 Umesh Prasad Yadav1
Umesh Prasad Yadav1 Tashvinder Singh1
Tashvinder Singh1 Pramit Kumar2
Pramit Kumar2 Praveen Sharma1Harsimrat Kaur1,3Sadhana Sharma2
Praveen Sharma1Harsimrat Kaur1,3Sadhana Sharma2 Sandeep Singh1*
Sandeep Singh1* Santosh Kumar2*Kapil Mehta4
Santosh Kumar2*Kapil Mehta4- 1Laboratory of Molecular Medicine, Department of Human Genetics and Molecular Medicine, Central University of Punjab, Bathinda, India
- 2Department of Biochemistry, All India Institute of Medical Sciences, Patna, India
- 3Desh Bhagat Dental College, Mandi Gobindgarh, India
- 4Department of Experimental Therapeutics, MD Anderson Cancer Centre, The University of Texas, Houston, TX, United States
Cancer stem cells (CSCs) are a small and elusive subpopulation of self-renewing cancer cells with remarkable ability to initiate, propagate, and spread the malignant disease. In addition, they exhibit increased resistance to anticancer therapies, thereby contributing to disease relapse. CSCs are reported to be present in many tumor types such as melanoma, sarcoma, mammary tumors, colon cancer and other solid tumors. These cells from different tumors show unique energetic and metabolic pathways. For example, CSCs from one type of tumor may predominantly use aerobic glycolysis, while from another tumor type may utilize oxidative phosphorylation. Most commonly these cells use fatty acid oxidation and ketone bodies as the main source of energy production. CSCs have a remarkable ability to reprogram their metabolism in order to survive under adverse conditions such as hypoxia, acidosis, and starvation. There is increasing interest to identify molecular targets that can be utilized to kill CSCs and to control their growth. In this review, we discuss how an understanding of the unique metabolism of CSCs from different tumors can offer promising strategies for targeting CSCs and hence to prevent disease relapse and to treat the metastatic disease.
Introduction
Cancer is the result of the accumulation of genetic and epigenetic changes that eventually lead to uncontrolled cell growth and the gain of invasive functions by cancer cells. Cancer cells from different tumors can exhibit different properties ranging from low to high metastatic potential, low to high cellular plasticity, and chemosensitivity to chemoresistance. Cancerous mass is itself extremely heterogeneous in terms of metabolism, proliferating ability, and morphology due to genetic and epigenetic variations in intra-tumor subpopulations (1). Cancer stem cells (CSCs) represent a small subpopulation of cancer cells within these heterogeneous tumors that are aggressive, undifferentiated, with self-renewal ability, sensitivity to ROS molecules, and are known for hyperactive metabolism. CSCs were first identified in AML in 1997 as a rare and phenotypically different subset of tumor cells. These cells are able to divide in immuno-compromised mice and to give rise to leukemic progenitor cells and then to differentiated tumor cells (2). It is now well-known that altered glucose (through aerobic glycolysis known as “Warburg effect”) and lipid metabolism (β-oxidation) is a characteristic feature of CSCs, deciding the fate of their progression and self-renewal. This altered metabolism is now considered an important hallmark of CSCs and targeting cancer metabolism is emerging as a crucial therapy (3–5). Pioneer work by the German physiologist, Otto Warburg, revealed that metabolic processes are exploited to meet increased energy demands for proliferation and survival. The highly proliferative cancerous state usually differs from normal metabolism by using high glucose uptake in the presence of oxygen (through glycolysis) producing biomass and lactate rather than relying on oxidative phosphorylation for energy (3). Cancer cells increase their glucose consumption with an equal rate of increased glucose supply, while glutamine utilization is also increased for macromolecule synthesis. Altered glucose metabolism by cancer cells is critical for their growth, and to respond to the environmental changes (6).
Oncometabolism
Metabolic rewiring is essential to meet the increased energy demands by cancer cells for their survival under stressful conditions and to generate metabolic intermediates to meet their rapid growth demands. These metabolic alterations could be the reason for higher proliferation, aggressive invasiveness, and chemo-resistant tumor cells (7–9). The importance of metabolic reprogramming was highlighted in a study showing that disallowing altered metabolic homeostasis using negative modulator of glucose metabolism, slowed the metabolic growth of triple-negative breast cancer (Negative for estrogen, progesterone, and epidermal growth factor 2 receptor) cells due to lowering of lactate production (9). Normal cells predominantly utilize glucose through glycolysis followed by subsequent metabolism of pyruvate via the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OxPhos) in mitochondria. But highly proliferative cancer cells need to adapt to cellular metabolism to provide regular support for the increased proliferation rate and rapidly generating higher amounts of ATP than non-proliferative cells. The normal proliferating cells such as regenerating hepatocyte or proliferative cells in culture media also utilize glucose anaerobically (10). The metabolic features of the proliferative cell either of normal or transformed differs from non-proliferative cells (11). The normal tissues cells switch their cellular metabolism for proliferation from OxPhos to aerobic glycolysis and once differentiated revert back to OxPhos. Cellular signaling helps in cell proliferation and in maintaining the undifferentiated state of cancer cells as well as in restructuring the metabolism during cancer cell proliferation. This high energy production metabolism is essential to fulfill the energy demands, maintain the increased demand for macromolecules, and tight regulation of the cellular redox status (12). Cancer cells meet their energy demand by metabolizing glucose to lactate via glycolysis in the presence of oxygen rather than mitochondrial OxPhos (13). The Warburg effect i.e., aerobic glycolysis is primarily found in malignant tumor cells for maintenance and survival for cancer cells due to impairment of mitochondrial function in the cancer cells. Studies show that in many cancers, mitochondria are functional and still the cells utilize aerobic glycolysis and show reverse Warburg effect when glycolysis is inhibited (14). The metabolic alterations of a cancer cell depend on various factors such as a change in oncogenes, tumor suppression genes, hypoxic microenvironment, mtDNA mutation, genetic factors, proliferation rate, and others (15). The reason for the decreased mitochondrial OxPhos could be deleted copies of mitochondrial pyruvate carrier (MPC) or its decreased expression. Indeed, forced expression of MPC-1 and MPC-2 increased the pyruvate oxidation (11, 16). Also, the glycolytic rate of ATP production is faster than OxPhos, but the energetic yield of glycolysis of 2 ATP is considerably lower in comparison to OxPhos, which produces 32 ATPs per molecule of glucose (17). For ATP generation, besides glycolysis, other important pathways involved are fatty acid oxidation and amino acid catabolism as shown in Figure 1.
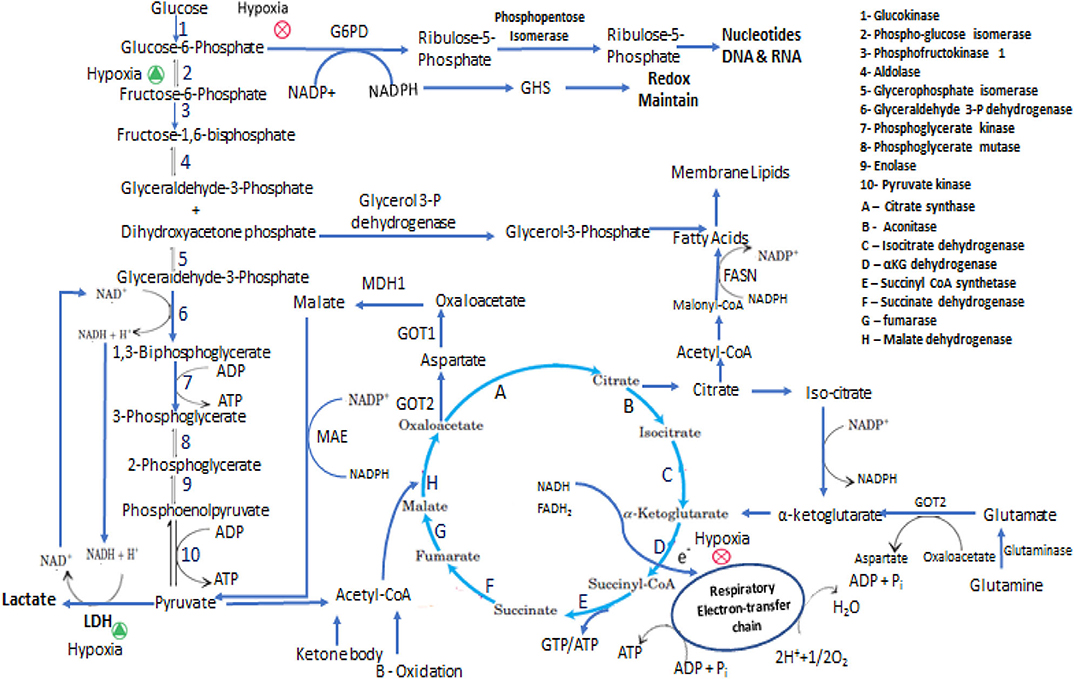
Figure 1. Representation of some of the metabolic adaptation and reprogramming of cancer stem cells. In the interconnected metabolic pathways of glucose, the key junctions are glycolysis and pentose phosphate pathway and central molecules are Glucose-6-phosphate, Pyruvate and Acetyl-CoA. In proliferative state cells metabolized Glucose mainly through PPP and perform less glycolysis and ATP has been produced from TCA cycle via respiratory electron-transfer chain by Acetyl-CoA coming either from pyruvate, Fatty acids and Amino acids oxidation. In Hypoxic condition PPP is inhibited and glucose metabolized through anaerobic glycolytic pathway. In presence of low glucose intermediate metabolites and ATP are produced mainly by glutaminolysis and fatty acids oxidation. CSCs synthesize fatty acids for membrane formation and oxidized this for energy production the equilibrium between anabolism and catabolism of fatty acids is maintain by NADPH and ATP requirement.
Glycolysis, TCA, and OxPhos are an integral component of biosynthesis pathways that are needed to produce metabolic intermediates e.g., acetyl-CoA serves as a substrate for both catabolic and anabolic processes (18). Cells metabolism varies according to cell type i.e., adipocytes undergo increased lipolysis to provide simple lipid for β-oxidation in tumor cells. Adipocytes play a supportive role in tumor progression and metastasis by providing nutrients (lipids in this case) and adipokines that facilitate tumor growth by metabolizing adipocytes (19). Other studies suggest the polymorphism of mitochondria among benign, low malignant and malignant ovarian tumor. For example, platinum-resistant sublines such as SKOW3/CDPP, SKOW3/CBP, A2780/CDDP, and A2780/CBP have lower expression of electron transport chain proteins such as ATP- α, PRDX3, PHB, ETF and ALDH as compared to platinum-sensitive cells SKOW3 and A2780 (20, 21). Lactic acid generated during glycolysis elevates NF-kB mediated IL-8 expression and enhances the tumor progression and angiogenesis (22). Chemokine such as IL-8 mediates tumor progression, angiogenesis, and metastasis in both omental adipocytes and endothelial cells (19, 22). Acidic pH is another important factor regulating enzymatic microenvironment of tumor cells. It is observed that acidic pH results in a significant increase in metastasis of both weak and strong melanoma cell lines A375P and C8161, respectively (23). Lactic acid produced in the cell decreases the pH level activating the MMPs and Cathepsin B, and subsequently increased degradation of collagen IV, enhancing matrix degradation and tumor invasion thus supporting the acidification mediated invasion hypothesis both in-vitro and in-vivo (24). The Warburg effect i.e., aerobic glycolysis is primarily found in malignant tumor cells in the presence of oxygen while some cancerous cells acquire glycolytic metabolic phenotype only because of the hypoxic environment (25). Besides the overwhelming described role of lactate in tumor energy metabolism (hyperactive glycolysis mostly due to hypoxic environment), the role of oxidative phosphorylation is still important for fulfilling energy demands, macromolecule biosynthesis in tumor cells (15, 26, 27). Now it is feasible to validate the inevitable role of different energy metabolic processes and their metabolic intermediates participating in macromolecule biosynthesis, cell survival, and supporting metastatic properties. Targeting CSC metabolism thus represents a promising approach to halt tumor growth and disease relapse by understanding their biology and designing novel therapeutic modalities (4, 28).
