 Xiaoying Sun
Xiaoying Sun Na Zhang
Na Zhang Chengqian Yin
Chengqian Yin Bo Zhu
Bo Zhu Xin Li
Xin Li- 1Department of Dermatology, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Institute of Dermatology, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
- 3Department of Cardiology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 4Department of Dermatology, Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States
- 5Department of Cancer Immunology and Virology, Dana-Farber Cancer Institute, Boston, MA, United States
Melanoma is the deadliest form of skin cancer, and nearly 90% of melanomas are believed to be caused by ultraviolet radiation (UVR), mainly from sunlight. UVR induces DNA damage, forming products such as cyclobutane pyrimidine dimers (CPD) and 6-4-pyrimidone photoproducts (6-4PP) in a wavelength-dependent manner and causes oxidative DNA damage. These DNA lesions lead to DNA mutations and contribute to the formation of melanoma. In this review, we discuss the protective role of melanocytes against UV-induced DNA damage and how genetic variations, including those in p53 and melanocortin-1 receptor (MC1R), or epigenetic histone modifications in melanocytes result in a tendency toward melanoma. We also provide a summary of prevention and treatment strategies against melanoma, including the most recent immunotherapies. Collectively, this work contributes to the understanding of the molecular pathogenesis of UV-induced melanoma.
UV and Melanomagenesis
Solar ultraviolet radiation (UVR) is considered to be the main etiological factor for skin cancer, including melanoma. UVR comprises ultraviolet C (UVC; 200–290 nm), ultraviolet B (UVB; 290–320 nm), and ultraviolet A (UVA; 320–400 nm) (1). UVR is the major environmental risk factor for melanoma development (2). UVB causes sunburn and damages the epidermis, thus playing a central role in the development of skin cancer, whereas UVA penetrates the skin deeper than UVB or UVC, but does not damage the epidermis considerably; UVC, however, penetrates the deeper layers of the skin limitedly (3) (Figure 1). The direct consequence of solar UV is generation of DNA photoproducts, mainly cyclobutane pyrimidine dimers (CPD) and pyrimidine 6–4-pyrimidone photoproducts (6-4PP). In addition, UV-induced reactive oxygen species (ROS) indirectly cause oxidative DNA damage (4–6). UV-induced damage to cells and tissues includes DNA mutations and altered DNA integrity, transcription profile, and protein modification, which result in the dysregulation of multiple oncogenes and tumor suppressor genes (7, 8).
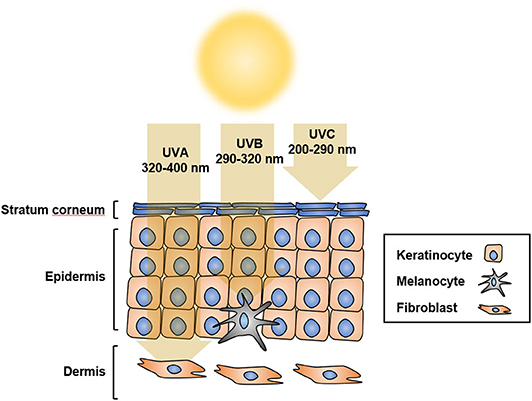
Figure 1. UV penetration into the skin. UV radiation has shorter wavelength than visible light, which makes it invisible to the naked eye. Based on different wavelengths, UV radiation is classified as UVA at 320 to 400 nm, UVB at 290 to 320 nm, or UVC at 200 to 290 nm. UVA penetrates the skin more deeply than UVB and UVC, but does not damage the epidermis considerably. UVB causes sunburn and damages the epidermis, thus playing a central role in the development of skin cancer. Most UVC does not reach the earth because of its short wavelength, and is absorbed by the ozone layer.
There are three types of skin cancers: basal cell carcinoma, squamous carcinoma, and melanoma. Basal cell carcinoma and squamous carcinoma, also known as non-melanoma skin cancer, account for nearly 98% of all skin cancer cases in the United States (9). Although melanoma accounts for the least number of cases of all skin cancers, because of its high metastatic potential and resistance to chemotherapy and radiation therapy, melanoma is responsible for the majority of skin cancer-related deaths (10). Compared to the incidence of non-melanoma skin cancers, the incidence of melanoma is increasing. In 2020, ~100,350 new cases of melanoma and 6,850 deaths are expected (11). Similar to other types of solid tumors, melanoma shows frequent alterations in MAPK and PI3K/PTEN signaling pathways, especially with 40–60% of cultured primary melanoma cells bearing activating BRAF mutations (12–20) (Figure 2). Constitutive activation of BRAF kinase through mutations is the most common way for melanoma to activate the MAPK pathway (20). The most common oncogenic mutation for BRAF kinase is the substitution of amino acid valine for glutamic acid at position 600 (V600E) accounting for nearly 90% of the BRAF mutations. In addition, there are other less common V600 mutations such as V600K, V600R, V600M, and V600D, as well as some non-V600 mutations such as K601E and D594N (21). Targeting the dominant activating mutation BRAF V600E, which activates the MAPK pathway leading to uncontrolled proliferation, with type I RAF inhibitor vemurafenib showed promising clinical benefits (22). However, specific inhibitors of PI3K/AKT were developed and assessed in clinical trials, providing translational potentials (23). However, after the initial tumor response, melanoma cells acquired resistance to the inhibitors, indicating secondary alterations or mutations (24). Intriguingly, mutated BRAF cooperates with alterations in PTEN/AKT to promote melanoma progression (25). Therefore, targeting both signaling pathways simultaneously was proposed for melanoma treatment (26).
