
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 07 July 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01105
This article is part of the Research Topic The Role of Epigenetic Modifications in Cancer Progression View all 18 articles
 Jiahua Lu1,2,3,4
Jiahua Lu1,2,3,4 Junjie Qian1,2,3,4
Junjie Qian1,2,3,4 Shengyong Yin1,2,3,4
Shengyong Yin1,2,3,4 Lin Zhou1,2,3,4
Lin Zhou1,2,3,4 Shusen Zheng1,2,3,4*Wu Zhang5,6*
Shusen Zheng1,2,3,4*Wu Zhang5,6*N6-Methyladenosine (m6A) is the most common RNA internal modification in eukaryotic cells. Its regulatory effects at the post-transcriptional level on both messenger RNAs (mRNAs) and noncoding RNAs have been widely studied; these include alternative splicing, stability, translation efficiency, nucleus export, and degradation. m6A modification is implicated in a series of physiological and pathological activities, such as embryonic stem cell differentiation, immunoregulation, adipogenesis, and cancer development. Recently, the significance of m6A methylation has been identified in both viral hepatitis and non-alcohol fatty liver disease (NAFLD), which are major risk factors in the development of hepatocellular carcinoma (HCC). Given the high incidence and mortality rate of HCC worldwide, it is of great importance to elucidate the mechanisms underlying HCC initiation and progression. m6A as an emerging research focus has great potential to facilitate the understanding of HCC, particularly from an etiological perspective. Thus, in this review, we summarize recent progress in understanding m6A modification related to viral hepatitis, NAFLD, and HCC, including their mechanisms and clinical applications.
Hepatocellular carcinoma (HCC) is the fourth leading cause of cancer-related death worldwide and accounts for more than 80% of primary liver cancers (1). Viral hepatitis and non-alcoholic fatty liver disease (NAFLD) are two significant risk factors for HCC development (2–4). The infection rates of hepatitis B virus (HBV) and hepatitis C virus (HCV) remain significant in high-risk areas, although vaccines and effective medicines have been designed for the prevention and treatment of viral hepatitis (5, 6). With the global epidemic of obesity, NAFLD has emerged as another nonnegligible force in HCC etiology (7). Due to asymptomatic disease progression, most patients have already advanced into liver cirrhosis or even HCC at the first diagnosis (3). Hence, these patients are not eligible for curative treatments such as surgical resection or liver transplantation, and instead are left with the exclusive choice of palliative therapies. Worse still, first-line-medicines such as sorafenib can only extend the overall survival of patients for another 3 months, and the response rate of the emerging programmed cell death ligand-1 (PD-L1) immune checkpoint inhibitor is lower than 20% in HCC patients (8, 9). Thus, it is of great urgency to develop novel therapies for the effective prevention, early diagnosis and precision treatment of HCC.
N6-methyladenosine (m6A) is a ubiquitous RNA internal modification at the posttranscriptional level; it was first identified in eukaryotic cells, and later found in prokaryotic cells and viruses (10–15). In 2012, the landscape of m6A modification was for the first time identified at the whole-transcriptome level (16, 17). Most m6A sites are enriched within a consensus motif of RRACH (R = G or A, A = m6A and H = A, C, or U), and are preferentially located around stop codons and within long internal exons. Regulators of m6A modification include “writers,” which are methyltransferases responsible for transferring the methyl group to the N6 position; “readers,” which are RNA-binding proteins that recognize specific m6A-modified positions to regulate RNA functions; and “erasers,” which are demethylases that mediate this reversible biological process (18–20).
m6A modification regulates the metabolic processes of both messenger RNAs (mRNAs) and noncoding RNAs (ncRNAs), which include structural stability, alternative splicing, translation efficacy, export, and decay (21–24). The reciprocal effects of m6A methylation with mRNAs or ncRNAs are associated with a series of physiological and pathological biological behaviors such as stem cell differentiation, immunoregulation and carcinogenesis (25–27). There is mounting evidence that m6A dysregulation is critically involved in HCC occurrence and development. For example, elevated m6A levels in HCC fuel inflammation and neovascularization in tumors by inhibiting m6A readers (28). In addition, a reduction of m6A modification promotes HCC metastasis in an m6A-dependent manner by modulating microRNA (miRNA) processing (29). Furthermore, some m6A regulators have shown clinical value as biomarkers or therapeutic targets in HCC (30). Therefore, in this review, we mainly summarize the potential mechanisms of m6A modification in HCC development from an etiological perspective, as well as its possible clinical applications, including as biomarkers and therapeutic targets.
The m6A regulators, including “writers,” “erasers,” and “readers,” cooperatively maintain the dynamic and reversible balance of m6A methylation (31) (Figure 1). “Writers” including methyltransferase-like 3 (METTL3), METTL14, and Wilm's tumor 1-associated protein (WTAP) comprise the major components of the methyltransferase complex within the nucleus, which is responsible for the m6A methylation process (20). METTL3 is the core catalytic enzyme for transferring methyl groups to N6 positions, while METTL14 is vital for recognizing and stabilizing the METTL3-METTL14 complex (32). WTAP binds the methyltransferase complex and recruits it toward mRNA targets (33). Other identified “writers” include METTL16, KIAA1429, zinc finger CCCH domain-containing protein 13 (ZC3H13), and RNA-binding motif protein 15 (RBM15) (20). It was not until 2011 that scientists discovered that m6A modification could be reverted by demethylase, which drew the attention of the broader academic community (34). Then it was discovered that the dynamic demethylation process could be achieved by “erasers” including AlkB homolog 5 (ALKBH5), and fat mass and obesity-associated protein (FTO). FTO was the first identified gene contributing to human obesity, and its polymorphisms are closely related to insulin resistance and metabolic diseases (35, 36). Two studies reported the identical increase in intracellular m6A methylation after knocking out two different “erasers” (34, 37). Moreover, the demethylation of FTO and ALKBH5 mainly affects m6A residues within the nucleus.
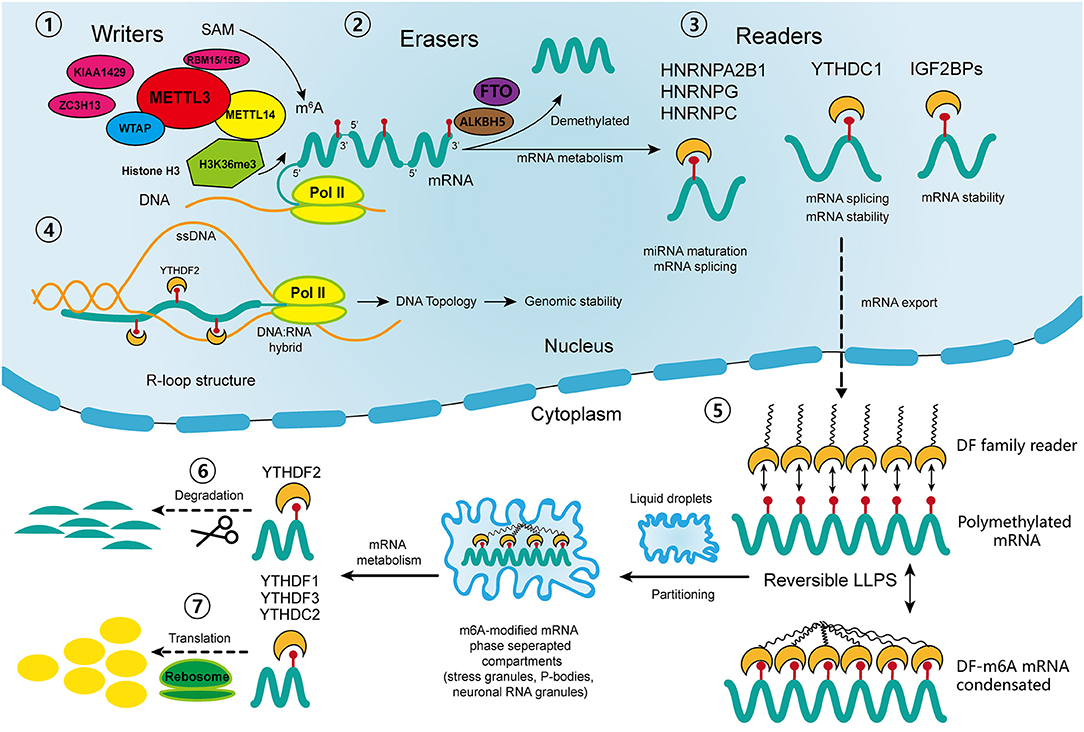
Figure 1. Schematic diagram of the process and biological function of RNA m6A methylation. The methyltransferase complex composed of METTL3, METTL14 and WTAP co-transcriptionally catalyzes the transfer of the methyl group from adenosylmethionine (SAM) onto the N6 position of adenosine; other “writers” include RBM15/15B, KIAA1429, and ZC3H13. With the guidance of histone H3K36me, the m6A sites are preferentially located near the 3′ terminus of the transcripts. Demethylases FTO and ALKBH5 reverse the m6A process. In nucleus, the “readers” of YTHDC1, IGF2BPs, HNRNPs participate in diverse RNA biological processes, including mRNA alternative splicing, stability, export, and miRNA maturation. The m6A-modified R-loop structures are implicated in the regulation of genomic stability. In cytoplasm, the m6A-modified mRNAs are targeted for regulation through a process called liquid-liquid phase separation (LLPS), which is mediated by YTHDFs. Then, other “readers” regulate the degradation and translation of mRNAs in the cytoplasm. ssDNA, single strand DNA; pol II: RNA polymerase II.