Heterogeneity of CSCs
Cancer is not a single disease but a group of diseases in which cells share some common features of abnormal cellular processes with extremely heterogeneous metabolic features in each type of cancer. Even within the same tumor, constituent cells are heterogeneous and metabolic phenotypes vary from one cell to another.Despite predominant aerobic glycolytic metabolism and elevated glycolytic enzymes, proliferating cancer cells have poor prognosis in various types of cancer (29). Some studies have reported both inter- and intratumor metabolic heterogeneity within the same type of tumors (30, 31). According to somatic mutation theory, cancer arises from somatic mutations in cells that undergo clonal selection followed by expansion and ultimately becoming malignant. All somatic mutations are not cancer drivers as most but some are passive. One study reported that the prevalence of the somatic mutation in a kinase gene in different types of tumors (lung, breast, colorectal, gastric, ovarian) does not show the mutation in 73 cases out of 210 cases (32). Somatic mutation analysis of NOTCH1, NOTCH2, NOTCH3, TP53, CDKN2A, and other genes by biopsy in normal eyelid epidermis exposed to ultraviolet light of four donors indicated that these driving mutations help in a positive selection over normal tissue for development of colonies which are non-malignant and non- invasive. These genes are often expressed in squamous cell carcinoma (SCC) and are mutated in other skin cancers also. The clones are genetically heterogeneous and the driver mutations transform cells into malignant phenotype. Although the CDKN2A gene is not associated with positive selection over normal tissue but has a positive impact on progression to advanced-stage disease. Similarly, many somatic mutations found in normal esophageal epithelium tissue could yield heterogeneous colonies that could lead to esophageal SCC in presence of driver mutation. The RNA sequencing of 29 normal tissues out of 6,700 tissue samples revealed multiple somatic variants (33). Studies conducted using deep genome sequencing, histopathological studies or molecular marker analyses revealed a surprising morphological, genetic and clinical heterogeneity of cancer cells that fluctuates within the tumor mass (34–36). There are two theories that explain the reason for heterogeneity; clonal variation and cancer stem cell theory; both vary with tumor subtypes. Clonal variations theory supports the role for genetic, epigenetic, and micro-environment changes that contribute to tumor heterogeneity where tumor cells differ in phenotypic and metabolic processes (37, 38). Whereas, the cancer stem cells theory supports the notion that transformed stem cells (sub populated part of the tumor mass) acquire the properties like high tumorigenic and malignant potential to generate differentiated tumor cell pools (39). The cancer therapeutics should be developed on the basis of tumor type and evaluating CSCs to identify the origin and reason for the problem that will help to find the solution (40). The tumor cells are phenotypically and functionally heterogeneous and this heterogeneity could be intra-tumor or inter-tumor. One such study conducted on 72 patients using FDG-PET analysis has quantified the intratumoral metabolic heterogeneity in primary cervix tumors indicating its relevance with tumor volume rather than tumor stage or histology (41). A similar study conducted on 93 patients with metabolic heterogeneity in cervical cancer using FDG-PET evaluated the significance of heterogeneity as a prognostic marker for predicting tumor relapse. Higher heterogeneity was seen in patients with poor survival and high disease relapse rates than in non-relapsed cancer patients (42). The CSCs contribute to this tumor heterogeneity within a single tumor mass and is responsible for reestablishing the same phenotypic heterogeneity (as in parent tumor) elsewhere upon serial passaging or transplanting in-vivo (43). Improved technology has uncovered additional features of heterogeneity of tumors including variations in cell surface markers, tumor growth, and response to therapy. There is strong evidence supporting that multiple tumor cell subpopulations exist within single cancer e.g., single subcutaneous tumor contained 6 clonal variations from slow-growing to fast-growing melanomas (44), karyotypic heterogeneity in fibrosarcoma (45), and genetic heterogeneity in mouse mammary tumor (46). Similarly, the tumor cell heterogeneity was reported in colon cancer (47) and other transplantable and inter-convertible solid tumors (48, 49). The heterogeneous tumors generally comprised of undifferentiated CSCs, supportive cells, differentiated tumor progenitors, and tumor-infiltrating cells (2, 50). In addition, revertible phenotypic heterogeneity in melanoma cells from the patient's sample without hierarchy (51) and cells contributing to variable tumorigenic frequency in SCID mice from single melanoma tumor has been observed (52). Furthermore, human glioma contains subpopulations of cells with morphological and karyotypic heterogeneity that further gets intensified upon clonal variation and microenvironment. These subpopulations of human glioma cells differed in morphological (fibrous, squamous, and astrocytes), chemosensitivity, and rate of tumor growth (53–55). While most of the studies point out the relationship between variations of phenotype and metabolic process, such is not always the case. Phenotypic heterogeneity of CSCs/clonal variation-derived differentiated tumorsare not necessarilyinvasive. These genes are often expressed in SCC and mutated in other skin cancersalso. The clones are genetically heterogenous and further driver mutation transform the cell into malignant. CDKN2A gene is not associated with positive selection over normal tissue but on advanced stage has positive selection impact. Similar observations found that mosaicism of somatic mutation found in physiological normal esophageal epithelium generating heterogenous colonies and driver mutations leading toesophageal SCC. Isolated tumor cells from metastatic colon cancer show self-renewal as well as tumor-initiating properties and maintain stemness up to several generations, resulting in homogeneity of tumor subpopulation (56). Within a tumor mass, microenvironmental stress-mediated cancer cell's response can result in heterogeneous gene expression; where some cells are more sensitive to these stresses than others. The malignant efficiency of heterogeneous tumor cells differs in terms of tumor initiation, invasiveness, metastasis, and chemo-resistance (57). The variable nature of cancer cells includes some highly active and self-renewal aggressive cells (referred to as CSCs) that are modulated by epigenetic factors such as DNA and histone modifications (DNA methylation, histone trimethylation, and mono acetylation), and chromatin modifiers (58, 59). CSCs represent a small and elusive subpopulation of cancer cells within a tumor mass with stem cell properties and are responsible for enhanced tumorigenesis, EMT-mediated metastasis, relapse, resistance to combinatorial treatment and variant epigenetic expression (60–63). CSCs divide to replenish the tumor cells pool in a symmetrical manner whereas asymmetrical cell division gives rise to non-CSCs that are less tumorigenic, less proliferative, poorly metastatic, and more differentiated. These differentiated non-CSCs tumor cells exhibit non-heterogeneous nature of the tumor where the majority of cells in the primary tumor do not show stemness properties. According to recent evidence, these CSCs and non-CSCs may exhibit inter-convertible plasticity (1, 64). The heterogeneity of CSCs and plasticity in differentiated tumor cells (non-CSCs) supports the inter-convertible feature of CSCs and heterogeneous tumor mass (64)On the basis of phenotypic and genetic heterogeneity, tumor cells are arranged in hierarchical order with the CSCs comes at the top while the most differentiated tumor cells at the bottom (65). Multiple CSCs have also been reported in many other cancers including prostate, lung, liver, pancreas, kidney, bladder, ovary, and brain etc. (66). Isolated pure normal human breast “stem cells” showed same marker expression as of breast tumors and more CSCs were isolated from undifferentiated grade 3 tumors than the differentiated grade 1 tumor. Taken together these observations suggest that CSCs give rise to more heterogeneous tumors than non-CSCs cancer cells (67). In ovarian cancer too, spheroid cells are known to have CSCs like properties with increased ALDH activity and show high tumorigenic and metastatic potential both in-vitro and in-vivo and are more resistant to cisplatin (68). Recent studies documented that most of the solid breast tumor cell lines expressing CD44 (basal-like cells), CD24 (luminal-like cells), PROCR, and ESA are highly tumorigenic and show EMT markers on their surface and play role metastasis, instead of commonly thought CD44+ /CD24–/low and ALDH expression. The studies also advocated that not only CSC marker signature in breast cancer cells is heterogeneous but many subsets of CSC exist that vary from patient to patient and may be related to the individual genetic makeup of the tumor (69).
CSC Markers and Circulating Cancer Stem Cells
CSCs from different cancer types can be isolated based on the presence of cell surface markers. CSCs express stemness markers such as CD133, CD34, CD24, CD44, CD166, and EpCAM, ESA, ALDH1on cell surfaces (66, 70). BCNU resistant subpopulation of Glioma cells exhibits stem cell-like properties, and express CD133, CD117, CD90, CD71, and CD45, also revealing tumorigenicity in vivo upon transfer to SCID mice (71). The markers expressed by these CSCs are required for their stemness, invasiveness, and tumorigenic properties and differ in different subtypes of cancers. For example, CD44+/CD24- in breast CSCs (72), ALDH1+ breast carcinoma (73); CD44 expression in prostate CSCs (74); CD133 (75), ALDH1(76), and CD44 (77) expression in lung CSCs; ALDH1expressing epithelial CSCs (78); while human glioblastoma expressing SSEA-1 (79), EGFR (80), CD44 and Id1 (81). These cancer stem cell markers are essential for their self -renewal, migratory ability, and tumorigenesis such as in the case of the role of CD133 and CXCR4 in pancreatic CSCs (82). The CXCR4 is inevitably responsible for tumor cell invasion to the site of metastasis and tumor cell mobility (pseudopodia formation and actin rearrangement) (83, 84). Brain tumor CSCs expressing CD133 antigen were able to develop tumors in-vivo in SCID mice while the CD133-negative brain tumor cells failed to establish tumors in these mice (85). All these findings suggest that CSCs exhibit particular kinds of stemness markers responsible for self-renewal and tumorigenicity. The therapeutic intervention targeting CSCs markers is still an undiscovered field of study. It not only could delay the cancer progression but may possibly kill CSCs having tumorigenic/stemness properties. There is a special circulating subset of CSCs known as circulating cancer stem cells (CCSCs). The CCSCs detected and analyzed by Raman imaging from four breast cancer subtypes, showed the expression of CD133 marker. Upon culture of CCSCs in breast cancer differential media these cells showed changes in the expression of cell surface markers such as Her2 and EGFR (beside CD133) suggesting their differentiation into Her2+ breast cancer. The CCSCs were endowed with self-renewal ability, tumorigenicity, differentiation, stemness, and metastatic property both in-vitro and in-vivo (86). Isolating CCSCs from a heterogeneous CSCs population from advanced-stage tumors may offer new insight into cancer metastasis and disease relapse. Poor prognosis and relapse with chemo-resistivity were reported in cervical cancer having chromosomal aberration and high genetic heterogeneity that generally developed during cancer progression (87). As in the case of cervical CSCs, traditional anticancer therapies do not work due to overexpression of drug efflux transporter i.e., ABCG2 in undifferentiated tumor subpopulation indicating its role in maintaining stemness (88). Liver metastatic colorectal cancer cells express high levels of various progenitor markers such as EpCAM, CD44, CD24, and CEA-CAM along with CDX1. Cells expressing these markers show a close correlation with stemness, disease progression, and susceptibility to chemotherapy however their continuous exposure to chemotherapy resulted in the development of drug resistance phenotype. These observations suggest that CSCs mediated heterogeneity of tumors is important for metastasis and chemoresistance and that failure to target these cells results in tumor relapse.
EMT and CSCs
The epithelial-to-mesenchymal transition (EMT) defines the transition of some tumor cells from differentiated to an undifferentiated state, linking it with cancer progression and metastasis. EMT-related transcription factors such as Wnt, TGF-β, Notch ligands regulate EMT mediated cancer progression, tumor cell plasticity and increased tumorigenicity (89, 90). Thus, EMT is an important factor regulating heterogeneity, metastatic ability, and plasticity of the tumor cells. Plasticity in breast CSCs can be decoded with interchangeable expression of two marker profiles i.e., CD24-CD44+ (mesenchymal type) and slightly higher tumorigenic potent ALDH+ (epithelial type). Breast cancer cells undergoing EMT show the stemness and tumorigenic properties which are regulated by the tumor microenvironment (91). EMT pathway is multi-step process, which starts with loss of epithelial markers and acquiring the mesenchymal characteristicsenabling them to metastasizetodistantorgansvia activation of proteases, degradation of extracellular matrix and formation of new blood vessels (angiogenesis). EMT is a fundamental first step in successful invasion of cancer cells from primary tumor to distant organs in response to sensing the low nutrient supplies at primary tumor site. In vitro induction of EMT in human mammary epithelial cells is associated with loss of epithelial marker proteins, gain of mesenchymal marker proteins, ability to form spheroids, anchorage independent growth and acquisition of stem cell-like phenotype (92, 93). Similarex-vivo results were reported in samples isolated from human breast carcinoma and normal mouse mammary stem cells with CD44high/CD24low expression (94). Human mammary epithelial cells induced simultaneously by Ras-MAPK and EMT pathway activation exhibited both tumorigenic and stemness properties. Combined ectopic expression of H-Rasv12 and TGF-β1 shortened the time taken to acquire mesenchymal phenotype in CD24+ cells (95). Activation of the Notch pathway resulted in increased NF-kB signaling and upregulated the expression of mesenchymal markers as observed in gemcitabine-resistant pancreatic cancer cells. Conversely, inhibition of the notch pathway or its downstream targets attenuated NF-kB activation, invasion, loss of epithelial markers and gain of mesenchymal markers in gemcitabine-resistant pancreatic cancer cells (96). Upregulated expression of EMT markers in hepatocellular carcinoma (HCC) is considered to be essential for their malignant phenotype as well as for transformation of non-differentiated malignant cell (97). Various transcription activators/repressors and miRNAs either stabilize or antagonize the EMT-induced stem cell traits and undifferentiated state in cancer cells. These miRNAs silence the protein expression of mRNA transcripts by guiding them for degradation while transcriptional repressors and activators downregulate or upregulate the gene transcriptional activity, respectively. ZEB-1 a transcriptional repressor of epithelial marker genes (e.g., E-cadherin) is an inducer of EMT and inhibits the expression of miR-200c and miR-141, the negative regulators of an EMT pathway. Thus, ZEB1 and ZEB2 are responsible for upregulating and downregulating the mesenchymal and epithelial phenotypes, respectively, while miR-200 family has just the opposite effect (98–102). Expression of miR-200 family genes is inhibited when cancer cells acquire the stem cell traits and its expression inhibits mammosphere formation. However, the transient downregulation of miR-200b had no observable effect on CSCs formation. MiR-200b inhibits the Suz12 and its regulated E-cadherin repression (103). Micro RNAs that inhibit stemness (such as miR-200a, miR-200b, miR-200c, miR-141, miR-429, miR-205, miR-203 and miR-183) do so by inhibiting the stem cell regulators such as ZEB1, ZEB2, SIP1, Sox2, Klf4 and hence repress the stemness maintenance and EMT induced CSCs formation (104, 105). EMT regulation is not restricted to the miRNA repression/activation or to the action of activators/repressors such as ZEB1, TGF-β1 and Ras-MAPK pathway; rather it is controlled by many transcriptional factors. Wnt/β-catenin signaling is a key positive regulator of EMT and CSCs formation. Indeed, the high Wnt/β-catenin signaling correlates with the progression of EMT, increased malignant phenotype and acquiring of stem cell traits (106–111). Disrupted Wnt signaling regulates the expression of β-catenin in colorectal cancer cells and is another example of transcriptional activators of oncogenes. Overexpressed β-catenin in cells undergoing EMT at the invasive front was observed in colorectal cancer while such expression was absent in normal colon epithelial cells present far from the invasion site (106). High canonical Wnt/β-catenin signaling in breast cancer demonstrates the action of β-catenin-TCF complex-mediated Snail1 activity and thus regulating EMT in Axin2 dependent pathway (107). PGE2 mediated C-terminal phosphorylation of β-catenin stabilizes it and upregulates Wnt signaling which is the conserved path for hematopoietic stem cell (HSC) formation, self-renewal, and for their sustenance (110). In yet another study, the prevalence of β-catenin expression in T-cell malignancy and non-Hodgkin lymphomas were analyzed and the activated Wnt signaling and highly accumulated β-catenin levels were observed in the nucleus in almost one-third of the tumor samples with some having gained the functional mutation in the β-catenin gene (109). HSCs with β-catenin deletion have difficulty in maintaining the prolonged growth and stemness ability without having any effect on differentiation to their subsequent lineage. In vivo transplantation of cells with β-catenin deletion and expressing BCR-ABL transcripts imparted hind limb paralysis but could not induce leukemic phenotype in mice, while cells expressing wild type β-catenin could successfully induce CML in mice. The loss of β-catenin attenuated the progression of CML and self-renewal ability of CML stem cells, thus β-catenin could be one of the few hallmarks of CSCs survival and tumorigenicity (108). Take together, these observations suggest that Wnt/β-catenin signaling has an important role in the development of tumorigenicity and stemness features of CSCs. Thus, we can conclude that EMT is a necessarily parallel path by multi-grades tumor cells or CSCs for cancer progression, and thus to acquire metastatic and stem cell-like properties.