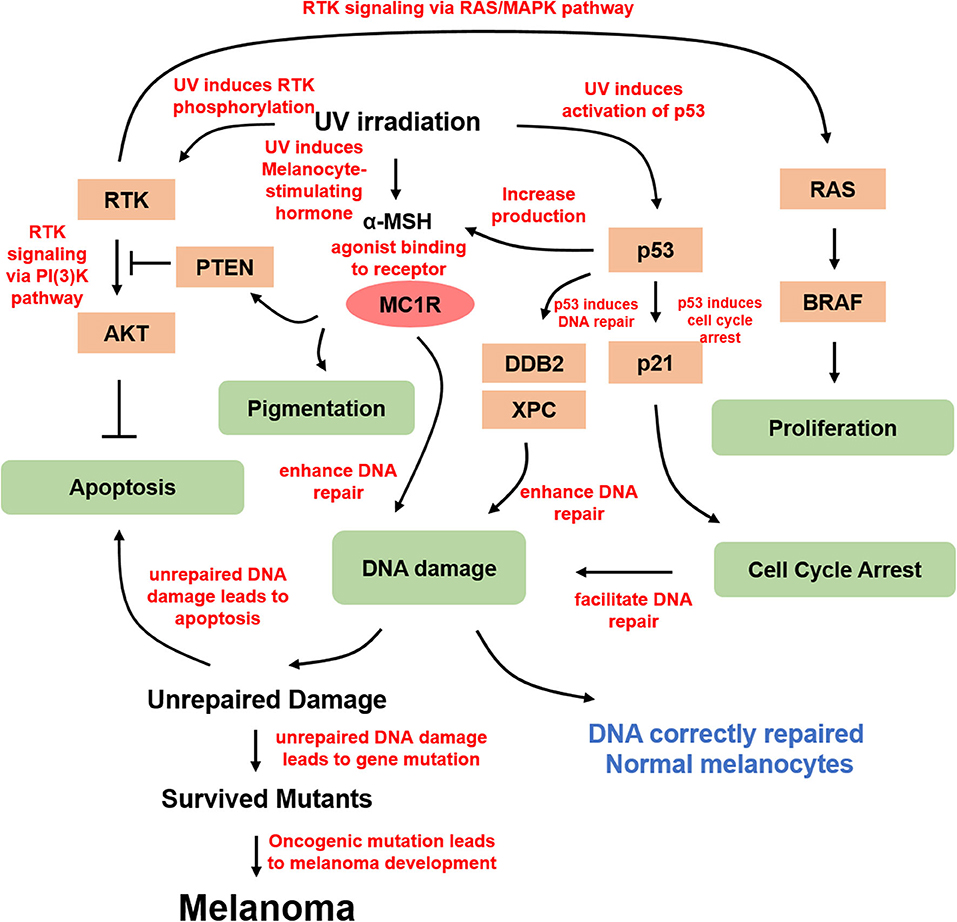
Figure 2. Molecular mechanism of UV-derived melanomagenesis. Direct UV irradiation results in DNA damage, typically in the form of CPD and 6-4PP, which are generated from UVB. α-MSH is the most important melanocortin stimulated by UV irradiation, thereby activating MC1R for melanogenesis and DNA damage repair. In addition, α-MSH/MC1R stabilizes PTEN upon UVB stimulation. UV irradiation also activates p53 and its downstream targets, including p21 in proliferating melanocytes arresting cell cycle at G1 or G2 prior to repair, and several genes from the xeroderma pigmentosum (XP) gene family involving XPC and damage-specific DNA binding protein 2 (DDB2), which is a product of XPE, stimulating nucleotide excision repair for efficient DNA damage repair. DNA damage-induced mutant melanocytes may constitute a pool of cells in which melanoma develops eventually. Moreover, UV irradiation directly activates RTKs, in turn activating several essential pathways, including anti-apoptotic signaling through AKT and proliferation signaling through MAPK.
The UV-induced DNA damage response pathway is modulated by the tumor suppressor p53, whose functional deletion drives UV-mediated mutagenesis in melanoma, squamous cell carcinoma, basal cell carcinoma, and actinic keratosis (27). Tumor suppressor p53 and its related gene products play important roles under different cellular conditions. Accumulation of p53, stimulated by UVR, is required for inducing cell cycle arrest, DNA repair, and apoptosis (28, 29). UV-irradiated p53 knockout mice show a higher incidence of skin tumor development (30). G1/G2 arrest is required for repairing UV-induced DNA damage (31, 32). P53 upregulation directly induces the expression of cyclin-dependent kinase (CDK) inhibitor p21, which mediates UV-induced DNA damage at G1 cell cycle arrest. In addition, the accumulation of p53 also activates several genes from the xeroderma pigmentosum (XP) gene family, including XPC and damage-specific DNA binding protein 2 (DDB2), which is a product of XPE, to stimulate nucleotide excision repair (NER) for efficient DNA damage repair (33). TP53/Trp53 was also shown to cooperate with BRAF V600E to induce melanoma in the presence of UVR (34). Moreover, p53 was found to potently stimulate the proopiomelanocortin (POMC) promoter and induce the generation of melanocortin peptides in response to UV, and research based on a transgenic knockout mouse model demonstrated that p53 loss results in the absence of the UV-tanning response (35). Therefore, p53 functions as a sensor and effector for UV-induced pigmentation, providing insight into the treatment of pathologic hyperpigmentation due to tanning response.
Melanocytes play an important photoprotective role in response to UV exposure via melanin synthesis. Melanocyte-produced melanin protects nuclear DNA from UV irradiation and reduces the generation of DNA damage (36). Melanin within melanosomes of melanocytes is transferred to keratinocytes where they cause tanning, which is a hallmark of UV exposure, mediated by multiple paracrine factors synthesized by keratinocytes (37–39). There are two major forms of melanin generated by melanocytes, eumelanin and pheomelanin. Eumelanin is dark brown or black, whereas pheomelanin is red or orange. It is known that pheomelanin is associated with type I/II skin, freckles, red hair, and an inability to tan (40). The diversity of skin pigmentation phenotypes among individuals from distinct ethnic groups is mainly due to the difference in the content of eumelanin and pheomelanin in melanocytes (41, 42). Studies show that skin cancer, including melanoma, is inversely related to skin pigmentation, with lower skin cancer incidence in individuals with dark skin and higher incidence in individuals with fair skin (43–47). Consistent with these findings, increased eumelanin content was observed to reduce the generation of DNA photoproducts following UV exposure (48).
UVR induces the synthesis and release of melanocortin peptides, including α-melanocyte-stimulating hormone (α-MSH), in both melanocytes and keratinocytes to activate melanocortin 1 receptor (MC1R) signaling (49, 50). MC1R is a highly conserved G protein-coupled receptor expressed on the surface of melanocytes (51). MC1R transduces extracellular signals mediated by melanocortins to downstream effectors including microphthalmia-associated transcription factor (MITF), to regulate skin pigmentation and control cell proliferation and apoptosis (52). In melanocytes, the activation of the MC1R signaling pathway stimulates tyrosinase (TYR) activity, which is the rate-limiting enzyme of melanin production (53). Therefore, MC1R plays a critical role in protecting the skin from UVR.
MC1R is identified as an important factor in preventing melanoma formation. The activation of MC1R by UVR induces adenylate cyclase, cAMP production, protein kinase A activation, melanin synthesis, and downstream UV protective genes such as MITF, tyrosinase, and TRP1 (54–56). Studies show that UVR can induce MSH expression, but fails to stimulate pigmentation in the absence of functional MC1R in red/blonde-haired MC1R transgenic mice. A cyclic AMP agonist forskolin was applied topically, and the resulting chemically induced pigmentation was protective against UV-induced cutaneous DNA damage and tumorigenesis (50). A systematic study on the contribution of MC1R to somatic mutations in sporadic melanoma found that individuals with germline disruptive variants in MC1R have a significantly higher somatic mutational load. This study is the first to report the role of germline MC1R variants in influencing the somatic mutational landscape of melanoma from human data (57). The MC1R mechanistic study showed that the MC1R–PTEN axis serves as a central regulator in response to UVB exposure in melanocytes, which reveals the molecular basis underlying the association between MC1R variants and melanomagenesis. The study indicated that MC1R variants are defective in association with PTEN following UV exposure, consequently failing to suppress the PI3K/AKT signaling pathway, and the MC1R deficiency-induced elevation in PI3K/AKT signaling drives oncogenic transformation in melanoma (58).
Individuals carrying MC1R variants, especially those associated with red hair color, fair skin, and poor tanning ability [red hair color (RHC) variants], have a higher risk of developing melanoma (59). However, how MC1R activity is modulated by UVR and why individuals with red hair are more prone to develop melanoma remain unclear. A recently published study used mouse models to demonstrate a potential MC1R-targeted intervention strategy to rescue loss-of-function MC1R for therapeutic benefits. Studies show that MC1R palmitoylation, primarily mediated by the protein acyltransferase ZDHHC13, is essential for activating MC1R signaling. The activated MC1R signaling increased pigmentation, UVB-induced G1 cell cycle arrest, and control of senescence both in vitro and in vivo (60, 61). Using transgenic mice expressing MC1R RHC variants, studies showed that pharmacological activation of palmitoylation rescues the functional defects of MC1R RHC variants and prevents melanomagenesis. The results highlight a central role for MC1R palmitoylation in pigmentation and protection against melanoma (61).
