 Yi-Dong Li1†Yong Mao2†Xing-Duo Dong1
Yi-Dong Li1†Yong Mao2†Xing-Duo Dong1 Zi-Ning Lei1
Zi-Ning Lei1 Yuqi Yang1Lizhu Lin3
Yuqi Yang1Lizhu Lin3 Charles R. Ashby Jr.1
Charles R. Ashby Jr.1 Dong-Hua Yang1
Dong-Hua Yang1 Ying-Fang Fan1,4*
Ying-Fang Fan1,4* Zhe-Sheng Chen1*
Zhe-Sheng Chen1*- 1Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John's University, Queens, NY, United States
- 2Department of Oncology, Affiliated Hospital of Jiangnan University, Wuxi, China
- 3Cancer Center, The First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, China
- 4Department of Hepatobiliary Surgery, Zhujiang Hospital, Southern Medical University, Guangzhou, China
In this study, we investigated the efficacy of methyl-cantharidimide (MCA), a cantharidin (CTD) analog, as an anticancer drug, in cancer cells overexpressing either ABCB1 or ABCG2 transporters and in cisplatin-resistant cancer cells. The results indicated that: (i) MCA was efficacious in the ABCB1-overexpressing cell line, KB-C2, and the ABCB1-gene-transfected cell line, HEK293/ABCB1 (IC50 from 6.37 to 8.44 mM); (ii) MCA was also efficacious in the ABCG2-overexpressing cell line, NCI-H460/MX20, and the ABCG2-gene-transfected cell lines, HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and the HEK293/ABCG2-482-T7 cell lines (IC50 from 6.37 to 9.70 mM); (iii) MCA was efficacious in the cisplatin resistant cancer cell lines, KCP-4 and BEL-7404/CP20 (IC50 values from 7.05 to 8.16 mM); (iv) MCA (up to 16 mM) induced apoptosis in both BEL-7404 and BEL-7404/CP20 cancer cells; (v) MCA arrested both BEL-7404 and BEL-7404/CP20 cancer cells in the G0/G1 phase of the cell cycle; (vi) MCA (8 mM) upregulated the expression level of the protein, unc-5 netrin receptor B (UNC5B) in HepG2 and BEL-7404 cancer cells. Overall, our results indicated that MCA's efficacy in ABCB1- and ABCG2-overexpressing and cisplatin resistant cancer cells is due to the induction of apoptosis and cell cycle arrest in the G0/G1 phase.
Introduction
Multidrug resistance (MDR) is one of the major factors that mediates the loss of efficacy of chemotherapy in cancer patients, increasing the frequency of treatment failure (1, 2). MDR can be defined resistance to a drug that produces resistance to other structurally and functionally different drugs that are distinct from the original drug (3). Mechanisms of MDR in cancer cells include: (1) an overexpression of specific ATP binding cassette (ABC) transporters; (2) increased metabolism or biotransformation of anticancer drugs; (3) increased DNA damage response and repair; (4) evasion and resistance to apoptosis; (5) mutations in the cellular targets for anticancer drugs; (6) sequestration of drugs by cellular organelles; (7) increased tolerability to the tumor microenvironment (4–11). The overexpression of efflux pump proteins known as ABC transporters is one of the most frequent mechanisms that mediates MDR, which are present on the cell membrane of certain MDR cancer cells (4, 5).
The family of ABC transporters is known as one of the largest protein families, which includes seven subfamilies from ABCA to ABCG (5, 12, 13). They are widely expressed in the intestines, blood-brain barrier (BBB), liver, placenta and kidneys (14, 15), and mediate the transport or efflux of their physiological substrates such as lipids, sterols, porphyrins, and xenobiotics (16). Among the ABC transporter sub-families, ABCB1 (multidrug resistance 1, MDR1; P-glycoprotein, P-gp), ABCG2 (breast cancer resistance protein, BCRP; mitoxantrone resistance, MXR), ABCC10 (multidrug resistance protein 7, MRP7), and ABCC1 (multidrug resistance protein 1, MRP1) play an important role in producing MDR in cancer cells (17–19). The overexpression of ABC transporters can significantly decrease the intracellular concentration of certain anticancer drugs, thereby decreasing their therapeutic efficacy (20). The expression level of ABC transporters has been shown to be significantly correlated with the level of malignancy progression and chemotherapy efficacy (21–25). Consequently, it is important to know if a chemotherapeutic drug is a substrate for a specific ABC transporter, as its overexpression will significantly attenuate or abolish the efficacy of the drug.