The most studied m6A “readers” are YTH domain-containing proteins, including YTHDF1-3 (located in the cytoplasm), YTHDC1 (in the nucleus), and YTHDC2 (in both) (19). YTHDF1 and YTHDF3 have coordinated functions that promote the translation of m6A-modified mRNAs, while YTHDF2 expedites mRNA degradation, and YTHDC1 takes part in mRNA alternative splicing and nuclear export (38–40). YTHDC2 acts paradoxically in either promoting translation or accelerating mRNA degradation (41). Recently, a novel cellular biological process called liquid-liquid phase separation (LLPS) was discovered, through which degradation, translation and splicing of m6A-modified mRNAs are regulated (42). Some low-complexity domains belonging to YTHDFs have the ability to interact with each other and to partition into liquid droplets within the cytoplasm (43). This process can be greatly enhanced by mRNA transcripts containing multiple m6A residues, which recruit and juxtapose YTHDF proteins, initiating the phase separation process. Subsequently, the mRNA-YTHDF complexes partition into endogenous phase-separated liquid droplets, such as stress granules, P-bodies, or neuronal RNA granules, thereby transforming into membraneless compartments, where their degradation, splicing, and transportation are regulated (42). In addition to the YTH reader proteins, there are some binding proteins regulated by m6A-induced structural changes called m6A “switches,” which include heterogeneous nuclear ribonucleoprotein C (HNRNPC), HNRNPG, HNRNPA2B1, and insulin-like growth factor 2 mRNA binding protein 1-3 (IGF2BP1-3) (44). HNRNPC and HNRNPG are involved in mRNA splicing, and HNRNPA2B1 participates in miRNA maturation (18). IGF2BPs function through the recruitment of RNA stabilizers, such as Hu antigen R (HuR), an RNA stabilizer, to maintain mRNA stability (45–47). However, the detailed mechanisms of their interactions with m6A-modified sites require further exploration.
In addition to the well-studied functions of m6A regulators, there are some newly identified mechanisms that participate in the regulation of m6A modification. Huang et al. discovered that histone H3 trimethylation at Lys36 (H3K36me3), one of the transcriptional markers, interacts with METTL14 to guide the methyltransferase complex to nascent RNA and to specific regions of mRNA transcripts (48). In that study, silence of either METTL14 or H3K36me greatly reduced the m6A levels throughout the transcriptome. Moreover, the overlapped sites of histone modification and m6A modification are preferentially located near the coding sequence (CDS) and 3′ terminus of the transcripts. This finding partially explained the site specificity of m6A modification in mRNA transcripts, and revealed another layer of regulatory mechanism involving crosstalk between m6A methylation and histone modification. Other recent studies have shown that m6A modification regulates not only the stability of mRNAs but also DNA structures. R-loops are nucleic acid structures consisting of three strands: an RNA: DNA hybrid and a single strand of unpaired DNA (49). They actively regulate genome dynamics and functions, including immunoglobulin class switching, transcription initiation and termination as well as genomic stability (49). Two independent studies have confirmed the existence of m6A modified-RNAs within R-loops. Abakir et al. found that YTHDF2 recognized m6A-modified R-loops and promoted their degradation (50). By contrast, Yang et al. observed a decreased R-loop levels upon METTL3 silencing, which suggests that m6A may promote R-loop formation (51). Although there are discrepancies between these two studies, they both indicate that m6A plays an important role in maintaining genomic integrity by modulating the accumulation of R-loops. Further, this modulation prevented the occurrence of a variety of diseases such as cancers, Kaposi's sarcoma and neurological disorders. In addition, Liu et al. identified METTL3-catalyzed m6A on chromosome-associated regulatory RNAs (carRNAs), such as enhancer RNAs, repeats RNAs and promoter-associated RNAs, and YTHDC1 promotes the degradation of these RNAs (52). Moreover, a reduction of m6A modification increases carRNA levels and promotes the open chromatin state and transcription. This finding suggests a regulatory effect of m6A on carRNAs, which in turn affects the chromatin state and transcription.
The regulators of m6A play a pleiotropic role in the modulation of HCC, and both mRNA and ncRNA participate in m6A-mediated biological processes in HCC (Figure 2). The m6A regulators reviewed in this article include the “writers” METTL3, METTL14, WTAP, and KIAA1429; the “eraser” FTO and the “readers” YTHDF1, YTHDF2 and IGF2BP1; their regulatory effects in HCC are summarized in Table 1.
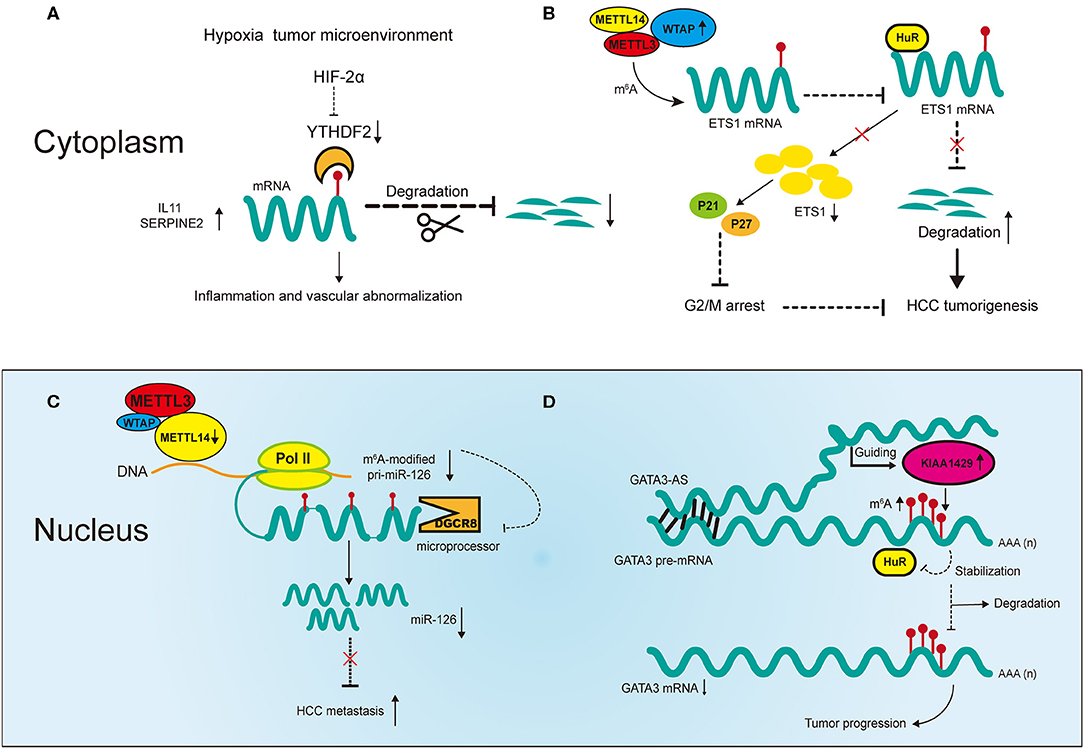
Figure 2. Partial m6A regulatory mechanisms involved in messenger RNAs (mRNAs) and noncoding RNAs (ncRNAs) in HCC. (A) The hypoxia tumor microenvironment negatively regulates YTHDF2 to promote cancer inflammation and vascular abnormalization. (B) Upregulated WTAP promotes HCC tumorigenesis by destabilizing its targeted mRNAs in an m6A-dependent manner. (C) Downregulated METTL14 promotes HCC metastasis by inhibiting microRNA 126 (miRNA) maturation in an m6A-dependent manner. (D) Upregulated KIAA1429 promotes HCC progression in an m6A- and lncRNA-dependent manner.
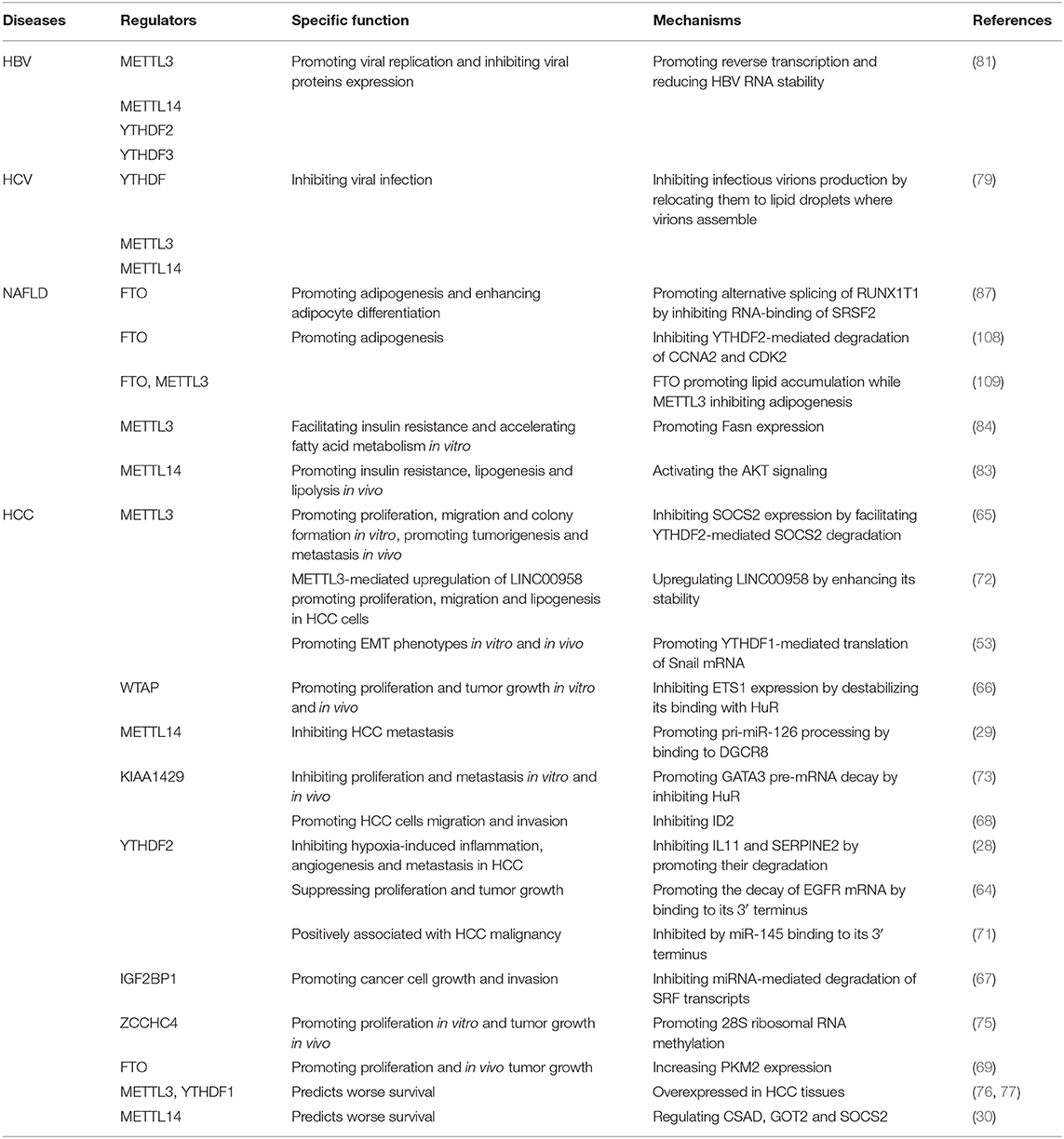
Table 1. Role of m6A regulators in HCC occurrence and development.