UV and Immunosuppression
Besides inducing melanomagenesis, UVR can suppress immunity in several ways, including inhibition of antigen presentation, the release of immunosuppressive cytokines, and apoptosis of immune cells. UVR-suppressed immunity contributes to the clearance of tumor cells.
Cutaneous immunity depends on the proper functioning of epidermal Langerhans cells (LCs), which are the main antigen-presenting cells (APCs) in the skin. UVR directly damages LCs with decreased cell numbers and inhibition of antigen-presenting function (62). UV-irradiated LCs lose the ability to stimulate T-helper 1 (Th1) cells in response to foreign antigens, and preferentially activate Th2 cells to promote suppressor T cell function (63). Spleen cells from mice treated with UVR fail to present antigen to Th1 cells. However, this failure could be reversed by injecting anti-IL-10 antibodies into these mice. In this rescue experiment, the antigen-presenting ability of LCs was restored and the LCs effectively activated the Th1 cells. Moreover, the administration of anti-IL-10 antibodies could significantly inhibit UVR-induced antigen presentation of LCs to Th2 cells. The repression may be mediated by suppressive cytokines, such as IL-4 and IL-10, released by the induced T suppressor cells (64, 65).
TNF-α is another UVR-modulated immunosuppressive cytokine (66, 67). Mice treated with anti-TNF-α antibody showed a significant decrease in LCs (68). The pro-inflammatory cytokine IL-12 played an important role in the activation of Th1 cells and blockade of Th2 cells. UVR exposure significantly reduced IL-12 expression, resulting in the suppression of Th1 and de-repression of Th2 cells (69). Another study reported similar results where treatment of mice with IL-12 strongly inhibited UV-induced suppressor T cells in vivo (70).
Skin cancer cells are highly antigenic and could be forcefully rejected by mice. However, UVR-treated mice fail to reject these cancer cells, suggesting that UV-induced immunosuppression promotes tumor growth and progression in vivo (71). UV-induced immunosuppression results from increased T suppressor cells or T regulatory cells, which enhances immune tolerance to tumor antigens (72). This study also suggests that regulatory T cells could be targeted as a vital effector to inhibit UVB-induced immunosuppression, thus enhancing anti-tumor immunity. Therefore, UVB-induced skin cancer is not only caused by UV-induced DNA lesions but also fueled by the generation and maintenance of an immunosuppressed tumor microenvironment (73).
Immunotherapies in Melanoma
The immune system plays a critical role in clearing neoplastic cells. Evading the immune system is crucial for tumor cell survival and proliferation. Treatment of the most deadly form of skin cancer, metastatic melanoma (74), has advanced tremendously in the last decade, especially targeted therapy and immunotherapy. Currently, several types of immunotherapies are being studied to treat melanoma (Table 1).
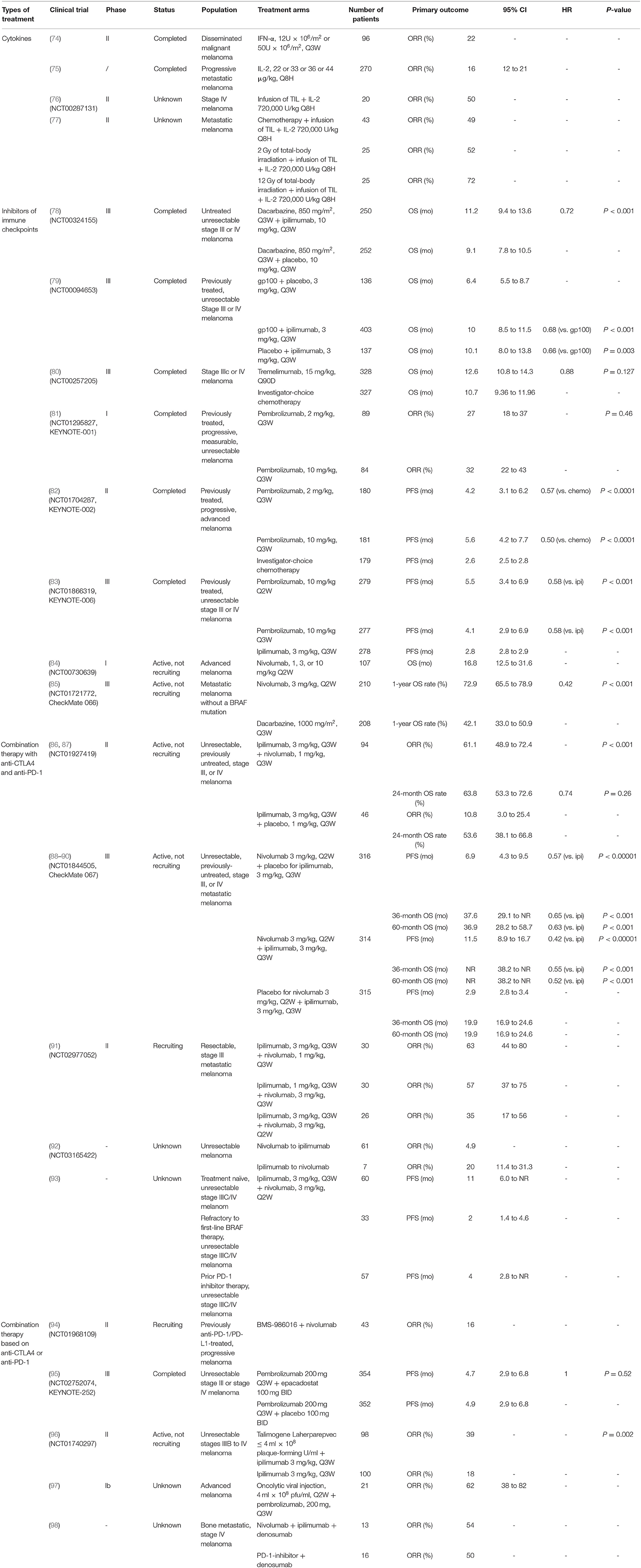
Table 1. Key findings of immunotherapies on melanoma.
Cytokines (Interferon-α and Interleukin-2)
One common method used to boost the immune system is by treatment with cytokines. For example, both interferon-α (IFN-α) and interleukin-2 (IL-2) are administered to melanoma patients. A clinical study found that IFN-α shrinks advanced melanomas with tumor response rates of about 22% and median times to progression and survival of 1.5 and 5 months (99). Intriguingly, tumor burden is found to affect the response. A higher potential of response is observed among patients who have a lower tumor burden. The different immune responses in patients with advanced tumors from those with lower tumor burden indicates different benefits and risk for recurrence and death in patients treated with IFN-α. This hypothesis opened the door for the initial evaluation of adjuvants with IFN-α in melanoma (100). Studies report that IFN-α has a significant effect on activating STAT signaling. The treatment with IFN-α was involved in the significant promotion of tumor-infiltrating dendritic cells (DCs) and T cells (101). In addition, the type I IFN signature is associated with an effector immune response to melanoma in both mouse models and human melanoma patients (75).