Metabolic Rewiring of Cancer Stem Cells
Energy metabolism is an important physiological function to support the survival of cells and is critical for cancer progression. Hepatocellular CSCs expressing CD133+ stemness marker showed increased glucose metabolism than CD133- cells while inhibiting glycolytic enzymes in CD133+ CSCs by siRNA reduced the expression of Sox2, Oct4, and Nanog genes important for stemness. Providing extracellular glucose to CD133- cells, increased their stemness property (112). Pancreatic CSCs expressing stemness markers exhibit its dependency on the non-canonical pathway of glutamine metabolism and displayed increased apoptosis and ROS- generation in response to glutamine deprivation from the cell cultures. The pancreatic CSCs were readily sensitized to radiotherapy, accumulated ROS- in response to inhibition of glutamine metabolism both in-vitro and in-vivo (113). In another study, pancreatic CSCs showed increased expression of pluripotent stem cell markers (CD133, SSEA-1, CD44, and CXCR4) in response to activation of Nodal/Activin signaling or expression of its downstream mediators. The Nodal/Activin signaling pathway is important for stemness properties as their inhibition resulted in the sensitization of pancreatic CSCs to gemcitabine and abrogated theirs in vivo tumorigenic potential (114). CSCs exhibit unique metabolic adaptation to physiological and metabolic stresses such as decreased energy source, hypoxia, pH of the microenvironment, etc., thus CSCs differ in metabolic processes when compared to the non-CSC differentiated tumor. Evidence reports the similarity between CSCs and normal stem cells in their ability to differentiate into more mature cells and to self-renewal. CSCs metabolism is predominantly glycolytic but depending on the tumor type can partly be dependent on OxPhos too (115). The preference of glycolytic metabolism in CSCs over OxPhos will not be totally correct as growing evidence suggests that OxPhos has an enormous effect on CSCs survivability (116). In one such study, it was suggested that glioblastoma CSCs rely on Imp2 regulated OxPhos for their energy demand, survivability, in vitro clonogenicity and tumorigenic properties. Depletion of the Imp2 impaired the OxPhos and subsequently resulted in the loss of stem cell properties in glioblastoma CSCs (117). Leukemia CSCs expressing CD34+/CD38- in a dormant state are characterized by low ROS- production and lower glycolysis dependency while Bcl-2 regulated OxPhos is the main source of energy. Inhibition of the Bcl-2 impaired the OxPhos pathway and subsequently eradicated the quiescent leukemia CSCs (118). There is metabolic difference between CSCs and normal stem cells and also between CSCs and differentiated tumor cells, so further studies are warranted to elucidate the role of preferred energy metabolic process over other metabolic processes in a particular type of CSCs (119). It has been reported in glioma cancer stem cell and progenitor cells that CSCs exhibit low glucose consumption, low lactate production, high ATP generation, and high mitochondrial oxidation for their energy demands than their differentiated glioma cell, suggesting OxPhos be the main source for metabolic and energy dependency. The factors that make the difference in metabolism include oxygen consumption rates, extracellular acidification rate, intracellular ATP level, glucose uptake, lactate production, pyruvate kinase M1 (PKM1) levels, and pyruvate kinase M2 (PKM2) expression, cell cycle duration (120). There are convincing reports demonstrating that pancreatic and lung CSCs too depends on OxPhos as the main source of energy production and cell survivability. High mitochondrial membrane potential, low mitochondrial DNA, low ATP and ROS-, lower oxygen consumption over glucose metabolism were reported in lung CSCs when compared to non-lung CSCs. Thus, mitochondrial metabolism is a significant measure of differentiating lung CSCs from non-lung CSCs and may be used to signify other CSCs types (121). Pancreatic CSCs with low metabolic plasticity are highly dependent on OxPhos and are sensitized to metformin drug when compared to primarily glycolytic insensitive non-CSCs (122). However, other investigators have reported that CSCs are more dependent on anaerobic glucose metabolism. The metabolism not only differs in differentiated and CSCs but also varies from their progeny. Breast CSCs predominantly use glycolytic metabolism with a high level of LDH-1 and PKM2 (anaerobic glycolytic enzymes) and low β-oxidation as its main metabolic feature; anaerobic glycolysis inhibitor, 2-deoxyglucose reduced the cell growth and survivability of breast CSCs (123). Human U87 glioblastoma CSCs isolated from xenograft modeled mice exhibited dependency on glycolytic metabolism along with low OxPhos and preference for hypoxia-mediated stemness and chemoresistance. In vitro inhibition of glycolysis induced cytotoxicity and retarded the tumor growth in mice (124). PKM2 plays an important role in anaerobic glycolysis, though is not essential for cancer cell survival or progression and is known to inhibit aerobic glycolysis (125, 126). However, in CSCs, which are more like non-dividing cancer cells, CSCs may require PKM2 for energy related functions as well as maintaining their status (126, 127).
Osteosarcoma CSCs are dependent on high glycolysis and low OxPhos for energy and survival when compared to non-CSCs MG63 cells. LDHA inhibition using sodium oxamate resulted in greater cytotoxicity against CSCs than the MG63 cells while low glucose induced increased fragmented mitochondrial morphology in contrast to network mitochondrial morphology in MG63 cells. These findings support the inability of CSCs mitochondria to become more active under glucose starvation (128). Breast CSCs showed expression of high glycolytic and low OxPhos related enzymes and fewer mitochondria when compared to non-tumorigenic cancer cells indicative of hyper glycolytic metabolism in these CSCs. DCA mediated increase in PDH enzyme (involved in OxPhos) expression in CSCs resulted in increased cytotoxicity in vitro and decreased tumor growth in vivo (129). Hypoxia-induced stemness and glycolytic dependency of breast CSCs is the main reason for chemoresistance in CSCs. However, glycolytic inhibitor along with combinatorial drugs under these conditions makes the CSCs more sensitive to conventional therapies. Leukemia CSCs exhibiting low FAO and high Myc expression with increased concentration of lactate, citrate, and succinate could be positively linked with CD133+ stemness marker as compared to non-cancer stem cells. High lactic acid production is correlated with hyperactive glycolysis in CD133+ cells, through the enzymatic activity of LDHA (112, 130). Radioresistant nasopharyngeal carcinoma CSCs and hypoxia resistant spheroid cells of ovarian cancer exhibiting stemness properties mainly follows the glycolytic pathway and use the byproduct amino acids in biosynthetic pathways rather than their complete oxidation and FA biosynthesis, respectively (68, 131). While the mitochondrial respiration was shut off in nasopharyngeal carcinoma CSCs, the mitochondrial activity and biogenesis were still active in expressing low-ROS and higher TFAM, POLG, and PGC-1a genes (131). The microenvironment of the cells gives clues about active metabolic programming. For example, Paneth cells with increased glycolytic metabolism that supports intestinal stem cells are known to be Lgr+ crypt base columnar cells (CBCs). Paneth cells are functional in the intestinal crypt and produce lactate which is converted into pyruvate to support mitochondrial OxPhos in CBCs having high mitochondrial activity and low redox burden (132). In general, stem cells such as hematopoietic stem cells and human mesenchymal stem cells mainly follow glycolytic metabolism to fulfill their energy demands and shift to other metabolic pathways upon their differentiation. Osteogenic induction of human mesenchymal stem cells is followed by increased mt-DNA copy number, OxPhos, and decrease in cytosolic ROS-, glycolytic metabolism, lactic acid formation while the accumulation of ROS- and increased OxPhos mitigates osteogenic differentiation of human mesenchymal stem cells (133). Glycolytic dependent and hypoxia-resistant HSCs showed low mitochondrial potential, low OxPhos, and high stemness properties and increased expression of HIF-1α both in vitro and in vivo (134). These findings suggest that CSCs can exhibit variable metabolic features depending on their origin and the microenvironment in which they metastasize. Metastatic competent 4T1 breast cancer cells expressed increased capacity for both glycolytic and oxidative metabolism compared to non-metastatic 67NR cells. For example, changes such as PDK-1 support glycolytic metabolism in liver cells but OxPhos metabolism in lung and bone cells (135). Hyperoxia mediated aerobic glycolysis downregulates ROS- in CSCs while in the absence of glucose and hypoxic conditions CSCs metabolism shifts to the mitochondrial respiration. These variations in glucose metabolisms may be influenced by the tumor microenvironment (136).
Glycolysis and Oxphos Correlation in Tumorigenicity
Many metabolic processes e.g., Glycolysis and OxPhos (primary), fatty acid oxidation (secondary) that are regulated by the microenvironment may vary in ATP production in order to fulfill the energy demands and to fuel the anabolic pathways for cancer cells growth (137). The glycolytic product pyruvate undergoes OxPhos under normal aerobic conditions but is converted to lactate by lactate dehydrogenase-A (LDH-A) under an anaerobic state and is transported to the extracellular fluid through MCT (subfamily of cell membrane transporters). These pathways are co-regulated to maintain energy balance. While glycolysis provides instant energy and is the main source for ATP under hypoxic environment, the energetic yield via ATP production is too low compared to ATP production via OxPhos pathway (15). Glycolysis is predominantly activated in tumor cells and aerobic glycolytic cells under hypoxic conditions but OxPhos too does not lose its ability to generate energy (ATP) required for tumor growth. OxPhos is rather suppressed by enhanced glycolysis. Suppression of one metabolic pathway in tumor cells is paved away by another metabolic process to be activated, and it is dependent on the tumor microenvironment. Tumor cells might have functional OxPhos (137–141) despite having multiple mtDNA mutations (142) due to large number of heterogenous genomic copies. The stroma adjacent to tumor cells exhibits high glycolysis which acts as a fuel for active OxPhos in epithelial tumor cells with functional mitochondria (143). Overall, factors such as nutrients, oxygen availability, tumor microenvironment, cell's energy demands, etc., determine the switch between different metabolic pathways.
Metabolism of Glucose in CSCS
As previously stated, glucose is one of the main sources of energy for both CSCs and differentiated tumor cells, providing instant energy but with a lower yield. Glycolysis serves two purposes, one it provides energy readily to the cell, and second, the glycolytic end-product (pyruvate) is involved in the biosynthesis of amino acids (through TCA cycle intermediates), and lipids (precursor for acetyl CoA). Thus, meeting the need of proliferating metastatic cancer cells, which require high energy and precursors for macromolecule biosynthesis in a very short time frame (3, 144). Increased glucose concentration and higher expression of glucose transporters such as GLUT and SGLT, increase glycolysis and is linked with increased viability of tumor cells and CSCs (145–147). There are two classes of glucose transporters which facilitate the transport of glucose by different mechanism.