Dysregulation of m6A methylation and abnormal expression of regulators are involved in various cancer functions, such as distant metastasis, cancer immunoregulation, tumor angiogenesis and cancer stem cell formation, by modulating the degradation, processing or translation of the downstream targeted RNAs. The epithelial mesenchymal transition (EMT), a critical process for cancer cell metastasis, is regulated by the m6A writer METTL3, not only in HCC but across diverse cancers. Lin et al. discovered that METTL3 is upregulated in HCC, promoting EMT by enhancing the m6A modification of Snail mRNA (53). Moreover, YTHDF1 mediates the m6A-increased translation of Snail mRNA, which promotes the HCC metastasis. Notably, METTL3 also participates in EMT in gastric cancer (GC), ovarian cancer and prostatic cancer (27, 54). Yue et al. found that m6A modification of zinc finger MYM-type containing 1 (ZMYM1), a downstream target of METTL3, enhanced its stability through the regulation of HuR, and that ZMYM1 represses E-cadherin promoter by recruiting the CtBP/LSD1/CoREST complex, thereby facilitating the EMT process and metastasis of GC (55). Intriguingly, Ma et al. reported that METTL14 suppresses HCC metastasis by regulating miRNA in an m6A-dependent manner (29). METTL14 also inhibits the progression and metastasis of colorectal cancer (CRC) by downregulating oncogenic lncRNA (56). Moreover, it can inhibit CRC cell growth via miR-375 /Yes-associated protein 1 (YAP1) pathway (57). These findings confirm the significant roles of m6A writers in regulating tumor metastasis and progression across diverse cancers.
It is noteworthy that, because m6A modification and the expression of m6A regulators are highly heterogeneous across various cancer types, the effects of m6A methylation may differ even in the same context. For instance, hypoxia in breast cancer stimulates hypoxia-inducible factor (HIF)-1α and HIF-2α- dependent expression of ALKBH5, which leads to the demethylation of NANOG mRNA and the induction of cancer stem cell phenotype (58). In non-small cell lung cancer (NSCLC), YTHDF1 is identified as a hypoxia adaptor, whose depletion inhibits tumor progression by regulating translation efficacy (59). However, in HCC, hypoxia induces the reduction of YTHDF2, which promoted tumor inflammation and angiogenesis (28). Additionally, YTHDF2 is recognized as an oncogene that is upregulated in lung cancer and acute myeloid leukemia (AML) to promote tumor initiation and growth, exhibiting a completely opposite function as in HCC (60, 61).
The functional implications of m6A in tumor immunology are drawing increasing attention. Yang et al. reported overexpression of FTO in melanoma, and that its knockdown increased the m6A level of critical tumorigenic genes such as PD-1, CXCR4, and SOX10, leading to increased mRNA degradation mediated by YTHDF2 (62). Moreover, silencing of FTO sensitized melanoma to anti-PD-1 treatment. Li et al. found that overexpression of WTAP is associated with poor prognosis in GC patients, which might be correlated with T lymphocyte infiltration (63). However, to date, no studies have explored the relationship between m6A and immunoregulation in HCC. Due to the low response rate of advanced HCC patients to anti-PD-L1 treatment, m6A might be a promising new target for the study of drug insensitivity and resistance in immunotherapies.
Overall, the above evidence indicates that dysregulated m6A modification plays an important role in cancer biology, and this epigenetic alteration is highly heterogeneous.
Two recent studies have revealed m6A mechanisms underlying the negative correlation between YTHDF2 and HCC under hypoxic conditions (28, 64). Hou et al. reported that reduced expression of YTHDF2 in HCC tumor tissues was accompanied by increases in m6A methylation and mRNA expression, and was associated with unfavorable survival outcomes and poor clinical classification (28). The study revealed that YTHDF2 inhibits HCC progression through m6A modification by decreasing the stability of interleukin 11 (IL11) and serpin family E member 2 (SERPINE2) mRNA, which are involved in tumor angiogenesis and inflammation activation respectively (Figure 2A). Moreover, repression of HCC by YTHDF2 was reversed by HIF-2α under hypoxic conditions. These findings reveal molecular mechanisms through which targeted HCC treatment may be utilized. Interestingly, Zhong et al. showed different mechanisms in HCC in terms of YTHDF2 reduction induced by hypoxia (64). YTHDF2 directly bound with the 3′ terminus of the EGFR m6A site to accelerate its decay, which impeded the MEK/ERK signaling and thereby suppressed HCC. In addition, the expression of YTHDF2 was repressed under hypoxic conditions in HCC. Although the study demonstrated the significant role of YTHDF2 in inhibiting HCC, whether it involves m6A modification requires validation in more experiments.
In addition to the direct regulatory effect of YTHDF2, there is evidence suggesting the cooperative function of m6A regulators in HCC. Chen et al. showed that METTL3 promotes HCC progression in a YTHDF2- and m6A-dependent manner (65). In that study, overexpression of METTL3 promoted tumor growth in CRISPR/dCas9-VP64 models in vivo and in vitro, indicating its positive correlation with HCC. Suppressor of cytokine signaling 2 (SOCS2), a direct downstream target of METTL3, inhibited tumor progression. The expression of SOCS2 was downregulated by METTL3 through m6A modification, in which YTHDF2 bound to the 3′ terminus of SOCS2 to facilitate its decay. Therefore, the m6A regulators METTL3 and YTHDF2 cooperatively promoted the development of HCC in an m6A-dependent manner. Another regulator of m6A, WTAP, has also been found to promote HCC progression through m6A modification (Figure 2B) (66). WTAP is significantly upregulated in HCC and this is associated with unfavorable survival outcomes (66). ETS proto-oncogene 1 (ETS1), a tumor suppressor in HCC, is negatively regulated by WTAP via m6A modification. Moreover, m6A modification of ETS1 simultaneously inhibits its binding with HuR, the RNA stabilizer, which promotes its degradation. In addition, WTAP promotes the G2/M phase transition via ETS1-p21/p27 signaling pathway to regulate the HCC cell cycle. Overall, these results confirm the significance of cooperative effects between m6A regulators themselves and RNA stabilizers in HCC progression.
As m6A research advances, new regulators are being identified that take part in the biological processes of tumors. IGF2BPs (including IGF2BP1/2/3) are newly identified m6A reader proteins that promote the stability and storage of targeted transcripts by recognizing the consensus motif GGAC (A = m6A) (45). In HCC cells, IGF2BP1 upregulates serum response factor (SRF) by binding with its 3′ terminus and promoting its m6A modification (67). It also inhibits the miRNA-mediated degradation of SRF transcripts. The dual regulations enhance the expression of SRF-targeted genes thereby facilitating HCC tumorigenesis. Strikingly, there are 35 genes regulated by SRF or IGF2BP1 in total, which in combination construct an oncogenic cellular network in HCC, making themselves valuable therapeutic targets. Recently, KIAA1429 and FTO were both found to be overexpressed in HCC, which predicted unfavorable prognoses in HCC patients (68, 69). KIAA1429 inhibits the expression of inhibitor of DNA binding 2 (ID2) by promoting its m6A methylation while FTO downregulates pyruvate kinase M1/2 (PKM2) by triggering its demethylation. Consequently, KIAA1429 and FTO together promote the tumorigenesis of HCC. However, these results need higher levels of evidence for validation.
Overall, these findings suggest that mRNA stability and degradation are the most common m6A regulatory function in HCC. We believe that with more in-depth research on m6A regulators, new mechanisms will soon be revealed.
The regulatory effects of ncRNAs including long noncoding RNAs (lncRNAs), microRNAs and circular RNAs (circRNAs) on m6A are nonnegligible in HCC. Recently, several studies revealed how the interplay between m6A modification and ncRNAs influences the biological behaviors of HCC.
METTL14 inhibits HCC metastasis by regulating primary miR-126 (pri-miR-126) processing in an m6A-dependent manner (Figure 2C) (29). The processing of pri-miRNAs is usually performed by microprocessor proteins such as DiGeorge syndrome chromosomal region 8 (DGCR8) (70). In this study, overexpression of METTL14 upregulated the expression of miR-126 by promoting pri-miR-126 binding with DGCR8, which facilitated its processing. The same study found that in metastatic HCC, decreased METTL14 led to the accumulation of unprocessed pri-miR-126. Moreover, the inhibition of metastasis was reversed by miR-126 inhibitor in METTL14-overexpressing HCC cells, indicating the significance of miR-126 as a downstream target of METTL14 to inhibit metastatic HCC. In another study, miR-145 regulated m6A levels in HCC cells through the modulation of reader protein YTHDF2 (71). Overexpression of miR-145 increased m6A methylation in HCC cells and this regulation was reversed by overexpression of YTHDF2. Moreover, the luciferase activities confirmed that miR-145 directly targeted the 3′ terminus of YTHDF2 mRNA, and in vitro experiments proved its inhibition of HCC cell proliferation. This is the first report in HCC that ncRNAs regulate m6A regulators to influence cancer development. However, it should be noted that the effects of YTHDF2 in that study contradict earlier reports, which we suspected could be due to tumor heterogeneity in HCC (28, 64).