Metastatic melanoma trials showed that IL-2 treatment shrinks advanced melanomas with an objective response rate of 16%, with a median duration of response of 8.9 months (76). Intriguingly, a combination of IL-2 treatment with an infusion of ex vivo expanded TIL after chemotherapy-induced lymphodepletion or total body radiotherapy increased the response rate to about 50–72% in metastatic melanoma patients (77, 102). IL-2 is produced by activated CD4+ T, CD8+ T cells, NK cells, and DCs (103). IL-2 boosts the effector lymphocyte immune response and plays an essential role for the IL-2 receptor in immunosuppressive regulatory T cells (104).
Inhibitors of Immune Checkpoints
To enhance anti-tumor immunity, the most successful immunotherapeutic strategy is the use of monoclonal antibodies to block immunoregulatory suppression in the tumor microenvironment.
As a member of the CD28:B7 immunoglobulin superfamily, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is normally expressed at low levels on naïve effector T cells and regulatory T cells (Tregs), but is strongly induced after activation. The upregulated CTLA-4 competes with CD28 for binding with B7, which suppresses T cell receptor signaling to inhibit immunity (105). The anti-CTLA-4 monoclonal antibodies, ipilimumab, and tremelimumab were used in clinical trials to blockade the CTLA-4 signaling, which causes T cell proliferation, activation, and infiltration. Amplification of T cell-mediated immunity and enhanced anti-tumor immune response in the tumor microenvironment were observed (78) (Figure 3). The clinical data show that ipilimumab is a promising drug in treating cancer. The overall survival (OS) was extended in the treated patients (11.2 vs. 9.1 months; HR 0.72; p < 0.001) (79). Moreover, the combination of ipilimumab with the gp100 peptide, a promising anti-tumor vaccine, has improved gp100 peptide benefits with the OS extending from 6.4 to 10.0 months (80). In addition, another ipilimumab clinical trial showed that the treatment improved median OS to 11.4 months (95% CI, 10.7 to 12.1 months), with a plateau at 21% in the survival curve beginning around 3 years. Tremelimumab also shows promising clinical activity in advanced melanoma when tested in a phase III clinical trial (A3671009). The OS is 12.6 months with a 1-year survival rate of more than 50%, compared to 10.7 months for chemotherapy (106).
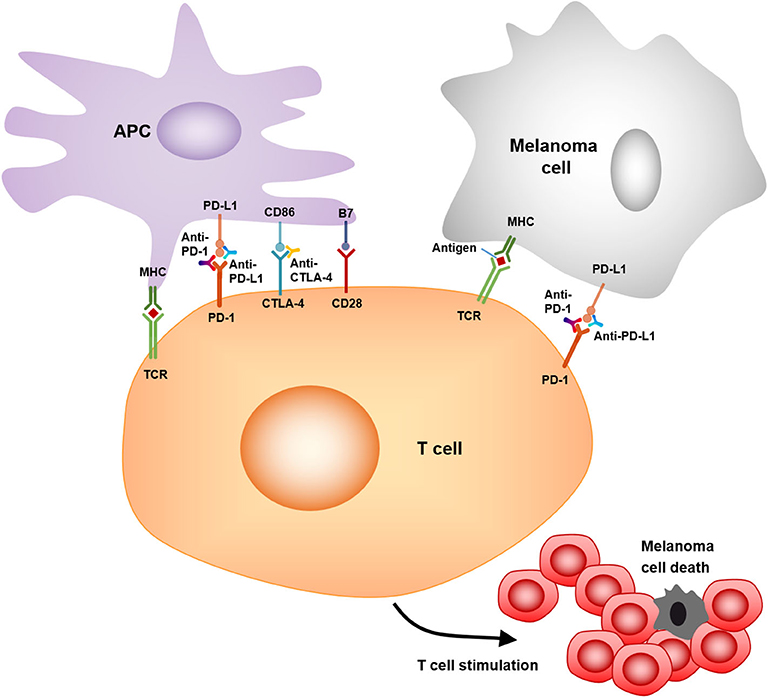
Figure 3. Mechanism of action of anti-PD-1, anti-PD-L1, and anti-CTLA4. PD-1/PD-L1, CTLA-4/CD86 binding inhibits T cell killing of melanoma cells. Blocking PD-1, PD-L1, or CTLA-4 allows T cell killing, APC–T cell interaction, and T cell stimulation (i.e., cytokine secretion, lysis, proliferation, and migration to melanoma) in a melanoma microenvironment.
Programmed cell death protein 1 (PD-1) and programmed cell death-ligand 1 (PD-L1) are also important immunotherapeutic targets for melanoma. PD-1/PD-L1 signaling has been shown to inhibit tumor effector CD8+ T cells. PD-L1 expression is observed in many tumors, including melanoma (107). High expression of PD-L1 on melanoma cells correlates with poor prognosis and low survival rate (108). The PD-1/PD-L1-induced immunosuppression in the microenvironment has resulted in tumor resistance to cytotoxic T cell response (81, 107). Clinical trials showed that treatment with pembrolizumab, an anti-PD-1 antibody, has promising benefits for cancer patients. The overall response rate was 18–43%, and these responses were robust, with OS of 69% at 1 year (82). Another phase II clinical trial with KEYNOTE-002 showed a higher 6-month progression-free survival compared to chemotherapy (38 vs. 16%) (83). Moreover, a phase III trial with ipilimumab and pembrolizumab showed that the overall response rate (ORR) was 33% (pembrolizumab) vs. 12% (ipilimumab). The OS rates for 1 year were 68–74% for pembrolizumab vs. 58% for ipilimumab. Therefore, pembrolizumab was superior to ipilimumab in this study (84).
As a fully human anti-PD-1 monoclonal antibody, nivolumab showed an OS of 17.3 to 20.3 months (85). Nivolumab became the second monoclonal antibody against PD-1 receptor to be approved by the FDA for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and a BRAF inhibitor (if BRAF V600 mutation-positive). The study of phase III trials on patients with metastatic melanoma showed an overall response rate of 32% for nivolumab treatment vs. 11% for chemotherapy (109).
Combination Immunotherapy in Melanoma
Combination Therapy With Anti-CTLA4 and Anti-PD-1
The emergence of combination immunotherapy has greatly improved the poor survival outcome of patients with advanced melanoma in the past with a median OS time of about 8 months, and a 5-year survival rate of about 10% from patients diagnosed with metastatic diseases (86). The results of a phase II trial (CheckMate 069) showed that the combination of nivolumab and ipilimumab demonstrated a statistically significant improvement in objective response rate and longer progression-free survival in treatment-naïve patients with BRAF wild-type melanoma compared with ipilimumab alone (87). The two-year OS rate was 63.8% (95% CI, 53.3–72.6) in the combination group and 53.6% (95% CI, 38.1–66.8) in the ipilimumab alone group (88). Recently, a phase III trial of treatment-naïve patients with advanced melanoma (CheckMate 067) showed that nivolumab in combination with ipilimumab leads to longer progression-free survival and a higher objective response rate than ipilimumab alone (89). The results of the 3-year and 5-year overall data reports for this trial were observed as follows. During the follow-up period of at least 36 months, the median OS time of the nivolumab plus ipilimumab group was not reached, and the median OS time of the nivolumab group was 37.6 months, while that of the ipilimumab group was 19.9 months. The overall 3-year survival rate was 58% in the nivolumab plus ipilimumab group, 52% in the nivolumab group, and 34% in the ipilimumab group, and the security data were not changed from the original report (90). During a follow-up of at least 60 months, the median OS time was more than 60.0 months in the nivolumab plus ipilimumab group, 36.9 months in the nivolumab group, and 19.9 months in the ipilimumab group. Compared with 26% in the ipilimumab group, the 5-year OS rate was 52% in the nivolumab plus ipilimumab group and 44% in the nivolumab group. Patients who received the nivolumab regimen had no significant loss of quality of life such as new late toxic effects (110).