Cantharidin (CTD) is a biologically active compound present in Mylabris phalerata, the Chinese blister beetle (26). CTD has been used in traditional Chinese medicine for more than 2000 years to treat many diseases, including cancer (27–32). Recent reports indicate that CTD has significant anticancer efficacy in different types cancer cells (33–39). However, the use of CTD in clinical cancer treatment is limited due its severe adverse and toxic effects (39). Therefore, several chemically modified CTD analogs have been synthesized to produce analogs that are less toxic than CTD, thereby increasing the likelihood that these compounds could be used to treat certain types of cancer (40). One of these analogs, 2,3a,7a-trimethyl-hexahydro-4,7-epoxido-isoindol-1,3-dion, or methyl-cantharidimide (MCA), has been shown to have anticancer efficacy but was less toxic than CTD (41, 42). Currently, however, MCA's anticancer efficacy in drug resistant cancer cells, as well as its mechanism of action, remain to be elucidated.
Therefore, in this study, we determined the efficacy of MCA in ABCB1- and ABCG2-overexpressing cancer cells and in HEK293 cells transfected with either ABCB1 or ABCG2 cDNA. In addition, we also determined the efficacy of MCA in the cisplatin-resistant cancer cell lines, KCP-4 and BEL-7404/CP20. Finally, we ascertained the effect of MCA on (1) apoptosis, (2) the cell cycle, and (3) gene expression.
Results
The Efficacy of Paclitaxel and MCA in KB-C2 Cells That Overexpressing ABCB1 Transporter and Parental KB-3-1 Cells
As shown in Figures 1A,B and Table 1, the efficacy of paclitaxel, a substrate of the ABCB1 transporter (43), was signfiicantly decreased (920-fold) in KB-C2 cells overexpressing the ABCB1 transporter, compared to the wild type parental cells, KB-3-1. However, the efficacy of MCA was not significantly different between KB-3-1 and KB-C2 cancer cells, indicating that MCA was efficacious in the ABCB1-overexpressing cell line, i.e., there was no resistance to MCA.
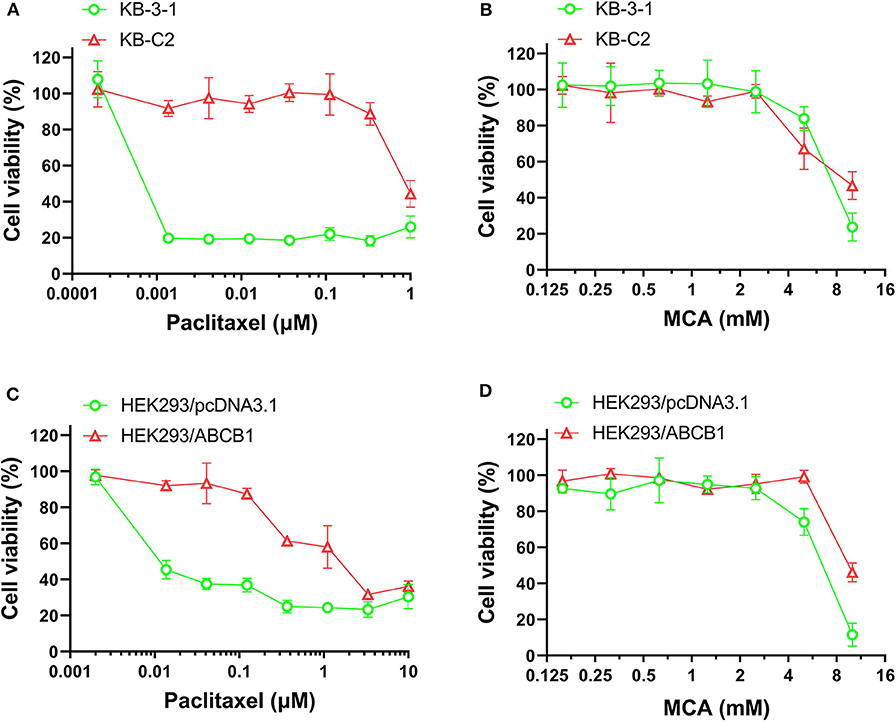
Figure 1. The viability of parental cells and ABCB1-overexpressing cell lines affected by MCA and paclitaxel. Concentration-dependent viability curves for the efficacy of (A) paclitaxel in KB-3-1 and KB-C2 cells; (B) MCA in KB-3-1 and KB-C2 cells; (C) paclitaxel in HEK293/pcDNA3.1 and HEK293/ABCB1 cell; (D) MCA in HEK293/pcDNA3.1 and HEK293/ABCB1 cells.