The GLUT transporters facilitate glucose uptake along the gradient while sodium-dependent SGLT transporters does against the gradient by (148–150). GLUT1 is primarily responsible for glucose uptake in tumor cells e.g., relieving glucose depleted prostate tumor cells from oxidative stress (151, 152). There are also some reports supporting the role of GLUT3 in glucose uptake for cancer progression e.g., in non-small cell lung metastasis and colorectal cancer (153, 154). The dependency of CSCs for high glycolytic and low OxPhos metabolism increases glucose uptake induced by glucose's own concentration. Reduced glucose concentration, genetic knockdown or pharmacological inhibition of GLUT1 attenuated the stemness properties and spheroid formation in pancreatic, ovarian, and glioblastoma CSCs without compromising the cell viability (155). Similarly, the reduced tumorigenic potential was observed when pancreatic CSCs were treated with WZB117 prior to their administration in immuno-compromised mice (155). High glucose concentration resulted in increased GLUT1 and GLUT3 mRNA expression that was associated with increased HIF-1α expression (156). Pentose phosphate pathway (PPP) and glycolysis are inter-connected to each other through glucose-6P, pyruvate, and acetyl CoA. Glucose-6P is derived from glucose by the catalytic action of hexokinase, the enzyme that represents the starting link between glycolysis and PPP (157). Glucose metabolism through PPP pathway is necessary for HIF-1α stabilization in glucose-dependent hypoxic tumor cells and subsequently supports angiogenesis (156). Glioma CSCs undergoing hypoxia show up-regulated mRNA expression for both the glycolytic and PPP genes but protein expression was limited to only glycolytic enzymes such as LDH-A and hexokinase-2, while downregulation of PPP enzymes such as glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase and transketolase like protein was observed. Acute hypoxia although upregulated migration ability but slowed down proliferation activity, whereas acute oxygenation had just the opposite effect i.e., decreased migration. Under both the conditions increase in apoptotic cells was observed. Rapidly dividing cells have an up-regulated PPP pathway whereas under acute hypoxic conditions pathway shifted toward glycolysis (158). The glycolytic byproduct, lactate is a potent metabolite for inducing angiogenesis and invasiveness in macrophages and vascularisation of endothelial cells (159). The MCT1 mediated lactate uptake increases HIF-1α expression in endothelial cells while blocking MCT1 reduces the VEGFR2 and HIF-1α expression in both Genetic knockdown of glycolytic and PPP enzymes showed the same metabolic correlation both in vitro and in vivo (160). Glucose metabolism and lactate production were seen in at half of the contact inhibited mouse neural progenitor cells while low glucose/glutamine stimulated the proliferation in fibroblastic cells. Equal PPP metabolic activity among contact inhibited and proliferating cells led to the flux of ribose phosphate into glycolysis and nucleotide biosynthesis, respectively. Inhibition of PPP pathway, on the other hand, induced more apoptotic effect in contact inhibited cells than fibroblastic cells (161). Thus the profound activity of glycolysis in a hypoxic environment makes it an essential metabolic process for tumor cells residing in the center of the tumor mass with the lower blood supply of oxygen and nutrients.HUVEC and BAEC cells (162). Proliferating and migrating glioblastoma cells differ in their metabolic nature of consuming glucose and its downstream pathways as seen in in vitro and in vivo studies. Migratory cells showed increased and decreased expression of glycolytic enzymes and PPP enzymes, respectively. In contrast, proliferating cells had higher PPP enzymes expression but low glycolysis metabolic expression.
Metabolism of Lipids in CSCS
Fatty acid synthase (FASN) is an intracellular enzyme that is involved in fatty acid synthesis by converting malonyl-CoA and acetyl-CoA into palmitate in three steps using NADPH. Domain I of FASN catalyzes the formation of carbon-carbon bond between malonyl-CoA and acetyl-CoA and subsequent reduction of the elongated fatty acid chain by domain II followed by thioesterase activity of domain III (163). CSCs play an important role in cancer progression and are often reported to have increased activity of FASN (164). These elongated FA chains are structural components of membranes and their de novo synthesis is highly activated in tumor cells compared to normal tissues (165, 166). There are reports supporting increased FASN activity and expression in various human cancers such as breast cancer (167–169), thyroid cancer (170), ovarian cancer (171), prostate cancer (172, 173), oral cancer (174–176), colorectal cancer (177), endometrial metastatic cancer (178) and in mesothelioma (179), renal cancer (180), and retinoblastoma (181, 182). All these studies, inevitably describe FASN as an important metabolic target in cancer therapeutics. Increased FASN expression plays a fundamental role in maintaining the stemness, invasiveness and tumor-forming abilities of Glioma CSCs. Inhibition of FASN in these cells reduced their invasiveness and spheroid forming ability along with the reduced expression of stemness markers such as CD133, FABP7, while increased expression of differentiation marker GFAP (183). NANOG, a transcriptional factor is responsible for promoting the stemness properties and it is preferentially relevant to the mitochondrial metabolism. It reduces the expression of mitochondrial OxPhos genes and elevates the expression of mitochondrial fatty acid oxidation genes in tumor-initiating cells/CSCs of hepatocellular carcinoma.Inhibition of OxPhos gene COX6A2 or COX15 reduced ROS- production whereas activation of the fatty acid oxidation (FAO) gene inhibited glucose utilization via OxPhos and also altered the anaplerotic reactions that help in maintaining the stemness of cells. Induction of the Oxphos gene and inhibition of FAO reduced the spheroid formation ability of cells (184). Pancreatic CSCs showed increased expression of glycolytic and PPP enzymes and immensely induced FASN expression while decreased TCA metabolic enzymes in comparison to pancreatic non-CSCs. Pharmacological inhibition through cerulenin and atorvastatin reduced the cell-viability and dramatically distorted the mesenchymal appearance and spheroid forming ability of pancreatic CSCs (185). Lipid droplets store the excess fatty acids in the cell and act as a reservoir to maintain cholesterol levels, triglyceride levels for cell membranes synthesis, and conserving the energy. Lipid droplets are the key regulators of CSCs metabolism besides their role in the storage and synthesis of inflammatory factors (186, 187). FASN hyperactivation in colorectal cancer cells leads to lipid droplets accumulation, increased FAO and reliability on aerobic glycolysis, suggesting the dependence of tumor cells on stored lipid forms for energy homeostasis (188). All these studies, unequivocally suggest that FASN is an important metabolic target in cancer therapeutics. Stearoyl-CoA-desaturase-1(SCD1) is an enzyme involved in fatty acid synthesis by converting saturated fatty acids into mono-unsaturated FA which serves as a substrate for other lipids and has conserved activity in brain and pancreas (189). SCD1 expression is elevated in CSCs and serves as a marker for poor prognosis of lung adenocarcinoma along with other stemness markers. Pharmacological inhibition of SCD1 resulted in increased susceptibility of lung CSCs to cisplatin-induced apoptosis in (190). Besides aerobic glycolysis and TCA cycle, the role of FAO in satisfying energy demands, chemo- and immune-resistance, stemness, and cancer progression could be demonstrated by analyzing the lipolytic phenotype of CSCs that ispredominantly dictated by the tumor microenvironment (191). Gonadal adipose tissue enriched in pro-inflammatory leukemic CSCs undergoes lipolysis to serve as repository of lipids needed by the leukemic CSCs to maintain stemness and survival. Leukemic-CSCs with increased FAO and upregulated CD36 (FA transporter) expression exhibit chemoresistance while in vivo and ex vivo studies with CD36 knockout cells showed decreased tumor size when injected in mice. These findings suggest the importance of CD36 along with FAO for phenotypic and metabolic distinction in comparison to non-leukemic CSCs (192). FAO has been reported to be a crucial metabolic pathway for breast CSCs as pharmacological inhibition of FAO, reduced the cell viability, ATP levels, and tumor-forming ability in breast CSCs. While JAK-STAT is the key signal transduction pathway responsible for FAO-mediated chemoresistance and tumorigenicity as inhibition of STAT3 reduced the β-oxidation in these CSCs (193). CSCs display high levels of carnitine palmitoyltransferase (CPT) which facilitates the transport of fatty acid from the cytosol to mitochondria for their oxidation (194). In CSCs, CPT1 serves as an anti-apoptotic molecule by interacting with BH3 family of proteins and acts as a cell survival factor (194, 195). This is interesting how CSCs simultaneously complete synthesis and oxidation of fatty acids. During energy depleted state, NADPH generation by the PPP is impaired and this results in decreased FA synthesis. Under these conditions mitochondrial generation of NADPH is important. FAO provides acetyl-CoA which enters into the TCA cycle to form citrate and malate the substrates for NADPH producing enzymes isocitrate dehydrogenase and malic, respectively. AMPK mediated increased β-oxidation and decreased fatty acid synthesis during energy stress maintains the NADPH levels in tumor cells, thus supporting their survivability (196). Cancer cells optimize their requirements for growth and proliferation by regulating the lipid anabolic and catabolic switch. NADPH and ATP demand play an important role to equilibrate the fatty acid synthesis and oxidation in cancer cells.
Mitochondria and Stemness in CSCS
Mitochondrion is the cell organelle involved in the generation of ATP. In addition, they are also involved in many cellular processes such as maintaining the redox state, generation of ROS-, maintaining cytosolic Ca++ level, helping the biosynthesis, and inducing apoptotic death of the cell. Multiple reports have suggested that, despite enhanced glycolysis, cancer cells can produce a significant fraction of their ATP via mitochondrial respiration (197–199). Metabolic plasticity is a characteristic of cancer cells and anaerobic glycolysis performed by cancer cells is not only due to mitochondrial dysfunctions. Mitochondria are necessary for the development and progression of cancer as removal of cancer cell mitochondria diminished cancer growth rate and tumorigenicity. When mtDNA is depleted from tumor cells (ρ° cells) they show reduced growth rate, poor colony-forming ability and considerably reduced tumor growth in mice (200–202). These observations suggest that functional mitochondria are required for the successful survival of cancer cells. Mitochondrial DNA mutations have been reported in many types of cancers such as renal adenocarcinoma, colon cancer cell, head, and neck cancer, breast cancer, prostate and bladder cancer, ovarian cancer, thyroid cancer and neuroblastoma (201, 203–207). MtDNA mutations are a risk factor in some cancer cell populations and are a positive selection in tumorigenesis. Mitochondrial mutations and reduced mtDNA cause stress in mitochondria and thus mitochondria reprogram itself. Studies in breast cancer cell line MCF-7 and normal mammary epithelial cells MCF-10A revealed that mtDNA mutation or reduced mtDNA induces the EMT and CSC phenotype resulting in increased migration and colonization at distant places. This contention was further supported by treatment of MCF-7 and MCF10A cells with ethidium bromide (50 ng/ml) up to 5 passages that resulted in an increased number and increased life span of mammospheres formed by reduced mtDNA.Moreover, flow cytometry analysis showed that cell surface stem cell markers (high CD44+ and low CD24−) are abundantly expressed in reduced mtDNA cells than normal and reverted cancer cells (201). Mutation or knockdown of mitochondrial transcription factor TFAM which is encoded by the nucleus and acts as the main transcription factor for mtDNA and also helps in mtDNA replication and packaging mitigated the tumorigenic potential of cancer cells (208). The mitochondrial morphology, its distribution, and mtDNA status in CSC vary when compared to the differentiated cancer cells or their normal counterparts. Other studies performed on lung CSCs A549 having high expression of CD34, CD133, c-kit, Twist1, Sox2, Oct4, NANOG, and Bmi show reduced mtDNA and higher Δψm in comparison to non-lung CSC. CSCs have high Δψm which suggest that the mitochondria play a role in cell differentiation, tumorigenesis and maintaining the stemness of cell (121). Mutations in oncogenes and mtDNA initiate the formation of cancer/CSC by altering the wave of transcription, which is overblown by the hypoxia and microenvironments, and the mitochondrial stress results in transcription to reprogram the cancer cell metabolism. Decreased mtDNA and mutation coordinates with a nucleus and reprograms the metabolism by sending mitochondrial retrograde signaling (208). Recent research supported that there might be a strong interplay between mitochondria and its role in stemness as mitochondria being an important player in providing energy for the maintenance of stemness. Overall it is necessary to explore the mitochondrial physiology in CSCs and its mechanism in promoting the therapy-resistance (209). While stemness induces suppression of mitochondrial biogenesis and maturity, it activates gene expression of glycolytic enzymes with increased substrate consumption and lactic acid production (210). It has been reported that CSCs from both ovarian adenocarcinoma and cervical SCC express stemness marker, low-ROS- levels, low lactate production, high amino acid intermediates of TCA when grown into spheroids (3D-culture) while opposite effects were seen in adherent tumor cells (2D-culture). These observations suggest that TCA and overall mitochondrial respiration guides and maintains the stemness properties of CSCs and their inhibition is activated upon differentiation, mediating the shifting of CSCs from OxPhos to glycolytic metabolism (211). Mitochondrial respiration and FAO are inter-linked where excess acetyl-CoA is used in TCA cycle. Ketones may too increase mitochondrial respiration and suppress ROS- production by its conversion into acetyl-CoA (212). The reduced ROS- level is favorable to CSCs survival under stress conditions and thus the utilization of ketone bodies may relieve CSCs from oxidative stress. While the ketone metabolism is active in the absence of nutrients, tumor cells and CSCs constitutively express it (213–217). Mitochondrial metabolic activity is also related to cell differentiation, as early passages of an adult primate stromal cell line have a higher OCR and a low ATP/mitochondrial DNA content compared with long-term cultured cells (218) but CD34+ hematopoietic stem cells have low mitochondrial OCR and mitochondrial mass (219). The HSC mitochondria play important roles in maintaining stemness and differentiation. However, whether the roles of CSC mitochondria are similar to HSC mitochondria or cancer cells, in general, is unknown. Two hypotheses on the origin of CSCs, both of which contribute to acute myeloid leukemia (220), have been proposed. One hypothesis supports that CSCs are derived from normal stem cells residing in various organs. Genetic mutations and epigenetic changes, which are crucial for the initiation and progression of tumor growth, accumulate in long-lived stem cells, and the transformation of stem cells into CSCs initiates the carcinogenesis. CSCs may also have a greater differentiation potential than normal stem cells. Another hypothesis assumes the existence of embryonic stem cell-like cells that transform into CSCs when they are exposed to damaging environmental factors. Additional differentiation and mutations of these cells may also contribute to the development of CSCs (221). As reported, ovarian CSCs show higher mitochondrial ROS- production and ΔΨm than non-CSCs. In addition, targeting mitochondrial biogenetics induced caspase-independent cell death in ovarian CSCs (222). In glioma CSCs, a higher mitochondrial reserve capacity was measured as compared to the differentiated cells (120). Glioblastoma CSCs also depend on OXPHOS for their energy production and survival (117). Besides, breast CSCs have higher ATP content compared to their differentiated progeny (223). Therefore, it is reasonable to believe that CSCs mitochondria show different roles and features depending on the cancer type and CSCs mitochondria differ from those of non-CSCs. Importantly, little information is available on the mitochondrial features related to energy metabolism and the ROS-/antioxidant enzyme system of CSCs in colon, stomach, liver, bone, and prostate cancer. Therefore, defining these features will be essential for developing a mitochondria-targeted therapeutic approach to facilitate the death of CSCs, and therefore, to reduce the risk of disease relapse and progression to refractory cancer.