Although both combination therapy and nivolumab monotherapy were more effective than ipilimumab, the frequency of grade 3/4 adverse events (AEs) induced by combination therapy with a dose of ipilimumab (3 mg/kg) plus nivolumab (1 mg/kg) once every 3 weeks was higher than that of nivolumab monotherapy. However, the difference in grade 3/4 AEs between the two groups did not translate into the reported difference in the clinical significance of health-related quality of life (HRQL), further supporting the clinical benefits of nivolumab monotherapy and the combination of nivolumab and ipilimumab in the treatment of advanced melanoma (91). For more extensive clinical application, the early observations of the OpACIN-NEO trial confirmed that two cycles of ipilimumab (1 mg/kg) plus nivolumab (3 mg/kg) once every 3 weeks intravenously could be used as a less toxic but equally effective dose plan for ipilimumab plus nivolumab (92). In addition to combination therapy, sequential therapy is also common in clinical applications. Another retrospective study describing the treatment patterns of nivolumab and ipilimumab observed that switching from nivolumab to ipilimumab was common in Japanese melanoma patients, and the independent factors of high neutrophil-to-lymphocyte ratio and high C-reactive protein before nivolumab treatment could predict the poor prognosis of progression-free survival (111).
To supplement the traditional survival endpoint, it is necessary to fully capture the result measurement method of immuno-oncology drug properties. The concept of treatment-free survival (TFS) is proposed, which can characterize the anti-tumor activity and refer to the period from the discontinuation of immune checkpoint inhibitor therapy caused by adverse events to the subsequent systemic treatment or death (112). TFS analysis of CheckMate 067 and 069 showed that patients with advanced melanoma treated with nivolumab plus ipilimumab had longer TFS and less toxicity than those who received nivolumab or ipilimumab (93).
Although the combination of ipilimumab and nivolumab is a highly effective systematic therapy for metastatic melanoma, patients with BRAF mutations who failed in previous target therapy of BRAF/MEK inhibitors showed less response, and the median progression-free survival was only 2.0 months (95% CI, 1.4–4.6) (113). The study of ipilimumab and nivolumab in patients with metastatic melanoma showed that soft tissue and lung metastasis had the highest lesional response rate (79 and 77%, respectively), while liver metastasis had the lowest (46%), suggesting that specific disease sites may have unique response patterns and drug resistance mechanisms, and personalized treatment should be allowed (94).
Combination Therapy Based on Anti-CTLA4 or Anti-PD-1
The early clinical trials of the new immunotherapy combination also proved that blocking PD-1 combined with other new immunoregulatory targets is safe and effective. Nivolumab combined with anti-lymphocyte activating gene 3 (LAG-3) antibody had an objective response in 31 patients with prior disease progression, with an objective effective rate of 16% and a disease control rate of 45% (95). The I/II phase trial of pembrolizumab and indoleamine 2-dioxygenase 1 (IDO1) inhibitor epacadostat has also shown preliminary efficacy, but the combined randomized III phase trial did not show any additional benefits compared with pembrolizumab alone (114). At present, several clinical trials (NCT01968109, NCT03743766, NCT02676869, and NCT03470922) are underway to combine PD-1 blocking with LAG-3 blocking or IDO1 blocking. In addition, phase I/II trials (NCT02817633, NCT02608268, NCT03099109, and NCT03066648) are being conducted to analyze the efficacy and safety of dual anti-T cell immunoglobulin and mucin 3 (MBG453) and anti-PD-1 (PDR001) effects.
The effectiveness of BRAF/MEK inhibitors has been explored to combine it with immunotherapy. However, despite the promising combination of vemurafenib (BRAF inhibitor) and ipilimumab, the first phase I study was discontinued early because of unexpectedly high hepatotoxicity (96). Other combinations that can improve the therapeutic effect are being explored. In a phase II study, the objective response rate of talimogene laherparepvec combined with ipilimumab was significantly higher, without additional safety issues (97). The combination of pembrolizumab with talimogene laherparepvec has also demonstrated safety and effectiveness in an early phase study (115), and a randomized phase III trial is underway (NCT02263508). Targeting the Wnt/β-catenin signaling pathway should be a high priority for combination therapy to improve the efficacy of anti-CTLA4 or anti-PD-1 agents (98). In addition, the combination of PD-1 inhibitor and denosumab that inhibits receptor activator of nuclear factor kappa-B ligand (RANKL), which promotes osteoclast formation and has an immunological effect, showed good therapeutic signals in patients with bone metastasis from melanoma (116). Although most melanoma immunotherapy evaluation trials exclude brain metastasis patients, it is commendable that local therapy (stereotactic radiosurgery, surgery, or laser interstitial thermal therapy) continues to play an important role in the treatment of melanoma patients with brain metastases receiving immunotherapy (117), and combination of radiotherapy and immunotherapy can even improve the survival rate (118).
Conclusions and Prospects
Exposure to the sun or UVR and the sensitivity of an individual's skin are risk factors for melanoma. In a skin protection behavior study, melanoma survivors were encouraged to use sunscreen and seek shade more frequently. The study stressed that melanoma survivors should always use sunscreen and seek shade to protect their health (119).
It remains significantly important to understand UV-associated skin pathology and identify therapeutic strategies to bypass resistance and increase the proportion of response in order to advance melanoma research. A few previously incurable metastatic melanomas now have potential cures. Complementary insights from melanoma and immunology studies are essential for the development of novel therapeutic strategies and combination rationales for melanoma treatment (120).
Immunotherapy, especially against CTLA-4 and PD-1/PD-L1 signaling to promote anti-tumor immunity, shows promising progression-free survival and recovery in many patients. However, acquired resistance is still observed and diagnosed in PD-1 antibody treatments (121, 122). In the Sunbelt Melanoma Trial with a high dose of interferon, the results showed no survival benefit for patients with a single positive sentinel lymph node (stage III) (123). Therefore, alternative strategies of PD-1/PD-L1 inhibition therapy are also required to overcome acquired resistance, provide more options in new therapeutic strategies, and eventually improve clinical achievements for patients with melanoma.
Author Contributions
XS, NZ, and XL conceived and wrote this manuscript. XL, CY, and BZ discussed the literature and contributed to the revision of the manuscript. All authors have read and approved the final manuscript.