More recently, m6A modification has been reported to be correlated with the upregulation of LINC00958 in HCC, which may promote proliferation, invasion, migration and lipogenesis of HCC cells in vitro (72). Moreover, the upregulation of LINC00958 may be regulated by METTL3 via the modulation of the stability of RNA in HCC cells, and high expression of both genes may predict poor prognosis in HCC. However, this only shows that METTL3 is positively related with LINC0958, and thus more specific experimental methods are needed for validation. KIAA1429 can also promote HCC development via the m6A-dependent modification of GATA binding protein 3 (GATA3), in which its antisense transcript-derived lncRNA called GATA3-AS plays an important role (Figure 2D) (73). KIAA1429 is significantly upregulated in HCC and predicts worse survival outcomes in HCC patients. Knockdown of KIAA1429 is negatively related with hepatoma growth both in vitro and in vivo. KIAA1429 promotes the decay of GATA3 precursor mRNA (pre-mRNA) through m6A modification, thereby promoting HCC progression. The RNA binding protein HuR increases the stability of GATA3 pre-mRNA by binding with the 3′ terminus of pre-mRNA. However, this binding is negatively regulated by KIAA1429 through m6A modification. On the other hand, the lncRNA GATA3-AS acts as a cis-acting element for KIAA1429 to guide m6A methylation on GATA3 pre-mRNA 3′ terminus. This KIAA1429/GATA3 regulatory axis in HCC development reflects the breakdown of m6A dynamic balance under pathological conditions.
In recent research, circRNA has been found to facilitate HCC progression indirectly through m6A modification. CircRNA_104075 promotes YAP-related HCC progression via hepatocyte nuclear factor 4 α (HNF4a) in HCC (74). It also acts as a competitive endogenous RNA (ceRNA) to increase YAP expression by absorbing miR-583-3p. Intriguingly, an m6A motif, AGACU, has been identified within the 3′ terminus of YAP, which may be critical for the interplay between YAP and miR-582-3p. Furthermore, the critical effects of m6A was confirmed by luciferase activities and RNA immunoprecipitation quantitative polymerase chain reaction (RIP-qPCR) analyses. However, the mechanisms underlying this m6A modification should be further validated.
Overall, in HCC, m6A modification regulates ncRNAs in diverse ways, such as through splicing, preprocessing, stability and decay, and in turn ncRNAs affect and participate in the m6A process. Therefore, the interactions between m6A and ncRNAs play an important role in HCC progression, and we believe that more regulatory mechanisms will be identified in the future.
Considering the diverse biological effects mediated by m6A modification in HCC, there may ample opportunity to devise effective treatments and targeted therapies for HCC patients.
Recently, progress has been made on experimental therapies targeting m6A-specific mechanisms. Not only novel medicines but also new m6A regulators are being identified. Hou et al. demonstrated that PT2385 has excellent efficacy in the treatment of HCC, and significantly reduces tumor volume in subcutaneous HCC tumor models (28). It can reverse the reduction of YTHDF2 regulated by HIF-2α, which attenuates tumorous inflammation and angiogenesis thereby suppressing HCC growth. Although the administration of PT2385 does not affect the localization of YTHDF2 and cannot take effect with YTHDF2 deficiency, it does provide an opportunity to inhibit hypoxia-induced HCC development. Zuo et al. invented a novel PEGylated PLGA nanoplatform (PLGA-PEG NP) encapsulating si-LINC00958 targeting LINC00958 for the treatment of HCC (72). In the study, it exhibited controllable release, excellent cellular drug uptake and precise tumor-targeting capacity. Moreover, tumor size was effectively reduced in PLGA-PEG (si-LINC00958) NPs-treated HCC-patient-derived xenograft (PDX) model, and the pathological results and blood examination exhibited no significant treatment-related toxicity in the mouse model. A new m6A methyltransferase, zinc finger CCHC-type containing 4 (ZCCHC4), which mainly takes part in the methylation of 28S ribosomal RNAs (rRNAs), has been identified both in vitro and in vivo in HCC (75). Silence of ZCCHC4 decreases m6A methylation in 28S rRNA and inhibits proliferation of CRISPR/cas9-edited HepG2 cells. In addition, it is overexpressed in HCC tumor tissues and its silence significantly inhibits tumor growth in a xenograft animal model, confirming its significant role in promoting HCC progression through m6A methylation. Although its detailed biological functions in HCC require more validation, it might be a potential novel target for HCC treatment.
The application of m6A in HCC is not limited to clinical treatment; it may also be applied to cancer diagnosis and prognostic prediction. Recent studies have shown that m6A regulators have significant potential to serve as biomarkers. METTL3 and YTHDF1 are both overexpressed in HCC in several studies, and thus may be useful biomarkers for survival prediction and clinical classification (76, 77). Another study identified three METTL14-related downstream genes including cysteine sulfinic acid decarboxylase (CSAD), glutamic-oxaloacetic transaminase 2 (GOT2), and SOCS2 using bioinformatics methods (30). They were included in a nomogram for the prediction of overall survival. Other overexpressed m6A regulators in HCC include WTAP, KIAA1429 and FTO, while downregulated enzymes include METTL14 and YTHDF2 (28, 66, 68, 69, 73).
Infection with hepatitis virus is the leading cause of HCC worldwide (3). There is a high rate of HBV infection in eastern Asia while HCV infection is more prevalent in developed countries (2, 5). The etiological connection between viral hepatitis and HCC enable us to intervene with the progression of disease for early prevention of HCC. HBV or HCV infection underlies the context of chronic liver inflammation, which would lead to liver cirrhosis and eventually HCC (3, 78). During the process of carcinogenesis, dysregulation of m6A may facilitate virus activity and disturb host immunity, causing persistent virus infection, shaping inflammatory microenvironment and consequently leading to HCC occurrence (Figure 2). For instance, depletion of certain m6A regulators in HCV can enhance viral activity, which increases virus titers without disturbing viral replication (79). Moreover, HCV infection affects host immunity in an m6A-dependent manner (80). m6A modification has also been observed in HBV, and is associated with virus reverse transcription (81). Thus, dysregulation of m6A in viral host cells is closely associated with the development of viral hepatitis, and may contribute to the occurrence of HCC.
NAFLD or non-alcohol steatohepatitis (NASH) is gaining increasing attention for its prevalence around the world and has become one of the leading etiologies in HCC (7). NAFLD is in fact a wide-range of chronic liver diseases, characterized by excessive accumulation of triglycerides in hepatocytes. It tends to progress from isolated hepatocyte triglyceride accumulation and steatosis (NAFL), to hepatic triglyceride accumulation with inflammation and hepatic injury (NASH), and eventually to liver fibrosis and HCC (82). NAFLD is strongly correlated with metabolic syndromes such as diabetes and obesity, and these risk factors interact to increase the risk for HCC occurrence in NAFLD patients. The pathogenesis of HCC in NAFLD patients is a complex process involving multiple mechanisms including lipogenesis, fat accumulation, insulin resistance, oxidative, and endoplasmic reticulum (ER) stress, inflammatory response and DNA damage (83–86) (Figure 2). Recent studies have indicated that m6A modification actively participates in these processes. The m6A demethylase FTO has been widely studied due to its established relationship with obesity, and it has been found to promote adipogenesis through multiple signaling pathways (87). Additionally, other m6A regulators such as METTL3 and METTL14 have been reported to participate in the regulation of insulin resistance (83, 84). These studies prove the significance of m6A modification in NAFLD progression, opening up an opportunity to prevent HCC in the future.
Gokhale et al. outlined the mechanisms of m6A methylation in regulating the cellular activity of HCV in host cells (79). Knockdown of METTL3 and METTL14 with small interfering RNA (siRNA) increases the production of infectious virions of HCV, which has also been observed in YTHDF reader proteins (79). This may suggest that HCV replication can be inhibited by m6A modification. In addition, m6A can directly modulate the generation of HVC virions as revealed by immunofluorescent staining that YTHDF reader proteins are directly localized to the lipid droplets on which HCV virions assemble (Figure 3A) (79). Gokhale et al. discovered new signaling pathways affected by m6A modification after HCV infection, and reported that the infection alters m6A modification of cellular mRNA (80); for example, increased m6A levels of ROI kinase 3 (RIOK3) promotes its translation while decreased m6A levels of cold inducible RNA binding protein (CIRBP) promotes its alternative splicing (80). RIOK3 may be translated into serine/threonine kinase, which may modulate antiviral pathways, and CIRBR encoding protein might bond RNA in the cellular stress response (88–90). Moreover, the innate immune response and endoplasmic reticulum (ER) stress can trigger this m6A methylation. However, it remains unclear whether the specific cellular changes involving those two mRNAs result from the innate immune response of the host to defend against infection or is normal cellular response to ensure robust replication of the HCV. Still, study of new m6A-modified transcripts could help better elucidate the dynamic changes that occur during viral infection and such materials may be potential antiviral-targets.