Mitochondrial studies using whole-cell approaches make it difficult to distinguish mitochondria-specific effects from those contributed by the nucleus. This gap can be filled by using trans mitochondrial cybrid models in order to investigate the mitochondria-regulated energy and cancer pathways (224–226). Cybrid models using moderately metastatic triple-negative breast cancer cell line (SUM159) as a background control for nuclei and mitochondria and comparing them with the benign breast cells (MCF-10A or A1N4), and highly metastatic (MDA-MB-231) breast cancer cells can offer a good working model for this purpose. Indeed, these cybrids show the tumor-like properties both in vitro and in vivo according to their mitochondrial origin. Mitochondria obtained from benign cells almost completely abolished the tumorigenic properties of SUM159 cells when tested in vitro and in vivo (226). Proteomic and mass spectrometry analysis revealed several proteins related to mitochondrial. Fatty acid oxidation is also up regulated in cybrids with mitochondria derived from MDA 231 metastatic breast cancer cells. Knockdown of enzyme essential for fatty acid oxidation carnitine palmitoyltransferase-1(CPT1) or carnitine palmitoyltransferase-2 (CPT2) by shRNA significantly inhibited the migration potential and wound healing potential of these cells (226). Breast cancer patient dataset (n = 1,302) with long-term clinical follow-up showed that high CPT1A mRNA expression in tumors promotes distant metastasis (227). Altogether, these results show the role of mitochondrial energy reprogramming in fatty acid oxidation in CSCs and its significance in regulating the driving the protein of a major cancer pathway via its posttranslational modification.
Metabolism of Glutamine in CSCs
Glutamine is a non-essential amino acid that plays a key role in energy and metabolic homeostasis during CSCs proliferation. It mediates these effects by regulating the consumption and uptake of other amino acids, hence maintaining mitochondrial redox potential and NADPH levels. Besides maintaining energy homeostasis, glutamine is the precursor of TCA cycle intermediates, nucleotides, proteins, and biosynthesis of other amino acids. CSCs from small cell lung carcinoma utilize glutamine actively in anaplerotic reaction in order to produce ATP for substrate-level phosphorylation. Complete inhibition of OxPhos and glycolysis by oligomycin and 2-DG in CSCs and non-CSCs caused more substrate-level phosphorylation in uPAR+ CSCs. Under respiratory distress conditions, substrate-level phosphorylation provides the necessary GTP/ATP to CSCs and this is more efficient under hypoxic conditions (228, 229). In vivo and ex vivo studies in OxPhos defective mtDNA mutated cybrid A6MT cells and wild type cells that have no mtDNA mutations, showed that Glutamine accelerates the proliferation of A6MT cell and its uptake was higher in mtDNA mutated cells than in wild type cells. Glutamine undergoes oxidative or reductive pathways depending on the severity of OxPhos defect. Isotopic C13-labeled glutamine supplemented in culture media for tracking the glutamine-glucose-αKG flux and metabolic fate of glutamine in A6MT cells suggested that glutamine enters the TCA cycle in the form αKG in a clockwise direction and provides GTP/ATP for substrate-level phosphorylation following its conversion into succinate as mentioned in Figure 1. Succinate is then converted into aspartate that migrates into the cytosol and subsequently converted into alanine and lactate. Published work from other laboratories have revealed similar results and suggest that Glutamine enters the TCA cycle and actively participates in anaplerotic reaction; the amount of lactate formed is less compared to that formed by glucose (230–232). These events are critical in order to maintain the pace of TCA cycle and to re-oxidize glycolytic NADH to generate NADPH which is a critical cofactor needed in reductive pathways such as during lipid synthesis as notified in Figure 1. Thus, glutamine has an important role in metabolic rewiring in OxPhos impaired cells to provide energy and other synthetics intermediates (233). Many researchers are attempting to target the glutamine-dependent self-renewal ability of CSCs as a novel therapeutic approach (234–236). For example, attempts to inhibit glutamine transamination in glioblastoma, blocking the increased glutamine regulated signaling pathways (mTOR pathway), or by targeting c-myc regulated glutamine uptake were shown to induce significant cytotoxicity to tumor cells (237, 238). Deamidation product of glutamine, along with cysteine and glycine, is an important component of glutathione that is needed to maintain the redox balance and NADP+/NADPH ratio (239). The dependency of cancer cells on glutamine metabolism for nitrogen and redox balance is correlated with their tumorigenic potential and tumor survival (240). mTOR is an important signaling pathway for glutamine metabolism and is differentially regulated by glutamine concentration of tumor cells. Colorectal CSCs grown in glutamine lacking media were more sensitive to metformin drug (relevant to mTOR pathway) while being resistant when media contained glutamine. Combinatorial treatment with metformin and glutaminase inhibitor, induced higher cytotoxic effect against colorectal CSCs than treatment with each drug individually. The colorectal CSCs had higher expression of glutamine transporter protein (ASCT2) than the non-CSCs cancer cells. Accordingly, the ASCT2 knockdown dramatically reduced the number of CD133+/CD44+ CSCs (241). In pancreatic CSCs, glutamine deficiency significantly decreased the stemness, self-renewal, and increased the ROS- production resulting in increased the apoptotic death of these cells (242). The uptake of extracellular proteins in pancreatic adenocarcinoma and bladder carcinoma cells, expressing oncogenic Ras, this mediated through macro-pinocytosis and to make glutamine via lysosomal degradation of these proteins (243). The α-ketoglutarate, an intermediate of TCA cycle, when supplemented to cell cultures as an alternate to Glutamine, may alter the cellular requirements for glutamine metabolism in order to synthesize nucleotides. CD34+/CD38– hematopoietic stem cells expressing high ASCT2 levels and glutamine metabolism show commitment to erythroid niche whereas abrogating glutamine metabolism leads to myeloid differentiation in-vitro. On the other hand, in-vivo antagonistic metabolism of glutamine and glucose regulates the differentiation of HSC between myeloid and erythroid (244)Glutamine is an important component in synthesis of glutathione (GSH), a tripeptide that serves as an intracellular antioxidant scavenger of ROS- and also is involved in DNA repair, activation of transcription factors, cell cycle regulation, and calcium homeostasis (245). Glutamine metabolizing enzymes such as glutamine synthetase and glutamate-oxaloacetate transaminase (GOT1 and GOT2) are highly expressed in CSCs (113). Glutamine metabolism varies with cancer types andgenerally proceeds through two pathways; acting as a mediator of anaplerotic flux through TCA cycle intermediates and a precursor for nucleotide synthesis (246). Incorporation of glucose carbon in glutamine and TCA cycle intermediates, non-dependency of tumor cells on glutamine was seen in non-small-cell lung carcinoma cells in-vivo. This phenomenon wasregulated by changes in the microenvironment and indicated glucose as a carbon source. These observations suggested glucose mediated glutamine synthesis and TCA cycle metabolite replenishment with the help of pyruvate carboxylase rather than dependency on glutamine anaplerosis (247). Glioblastoma expressing aerobic glycolysis and active TCA cycle predominantly depended on glucose-mediated lipogenesis and on anaplerotic glutamine metabolism to regenerate TCA cycle intermediates for NADPH production. Generally, low reliability of TCA mediated metabolites and high preference for glutamine is seen in proliferating cells than in contact inhibited fibroblasts. These findings support the contention that contact inhibition may induce mild anapleroticresponse in favor to glutamine metabolism and strong anapleroticresponse in favorto pyruvate-oxaloacetate flux (161). Myc mediated glutamine catabolism to α-ketoglutarate, replenishes the reduced TCA cycle metabolite pool in response to hypoxic stress and low pyruvate availabilityfor TCA cycle. Generated α-ketoglutarate undergoes reverse TCA cycle to form citrate, which is exported to the cytosol to form oxaloacetate and Acetyl-CoA for lipid synthesis and redox homeostasis of cell (248). Glutamine metabolism is undoubtedly a major metabolic pathway in CSCs and could be a promising therapeutic target toblock its functions and aidingthe replenishment of metabolites for energy and redox homeostasis.
Conclusion: CSCS Eradication by Targeting its Metabolism
It is important to understand the exact nature of factors in heterogeneous tumor mass that fuel the tumorigenic growth, drug refractoriness, and metastasis of less differentiated tumor cells to distant organs. It is also clear that CSCs play a major role in replenishing the tumor pool and as a source of differentiated tumor cells. The immortal nature of CSCs might be the reason why the tumors relapse even after most of the tumor mass is removed or eradicated (249). Oxphos and glycolysis remains the primary energy generation mechanisms for CSCs while metabolism of ktone bodies and fatty acids also contributed significantly. This may vary for each CSC type depending on the primary tumor from which these CSCs have developed. Yuan et al. has shown that multiple mutations in mtDNA may render compromised OxPhos function (142) thus pushing the cell toward glycylysis. On the other hand, several studies have shown that glycolytic tumors can not metabolize fatty acid and ketone bodies (230, 250, 251) for energy production. Individual mitochodnria are genetically heterogenous due to large copy number of the genome, each copy may have different mutations. This may overall produce almost all mitochondrial functional proteins, albeit in very low levels, thus enabling OxPhos to a smaller extent, thus explaining the partial contribution of energy by multiple mechanisms.
Metastatic and self-renewal property of CSCs (while being more quiescent) is tightly regulated by the mitochondrial respiration and glycolysis, while the ATP generation and fulfilling energy demandsare secondary events. While Myc suppression induced the sensitivity of CSCs to metformin, overexpression of Myc has just opposite effects making cancer cells behave like chemo-resistant pancreatic adenocarcinoma CSCs. Drugs targeting the OxPhos could be the primary aspect of therapeutic intervention in pancreatic CSCs as some drugs exclusively target the CSCs metabolism while showing no effect on non-CSCs (122). A rationale reasoning for metabolic immuned CSCs is still unknown where CSCs evade the immune response and may confuse the immune cells to distinguish itself from normal stem cells. Genomic analyses, mRNA profiling and mutational analysis of breast cancer (252), ovarian cancer (253), lung cancer (254), glioblastoma cancer (255), prostate cancer (256), gastric cancer (257), B-cell lymphoma (258), acute lymphoid leukemia (259), metastatic cancers (260), and melanomas (261) has been performed using sophisticated techniques such as MSK-IMPACT, next-generation sequencing, and whole-genome sequencing. These studies have provided a detailed and perspective view of the diverse genetic and epigenetic nature of different mutated genes in different tumor types that may be involved in the regulation of metabolic processes and proliferating efficiency based on their origin and microenvironment in which they reside. Future efforts to elucidate the mechanisms responsible for genetic, epigenetic and micro environment-induced changes in CSCs that regulate tumor progression and chemoresistance might offer therapeutic opportunities for successful intervention to block the progression of cancer to untreatable disease. Alternative approaches should be taken, while focusing on similarities between different CSCs. For example, common metabolic features that dictate the tumorigenic potential and stemness of CSCs should be targeted. Targeting of metabolic wiring should be investigatedduring two transitional states; (a) from normal stem cells to cancer stem cells, and (b) from cancer stem cells todifferentiated tumor cells, rather than using chemo- and radiation therapiestokillthe whole tumor mass that results in major side effects. While we have no knowledge on the metabolic adaptations that take placeduring normal stem cell to CSCs transition, only a handful studies have been done to understand the transition of CSCs to differentiated tumor cells.
Author Contributions
KM, SK, and SSi organized topics and contributed in writing the manuscript. UY, TS, PS, and PK collected the literature and contributing in write up. UY, TS, PS, SSh, and HK helped in revising the manuscript. UY and PS designed the figure. All authors contributed to the article and approved the submitted version.
Funding
UY and TS received Senior and Junior Research Fellowship, respectively, from the CSIR. PS and SSi thank the financial support from DST SERB EMR (SR/SO/AS-31/2014). SK thanks the DST SERB for Ramanujan Fellowship Award and for funding the project (DST/SERB SB/S2/RJN-103/2015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Central University of Punjab and AIIMS Patna for providing all the facilities.
Abbreviations
AML, Acute myeloid leukemia; CAM, cell adhesion molecule; CBC, Crypt base columnar; CD, Cluster of differentiation; CPT, Carnitine palmitoyl transferase; CSC, Cancer Stem Cell; CCSC, Circulating cancer stem cells; DCA, Dichloroacetic acid; EMT, Epithelial mesenchymal transition; ESCC, Esophageal squamous cell carcinoma; FAO, Fatty acid oxidation; FANS, Fatty acid synthase; HSC, Hematopoietic stem cells; MPC, Mitochondria pyruvate carrier; OxPhos, Oxidative phosphorylation; PDH, Pyruvate dehydrogenase; PK, Pyruvate kinase; PPP, Pentose phosphate pathway; FDG-PET, Fluorodeoxyglucose-Positron emission tomography; SCC, Squamous cell carcinoma; SCD1, Stearoyl-CoA-desaturase-1; SGLT, Sodium-glucose linked transporter.
References
1. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. (2013) 59:168–79. doi: 10.1373/clinchem.2012.184655
2. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
3. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
4. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. (2013) 12:829–46. doi: 10.1038/nrd4145
5. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Sacca M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. (2018) 37:2367–78. doi: 10.1038/s41388-018-0141-3
6. Sauer LA, Stayman JW, Dauchy RT. Amino acid, glucose, and lactic acid utilization in vivo by rat tumors. Cancer Res. (1982) 42:4090–7.