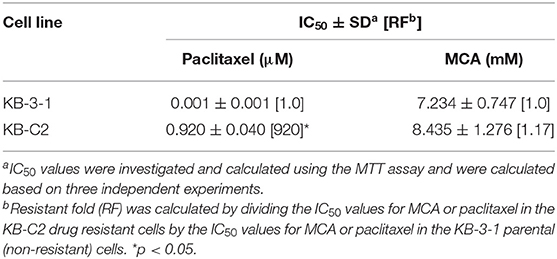
Table 1. The efficacy of paclitaxel and MCA in KB-3-1 cells overexpressing the ABCB1 transporter and KB-C2 cancer cells.
The Efficacy of Paclitaxel in HEK293 Cells Transfected With the ABCB1 Gene and in HEK293 Cells Transfected With an Empty Vector
Since the resistance of KB-C2 could be affected by mutiple factors, we determined the efficacy of paclitaxel and MCA in the with ABCB1-gene-transfected cell line, HEK293/ABCB1, whose resistance was due to the overexpression of the ABCB1 transporter. As shown in Figures 1C,D, and Table 2, the efficacy of paclitaxel in HEK293 cells tranfected with the ABCB1 gene was sigficantly decreased (135.6-fold) compared to HEK293 cells that did not have the ABCB1 gene (i.e., empty vector cells). These results indicate that the HEK293 cells transfected with the ABCB1 gene overexpressed the ABCB1 transporter, although to a lesser extent, than the colchicine-selected KB-C2 cell line. Similar to the results obtained with the KB-C2 and KB-3-1 cell lines, the efficacy of MCA was not signfiicantly alterd in the HEK293 cells overexpressing the ABCB1 transporter compared to the non-overexpressing empty DNA vector transfected HEK293 cells.
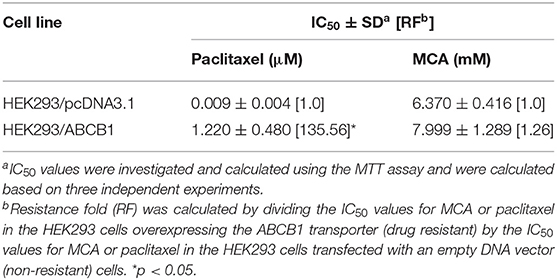
Table 2. The efficacy of paclitaxel and MCA in HEK293 cells overexpressing the ABCB1 transporter and in HEK293 cells transfected with an empty DNA vector.
The Efficacy of Mitoxantrone and MCA in NCI-H460/MX20 Cells Overexpressing the ABCG2 Transporter and Parental NCI-H460 Cancer Cells
As shown in Figures 2A,B and Table 3, the efficacy of mitoxantrone, a substrate of ABCG2 transporter (44), was significantly decreased (22-fold) in mitoxantrone-selected NCI-H460/MX20 cancer cells overexpressing the ABCG2 transporter, compared to its parental cell line, NCI-H460. In contrast, the efficacy of MCA was not significantly different (1.14-fold) in the NCI-H460 and NCI-H460/MX20 cancer cells, demonstrated that ABCG2-overexpressing cell line did not confer resistance to MCA.