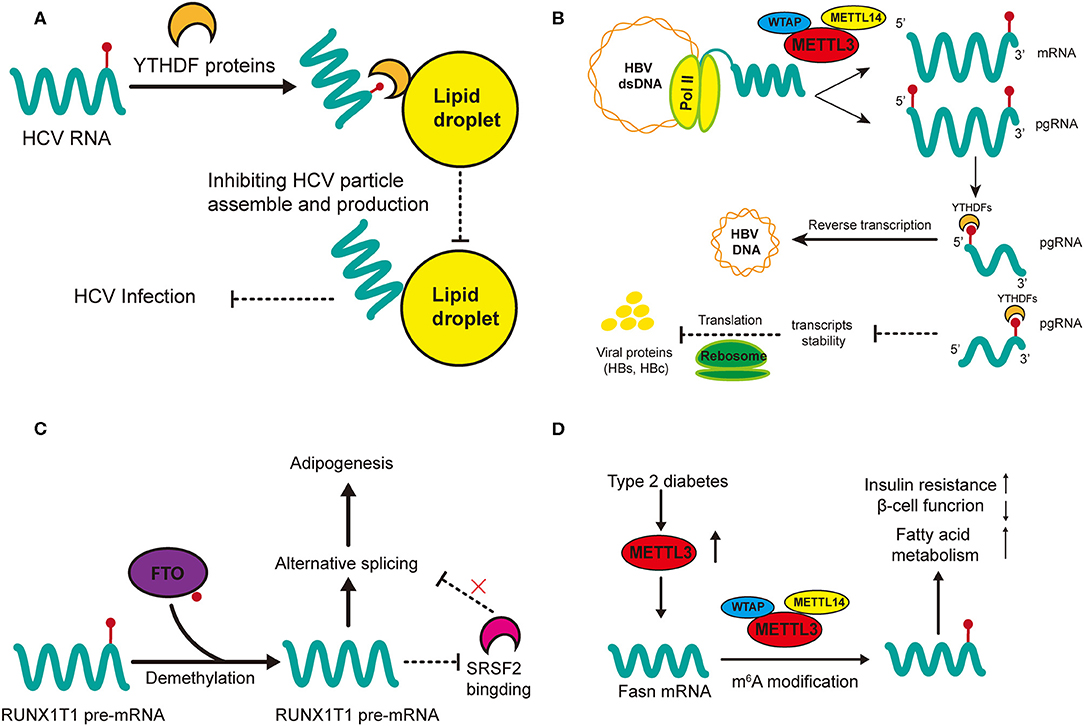
Figure 3. Regulatory functions of m6A in HCC-related hepatic diseases. m6A regulation in HCV (A) and HBV (B) affect viral activities (infection and replication) while m6A modification in non-alcohol fatty liver disease (NAFLD) promotes adipogenesis (C) and insulin resistance (D). (A) m6A-modified HCV RNAs are recruited by YTHDF proteins onto lipid droplets to impede the assembly of unmodified RNAs, thus inhibiting the production of HCV infectious particles. (B) m6A modification at the 5′ terminus of the pregenomic RNA (pgRNA) is required for HBV reverse transcription while m6A at the 3′ terminus of pgRNA lead to destabilization of HBV transcripts. (C) FTO promotes the demethylation of RUNX1T1 pre-mRNA; this inhibits SRSF2 binding and alters the alternative splicing of pre-mRNA, therefore promoting adipogenesis. (D) The expression of METTL3 is upregulated in type 2 diabetes. Upregulated METTL3 inhibits hepatic insulin sensitivity through m6A modification of Fasn mRNA and facilitates fatty acid metabolism.
m6A modification is vital in the regulation of HBV activity (81). Although HBV is a double-stranded DNA (dsDNA) virus, the extension of its life (replication) is fulfilled by RNA transcripts (91). Imam et al. was the first time to identify m6A in HBV-infected cell lines and liver tissues of chronic HBV patients (Figure 3B) (81). They also reported the determinant m6A motif DRACH (D = A, G, or U and H = A, C, or U) within the epsilon loop near the 3′ terminus of HBV mRNA, and that the m6A site is located at A1907 (1905-1909, GGACA) (81). Knockdown of the m6A regulatory enzymes (YTHDF2, YTHDF3, METTL3, and METTL14) may increase the expression of HBV proteins (HBc and HBs), by reducing the stability of HBV RNA. However, m6A at the 5′ stem loop increases reverse transcription and silencing of MTEEL3 as well as METTL14 reduces reverse transcription (81). Thus, it is obvious that the effects of m6A in regulating HBV replication or infection is two-sided, which suggests that m6A modification in viral metabolism is more of a temporal and transient modulation to adapt to the environment of host cells.
Antiviral treatment is indispensable in HCC patients with viral hepatitis, not only for the inhibition of viral replication but also for the prevention of tumor progression (92, 93). The universal presence of m6A in various viruses makes it a potential target for a wide range of viral infection including HBV and HCV.
Interferons (IFNs) have been used in HCC patients with chronic viral hepatitis to impede virus infection and replication, which can effectively prevent the occurrence, recurrence and metastasis of HCC (94–96). Recent studies have shown that, one IFN-stimulated genes (ISG), ISG20, selectively recognize and promotes degradation of m6A-modified HBV transcripts (97). In that study, ISG20-mediated decay of specific HBV RNAs was inhibited by the knockdown of YTHDF2 reader proteins or methyltransferases. Moreover, this ISG20-mediated RNA decay occurred only at m6A sites of A1907 and combined with YTHDF2 reader protein to form a complex (97). These results lead to the assumption that any degradation process of virus transcripts can be regulated by the combination of YTHDF2 protein and its recognition at the epsilon stem loop (A1907), therefore opening a new gate for the treatment of HCC patients infected with hepatitis virus. There is evidence that 3-deazaadenosine (DAA), the S-adenosylhomocysteine (SAC) hydrolase inhibitor, could serve as a broad-spectrum antiviral (98). The inhibition of SAC hydrolase results in the accumulation of SAC, therefore impairing the generation of S-adenosylmethionine (SAM), from which METTL3 acquire methyl groups for m6A methylation (98). In several studies, DAA has been found to inhibit the replication of diverse viruses by indirectly targeting m6A modification, and the compound has demonstrated no detectable toxicity (99–102). After more in vivo validation, DAA could be a promising medicine for the treatment of viral hepatitis and the prevention of HCC.
In addition to currently available medicines that inhibit viral activity through m6A mechanisms, there may be a relationship between specific structural overlaps of viral genome and consensus sequences of m6A modification (103). The guanine-based putative-quadruplex-forming sequences (PQSs) is a conserved secondary structure in viral RNA, which can fold into guanine-quadruplexes (G4s) to regulate viral replication and inhibit protein translation in flaviviruses (79, 104, 105). If viruses can transform PQSs into G4s to counteract surveillance by host cells, G4s could be a potential antiviral target (106, 107). In one study, during the matching of viral PQS and m6A sequencing data, it was found that some sites of m6A occur within the loops of PQSs (103). In addition, mutation data have confirmed some overlapped m6A site are vital for HBV replication. Therefore, it is reasonable to propose that the colocalization of m6A and PQSs in HBV RNA might result from the topological control of m6A methylation, which could generate synergistic effects if it becomes a therapeutic target. However, more sequencing data of m6A is needed for further validation.
m6A may play an important role in the metabolism of adipose tissues (85). Significant differences in m6A methylation landscape have been observed between NAFLD mice induced through a high-fat diet and normal mice (85). In addition, differential m6A genes are highly expressed in pathways associated with lipid metabolism (85). FTO has been widely studied since its relationship with obesity was discovered (35). Zhao et al. first demonstrated that demethylase FTO promotes the process of adipogenesis by reversing m6A modification (87). Silence of FTO can promote m6A modification, which strengthens the binding capacity of serine and arginine rich splicing factor 2 (SRSF2) with targeted exon Runt-related transcription factor 1 (RUNX1T1) (Figure 3C). This alters the alternative splicing of adipogenesis regulatory factor RUNX1T1, thereby slowing the differentiation of preadipocytes and adipogenesis (87). This positive regulation of FTO in lipogenesis has also been confirmed in other cellular networks (108, 109). FTO knockdown lowers the expression of cellular cycle regulators cyclin A2 (CCNA2) and cyclin-dependent kinase 2 (CDK2) but increases their m6A modification (108). In the meantime, their m6A-modified mRNAs are recognized and degraded by YTHDF2, resulting in suppressed adipogenesis. In addition, both FTO knockdown and METTL3 overexpression can decrease the accumulation of lipids by increasing mRNA m6A methylation (109). There are many other studies on the potential role of FTO in NAFLD, which we do not describe in detail here, because their mechanisms may not involve m6A modification or the methods used for m6A examination are not specific (110–112). In general, FTO and its related signaling could be a prospective research target in the treatment of NAFLD, and the prevention of NAFLD-related HCC.
Another major mechanism of NAFLD progression is insulin resistance (Figure 3D) (86). Intracellular METTL3 and m6A methylation in type 2 diabetes patients are positively related with insulin resistance (HOMA-β) and negatively related with β-cell function (84). METTL3 knockout in the liver of mice inhibits m6A modification and decreases the intracellular level of fatty acid synthase (Fasn). However, the depletion of METTL3 improves insulin sensitivity and inhibits fatty acid synthesis (84). These results indicate that insulin resistance is mediated by METTL3 through m6A modification of Fasn mRNA and it may accelerate the metabolism of fatty acids. Interestingly, another study reported that depletion of METTL14 in β cells in mice fed a high-fat diet led to seemingly paradoxical effects: insulin sensitivity increased while insulin secretion decreased (83). In that study, knockout of METTL14 lowered insulin secretion, increased insulin sensitivity, inhibited lipogenesis and enhanced lipolysis. Insulin sensitivity was enhanced due to the activation of AKT signaling and decreased gluconeogenesis; we speculate that the decrease in lipid accumulation might have resulted from the enhanced insulin signaling in the liver (83). All in all, METTL14 in β cells is positively correlated with lipogenesis and insulin secretion but negatively related with insulin sensitivity. Its function in insulin resistance and lipo-metabolism needs further validation in hepatocytes. However, METTL14 has great potential in the treatment of NAFLD, especially regarding to its multiple effects on insulin and lipid metabolism.
Although the number of people diagnosed with NAFLD is large, only a minority will fall into progressive liver disease or liver cancers (113). Thus, it is important to identify patients who are more likely to progress to HCC and conduct timely interventions.
The regulators of m6A methylation including METTL3, METTL14, YTHDF2, and FTO are potential targets based on their significant roles in the pathogenesis of NAFLD. For instance, dietary branched-chain amino acids (BCAA) can effectively regulate lipid metabolism in weanling piglets via m6A modification (114). In one study, high- and low-dose feeding of BCAA was associated with significantly lower levels of fatty acid synthesis compared to normal-dose feeding, due to the upregulation of lipolysis genes and downregulation of lipogenic genes. Moreover, a lower level of m6A methylation was identified with downregulation of METTL3 and FTO in the livers of the high-BCAA group (114). The results indicated that a dietary with high BCAAs can decrease lipid accumulation through the regulation of m6A modification.