Funding
This study was supported by the National Key Research and Development Program of China (Grant no. 2018YFC1705301), the NSFC of China (Grant no. 81874470), Shanghai Science and Technology Innovation Action Plan Sailing Plan Project (Grant no. 20YF1450400), and Shanghai 13th five-year Plan Clinical Key Department of Dermatology of Traditional Chinese Medicine (Grant no. shslczdzk05001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Slominski A, Pawelek J. Animals under the sun: effects of ultraviolet radiation on mammalian skin. Clin Dermatol. (1998) 16:503–15. doi: 10.1016/S0738-081X(98)00023-6
2. Garibyan L, Fisher D. E. How sunlight causes melanoma. Curr Oncol Rep. (2010) 12:319–26. doi: 10.1007/s11912-010-0119-y
3. Anna B, Blazej Z, Jacqueline G, Andrew CJ, Jeffrey R, Andrzej S. Mechanism of UV-related carcinogenesis and its contribution to nevi/melanoma. Expert Rev Dermatol. (2007) 2:451–69. doi: 10.1586/17469872.2.4.451
4. Lyamichev VI, Frank-Kamenetskii MD, Soyfer VN. Protection against UV-induced pyrimidine dimerization in DNA by triplex formation. Nature. (1990) 344:568–70. doi: 10.1038/344568a0
5. Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis. (1997) 18:2379–84. doi: 10.1093/carcin/18.12.2379
6. Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature. (2012) 491:449–53. doi: 10.1038/nature11624
7. Brenner M, Degitz K, Besch R, Berking C. Differential expression of melanoma-associated growth factors in keratinocytes and fibroblasts by ultraviolet A and ultraviolet B radiation. Br J Dermatol. (2005) 153:733–9. doi: 10.1111/j.1365-2133.2005.06780.x
8. Jhappan C, Noonan FP, Merlino G. Ultraviolet radiation and cutaneous malignant melanoma. Oncogene. (2003) 22:3099–112. doi: 10.1038/sj.onc.1206450
9. Force USPST, Bibbins-Domingo K, Grossman DC, Curry SJ, Davidson KW, Ebell M, et al. Screening for skin cancer: US preventive services task force recommendation statement. JAMA. (2016) 316:429–35. doi: 10.1001/jama.2016.8465
10. Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol. (2009) 4, 551–79. doi: 10.1146/annurev.pathol.3.121806.151541
11. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
12. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
13. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. (2007) 445:851–7. doi: 10.1038/nature05661
14. Zhu B, Zhang M, Williams EM, Keller C, Mansoor A, Davie JK. TBX2 represses PTEN in rhabdomyosarcoma and skeletal muscle. Oncogene. (2016) 35:4212–24. doi: 10.1038/onc.2015.486
15. Zhu B, Davie JK. New insights into signalling-pathway alterations in rhabdomyosarcoma. Br J Cancer. (2015) 112:227–31. doi: 10.1038/bjc.2014.471
16. Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. (2017) 542:484–8. doi: 10.1038/nature21357
17. Lombard DB, Cierpicki T, Grembecka J. Combined MAPK pathway and HDAC inhibition breaks melanoma. Cancer Discov. (2019) 9:469–71. doi: 10.1158/2159-8290.CD-19-0069
18. Leung GP, Feng T, Sigoillot FD, Geyer FC, Shirley MD, Ruddy DA, et al. Hyperactivation of MAPK signaling is deleterious to RAS/RAF-mutant melanoma. Mol Cancer Res. (2019) 17:199–211. doi: 10.1158/1541-7786.MCR-18-0327
19. Vashisht Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J, et al. A Novel mitochondrial inhibitor blocks MAPK pathway and overcomes MAPK inhibitor resistance in melanoma. Clin Cancer Res. (2019) 25:6429–42. doi: 10.1158/1078-0432.CCR-19-0836
20. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. (2014) 120:3446–56. doi: 10.1002/cncr.28864
21. Ihle MA, Fassunke J, Konig K, Grunewald I, Schlaak M, Kreuzberg N, et al. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer. (2014) 14:13. doi: 10.1186/1471-2407-14-13
22. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. (2010) 363:809–19. doi: 10.1056/NEJMoa1002011
23. Niessner H, Schmitz J, Tabatabai G, Schmid AM, Calaminus C, Sinnberg T, et al. PI3K Pathway inhibition achieves potent antitumor activity in melanoma brain metastases in vitro and in vivo. Clin Cancer Res. (2016) 22:5818–28. doi: 10.1158/1078-0432.CCR-16-0064
24. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. (2014) 4:80–93. doi: 10.1158/2159-8290.CD-13-0642
25. Goel VK, Lazar AJ, Warneke CL, Redston MS, Haluska FG. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol. (2006) 126:154–60. doi: 10.1038/sj.jid.5700026
26. Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. (2006) 5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084
27. Moshinsky DJ, Wogan GN. UV-induced mutagenesis of human p53 in a vector replicated in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. (1997) 94:2266–71. doi: 10.1073/pnas.94.6.2266
28. Smith ML, Ford JM, Hollander MC, Bortnick RA, Amundson SA, Seo YR, et al. p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol. (2000) 20:3705–14. doi: 10.1128/.20.10.3705-3714.2000
29. Decraene D, Agostinis P, Pupe A, de Haes P, Garmyn M. Acute response of human skin to solar radiation: regulation and function of the p53 protein. J Photochem Photobiol B. (2001) 63:78–83. doi: 10.1016/S1011-1344(01)00204-4
30. Jiang W, Ananthaswamy HN, Muller HK, Ouhtit A, Bolshakov S, Ullrich SE, et al. UV irradiation augments lymphoid malignancies in mice with one functional copy of wild-type p53. Proc Natl Acad Sci USA. (2001) 98:9790–5. doi: 10.1073/pnas.171066498
31. Pavey S, Russell T, Gabrielli B. G2 phase cell cycle arrest in human skin following UV irradiation. Oncogene. (2001) 20:6103–10. doi: 10.1038/sj.onc.1204707
32. Bissonnette N, Hunting DJ. p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene. (1998) 16:3461–9. doi: 10.1038/sj.onc.1201899
33. Drigeard Desgarnier MC, Rochette Patrick J. Enhancement of UVB-induced DNA damage repair after a chronic low-dose UVB pre-stimulation. DNA Repair. (2018) 63, 56–62. doi: 10.1016/j.dnarep.2018.01.008
34. Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature. (2014) 511:478–82. doi: 10.1038/nature13298
35. Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. (2007) 128:853–64. doi: 10.1016/j.cell.2006.12.045
36. Kobayashi N, Nakagawa A, Muramatsu T, Yamashina Y, Shirai T, Hashimoto MW, et al. Supranuclear melanin caps reduce ultraviolet induced DNA photoproducts in human epidermis. J Invest Dermatol. (1998) 110:806–10. doi: 10.1046/j.1523-1747.1998.00178.x
37. Im S, Moro O, Peng F, Medrano EE, Cornelius J, Babcock G, et al. Activation of the cyclic AMP pathway by alpha-melanotropin mediates the response of human melanocytes to ultraviolet B radiation. Cancer Res. (1998) 58:47–54.
38. Tada A, Suzuki I, Im S, Davis MB, Cornelius J, Babcock G, et al. Endothelin-1 is a paracrine growth factor that modulates melanogenesis of human melanocytes and participates in their responses to ultraviolet radiation. Cell Growth Differ. (1998) 9:575–84.