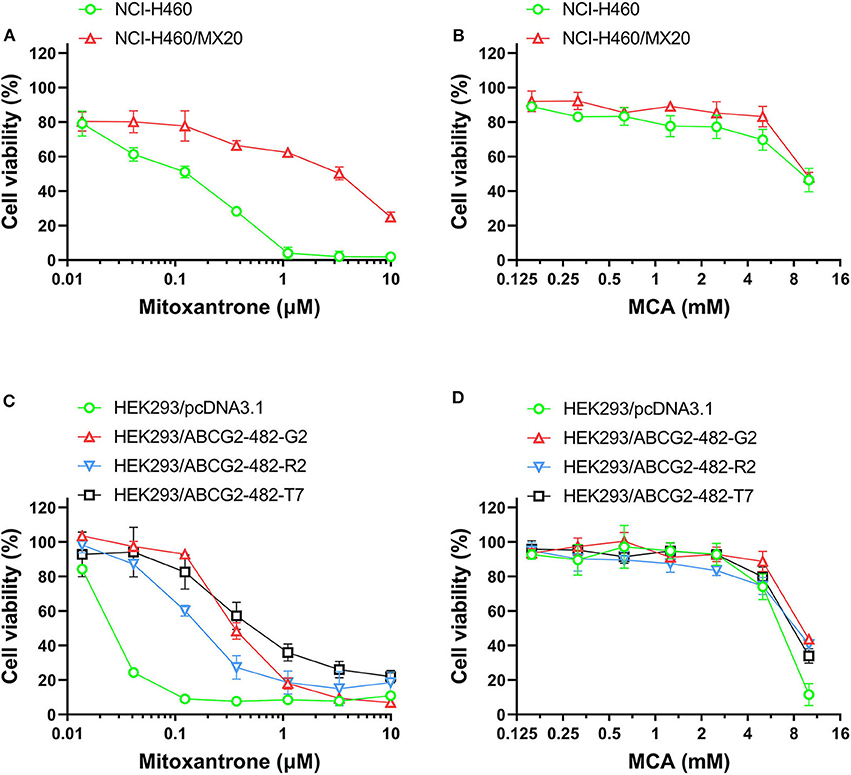
Figure 2. The viability of parental cells and ABCG2-overexpressing cell lines affected by MCA and mitoxantrone. The concentration-dependent viability data for the efficacy of: (A) mitoxantrone efficacy in NCI-H460 and NCI-H460/MX20 cells; (B) MCA in NCI-H460 and NCI-H460/MX20 cells; (C) mitoxantrone in HEK293/pcDNA3.1, HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and HEK293/ABCG2-482-T7 cells; and (D) MCA efficacy in HEK293/pcDNA3.1, HEK293/ABCG2-482-G2, HEK293/ABCG2-482-R2, and HEK293/ABCG2-482-T7.
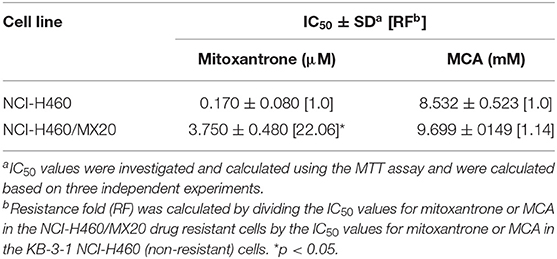
Table 3. The efficacy of mitoxantrone and MCA in NCI-H460/MX20 cells overexpressing the ABCG2 transporter and NCI-460 parental cells.
The Efficacy of Mitoxantronre in HEK293 ABCG2 Gene Transfected Cells and in Empty DNA Vector Transfected HEK293 Cells
As shown in Figures 2C,D and Table 4, the efficacy of mitoxantrone was significantly decreased in the ABCG2-gene-transfected cell lines HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, HEK293/ABCG2-482-T7 by 9.32-, 14.92-, and 19.49-fold, respectively, compared to the empty vector transfected cell line, HEK293/pcDNA3.1. However, the efficacy of MCA in the ABCG2-gene-transfected cells was not significantly different than the non-overexpressing empty vector transfected cells.
