2009) 129:148–54. doi: 10.1038/jid.2008.205
110. Davis DW, Weidner DA, Holian A, McConkey DJ. Nitric oxide-dependent activation of p53 suppresses bleomycin-induced apoptosis in the lung. J Exp Med. (2000) 192:857–69. doi: 10.1084/jem.192.6.857
111. Xu W, Liu L, Smith GC, Charles l G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat Cell Biol. (2000) 2:339–45. doi: 10.1038/35014028
112. Socco S, Bovee RC, Palczewski MB, Hickok JR, Thomas DD. Epigenetics: the third pillar of nitric oxide signaling. Pharmacol Res. (2017) 121:52–8. doi: 10.1016/j.phrs.2017.04.011
113. Shinyashiki M, Chiang KT, Switzer CH, Gralla EB, Valentine JS, Thiele DJ, et al. The interaction of nitric oxide (NO) with the yeast transcription factor Ace1: a model system for NO-protein thiol interactions with implications to metal metabolism. Proc Natl Acad Sci USA. (2000) 97:2491–6. doi: 10.1073/pnas.050586597
114. Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res. (1996) 24:2236–42. doi: 10.1093/nar/24.12.2236
115. Spiecker M, Peng HB, Liao JK. Inhibition of endothelial vascular cell adhesion molecule-1 expression by nitric oxide involves the induction and nuclear translocation of IkappaBalpha. J Biol Chem. (1997) 272:30969–74. doi: 10.1074/jbc.272.49.30969
116. Umansky V, Hehner SP, Dumont A, Hofmann TG, Schirrmacher V, Droge W, et al. Co-stimulatory effect of nitric oxide on endothelial NF-kappaB implies a physiological self-amplifying mechanism. Eur J Immunol. (1998) 28:2276–82. doi: 10.1002/(SICI)1521-4141(199808)28:08<2276::AID-IMMU2276>3.0.CO;2-H
117. Wang S, Wang W, Wesley RA, Danner RL. A Sp1 binding site of the tumor necrosis factor alpha promoter functions as a nitric oxide response element. J Biol Chem. (1999) 274:33190–3. doi: 10.1074/jbc.274.47.33190
118. Rupprecht HD, Akagi Y, Keil A, Hofer G. Nitric oxide inhibits growth of glomerular mesangial cells: role of the transcription factor EGR-1. Kidney Int. (2000) 57:70–82. doi: 10.1046/j.1523-1755.2000.00828.x
119. Vasudevan D, Bovee RC, Thomas DD. Nitric oxide, the new architect of epigenetic landscapes. Nitric Oxide. (2016) 59:54–62. doi: 10.1016/j.niox.2016.08.002
120. Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem. (2013) 288:16004–15. doi: 10.1074/jbc.M112.432294
Keywords: melanin chemiexcitation, reactive nitrogen species, reactive carbonyl stress, apoptosis inhibition, drug resistance
Citation: Premi S (2020) Role of Melanin Chemiexcitation in Melanoma Progression and Drug Resistance. Front. Oncol. 10:1305. doi: 10.3389/fonc.2020.01305
Received: 05 May 2020; Accepted: 23 June 2020;
Published: 06 August 2020.
Edited by:
Sami Naidu, Indiana University, United StatesReviewed by:
John August D'Orazio, University of Kentucky, United StatesGeorg Wondrak, University of Arizona, United States
Copyright © 2020 Premi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjay Premi, c2FuamF5LnByZW1pQG1vZmZpdHQub3Jn