The protective effects of betaine (trimethylglycine) on liver, including alleviation of impaired liver function and decreased lipid accumulation, have long been recognized (115, 116). Recently, it was confirmed that these protective effects are mediated by FTO through m6A modification (117). Betaine promotes lipolysis and lipid oxidative by reversing the hypomethylated-mRNA and overexpressed FTO in adipocytes of mice fed with high fat diet (118). In addition, this reversion requires the existence of AMP-activated protein kinase α1 subunit (AMPKα1) (118). Another study reported that lipopolysaccharide (LPS)-induced liver function and lipid metabolism disorder can be alleviated by curcumin through the upregulation of m6A (119). Furthermore, curcumin supplementation affected the expression of major m6A regulators, indicating its powerful hepatoprotective effect (119). There are plenty of other compounds such as exenatide and entacapone that directly target m6A regulator, that worth more researching in the future (120, 121).
In summary, m6A modification plays an important role in the occurrence and development of HCC. Viral hepatitis and NAFLD are major risk factors for HCC, which shows a significant relationship with m6A methylation. In HCV and HBV, m6A regulates the infection and replication of virus. In NAFLD, it participates in insulin resistance and the accumulation of lipids. Therapeutic experiments regarding m6A have been conducted on two diseases, which will greatly promote the prevention of HCC. However, the chronic inflammation that accompanies these ailments will lead to liver fibrosis and/or cirrhosis, whose pathogeneses regarding m6A warrant further study. In HCC, m6A regulators specifically regulate mRNA splicing, translation, and degradation, while ncRNAs interact with m6A, leading to changes in gene expression; consequently, mRNAs and ncRNAs both regulate the progression of HCC. Therefore, in terms of the diversity of m6A in HCC, m6A-related molecules are potential biomarkers for early diagnosis and prospective therapeutic targets for clinical treatment. Furthermore, in the age of precision medicine, m6A-related therapies could be tailored based on the etiology and pathogenesis of HCC patients, which will optimize treatments and their benefits.
JL wrote the first draft of the manuscript. JQ conceived the idea of the manuscript. LZ and SY designed the structure of the manuscript. SZ and WZ revised, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
This work was supported by the National S&T Large Project (No. 2018ZX10301201) and the National Science Foundation of China (Nos. 81721091 and 2017ZX10203205).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer XH declared a shared affiliation, with no collaboration, with the authors to the handling editor at the time of review.
HCC, Hepatocellular carcinoma; NAFLD, Non-alcohol fatty liver disease; HBV, Hepatitis B virus; HCV, Hepatitis C virus; PD-L1, Programmed cell death ligand-1; m6A, N6-methyladenosine; mRNAs, Messenger RNAs; ncRNAs, Noncoding RNAs; METTL3, Methyltransferase-like 3; METTL14, Methyltransferase-like 14; WTAP, Wilm's tumor 1-associated protein; ZC3H13, Zinc finger CCCH domain-containing protein 13; ALKBH5, AlkB homolog 5; FTO, Fat mass and obesity-associated protein; YTHDC1, YTH domain-containing 1; YTHDF2, YTH N6-methyladenosine RNA-binding protein; RBM15, RNA-binding motif protein 15; HNRNPC, Heterogeneous nuclear ribonucleoprotein C; RBM15, RNA-binding motif protein 15; siRNA, Small interfering RNA; RIOK3, ROI kinase 3; CIRBP, Cold inducible RNA binding protein; dsDNA, Double-stranded DNA; IFNs, Interferons; ISGs, IFN-stimulated genes; DAA, 3-deazaadenosine; SAC, S-adenosylhomocysteine; SAM, S-adenosylmethionine; PQSs, Putative quadruplex-forming sequences; G4s, Guanine-quadruplexes; SNP, Single nucleotide polymorphism; NASH, Non-alcoholic steatohepatitis; SRSF2, Serine and arginine rich splicing factor 2; RUNX1T1, Runt-related transcription factor 1; CCNA2, Cellular cycle regulators cyclin A2; CDK2, Cyclin-dependent kinase 2; Fasn, Fatty acid synthase; BCAA, Branched-chain amino acids; AMPKα1, AMP-activated protein kinase α1 subunit; LPS, Lipopolysaccharide; ZCCHC4, Zinc finger CCHC-type containing 4; IL11, interleukin 11; SERPINE2, Serpin family E member 2; HIF-2α, Hypoxia-inducible factor 2α; SOCS2, Suppressor of cytokine signaling 2; ETS1, ETS proto-oncogene 1; HuR, Hu-antigen R; IGF2BP1, Insulin-like growth factor 2 mRNA-binding proteins; SRF, Serum response factor; ID2, DNA binding 2; PKM2, Pyruvate kinase M1/2; Me-RIP-PCR, Methylated- RNA immunoprecipitation- polymerase chain reaction; lincRNAs, Long noncoding RNAs; miRNAs, MicroRNAs; CircRNAs, circular RNAs; pri-miRNA, primary miRNA; DGCR8, DiGeorge syndrome chromosomal region 8; GATA3, GATA binding protein 3; pre-mRNA, Precursor mRNA; YAP, Yes-associated protein; CeRNA, Competitive endogenous RNA; PDX, Patient derived xenograft; PLGA-PEG NP, PEGylated PLGA nanoplatform; rRNAs, Ribosomal RNAs; CSAD, Cysteine sulfinic acid decarboxylase; GOT2, Glutamic-oxaloacetic transaminase 2; LLPS, Liquid-liquid phase separation; H3K36me3, H3 trimethylation at Lys36; carRNA, Chromosome-associated regulatory RNA; EMT, Epithelial mesenchymal transition; GC, Gastric cancer; ZMYM1, Zinc finger MYM-type containing 1; CRC, Colorectal cancer; NSCLC, Non-small cell lung cancer; AML, Acute myeloid leukemia.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: gLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
3. Villanueva A. Hepatocellular carcinoma. N Engl J Med. (2019) 380:1450–62. doi: 10.1056/NEJMra1713263
4. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. (2018) 391:1301–14. doi: 10.1016/S0140-6736(18)30010-2
5. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. (2012) 142:1264–73 e1. doi: 10.1053/j.gastro.2011.12.061
6. Global Burden of Disease Cancer C, Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T, Alizadeh-Navaei R, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-Adjusted life-Years for 29 cancer groups, 1990 to 2016: a Systematic analysis for the global burden of disease study. JAMA Oncol. (2018) 4:1553–68. doi: 10.1001/jamaoncol.2018.2706
7. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From nASH to hCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. (2019) 16:411–28. doi: 10.1038/s41575-019-0145-7
8. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. (2017) 389:2492–502. doi: 10.1016/S0140-6736(17)31046-2
9. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. (2018) 391:1163–73. doi: 10.1016/S0140-6736(18)30207-1
10. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A rNA methylation. Nat Rev Genet. (2014) 15:293–306. doi: 10.1038/nrg3724
11. Jia G, Fu Y, He C. Reversible rNA adenosine methylation in biological regulation. Trends Genet. (2013) 29:108–15. doi: 10.1016/j.tig.2012.11.003
12. Deng X, Chen K, Luo GZ, Weng X, Ji Q, Zhou T, et al. Widespread occurrence of n6-methyladenosine in bacterial mRNA. Nucleic Acids Res. (2015) 43:6557–67. doi: 10.1093/nar/gkv596
13. Sommer S, Salditt-Georgieff M, Bachenheimer S, Darnell JE, Furuichi Y, Morgan M, et al. The methylation of adenovirus-specific nuclear and cytoplasmic rNA. Nucleic Acids Res. (1976) 3:749–65. doi: 10.1093/nar/3.3.749
14. Kennedy EM, Courtney DG, Tsai K, Cullen BR. Viral epitranscriptomics. J Virol. (2017) 91. doi: 10.1128/JVI.02263-16
15. Yadav PK, Rajasekharan R. The m(6)A methyltransferase ime4 epitranscriptionally regulates triacylglycerol metabolism and vacuolar morphology in haploid yeast cells. J Biol Chem. (2017) 292:13727–44. doi: 10.1074/jbc.M117.783761
16. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A rNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112
17. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' uTRs and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
18. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent rNA structural switches regulate rNA-protein interactions. Nature. (2015) 518:560–4. doi: 10.1038/nature14234
19. Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-Binding proteins. Trends Cell Biol. (2018) 28:113–27. doi: 10.1016/j.tcb.2017.10.001
20. Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. (2017) 33:319–42. doi: 10.1146/annurev-cellbio-100616-060758
21. Yang D, Qiao J, Wang G, Lan Y, Li G, Guo X, et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. (2018) 46:3906–20. doi: 10.1093/nar/gky130
22. Liu Z, Zhang J. Human c-to-U coding rNA editing is largely nonadaptive. Mol Biol Evol. (2018) 35:963–9. doi: 10.1093/molbev/msy011
23. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-Wide maps of m6A circRNAs identify widespread and cell-Type-Specific methylation patterns that are distinct from mRNAs. Cell Rep. (2017) 20:2262–76. doi: 10.1016/j.celrep.2017.08.027
24. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase mETTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
25. Lin S, Gregory RI. Methyltransferases modulate rNA stability in embryonic stem cells. Nat Cell Biol. (2014) 16:129–31. doi: 10.1038/ncb2914
26. Shulman Z, Stern-Ginossar N. The rNA modification n(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol. (2020) doi: 10.1038/s41590-020-0650-4
27. Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of aXL translation and epithelial to mesenchymal transition. Gynecol Oncol. (2018) 151:356–65. doi: 10.1016/j.ygyno.2018.09.015
28. Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. (2019) 18:163. doi: 10.1186/s12943-019-1082-3
29. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating n(6) -methyladenosine-dependent primary microRNA processing. Hepatology. (2017) 65:529–43. doi: 10.1002/hep.28885
30. Li Z, Li F, Peng Y, Fang J, Zhou J. Identification of three m6A-related mRNAs signature and risk score for the prognostication of hepatocellular carcinoma. Cancer Medicine. (2020) doi: 10.1002/cam4.2833
31. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m(6)A rNA methylation in cancer. J Hematol Oncol. (2018) 11:48. doi: 10.1186/s13045-018-0590-8
32. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of n(6)-adenosine methylation by the mETTL3-METTL14 complex. Nature. (2016) 534:575–8. doi: 10.1038/nature18298
33. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian wTAP is a regulatory subunit of the rNA n6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
34. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear rNA is a major substrate of the obesity-associated fTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
35. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the fTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. (2007) 316:889–94. doi: 10.1126/science.1141634
36. Wahlen K, Sjolin E, Hoffstedt J. The common rs9939609 gene variant of the fat mass- and obesity-associated gene fTO is related to fat cell lipolysis. J Lipid Res. (2008) 49:607–11. doi: 10.1194/jlr.M700448-JLR200
37. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian rNA demethylase that impacts rNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
38. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader yTHDC1 regulates mRNA splicing. Mol Cell. (2016) 61:507–19. doi: 10.1016/j.molcel.2016.01.012
39. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of n(6)-methyladenosine methylated mRNAs. Elife. (2017) 6. doi: 10.7554/eLife.31311
40. Liao S, Sun H, Xu C. YTH domain: a Family of n(6)-methyladenosine (m(6)A) readers. Genomics Proteomics Bioinformatics. (2018) 16:99–107. doi: 10.1016/j.gpb.2018.04.002
41. Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an n(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. (2017) 27:1115–27. doi: 10.1038/cr.2017.99
42. Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, et al. m(6)A enhances the phase separation potential of mRNA. Nature. (2019) 571:424–8. doi: 10.1038/s41586-019-1374-1
43. Gao Y, Pei G, Li D, Li R, Shao Y, Zhang QC, et al. Multivalent m(6)A motifs promote phase separation of yTHDF proteins. Cell Res. (2019) 29:767–9. doi: 10.1038/s41422-019-0210-3
44. Zhao Y, Shi Y, Shen H, Xie W. m(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. (2020) 13:35. doi: 10.1186/s13045-020-00872-8
45. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of rNA n(6)-methyladenosine by iGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
46. Zhou KI, Shi H, Lyu R, Wylder AC, Matuszek Z, Pan JN, et al. Regulation of co-transcriptional pre-mRNA splicing by m(6)A through the low-Complexity protein hnRNPG. Mol Cell. (2019) 76:70–81 e9. doi: 10.1016/j.molcel.2019.07.005
47. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-Dependent nuclear rNA processing events. Cell. (2015) 162:1299–308. doi: 10.1016/j.cell.2015.08.011
48. Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone h3 trimethylation at lysine 36 guides m(6)A rNA modification co-transcriptionally. Nature. (2019) 567:414–9. doi: 10.1038/s41586-019-1016-7
49. Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. (2015) 16:583–97. doi: 10.1038/nrg3961
50. Abakir A, Giles TC, Cristini A, Foster JM, Dai N, Starczak M, et al. N(6)-methyladenosine regulates the stability of rNA:DNA hybrids in human cells. Nat Genet. (2020) 52:48–55. doi: 10.1038/s41588-019-0549-x
51. Yang X, Liu QL, Xu W, Zhang YC, Yang Y, Ju LF, et al. m(6)A promotes r-loop formation to facilitate transcription termination. Cell Res. (2019) 29:1035–8. doi: 10.1038/s41422-019-0235-7
52. Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, et al. N (6)-methyladenosine of chromosome-associated regulatory rNA regulates chromatin state and transcription. Science. (2020) 367:580–6. doi: 10.1126/science.aay6018
53. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of snail. Nat Commun. (2019) 10:2065. doi: 10.1038/s41467-019-09865-9
54. Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin beta1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am J Cancer Res. (2020) 10:1012–25.
55. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated n6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. (2019) 18:142. doi: 10.1186/s12943-019-1065-4
56. Yang X, Zhang S, He C, Xue P, Zhang L, He Z, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding rNA xIST. Mol Cancer. (2020) 19:46. doi: 10.1186/s12943-020-1146-4
57. Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L, et al. METTL14 suppresses cRC progression via regulating n6-Methyladenosine-Dependent primary miR-375 processing. Mol Ther. (2020) 28:599–612. doi: 10.1016/j.ymthe.2019.11.016
58. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by hIF-dependent and aLKBH5-mediated m(6)A-demethylation of nANOG mRNA. Proc Natl Acad Sci U S A. (2016) 113:E2047–56. doi: 10.1073/pnas.1602883113
59. Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun. (2019) 10:4892. doi: 10.1038/s41467-019-12801-6
60. Sheng H, Li Z, Su S, Sun W, Zhang X, Li L, et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis. (2019) doi: 10.1093/carcin/bgz152. [Epub ahead of print].
61. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the rNA m(6)A reader yTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. (2019) 25:137–48 e6. doi: 10.1016/j.stem.2019.03.021
62. Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. m(6)A mRNA demethylase fTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. (2019) 10:2782. doi: 10.1038/s41467-019-10669-0
63. Li H, Su Q, Li B, Lan L, Wang C, Li W, et al. High expression of wTAP leads to poor prognosis of gastric cancer by influencing tumour-associated t lymphocyte infiltration. J Cell Mol Med. (2020) 24:4452–65. doi: 10.1111/jcmm.15104
64. Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the eGFR mRNA in hepatocellular carcinoma. Cancer Lett. (2019) 442:252–61. doi: 10.1016/j.canlet.2018.11.006
65. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA n6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through yTHDF2-dependent posttranscriptional silencing of sOCS2. Hepatology. (2018) 67:2254–70. doi: 10.1002/hep.29683
66. Chen Y, Peng C, Chen J, Chen D, Yang B, He B, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of eTS1. Mol Cancer. (2019) 18:127. doi: 10.1186/s12943-019-1053-8
67. Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, et al. IGF2BP1 promotes sRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. (2019) 47:375–90. doi: 10.1093/nar/gky1012
68. Cheng X, Li M, Rao X, Zhang W, Li X, Wang L, et al. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of iD2 mRNA. Onco Targets Ther. (2019) 12:3421–8. doi: 10.2147/OTT.S180954
69. Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. m6A demethylase fTO promotes hepatocellular carcinoma tumorigenesis via mediating pKM2 demethylation. Am J Transl Res. (2019) 11:6084–92.
70. Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The drosha-DGCR8 complex in primary microRNA processing. Genes Dev. (2004) 18:3016–27. doi: 10.1101/gad.1262504
71. Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, et al. MicroRNA-145 modulates n(6)-Methyladenosine levels by targeting the 3'-Untranslated mRNA region of the n(6)-Methyladenosine binding yTH domain family 2 protein. J Biol Chem. (2017) 292:3614–23. doi: 10.1074/jbc.M116.749689
72. Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of lINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. (2020) 13:5. doi: 10.1186/s13045-019-0839-x
73. Lan T, Li H, Zhang D, Xu L, Liu H, Hao X, et al. KIAA1429 contributes to liver cancer progression through n6-methyladenosine-dependent post-transcriptional modification of gATA3. Mol Cancer. (2019) 18:186. doi: 10.1186/s12943-019-1106-z
74. Zhang X, Xu Y, Qian Z, Zheng W, Wu Q, Chen Y, et al. circRNA_104075 stimulates yAP-dependent tumorigenesis through the regulation of hNF4a and may serve as a diagnostic marker in hepatocellular carcinoma. Cell Death Dis. (2018) 9:1091. doi: 10.1038/s41419-018-1132-6
75. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase zCCHC4 mediates ribosomal rNA methylation. Nat Chem Biol. (2019) 15:88–94. doi: 10.1038/s41589-018-0184-3
76. Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J, et al. Overexpression of yTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. (2018) 21:859–68. doi: 10.3233/CBM-170791
77. Zhou Y, Yin Z, Hou B, Yu M, Chen R, Jin H, et al. Expression profiles and prognostic significance of rNA n6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res. (2019) 11:3921–31. doi: 10.2147/CMAR.S191565
79. Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, et al. N6-Methyladenosine in flaviviridae viral rNA genomes regulates infection. Cell Host Microbe. (2016) 20:654–65. doi: 10.1016/j.chom.2016.09.015
80. Gokhale NS, McIntyre ABR, Mattocks MD, Holley CL, Lazear HM, Mason CE, et al. Altered m(6)A modification of specific cellular transcripts affects flaviviridae infection. Mol Cell. (2020) 77:542–55 e8. doi: 10.1016/j.molcel.2019.11.007
81. Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY, et al. N6-methyladenosine modification of hepatitis b virus rNA differentially regulates the viral life cycle. Proc Natl Acad Sci USA. (2018) 115:8829–34. doi: 10.1073/pnas.1808319115
82. Younes R, Bugianesi E. Should we undertake surveillance for hCC in patients with nAFLD? J Hepatol. (2018) 68:326–34. doi: 10.1016/j.jhep.2017.10.006
83. Liu J, Luo G, Sun J, Men L, Ye H, He C, et al. METTL14 is essential for beta-cell survival and insulin secretion. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2138–48. doi: 10.1016/j.bbadis.2019.04.011
84. Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 inhibits hepatic insulin sensitivity via n6-methyladenosine modification of fasn mRNA and promoting fatty acid metabolism. Biochem Biophys Res Commun. (2019) 518:120–6. doi: 10.1016/j.bbrc.2019.08.018
85. Luo Z, Zhang Z, Tai L, Zhang L, Sun Z, Zhou L. Comprehensive analysis of differences of n(6)-methyladenosine rNA methylomes between high-fat-fed and normal mouse livers. Epigenomics. (2019) 11:1267–82. doi: 10.2217/epi-2019-0009
86. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of nAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
87. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of n6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. (2014) 24:1403–19. doi: 10.1038/cr.2014.151
88. Takashima K, Oshiumi H, Takaki H, Matsumoto M, Seya T. RIOK3-mediated phosphorylation of mDA5 interferes with its assembly and attenuates the innate immune response. Cell Rep. (2015) 11:192–200. doi: 10.1016/j.celrep.2015.03.027
89. Feng J, De Jesus PD, Su V, Han S, Gong D, Wu NC, et al. RIOK3 is an adaptor protein required for iRF3-mediated antiviral type i interferon production. J Virol. (2014) 88:7987–97. doi: 10.1128/JVI.00643-14
90. Liao Y, Tong L, Tang L, Wu S. The role of cold-inducible rNA binding protein in cell stress response. Int J Cancer. (2017) 141:2164–73. doi: 10.1002/ijc.30833
91. Seeger C, Mason WS. Hepatitis b virus biology. Microbiol Mol Biol Rev. (2000) 64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000
92. Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, El-Serag HB. Risk of hepatocellular cancer in hCV patients treated with direct-Acting antiviral agents. Gastroenterology. (2017) 153:996–1005 e1. doi: 10.1053/j.gastro.2017.06.012
93. Papatheodoridis GV, Chan HL, Hansen BE, Janssen HL, Lampertico P. Risk of hepatocellular carcinoma in chronic hepatitis b: assessment and modification with current antiviral therapy. J Hepatol. (2015) 62:956–67. doi: 10.1016/j.jhep.2015.01.002
94. Ren H, Huang Y. Effects of pegylated interferon-alpha based therapies on functional cure and the risk of hepatocellular carcinoma development in patients with chronic hepatitis b. J Viral Hepat. (2019) 26 Suppl 1:5–31. doi: 10.1111/jvh.13150
95. Teng W, Jeng WJ, Yang HI, Chen WT, Hsieh YC, Huang CH, et al. Interferon is superior to direct acting antiviral therapy in tertiary prevention of early recurrence of hepatocellular carcinoma. Cancers (Basel). (2019) 12:23. doi: 10.3390/cancers12010023
96. Miyake Y, Takaki A, Iwasaki Y, Yamamoto K. Meta-analysis: interferon-alpha prevents the recurrence after curative treatment of hepatitis c virus-related hepatocellular carcinoma. J Viral Hepat. (2010) 17:287–92. doi: 10.1111/j.1365-2893.2009.01181.x
97. Imam H, Kim GW, Mir SA, Khan M, Siddiqui A. Interferon-stimulated gene 20 (ISG20) selectively degrades n6-methyladenosine modified hepatitis b Virus transcripts. PLoS Pathog. (2020) 16:e1008338. doi: 10.1371/journal.ppat.1008338
98. Bader JP, Brown NR, Chiang PK, Cantoni GL. 3-Deazaadenosine, an inhibitor of adenosylhomocysteine hydrolase, inhibits reproduction of Rous sarcoma virus and transformation of chick embryo cells. Virology. (1978) 89:494–505. doi: 10.1016/0042-6822(78)90191-5
99. Wyde PR, Ambrose MW, Meyer HL, Zolinski CL, Gilbert BE. Evaluation of the toxicity and antiviral activity of carbocyclic 3-deazaadenosine against respiratory syncytial and parainfluenza type 3 viruses in tissue culture and in cotton rats. Antiviral Res. (1990) 14:215–25. doi: 10.1016/0166-3542(90)90003-p
100. Bray M, Driscoll J, Huggins JW. Treatment of lethal ebola virus infection in mice with a single dose of an s-adenosyl-L-homocysteine hydrolase inhibitor. Antiviral Res. (2000) 45:135–47. doi: 10.1016/s0166-3542(00)00066-8
101. Mayers DL, Mikovits JA, Joshi B, Hewlett IK, Estrada JS, Wolfe AD, et al. Anti-human immunodeficiency virus 1 (HIV-1) activities of 3-deazaadenosine analogs: increased potency against 3'-azido-3'-deoxythymidine-resistant hIV-1 strains. Proc Natl Acad Sci USA. (1995) 92:215–9. doi: 10.1073/pnas.92.1.215
102. de Clercq E, Montgomery JA. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antiviral Res. (1983) 3:17–24. doi: 10.1016/0166-3542(83)90011-6
103. Fleming AM, Nguyen NLB, Burrows CJ. Colocalization of m6A and g-Quadruplex-Forming sequences in viral rNA (HIV, zika, hepatitis b, and sV40) suggests topological control of adenosine n6-Methylation. ACS Central Science. (2019) 5:218–28. doi: 10.1021/acscentsci.8b00963
104. Wang SR, Min YQ, Wang JQ, Liu CX, Fu BS, Wu F, et al. A highly conserved g-rich consensus sequence in hepatitis c virus core gene represents a new anti-hepatitis c target. Sci Adv. (2016) 2:e1501535. doi: 10.1126/sciadv.1501535
105. Jaubert C, Bedrat A, Bartolucci L, Di Primo C, Ventura M, Mergny JL, et al. RNA synthesis is modulated by g-quadruplex formation in hepatitis c virus negative rNA strand. Sci Rep. (2018) 8:8120. doi: 10.1038/s41598-018-26582-3
106. Saranathan N, Vivekanandan P. G-Quadruplexes: more than just a kink in microbial genomes. Trends Microbiol. (2019) 27:148–63. doi: 10.1016/j.tim.2018.08.011
107. Cammas A, Millevoi S. RNA g-quadruplexes: emerging mechanisms in disease. Nucleic Acids Res. (2017) 45:1584–95. doi: 10.1093/nar/gkw1280
108. Wu R, Liu Y, Yao Y, Zhao Y, Bi Z, Jiang Q, et al. FTO regulates adipogenesis by controlling cell cycle progression via m(6)A-YTHDF2 dependent mechanism. Biochim Biophys Acta Mol Cell Biol Lipids. (2018) 1863:1323–30. doi: 10.1016/j.bbalip.2018.08.008
109. Wang X, Zhu L, Chen J, Wang Y. mRNA m(6)A methylation downregulates adipogenesis in porcine adipocytes. Biochem Biophys Res Commun. (2015) 459:201–7. doi: 10.1016/j.bbrc.2015.02.048
110. Chen X, Luo Y, Jia G, Liu G, Zhao H, Huang Z. FTO promotes adipogenesis through inhibition of the wnt/beta-catenin signaling pathway in porcine intramuscular preadipocytes. Anim Biotechnol. (2017) 28:268–74. doi: 10.1080/10495398.2016.1273835
111. Merkestein M, Laber S, McMurray F, Andrew D, Sachse G, Sanderson J, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun. (2015) 6:6792. doi: 10.1038/ncomms7792
112. Guo J, Ren W, Li A, Ding Y, Guo W, Su D, et al. Fat mass and obesity-associated gene enhances oxidative stress and lipogenesis in nonalcoholic fatty liver disease. Dig Dis Sci. (2013) 58:1004–9. doi: 10.1007/s10620-012-2516-6
113. White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. (2012) 10:1342–59 e2. doi: 10.1016/j.cgh.2012.10.001
114. Heng J, Wu Z, Tian M, Chen J, Song H, Chen F, et al. Excessive bCAA regulates fat metabolism partially through the modification of m(6)A rNA methylation in weanling piglets. Nutr Metab (Lond). (2020) 17:10. doi: 10.1186/s12986-019-0424-x
115. Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. (2003) 124:1488–99. doi: 10.1016/s0016-5085(03)00276-2
116. Zhang L, Qi Y, Z AL, Liu S, Zhang Z, Zhou L. Betaine increases mitochondrial content and improves hepatic lipid metabolism. Food Funct. (2019) 10:216–23. doi: 10.1039/c8fo02004c
117. Chen J, Zhou X, Wu W, Wang X, Wang Y. FTO-dependent function of n6-methyladenosine is involved in the hepatoprotective effects of betaine on adolescent mice. J Physiol Biochem. (2015) 71:405–13. doi: 10.1007/s13105-015-0420-1
118. Zhou X, Chen J, Chen J, Wu W, Wang X, Wang Y. The beneficial effects of betaine on dysfunctional adipose tissue and n6-methyladenosine mRNA methylation requires the aMP-activated protein kinase alpha1 subunit. J Nutr Biochem. (2015) 26:1678–84. doi: 10.1016/j.jnutbio.2015.08.014
119. Lu N, Li X, Yu J, Li Y, Wang C, Zhang L, et al. Curcumin attenuates lipopolysaccharide-Induced hepatic lipid metabolism disorder by modification of m(6) a RNA methylation in piglets. Lipids. (2018) 53:53–63. doi: 10.1002/lipd.12023
120. Li S, Wang X, Zhang J, Li J, Liu X, Ma Y, et al. Exenatide ameliorates hepatic steatosis and attenuates fat mass and fTO gene expression through pI3K signaling pathway in nonalcoholic fatty liver disease. Braz J Med Biol Res. (2018) 51:e7299. doi: 10.1590/1414-431x20187299
Keywords: m6A methylation, hepatocellular carcinoma, molecular mechanism, cancer etiology, viral hepatitis, non-alcohol fatty liver disease
Citation: Lu J, Qian J, Yin S, Zhou L, Zheng S and Zhang W (2020) Mechanisms of RNA N6-Methyladenosine in Hepatocellular Carcinoma: From the Perspectives of Etiology. Front. Oncol. 10:1105. doi: 10.3389/fonc.2020.01105
Received: 23 February 2020; Accepted: 02 June 2020;
Published: 07 July 2020.
Edited by:
Hailong Pei, Soochow University, ChinaReviewed by:
Dali Han, Beijing Institute of Genomics (CAS), ChinaCopyright © 2020 Lu, Qian, Yin, Zhou, Zheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shusen Zheng, c2h1c2VuemhlbmdAemp1LmVkdS5jbg==; Wu Zhang, emhhbmd3dTUxNkB6anUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.