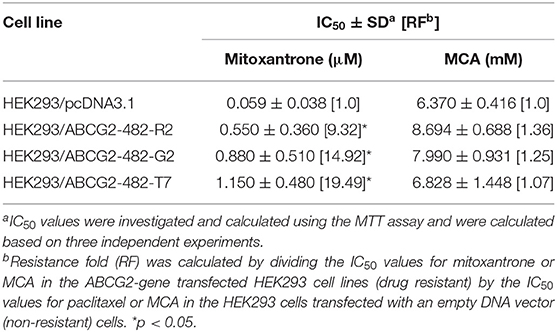
Table 4. The efficacy of mitoxantrone and MCA in the ABCG2-gene-transfected HEK293 cell lines and the HEK293 cells transfected with an empty DNA vector.
The Efficacy of Cisplatin and MCA in Cisplatin—Resistant Cancer Cells
We also determined the anticancer efficacy of MCA in the cisplatin-resistant cancer cell lines, BEL-7404/CP20 and KCP-4. The efficacy of cisplatin was significantly decreased in BEL-7404/CP20 and KCP-4 cells, compared to the parental cell lines, KB-3-1 (9.77-fold) and BEL-7404 (6.27-fold, respectively) (Figure 3 and Table 5). However, there was no significant difference in the anticancer efficacy of MCA in any of the cisplatin-resistant or parental cancer cell lines (Figure 3 and Table 5).
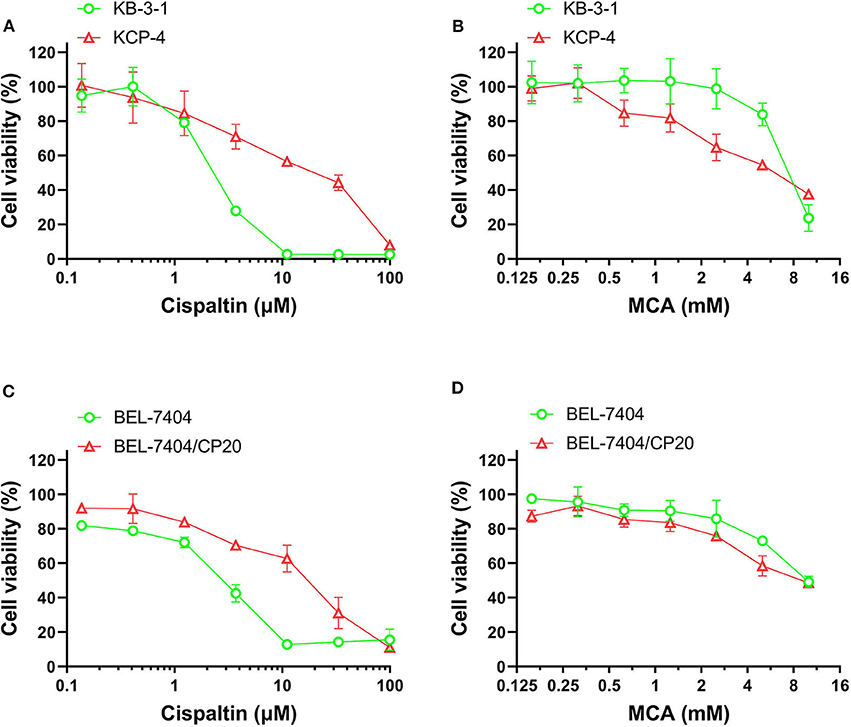
Figure 3. The effect of MCA and cisplatin on the viability of parental cells and cisplatin—resistant cell lines. Concentration-dependent viability curves for the efficacy of; (A) cisplatin in KB-3-1 and KCP-4 cells; (B) MCA in KB-3-1 and KCP-4 cells; (C) cisplatin in BEL-7404 and BEL-7404/CP20 cells; and (D) MCA in BEL-7404 and BEL-7404/CP20 cells.