7. Dumas JF, Brisson L, Chevalier S, Mahéo K, Fromont G, Moussata D, et al. Metabolic reprogramming in cancer cells, consequences on pH and tumour progression: integrated therapeutic perspectives with dietary lipids as adjuvant to anticancer treatment. In: Seminars in Cancer Biology. Elsevier (2017). p. 90–110. doi: 10.1016/j.semcancer.2017.03.004
8. Buono R, Longo VD. Starvation, stress resistance, and cancer. Trends Endocrinol. Metab. (2018) 29:271–80. doi: 10.1016/j.tem.2018.01.008
9. Kim H, Dong J, Xu J, Li D, Zheng Z, Ye N, et al. Abstract P1-02-08: reprogramming glucose metabolism and energy production in breast cancer Cells. In: San Antonio Breast Cancer Symposium. San Antonio, TX: AACR (2018). doi: 10.1158/1538-7445.SABCS17-P1-02-08
10. McKeehan WL. Glycolysis, glutaminolysis and cell proliferation. Cell Biol. Int. Rep. (1982) 6:635–50. doi: 10.1016/0309-1651(82)90125-4
11. Formentini L, Martínez-Reyes I, Cuezva JM. The mitochondrial bioenergetic capacity of carcinomas. IUBMB Life. (2010) 62:554–60. doi: 10.1002/iub.352
12. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11. doi: 10.1038/nrclinonc.2016.60
13. Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
14. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. (2010) 9:3506–14. doi: 10.4161/cc.9.17.12731
15. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation. Oncol Lett. (2012) 4:1151–7. doi: 10.3892/ol.2012.928
16. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras G12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. (2012) 22:399–412. doi: 10.1038/cr.2011.145
17. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
18. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
19. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. (2011) 17:1498–1503. doi: 10.1038/nm.2492
20. Ishioka SI, Sagae S, Ito E, Kudo R. Ultrastructural study of benign, low-malignant potential (LMP), and malignant ovarian tumors. Med Electron Microsc. (2004) 37:37–44. doi: 10.1007/s00795-003-0189-0
21. Dai Z, Yin J, He H, Li W, Hou C, Qian X, et al. Mitochondrial comparative proteomics of human ovarian cancer cells and their platinum-resistant sublines. Proteomics. (2010) 10:3789–99. doi: 10.1002/pmic.200900685
22. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. (2011) 71:2550–60. doi: 10.1158/0008-5472.CAN-10-2828
23. Martínez-Zaguilán R, Seftor EA, Seftor RE, Chu Y-W, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis. (1996) 14:176–86. doi: 10.1007/BF00121214
24. Rothberg JM, Bailey KM, Wojtkowiak JW, Ben-Nun Y, Bogyo M, Weber E, et al. Acid-mediated tumor proteolysis: contribution of cysteine cathepsins. Neoplasia. (2013) 15:1125–37. doi: 10.1593/neo.13946
25. Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. (2004) 313:459–65. doi: 10.1016/j.bbrc.2003.11.136
26. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. (2002) 277:30409–12. doi: 10.1074/jbc.R200006200
27. Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. (2015) 3:1. doi: 10.1186/s40170-015-0128-2
28. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. (2013) 4:e532. doi: 10.1038/cddis.2013.60
29. Yu M, Chen S, Hong W, Gu Y, Huang B, Lin Y, et al. Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J Cancer Res Clin Oncol. (2019) 145:967–99. doi: 10.1007/s00432-019-02847-w
30. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell. (2016) 164:681–94. doi: 10.1016/j.cell.2015.12.034
31. Dai Z, Locasale JW. Metabolic pattern formation in the tumor microenvironment. Mol Syst Biol. (2017) 13:915. doi: 10.15252/msb.20167518
32. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. (2007) 446:153–8. doi: 10.1038/nature05610
33. Yizhak K, Agust F, Kim J, Hess JM, Kubler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. (2019) 364:eaaw0726. doi: 10.1126/science.aaw0726
34. Büttner J, Jöhrens K, Klauschen F, Hummel M, Lenze D, Saeger W, et al. Intratumoral morphological heterogeneity can be an indicator of genetic heterogeneity in colorectal cancer. Exp Mol Pathol. (2018) 104:76–81. doi: 10.1016/j.yexmp.2018.01.007
35. Lenz G, Onzi GR, Pereira LC, Breitenbach K, Leonardo BG, Dalsin E. Abstract IA09: The Dynamics of Cancer Cell Heterogeneity. In: Translational Cancer Medicine. São Paulo: AACR (2018). doi: 10.1158/1557-3265.TCM17-IA09
36. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. (2018) 172:373–86 e310. doi: 10.1016/j.cell.2017.11.010
37. Nowell PC. The clonal evolution of tumor cell populations. Science. (1976) 194:23–8. doi: 10.1126/science.959840
38. Greaves M, Maley CC. Clonal evolution in cancer. Nature. (2012) 481:306–13. doi: 10.1038/nature10762
39. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414:105–11. doi: 10.1038/35102167
40. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. (2009) 138:822–9. doi: 10.1016/j.cell.2009.08.017
41. Kidd EA, Grigsby PW. Intratumoral metabolic heterogeneity of cervical cancer. Clin Cancer Res. (2008) 14:5236–41. doi: 10.1158/1078-0432.CCR-07-5252
42. Chong GO, Lee WK, Jeong SY, Park SH, Lee YH, Lee SW, et al. Prognostic value of intratumoral metabolic heterogeneity on F-18 fluorodeoxyglucose positron emission tomography/computed tomography in locally advanced cervical cancer patients treated with concurrent chemoradiotherapy. Oncotarget. (2017) 8:90402–12. doi: 10.18632/oncotarget.18769
43. Konrad CV, Murali R, Varghese BA, Nair RJC. The role of cancer stem cells in tumor heterogeneity and resistance to therapy. Can J Physiol Pharmacol. (2016) 95:1–15. doi: 10.1139/cjpp-2016-0079
44. Gray JM, Pierce GB Jr. Relationship between growth rate and differentiation of melanoma in vivo. J Natl Cancer Inst. (1964) 32:1201–11. doi: 10.1093/jnci/32.6.1201
45. Mitelman F, Mark J, Levan G, Levan AJS. Tumor etiology and chromosome pattern. Science. (1972) 176:1340–1. doi: 10.1126/science.176.4041.1340
46. Dexter DL, Kowalski HM, Blazar BA, Fligiel Z, Vogel R, Heppner GH, et al. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. (1978) 38:3174–81.
47. Dexter DL, Spremulli EN, Fligiel Z, Barbosa JA, Vogel R, VanVoorhees A, et al. Heterogeneity of cancer cells from a single human colon carcinoma. Am J Med. (1981) 71:949–56. doi: 10.1016/0002-9343(81)90312-0
48. Klein G, Klein E. Conversion of solid neoplasms into ascites tumors. Ann N Y Acad Sci. (1956) 63:640–61. doi: 10.1111/j.1749-6632.1956.tb50883.x
49. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
50. Meacham CE, Morrison SJJN. Tumour heterogeneity and cancer cell plasticity. Nature. (2013) 501:328–37. doi: 10.1038/nature12624
51. Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. (2010) 18:510–23. doi: 10.1016/j.ccr.2010.10.012
52. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJJN. Efficient tumour formation by single human melanoma cells. Nature. (2008) 456:593–8. doi: 10.1038/nature07567
53. Shapiro JR, Yung W-KA, Shapiro WR. Isolation, karyotype, and clonal growth of heterogeneous subpopulations of human malignant gliomas. Cancer Res. (1981) 41:2349–59.
54. Yung W-KA, Shapiro JR, Shapiro WR. Heterogeneous chemosensitivities of subpopulations of human glioma cells in culture. Cancer Res. (1982) 42:992–8.
55. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. (2017) 16:41. doi: 10.1186/s12943-017-0600-4
56. Odoux C, Fohrer H, Hoppo T, Guzik L, Stolz DB, Lewis DW, et al. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. (2008) 68:6932–41. doi: 10.1158/0008-5472.CAN-07-5779
57. Cajal SRY, Castellvi J, Hümmer S, Peg V, Pelletier J, Sonenberg N. Beyond molecular tumor heterogeneity: protein synthesis takes control. Oncogene. (2018) 37:2490–501. doi: 10.1038/s41388-018-0152-0
58. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. (2013) 153:38–55. doi: 10.1016/j.cell.2013.03.008
59. Deshmukh A, Binju M, Arfuso F, Newsholme P, Dharmarajan A. Role of epigenetic modulation in cancer stem cell fate. Int J Biochem Cell Biol. (2017) 90:9–16. doi: 10.1016/j.biocel.2017.07.003
60. Clarke MF, Hass A, Medicine M. Cancer Stem Cells. In: Reviews in Cell Biology and Molecular Medicine, RA Meyers, editor. (2006). p. 221–41. doi: 10.1002/3527600906.mcb.200300130
61. Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. Semin Cancer Biol. (2012) 22:396–403. doi: 10.1016/j.semcancer.2012.04.001
62. Gruber W, Scheidt T, Aberger F, Huber CG. Understanding cell signaling in cancer stem cells for targeted therapy–can phosphoproteomics help to reveal the secrets? Cell Commun Signal. (2017) 15:12. doi: 10.1186/s12964-017-0166-1
63. Azizi E, Granger J, Harouaka R, Luther TK, Wicha MS. Stem cells in breast development and cancer. In: The Breast, 5th Edn. Philadelphia, PA: Elsevier (2018). p. 308–14 e302. doi: 10.1016/B978-0-323-35955-9.00023-4
64. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. (2012) 22:457–72. doi: 10.1038/cr.2012.13
65. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. (2008) 8:755–68. doi: 10.1038/nrc2499
66. Kaur G, Sharma P, Dogra N, Singh S. Eradicating cancer stem cells: concepts, issues, and challenges. Curr Treat Options Oncol. (2018) 19:20. doi: 10.1007/s11864-018-0533-1
67. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. (2010) 140:62–73. doi: 10.1016/j.cell.2009.12.007
68. Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE. (2014) 9:e84941. doi: 10.1371/journal.pone.0084941
69. Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE. (2009) 4:e8377. doi: 10.1371/journal.pone.0008377
70. Yang M, Liu P, Huang PJTB. Cancer stem cells, metabolism, and therapeutic significance. BMB Rep. (2016) 37:5735–42. doi: 10.1007/s13277-016-4945-x
71. Kang M-K, Kang S-K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. (2007) 16:837–48. doi: 10.1089/scd.2007.0006
72. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
73. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
74. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. (2011) 17:211. doi: 10.1038/nm.2284
75. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. (2008) 15:504–14. doi: 10.1038/sj.cdd.4402283
76. Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. (2010) 70:9937–48. doi: 10.1158/0008-5472.CAN-10-0881
77. Leung EL-H, Fiscus RR, Tung JW, Tin VP-C, Cheng LC, Sihoe AD-L, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE. (2010) 5:e14062. doi: 10.1371/journal.pone.0014062
78. Deng S, Yang X, Lassus H, Liang S, Kaur S, Ye Q, et al. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS ONE. (2010) 5:e10277. doi: 10.1371/journal.pone.0010277
79. Son MJ, Woolard K, Nam D-H, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. (2009) 4:440–52. doi: 10.1016/j.stem.2009.03.003
80. Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi LS, et al. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. (2010) 70:7500–13. doi: 10.1158/0008-5472.CAN-10-2353
81. Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, et al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell. (2010) 18:655–68. doi: 10.1016/j.ccr.2010.10.023
82. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. (2007) 1:313–23. doi: 10.1016/j.stem.2007.06.002
83. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. (2001) 410:50–6. doi: 10.1038/35065016
84. Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol. (2012) 14:171–9. doi: 10.1016/j.semcancer.2003.10.003
85. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. (2004) 432:396–401. doi: 10.1038/nature03128
86. Cho HY, Hossain MK, Lee JH, Han J, Lee HJ, Kim KJ, et al. Selective isolation and noninvasive analysis of circulating cancer stem cells through raman imaging. Biosens Bioelectron. (2018) 102:372–82. doi: 10.1016/j.bios.2017.11.049
87. Cooke S, Temple J, Macarthur S, Zahra M, Tan L, Crawford R, et al. Intra-tumour genetic heterogeneity and poor chemoradiotherapy response in cervical cancer. Br J Cancer. (2011) 104:361–8. doi: 10.1038/sj.bjc.6605971
88. Huang R, Rofstad EK. Cancer stem cells (CSCs), cervical CSCs and targeted therapies. Oncotarget. (2017) 8:35351–67. doi: 10.18632/oncotarget.10169
89. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. (2016) 35:645–54. doi: 10.1007/s10555-016-9648-7
90. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. (2018) 18:128–34. doi: 10.1038/nrc.2017.118
91. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
92. Kumar A, Xu J, Brady S, Gao H, Yu D, Reuben J, Mehta K. Tissue transdlutaminase prometes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS ONE. 5:e13390. doi: 10.1371/journal.pone.0013390
93. Kumar A, Xu J, Sung B, Kumar S, Yu D, Aggarwar B, et al. Evidance that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transtion in mammary epithelial cells. Breast Cancer Res. (2012) 14:R4. doi: 10.1186/bcr3085
94. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
95. Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux AJP, et al. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. (2008) 3:e2888. doi: 10.1371/journal.pone.0002888
96. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. (2009) 69:2400–7. doi: 10.1158/0008-5472.CAN-08-4312
97. Niu R, Zhang L, Xi G, Wei X, Yang Y, Shi Y, et al. Up-regulation of twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. (2007) 26:385–94.
98. Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. (2008) 68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942
99. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. (2008) 9:582–9. doi: 10.1038/embor.2008.74
100. Korpal M, Lee ES, Hu G, Kang YJ. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. (2008) 283:14910–4. doi: 10.1074/jbc.C800074200
101. Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. (2008) 22:894–907. doi: 10.1101/gad.1640608
102. Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. (2009) 28:151–66. doi: 10.1007/s10555-008-9179-y
103. Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl KJM, et al. (2010). Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 39:761–72. doi: 10.1016/j.molcel.2010.08.013
104. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. (2008) 10:593–601. doi: 10.1038/ncb1722
105. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. (2009) 11:1487–95. doi: 10.1038/ncb1998
106. Brabletz T, Jung A, Hermann K, Günther K, Hohenberger W, Kirchner T, et al. Nuclear overexpression of the oncoprotein β-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract. (1998) 194:701–4. doi: 10.1016/S0344-0338(98)80129-5
107. Yook JI, Li X-Y, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt–Axin2–GSK3β cascade regulates snail1 activity in breast cancer cells. Nat Cell Biol. (2006) 8:1398–406. doi: 10.1038/ncb1508
108. Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, et al. Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. (2007) 12:528–41. doi: 10.1016/j.ccr.2007.11.003
109. Groen RW, Oud ME, Schilder-Tol EJ, Overdijk MB, Ten Berge D, Nusse R, et al. Illegitimate WNT pathway activation by β-catenin mutation or autocrine stimulation in T-cell malignancies. Cancer Res. (2008) 68:6969–77. doi: 10.1158/0008-5472.CAN-08-1322
110. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. (2009) 136:1136–47. doi: 10.1016/j.cell.2009.01.015
111. Vermeulen L, Felipe De Sousa EM, Van Der Heijden M, Cameron K, De Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. (2010) 12:468–76. doi: 10.1038/ncb2048
112. Song K, Kwon H, Han C, Zhang J, Dash S, Lim K, et al. Active glycolytic metabolism in CD133 (+) hepatocellular cancer stem cells: regulation by MIR-122. Oncotarget. (2015) 6:40822–35. doi: 10.18632/oncotarget.5812
113. Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. (2015) 6:31151–63. doi: 10.18632/oncotarget.5150
114. Lonardo E, Hermann PC, Mueller M-T, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. (2011) 9:433–46. doi: 10.1016/j.stem.2011.10.001
115. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. (2016) 114:1305–12. doi: 10.1038/bjc.2016.152
116. Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant SJF. Cancer stem cell metabolism and potential therapeutic targets. Front Oncol. (2018) 8:203. doi: 10.3389/fonc.2018.00203
117. Janiszewska M, Suvà ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. (2012) 26:1926–44. doi: 10.1101/gad.188292.112
118. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
119. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. (2016) 18:55. doi: 10.1186/s13058-016-0712-6
120. Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic Cells. Proc Natl Acad Sci USA. (2011) 108:16062–7. doi: 10.1073/pnas.1106704108
121. Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW, et al. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer. (2011) 129:820–31. doi: 10.1002/ijc.25944
122. Sancho P, Burgos-Ramos E, Tavera A, Kheir TB, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. (2015) 22:590–605. doi: 10.1016/j.cmet.2015.08.015
123. Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. (2014) 5:e1336. doi: 10.1038/cddis.2014.285
124. Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, et al. Metabolic alterations in highly tumorigenic glioblastoma cells preference for hypoxia and high dependency on glycolysis. J Biol Chem. (2011) 286:32843–53. doi: 10.1074/jbc.M111.260935
125. Koppenol WH, Bound PL. The Warburg effect and metabolic efficiency: re-crunching the numbers. Science. (2009) 324:1029–33.
126. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. (2013) 155:397–409. doi: 10.1016/j.cell.2013.09.025
127. Guo CY, Yan C, Luo L, Goto S, Urata Y, Xu JJ, et al. Enhanced expression of PKM2 associates with the biological properties of cancer stem cells from A549 human lung cancer cells. Oncol. Rep. (2017) 37:2161–6. doi: 10.3892/or.2017.5438
128. Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, et al. Energy metabolism characterization of a novel cancer stem cell-l ike line 3 AB-OS. J Cell Biochem. (2014) 115:368–79. doi: 10.1002/jcb.24671
129. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, et al. Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells. (2014) 32:1734–45. doi: 10.1002/stem.1662
130. Hur W, Ryu JY, Kim HU, Hong SW, Lee EB, Lee SY, et al. Systems approach to characterize the metabolism of liver cancer stem cells expressing CD133. Sci Rep. (2017) 7:45557. doi: 10.1038/srep45557
131. Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. (2015) 14:86–98. doi: 10.4161/15384101.2014.974419
132. Rodríguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. (2017) 543:424–7. doi: 10.1038/nature21673
133. Chen CT, Shih YRV, Kuo TK, Lee OK, Wei YH. (2008). Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 26:960–68. doi: 10.1634/stemcells.2007-0509
134. Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. (2010) 7:380–90. doi: 10.1016/j.stem.2010.07.011
135. Dupuy F, Tabariès S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. (2015) 22:577–89. doi: 10.1016/j.cmet.2015.08.007
136. Luo M, Wicha MS. Metabolic plasticity of cancer stem cells. Oncotarget. (2015) 6:35141–2. doi: 10.18632/oncotarget.6177
137. De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem. J. (2018) 475:1611–34. doi: 10.1042/BCJ20170164
138. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. (2007) 274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
139. Koppenol WH, Bounds PL, Dang CV. Otto warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. (2011) 11:325–37. doi: 10.1038/nrc3038
140. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. (2018) 24:2482–90. doi: 10.1158/1078-0432.CCR-17-3070
141. Pascual G, Domínguez D, Benitah SA. The contributions of cancer cell metabolism to metastasis. Dis Models Mech. (2018) 11:dmm032920. doi: 10.1242/dmm.032920
142. Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet. (2020) 52:342–52. doi: 10.1038/s41588-019-0557-x
143. Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe R, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. (2011) 10:4047–64. doi: 10.4161/cc.10.23.18151
144. Garber AJ, Karl I, Kipnis D. Alanine and glutamine synthesis and release from skeletal muscle. II the precursor role of amino acids in alanine and glutamine synthesis. J. Biol. Chem. (1976) 251:836–43.
145. Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. (2005) 202:654–62. doi: 10.1002/jcp.20166
146. Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to warburg hypothesis and beyond. Pharmacol Ther. (2009) 121:29–40. doi: 10.1016/j.pharmthera.2008.09.005
147. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. (2010) 1805:141–52. doi: 10.1016/j.bbcan.2010.01.005
148. Crane RK. The restrictions on possible mechanisms of intestinal transport of sugars. In: Membrane Transport and Metabolism. Proceedings of a Symposium held in Prague, August 22-27, 1960. Prague: Czech Academy of Sciences (1961). p. 439–49.
149. Oka Y, Asano T, Shibasaki Y, Lin J-L, Tsukuda K, Katagiri H, et al. C-terminal truncated glucose transporter is locked into an inward-facing form without transport activity. Nature. (1990) 345:550. doi: 10.1038/345550a0
150. Hebert DN, Carruthers A. Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J. Biol. Chem. (1992) 267:23829–38.
151. Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. (2012) 24:650. doi: 10.1097/CCO.0b013e328356da72
152. Gonzalez-Menendez P, Hevia D, Alonso-Arias R, Alvarez-Artime A, Rodriguez-Garcia A, Kinet S, et al. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol. (2018) 17:112–27. doi: 10.1016/j.redox.2018.03.017
153. Ha T-K, Her N-G, Lee M-G, Ryu B-K, Lee J-H, Han J, et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. (2012) 72:4097–109. doi: 10.1158/0008-5472.CAN-12-0448
154. Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC, et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. (2014) 2:11. doi: 10.1186/2049-3002-2-11
155. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. (2015) 6:651–61. doi: 10.18632/oncotarget.2892
156. Osada-Oka M, Hashiba Y, Akiba S, Imaoka S, Sato T. Glucose is necessary for stabilization of hypoxia-inducible factor-1α under hypoxia: contribution of the pentose phosphate pathway to this stabilization. FEBS Lett. (2010) 584:3073–9. doi: 10.1016/j.febslet.2010.05.046
157. Cho ES, Cha YH, Kim HS, Kim NH, Yook JI. The pentose phosphate pathway as a potential target for cancer therapy. Biomol Ther. (2018) 26:29–38. doi: 10.4062/biomolther.2017.179
158. Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. (2013) 126:763–80. doi: 10.1007/s00401-013-1173-y
159. Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, et al. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid Redox Signal. (2007) 9:1115–24. doi: 10.1089/ars.2007.1674
160. Kathagen-Buhmann A, Schulte A, Weller J, Holz M, Herold-Mende C, Glass R, et al. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. (2016) 18:1219–29. doi: 10.1093/neuonc/now024
161. Lemons JM, Feng X-J, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, et al. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. (2010) 8:e1000514. doi: 10.1371/journal.pbio.1000514
162. Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE. (2012) 7:e33418. doi: 10.1371/journal.pone.0033418
163. Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. (1989) 28:4523–30. doi: 10.1021/bi00437a001
164. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
165. Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. (2000) 16:202–8. doi: 10.1016/S0899-9007(99)00266-X
166. Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. (2009) 100:1369–72. doi: 10.1038/sj.bjc.6605007
167. Bobrow L, Happerfield L, Pasternack G, Smith P, Owens A. The expression of fatty acid synthase in primary breast cancer: is it an independent prognostic indicator. Breast Cancer Res Treat. (1993) 25:653–8.
168. Jensen V, Ladekarl M, Holm-Nielsen P, Melsen F, Sœrensen FB. The prognostic value of oncogenic antigen 519 (OA-519) expression and proliferative activity detected by antibody MIB-I in node-negative breast cancer. J. Pathol. (1995) 176:343–52. doi: 10.1002/path.1711760405
169. Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. (1996) 77:474–82. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K
170. Vlad L, Axiotis C, Merino M, Green W. Fatty acid synthase is highly expressed in aggressive thyroid tumors. In: Laboratory Investigation. Philadelphia, PA: Lippincott Williams & Wilkins. (1999). p. 70A
171. Gansler TS, Hardman WIII, Hunt DA, Schaffel S, Hennigar RA. Increased expression of fatty acid synthase (OA-519) in ovarian neoplasms predicts shorter survival. Hum Pathol. (1997) 28:686–92. doi: 10.1016/S0046-8177(97)90177-5
172. Epstein JI, Carmichael M, Partin AW. OA-519 (fatty acid synthase) as an independent predictor of pathologic stage in adenocarcinoma of the prostate. Urology. (1995) 45:81–6. doi: 10.1016/S0090-4295(95)96904-7
173. Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Hum Pathol. (1996) 27:917–21. doi: 10.1016/S0046-8177(96)90218-X
174. Krontiras H, Roye GD, Beenken SE, Myers RB, Mayo MS, Peters GE, et al. Fatty acid synthase expression is increased in neoplastic lesions of the oral tongue. Head Neck. (1999) 21:325–9. doi: 10.1002/(SICI)1097-0347(199907)21:4<325::AID-HED6>3.0.CO;2-P
175. Agostini M, Silva SD, Zecchin KG, Coletta RD, Jorge J, Loda M, et al. Fatty acid synthase is required for the proliferation of human oral squamous carcinoma cells. Oral Oncol. (2004) 40:728–35. doi: 10.1016/j.oraloncology.2004.01.011
176. Zhang Y, Guo C, Yu G. A pilot study of fatty acid metabolism in oral squamous cell carcinoma. Int J Oral Maxillofac Surg. (2005) 34:78–81. doi: 10.1016/j.ijom.2004.03.001
177. Rashid A, Pizer ES, Moga M, Milgraum LZ, Zahurak M, Pasternack GR, et al. Elevated expression of fatty acid synthase and fatty acid synthetic activity in colorectal neoplasia. Am J Pathol. (1997) 150:201.
178. Pizer ES, Lax SF, Kuhajda FP, Pasternack GR, Kurman RJ. Fatty acid synthase expression in endometrial carcinoma: correlation with cell proliferation and hormone receptors. Cancer. (1998) 83:528–37. doi: 10.1002/(SICI)1097-0142(19980801)83:3<528::AID-CNCR22>3.0.CO;2-X
179. Gabrielson EW, Pinn ML, Testa JR, Kuhajda FP. Increased fatty acid synthase is a therapeutic target in mesothelioma. Clin. Cancer Res. (2001) 7:153–7.
180. Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M. Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J Urol. (2008) 180:729–36. doi: 10.1016/j.juro.2008.03.186
181. Camassei FD, Cozza R, Acquaviva A, Jenkner A, Ravà L, Gareri R, et al. Expression of the lipogenic enzyme fatty acid synthase (FAS) in retinoblastoma and its correlation with tumor aggressiveness. Invest Ophthalmol Vis Sci. (2003) 44:2399–403. doi: 10.1167/iovs.02-0934
182. Vandhana S, Deepa P, Jayanthi U, Biswas J, Krishnakumar S. Clinico-pathological correlations of fatty acid synthase expression in retinoblastoma: an Indian cohort study. Exp. Mol. Pathol. (2011) 90:29–37. doi: 10.1016/j.yexmp.2010.11.007
183. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE. (2016) 11:e0147717. doi: 10.1371/journal.pone.0147717
184. Chen C-L, Kumar DBU, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. (2016) 23:206–19. doi: 10.1016/j.cmet.2015.12.004
185. Brandi J, Dando I, Dalla Pozza E, Biondani G, Jenkins R, Elliott V, et al. Proteomic analysis of pancreatic cancer stem cells: functional role of fatty acid synthesis and mevalonate pathways. J Proteomics. (2017) 150:310–22. doi: 10.1016/j.jprot.2016.10.002
186. Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J, et al. An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. (2017) 2017:1656053. doi: 10.1155/2017/1656053
187. Cruz AL, Barreto EDA, Fazolini NP, Viola JP, Bozza PT. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. (2020) 11:105. doi: 10.1038/s41419-020-2297-3
188. Zaytseva YY, Harris JW, Mitov MI, Kim JT, Butterfield DA, Lee EY, et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget. (2015) 6:18891–904. doi: 10.18632/oncotarget.3783
189. Castro LFC, Wilson JM, Gonçalves O, Galante-Oliveira S, Rocha E, Cunha I. The evolutionary history of the stearoyl-CoA desaturase gene family in vertebrates. BMC Evol Biol. (2011) 11:132. doi: 10.1186/1471-2148-11-132
190. Pisanu ME, Noto A, De Vitis C, Morrone S, Scognamiglio G, Botti G, et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. (2017) 406:93–104. doi: 10.1016/j.canlet.2017.07.027
191. Ma Y, Temkin SM, Hawkridge AM, Guo C, Wang W, Wang X-Y, et al. Fatty acid oxidation: an emerging facet of metabolic transformation in cancer. Cancer Lett. (2018) 435:92–100. doi: 10.1016/j.canlet.2018.08.006
192. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
193. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. (2018) 27:136–50 e135. doi: 10.1016/j.cmet.2017.11.001
194. Paumen MB, Ishida Y, Han H, Muramatsu M, Eguchi Y, Tsujimoto Y, et al. Direct interaction of the mitochondrial membrane protein carnitine palmitoyltransferase I with Bcl-2. Biochem Biophys Res Commun. (1997) 231:523–5. doi: 10.1006/bbrc.1997.6089
195. Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MAB, et al. tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1. Cell Death Differ. (2005) 12:603–13. doi: 10.1038/sj.cdd.4401636
196. Jeon S-M, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. (2012) 485:661–5. doi: 10.1038/nature11066
197. LeBleu VS, O'Connell JT, Herrera KNG, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. (2014) 16:992–1003. doi: 10.1038/ncb3039
198. Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci. USA. (2015) 112:8638–43. doi: 10.1073/pnas.1500722112
199. Lu J, Tan M, Cai Q. The warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. (2015) 356:156–64. doi: 10.1016/j.canlet.2014.04.001
200. King MP, Attardi G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell. (1988) 52:811–9. doi: 10.1016/0092-8674(88)90423-0
201. Copeland WC, Wachsman JT, Johnson F, Penta JS. Mitochondrial DNA alterations in cancer. Cancer Invest. (2002) 20:557–69. doi: 10.1081/CNV-120002155
202. Arya RK, Singh A, Yadav NK, Cheruvu SH, Hossain Z, Meena S, et al. Anti-breast tumor activity of eclipta extract in-vitro and in-vivo: novel evidence of endoplasmic reticulum specific localization of Hsp60 during apoptosis. Sci Rep. (2015) 5:18457. doi: 10.1038/srep18457
203. Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. (2002) 360:1323–5. doi: 10.1016/S0140-6736(02)11310-9
204. Brandon M, Baldi P, Wallace D. Mitochondrial mutations in cancer. Oncogene. (2006) 25:4647–62. doi: 10.1038/sj.onc.1209607
205. Gasparre G, Hervouet E, de Laplanche E, Demont J, Pennisi LF, Colombel M, et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet. (2008) 17:986–95. doi: 10.1093/hmg/ddm371
206. Pereira L, Soares P, Máximo V, Samuels DC. Somatic mitochondrial DNA mutations in cancer escape purifying selection and high pathogenicity mutations lead to the oncocytic phenotype: pathogenicity analysis of reported somatic mtDNA mutations in tumors. BMC Cancer. (2012) 12:53. doi: 10.1186/1471-2407-12-53
207. Lleonart ME, Grodzicki R, Graifer DM, Lyakhovich A. Mitochondrial dysfunction and potential anticancer therapy. Med Res Rev. (2017) 37:1275–98. doi: 10.1002/med.21459
208. Guha M, Srinivasan S, Ruthel G, Kashina A, Carstens R, Mendoza A, et al. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene. (2014) 33:5238–50. doi: 10.1038/onc.2013.467
209. Loureiro R, Mesquita KA, Magalhaes-Novais S, Oliveira PJ, Vega-Naredo I. Mitochondrial biology in cancer stem cells. In: Seminars in Cancer Biology. Elsevier (2017). p. 18–28. doi: 10.1016/j.semcancer.2017.06.012
210. Kim EJ, Jin X, Kim OR, Ham SW, Park S-H, Kim H. Glioma stem cells and their non-stem differentiated glioma cells exhibit differences in mitochondrial structure and function. Oncol Rep. (2018) 39:411–6. doi: 10.3892/or.2017.6075
211. Sato M, Kawana K, Adachi K, Fujimoto A, Yoshida M, Nakamura H, et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget. (2016) 7:33297–33305. doi: 10.18632/oncotarget.8947
212. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. (2007) 145:256–64. doi: 10.1016/j.neuroscience.2006.11.065
213. Sato K, Kashiwaya Y, Keon C, Tsuchiya N, King M, Radda G, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. (1995) 9:651–8. doi: 10.1096/fasebj.9.8.7768357
214. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF Jr. Ketone bodies, potential therapeutic uses. IUBMB Life. (2001) 51:241–7. doi: 10.1080/152165401753311780
215. Kim DY, Davis LM, Sullivan PG, Maalouf M, Simeone TA, Brederode J, et al. Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem. (2007) 101:1316–26. doi: 10.1111/j.1471-4159.2007.04483.x
216. Abdelwahab MG, Lee SH, O'Neill D, Thompson RJ, Duff HJ, Sullivan PG, et al. Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS ONE. (2015) 10:e0119316. doi: 10.1371/journal.pone.0119316
217. Ozsvari B, Sotgia F, Simmons K, Trowbridge R, Foster R, Lisanti MP. Mitoketoscins: novel mitochondrial inhibitors for targeting ketone metabolism in cancer stem cells (CSCs). Oncotarget. (2017) 8:78340–50. doi: 10.18632/oncotarget.21259
218. Lonergan T, Brenner C, Bavister B. Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J Cell Physiol. (2006) 208:149–53. doi: 10.1002/jcp.20641
219. Piccoli C, Ria R, Scrima R, Cela O, D'Aprile A, Boffoli D, et al. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells novel evidence of the occurrence of NAD (P) H oxidase activity. J Biol Chem. (2005) 280:26467–76. doi: 10.1074/jbc.M500047200
220. Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8; 21 chromosomal translocation. Proc Natl Acad Sci USA. (2000) 97:7521–6. doi: 10.1073/pnas.97.13.7521
221. Kucia M, Ratajczak M. Stem cells as a two edged sword-from regeneration to tumor formation. J Physiol. Pharmacol. (2006) 57:5–16.
222. Alvero AB, Montagna MK, Holmberg JC. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. (2011) 10:1385–93. doi: 10.1158/1535-7163.MCT-11-0023
223. Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, et al. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat. (2014) 146:525–34. doi: 10.1007/s10549-014-3051-2
224. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. (2008) 320:661–4. doi: 10.1126/science.1156906
225. Kaipparettu BA, Ma Y, Wong LJC. Functional effects of cancer mitochondria on energy metabolism and tumorigenesis: utility of transmitochondrial cybrids. Ann N Y Acad Sci. (2010) 1201:137–46. doi: 10.1111/j.1749-6632.2010.05621.x
226. Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. (2016) 14:2154–65. doi: 10.1016/j.celrep.2016.02.004
227. Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. (2012) 335:348–53. doi: 10.1126/science.1212728
228. Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS ONE. (2016) 11:e0154576. doi: 10.1371/journal.pone.0154576
229. Flores RE, Brown AK, Taus L, Khoury J, Glover F, Kami K, et al. Mycoplasma infection and hypoxia initiate succinate accumulation and release in the VM-M3 cancer cells. Biochim Biophys Acta. (2018) 1859:975–83. doi: 10.1016/j.bbabio.2018.03.012
230. Ta NL, Seyfried TN. Influence of serum and hypoxia on incorporation of [14 C]-d-glucose or [14 C]-l-glutamine into lipids and lactate in murine glioblastoma cells. Lipids. (2015) 50:1167–84. doi: 10.1007/s11745-015-4075-z
231. Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chaing GG, Zeev AR, et al. Comparative metabolic flux profiling of melanoma cell lines beyond the warburg effect. J. Biol. Chem. (2011) 286:42626–34. doi: 10.1074/jbc.M111.282046
232. Portairs JC, Voisin P, Merle M, Canioni P. Glucose and glutamine metabolism in C6 glioma cells studied by carbon 13 NMR. Biochimie. (1996) 78:155–64 doi: 10.1016/0300-9084(96)89500-9
233. Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. (2018) 27:1007–25 e1005. doi: 10.1016/j.cmet.2018.03.002
234. Zhang Y, Zhang SX, Trivedi J, Toll AD, Brahmer J, Hales R, et al. Pleural fluid secondary to pulmonary cryptococcal infection: a case report and review of the literature. BMC Infect Dis. (2019) 19:710. doi: 10.1186/s12879-019-4343-2
235. Reckzeh ES, Karageorgis G, Schwalfenberg M, Ceballos J, Stroet MC, Binici A, et al. Inhibition of glucose transporters and glutaminase synergistically impairs tumor cell growth. Cell Chem Biol. (2019) 26:1214–28. doi: 10.1016/j.chembiol.2019.06.005
236. Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun. Biol. (2019) 2:200. doi: 10.1038/s42003-019-0455-x
237. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. (2010) 29:313. doi: 10.1038/onc.2009.358
238. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
239. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. (2013) 13:572–83. doi: 10.1038/nrc3557
240. Medina MA, Sánchez-Jiménez F, Márquez J, Quesada AR, de Castro Núñez I. Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem. (1992) 113:1–15. doi: 10.1007/BF00230880
241. Kim JH, Lee KJ, Seo Y, Kwon J-H, Yoon JP, Kang JY, et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci Rep. (2018) 8:13111. doi: 10.1038/s41598-018-29895-5
242. Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. (2015) 75:544–53. doi: 10.1158/0008-5472.CAN-14-2211
243. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. (2013) 497:633–7. doi: 10.1038/nature12138
244. Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell. (2014) 15:169–84. doi: 10.1016/j.stem.2014.06.002
245. Todorova VK, Harms SA, Kaufmann Y, Luo S, Luo KQ, Babb K, et al. Effect of dietary glutamine on tumor glutathione levels and apoptosis-related proteins in DMBA-induced breast cancer of rats. Breast Cancer Res Treat. (2004) 88:247–56. doi: 10.1007/s10549-004-0783-4
246. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. (2017) 3:169–80. doi: 10.1016/j.trecan.2017.01.005
247. Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. (2016) 23:517–28. doi: 10.1016/j.cmet.2016.01.007
248. Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med. (2011) 17:641–9. doi: 10.1016/j.molmed.2011.06.001
249. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. (2018) 25:20. doi: 10.1186/s12929-018-0426-4
250. Bartmann C, Raman SJ, Floter J, Schulze A, Bahlke K, Willingstofer J, et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. (2018) 6:8. doi: 10.1186/s40170-018-0180-9
251. Lin L, Ding Y, Wang Y, Wang Z, Yin XG, Shen H. Functional lipidomics: palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. (2017) 66:432–48. doi: 10.1002/hep.29033
252. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
253. Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. (2011) 474:609–15. doi: 10.1038/nature10166
254. Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
255. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. (2008) 321:1807–12. doi: 10.1126/science.1164382
256. Walter BA, Valera VA, Pinto PA, Merino MJ. Comprehensive microRNA profiling of prostate cancer. J Cancer. (2013) 4:350–7. doi: 10.7150/jca.6394
257. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. (2014) 46:573–82. doi: 10.1038/ng.2983
258. Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. (2002) 8:68–74. doi: 10.1038/nm0102-68
259. Mancini M, Scappaticci D, Cimino G, Nanni M, Derme V, Elia L, et al. A comprehensive genetic classification of adult acute lymphoblastic leukemia (ALL): analysis of the GIMEMA 0496 protocol. Blood. (2005) 105:3434–41. doi: 10.1182/blood-2004-07-2922
260. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703. doi: 10.1038/nm.4333
Keywords: cancer stem cell, self-renewal, metabolism, oxidative phosphorylation, anaerobic respiration
Citation: Yadav UP, Singh T, Kumar P, Sharma P, Kaur H, Sharma S, Singh S, Kumar S and Mehta K (2020) Metabolic Adaptations in Cancer Stem Cells. Front. Oncol. 10:1010. doi: 10.3389/fonc.2020.01010
Received: 14 January 2020; Accepted: 21 May 2020;
Published: 25 June 2020.
Edited by:
Rafael Moreno-Sánchez, Instituto Nacional de Cardiologia Ignacio Chavez, MexicoReviewed by:
Sara Rodriguez-Enriquez, Instituto Nacional de Cardiología, MexicoThomas N. Seyfried, Boston College, United States
Peter Bai, University of Debrecen, Hungary
Copyright © 2020 Yadav, Singh, Kumar, Sharma, Kaur, Sharma, Singh, Kumar and Mehta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandeep Singh, c2FuZGVlcHNpbmdoODImI3gwMDA0MDtjdXAuZWR1Lmlu; Santosh Kumar, c2FudG9zaG5jY3MmI3gwMDA0MDtnbWFpbC5jb20=