39. Gilchrest BA, Park HY, Eller MS, Yaar M. Mechanisms of ultraviolet light-induced pigmentation. Photochem Photobiol. (1996) 63:1–10. doi: 10.1111/j.1751-1097.1996.tb02988.x
40. Nasti TH, Timares L. MC1R, eumelanin and pheomelanin: their role in determining the susceptibility to skin cancer. Photochem Photobiol. (2015) 91:188–200. doi: 10.1111/php.12335
41. Hennessy A, Oh C, Diffey B, Wakamatsu K, Ito S, Rees J. Eumelanin and pheomelanin concentrations in human epidermis before and after UVB irradiation. Pigment Cell Res. (2005) 18:220–3. doi: 10.1111/j.1600-0749.2005.00233.x
42. Wakamatsu K, Kavanagh R, Kadekaro AL, Terzieva S, Sturm RA, Leachman S, et al. Diversity of pigmentation in cultured human melanocytes is due to differences in the type as well as quantity of melanin. Pigment Cell Res. (2006) 19:154–62. doi: 10.1111/j.1600-0749.2006.00293.x
43. Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet. (2009) 41:920–5. doi: 10.1038/ng.411
44. Naeyaert JM, Eller M, Gordon PR, Park HY, Gilchrest BA. Pigment content of cultured human melanocytes does not correlate with tyrosinase message level. Br J Dermatol. (1991) 125:297–303. doi: 10.1111/j.1365-2133.1991.tb14161.x
45. Halder RM, Bridgeman-Shah S. Skin cancer in African Americans. Cancer. (1995) 75, (2. Suppl):667–73. doi: 10.1002/1097-0142(19950115)75:2+<667::AID-CNCR2820751409>3.0.CO;2-I
46. Kricker A, Armstrong BK, McMichael AJ. Skin cancer and ultraviolet. Nature. (1994) 368:594. doi: 10.1038/368594b0
47. Sober AJ. Cutaneous melanoma: opportunity for cure. CA Cancer J Clin. (1991) 41:197–9. doi: 10.3322/canjclin.41.4.197
48. Smit NP, Vink AA, Kolb RM, Steenwinkel MJ, van den Berg PT, van Nieuwpoort F, et al. Melanin offers protection against induction of cyclobutane pyrimidine dimers and 6-4 photoproducts by UVB in cultured human melanocytes. Photochem Photobiol. (2001) 74:424–30. doi: 10.1562/0031-8655(2001)074<0424:MOPAIO>2.0.CO;2
49. Chakraborty AK, Funasaka Y, Slominski A, Ermak G, Hwang J, Pawelek JM, et al. Production and release of proopiomelanocortin (POMC) derived peptides by human melanocytes and keratinocytes in culture: regulation by ultraviolet B. Biochim Biophys Acta. (1996) 1313:130–8. doi: 10.1016/0167-4889(96)00063-8
50. D'Orazio JA, Nobuhisa T, Cui R, Arya M, Spry M, Wakamatsu K, et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature. (2006) 443:340–4. doi: 10.1038/nature05098
51. Eves PC, Haycock JW. Melanocortin signalling mechanisms. Adv Exp Med Biol. (2010) 681:19–28. doi: 10.1007/978-1-4419-6354-3_2
52. Garcia-Borron JC, Abdel-Malek Z, Jimenez-Cervantes C. MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation. Pigment Cell Melanoma Res. (2014) 27:699–720. doi: 10.1111/pcmr.12257
53. Scherer D, Kumar R. Genetics of pigmentation in skin cancer–a review. Mutat Res. (2010) 705:141–53. doi: 10.1016/j.mrrev.2010.06.002
54. Hearing VJ, Tsukamoto K. Enzymatic control of pigmentation in mammals. FASEB J. (1991) 5:2902–9. doi: 10.1096/fasebj.5.14.1752358
55. Abdel-Malek Z, Swope VB, Suzuki I, Akcali C, Harriger MD, Boyce ST, et al. Mitogenic and melanogenic stimulation of normal human melanocytes by melanotropic peptides. Proc Natl Acad Sci USA. (1995) 92:1789–93. doi: 10.1073/pnas.92.5.1789
56. Busca R, Ballotti R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. (2000) 13:60–9. doi: 10.1034/j.1600-0749.2000.130203.x
57. Robles-Espinoza CD, Roberts ND, Chen S, Leacy FP, Alexandrov LB, Pornputtapong N, et al. Germline MC1R status influences somatic mutation burden in melanoma. Nat Commun. (2016) 7:12064. doi: 10.1038/ncomms12064
58. Cao J, Wan L, Hacker E, Dai X, Lenna S, Jimenez-Cervantes C, et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol Cell. (2013) 51:409–22. doi: 10.1016/j.molcel.2013.08.010
59. Cust AE, Goumas C, Holland EA, Agha-Hamilton C, Aitken JF, Armstrong BK, et al. MC1R genotypes and risk of melanoma before age 40 years: a population-based case-control-family study. Int J Cancer. (2012) 131:E269–81. doi: 10.1002/ijc.27357
60. Garcia-Borron JC, Jimenez-Cervantes C. Sticky fingers at work: palmitoylation-dependent MC1R activation. Pigment Cell Melanoma Res. (2018) 31:238–240. doi: 10.1111/pcmr.12659
61. Chen S, Zhu B, Yin C, Liu W, Han C, Chen B, et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature. (2017) 549:399–403. doi: 10.1038/nature23887
62. Granstein RD, Matsui MS. UV radiation-induced immunosuppression and skin cancer. Cutis. (2004) 74:(5 Suppl):4–9.
63. Shreedhar VK, Pride MW, Sun Y, Kripke ML, Strickland FM. Origin and characteristics of ultraviolet-B radiation-induced suppressor T lymphocytes. J Immunol. (1998) 161:1327–35.
64. Schmitt DA, Owen-Schaub L, Ullrich SE. Effect of IL-12 on immune suppression and suppressor cell induction by ultraviolet radiation. J Immunol. (1995) 154:5114–20.
65. Rivas JM, Ullrich SE. The role of IL-4, IL-10, and TNF-alpha in the immune suppression induced by ultraviolet radiation. J Leukoc Biol. (1994) 56:769–75. doi: 10.1002/jlb.56.6.769
66. Fourtanier A, Gueniche A, Compan D, Walker SL, Young AR. Improved protection against solar-simulated radiation-induced immunosuppression by a sunscreen with enhanced ultraviolet A protection. J Invest Dermatol. (2000) 114:620–7. doi: 10.1046/j.1523-1747.2000.00946.x
67. Baron ED, Fourtanier A, Compan D, Medaisko C, Cooper KD, Stevens SR. High ultraviolet A protection affords greater immune protection confirming that ultraviolet A contributes to photoimmunosuppression in humans. J Invest Dermatol. (2003) 121:869–75. doi: 10.1046/j.1523-1747.2003.12485.x
68. Moodycliffe AM, Kimber I, Norval M. Role of tumour necrosis factor-alpha in ultraviolet B light-induced dendritic cell migration and suppression of contact hypersensitivity. Immunology. (1994) 81:79–84.
69. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. (2015) 22:237–46. doi: 10.1038/cdd.2014.134
70. Schmitt DA, Walterscheid JP, Ullrich SE. Reversal of ultraviolet radiation-induced immune suppression by recombinant interleukin-12: suppression of cytokine production. Immunology. (2000) 101:90–6. doi: 10.1046/j.1365-2567.2000.00084.x
71. Strickland FM, Sun Y, Darvill A, Eberhard S, Pauly M, Albersheim P. Preservation of the delayed-type hypersensitivity response to alloantigen by xyloglucans or oligogalacturonide does not correlate with the capacity to reject ultraviolet-induced skin tumors in mice. J Invest Dermatol. (2001) 116:62–8. doi: 10.1046/j.1523-1747.2001.00160.x
72. Fisher MS, Kripke ML. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science. (1982) 216:1133–4. doi: 10.1126/science.6210958
73. Yarosh DB. DNA repair, immunosuppression, and skin cancer. Cutis. (2004) 74 (5 Suppl):10–3. doi: 10.1111/j.0105-1873.2004.0459l.x
74. Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer. (2016) 16:345–58. doi: 10.1038/nrc.2016.37
75. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
76. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. (1999) 17:2105–16. doi: 10.1200/JCO.1999.17.7.2105
77. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2010) 16:2646–55. doi: 10.1158/1078-0432.CCR-10-0041
78. Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. (2006) 18:206–13. doi: 10.1016/j.coi.2006.01.011
79. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
80. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
81. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. (2004) 64:1140–5. doi: 10.1158/0008-5472.CAN-03-3259
82. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. (2014) 384:1109–17. doi: 10.1016/S0140-6736(14)60958-2
83. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. (2015) 16:908–18. doi: 10.1016/S1470-2045(15)00083-2
84. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
85. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
86. Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. (2011) 16:5–24. doi: 10.1634/theoncologist.2010-0190
87. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
88. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. (2016) 17:1558–68. doi: 10.1016/S1470-2045(16)30366-7
89. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMc1509660
90. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
91. Schadendorf D, Larkin J, Wolchok J, Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Health-related quality of life results from the phase III CheckMate 067 study. Eur J Cancer. (2017) 82, 80–91. doi: 10.1016/j.ejca.2017.05.031
92. Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. (2019) 20:948–60. doi: 10.1016/S1470-2045(19)30151-2
93. Regan MM, Werner L, Rao S, Gupte-Singh K, Hodi FS, Kirkwood JM, et al. Treatment-free survival: a novel outcome measure of the effects of immune checkpoint inhibition-A pooled analysis of patients with advanced melanoma. J Clin Oncol. (2019) 37:3350–8. doi: 10.1200/JCO.19.00345
94. Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, et al. Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy. Cancer. (2020) 126:86–97. doi: 10.1002/cncr.32522
95. Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J Clin Oncol. (2017) 35 (15 Suppl):9520. doi: 10.1200/JCO.2017.35.15_suppl.9520
96. Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. (2013) 368:1365–6. doi: 10.1056/NEJMc1302338
97. Chesney J, Puzanov I, Collichio F, Singh P, Milhem MM, Glaspy J, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. (2018) 36:1658–67. doi: 10.1200/JCO.2017.73.7379
98. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-Catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
99. Creagan ET, Ahmann DL, Frytak S, Long HJ, Itri LM. Recombinant leukocyte A interferon (rIFN-alpha A) in the treatment of disseminated malignant melanoma. Analysis of complete and long-term responding patients. Cancer. (1986) 58:2576–8. doi: 10.1002/1097-0142(19861215)58:12<2576::AID-CNCR2820581203>3.0.CO;2-6
100. Tawbi HA, Beumer JH, Tarhini AA, Moschos S, Buch SC, Egorin MJ, et al. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: a phase I/II study and pharmacokinetic analysis. Ann Oncol. (2013) 24:1112–9. doi: 10.1093/annonc/mds591
101. Moschos SJ, Edington HD, Land SR, Rao UN, Jukic D, Shipe-Spotloe J, et al. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol. (2006) 24:3164–71. doi: 10.1200/JCO.2005.05.2498
102. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
103. Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, et al. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. (2001) 2:882–8. doi: 10.1038/ni0901-882
104. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
105. Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. (1996) 4:535–43. doi: 10.1016/S1074-7613(00)80480-X
106. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. (2013) 31:616–22. doi: 10.1200/JCO.2012.44.6112
107. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
108. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. (2010) 116:1757–66. doi: 10.1002/cncr.24899
109. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
110. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2019) 381:1535–46. doi: 10.1056/NEJMoa1910836
111. Tsutsumida A, Fukushima S, Yokota K, Yoshikawa S, Yamasaki O, Tanemura A, et al. Japanese real-world study of sequential nivolumab and ipilimumab treament in melanoma. J Dermatol. (2019) 46:947–55. doi: 10.1111/1346-8138.15073
112. Sun X, Roudi R, Dai T, Chen S, Fan B, Li H, et al. Immune-related adverse events associated with programmed cell death protein-1 and programmed cell death ligand 1 inhibitors for non-small cell lung cancer: a PRISMA systematic review and meta-analysis. BMC Cancer. (2019) 19:558. doi: 10.1186/s12885-019-5701-6
113. Mason R, Dearden HC, Nguyen B, Soon JA, Smith JL, Randhawa M, et al. Combined ipilimumab and nivolumab first-line and after BRAF-targeted therapy in advanced melanoma. Pigment Cell Melanoma Res. (2020) 33:358–65. doi: 10.1111/pcmr.12831
114. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
115. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. (2017) 170:1109–19.e10. doi: 10.1016/j.cell.2017.08.027
116. Angela Y, Haferkamp S, Weishaupt C, Ugurel S, Becker JC, Oberndorfer F, et al. Combination of denosumab and immune checkpoint inhibition: experience in 29 patients with metastatic melanoma and bone metastases. Cancer Immunol Immunother. (2019) 68:1187–94. doi: 10.1007/s00262-019-02353-5
117. Qian JM, Yu JB, Mahajan A, Goldberg SB, Kluger HM, Chiang VLS. Frequent use of local therapy underscores need for multidisciplinary care in the management of patients with melanoma brain metastases treated with PD-1 inhibitors. Int J Radiat Oncol Biol Phys. (2019) 105:1113–8. doi: 10.1016/j.ijrobp.2019.08.053
118. Stokes WA, Binder DC, Jones BL, Oweida AJ, Liu AK, Rusthoven CG, et al. Impact of immunotherapy among patients with melanoma brain metastases managed with radiotherapy. J Neuroimmunol. (2017) 313:118–22. doi: 10.1016/j.jneuroim.2017.10.006
119. Mayer D, Layman A, Carlson J. Sun-protection behaviors of melanoma survivors. J Am Acad Dermatol. (2012) 66:e9–10. doi: 10.1016/j.jaad.2010.10.002
120. Lo JA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. (2014) 346:945–9. doi: 10.1126/science.1253735
121. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
122. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. (2016) 315:1600–9. doi: 10.1001/jama.2016.4059
123. McMasters KM, Egger ME, Edwards MJ, Ross MI, Reintgen DS, Noyes RD, et al. Final results of the sunbelt melanoma trial: a multi-institutional prospective randomized phase III study evaluating the role of adjuvant high-dose interferon alfa-2b and completion lymph node dissection for patients staged by sentinel lymph node biopsy. J Clin Oncol. (2016) 34:1079–86. doi: 10.1200/JCO.2015.63.3776
Keywords: ultraviolet radiation, melanomagenesis, melanoma, immunotherapy, cytotoxic T-lymphocyte-associated protein 4, programmed cell death protein 1
Citation: Sun X, Zhang N, Yin C, Zhu B and Li X (2020) Ultraviolet Radiation and Melanomagenesis: From Mechanism to Immunotherapy. Front. Oncol. 10:951. doi: 10.3389/fonc.2020.00951
Received: 01 March 2020; Accepted: 14 May 2020;
Published: 02 July 2020.
Edited by:
Suzie Chen, Rutgers, The State University of New Jersey, United StatesReviewed by:
Yewseok Suh, High Point University, United StatesChandra K. Singh, University of Wisconsin-Madison, United States
Copyright © 2020 Sun, Zhang, Yin, Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Li, MTM2NjE5NTYzMjZAMTYzLmNvbQ==
†These authors have contributed equally to this work