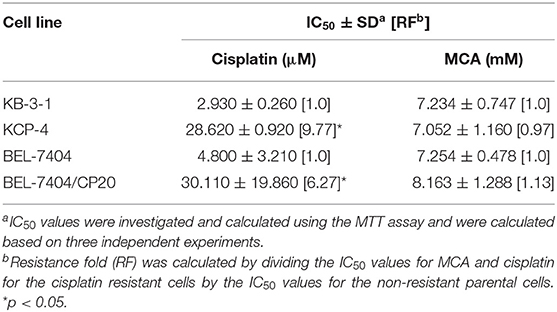
Table 5. The efficacy of cisplatin and MCA in the cisplatin-resistant cell lines, KCP-4 and BEL-7404/CP20, and their respective parental cells lines, KB-3-1 and BEL-7404.
MCA Induced Apoptosis in BEL-7404 and BEL-7404/CP20 Cells
To determine if the anticancer efficacy of MCA was due to the induction of apoptosis, BEL-7404 and BEL-7404/CP20 cells were incubated with MCA (4, 8, and 16 mM) for 72 h. As shown in Figures 4A,B, the total apoptotic rate was significantly increased in in both cell lines after incubation with 16 mM of MCA. We also determined if the apoptotic-inducing effect of MCA was time-dependent by incubating BEL-7404 and BEL-7404/CP20 cells with 16 mM of MCA for 24, 48, or 72 h (Figures 4C,D). The apoptotic rate was significantly increased in BEL-7404 and BEL-7404/CP20 cells incubated with 16 mM of MCA for 48 and 72 h compared to cells incubated with vehicle.
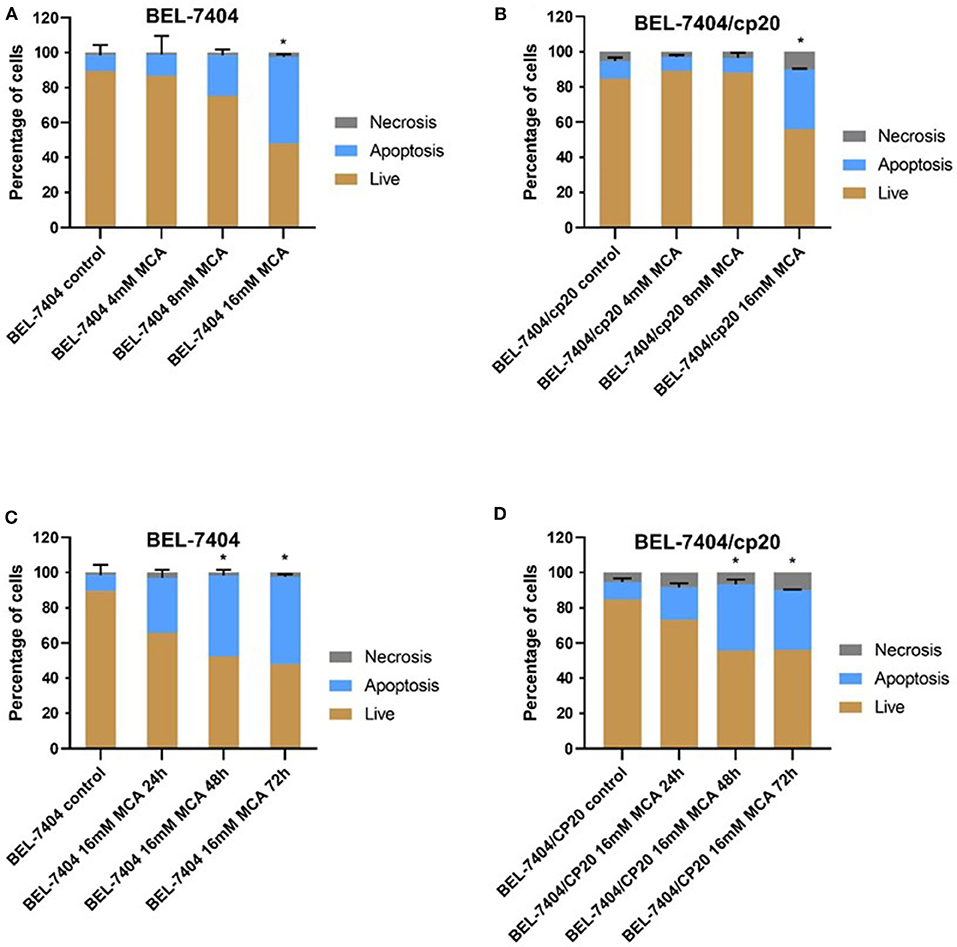
Figure 4. The effect of MCA on the apoptosis of BEL-7404 and BEL-7404/CP20 cells. BEL-7404 (A) and BEL-7404/CP20 (B) cells were incubated with MCA (4, 8, or 16 mM) for 72 h before test. We also incubated BEL-7404 (C) and BEL-7404/CP20 cells (D) with 16 mM of MCA for 24, 48, and 72 h. *p < 0.05 compared with the control group.
MCA Arrested BEL-7404 and BEL-7404/CP20 Cancer Cells in the G0/G1 Phase
To elucidate the mechanism of MCA's anticancer efficacy, we determined the effect of MCA on the progression of the cell cycle in BEL-7404 and BEL-7404/CP20 cells. The progression of the cell cycle from the G0/G1 to S phase was blocked following the incubation of BEL-7404 and BEL-7404/CP20 cells with MCA (5 mM) (Figure 5 and Table 6). The percentage of cells in the G0/G1 phase incubated with MCA were significantly greater than BEL-7404 and BEL-7404/CP20 cells incubated with vehicle (p < 0.05). However, there was no significant difference in the percentage of cells in the G2 and S phases between MCA and vehicle in both cell lines.
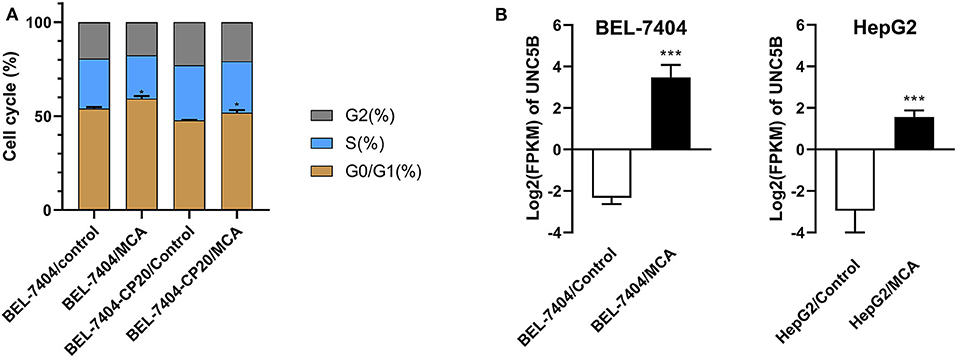
Figure 5. (A) The effect of MCA on the cell cycle of BEL-7404 and BEL-7404/CP20 cells. BEL-7404 and BEL-7404/CP20 cells were incubated with 5 mM of MCA for 48 h before test. *p < 0.05 compared with the control group. (B) The effect of MCA on UNC5B expression in BEL-7404 and HepG2 cells. The values of log2 fragments per kilobase million (FPKM) indicated the expression levels of UNC5B in BEL-7404 and HepG2 cells (***P < 0.001).
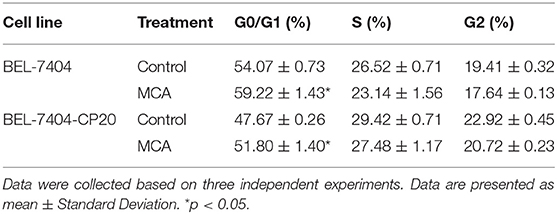
Table 6. The effect of 5 mM of MCA on the cell cycle of BEL-7404 and BEL-7404/CP20 cell lines.
MCA Induced the Expression of UNC5B in BEL-7404 and HepG2 Cells
To identify the underlying target and mechanism of MCA-induced apoptosis of the cancer cells used in our study, we incubated two hepatic cancer cell lines, BEL-7404 and HepG2, with 8 mM of MCA for 72 h, and then performed a differentially expressed genes (DEGs) analysis. The results indicated that the protein, UNC5B (p53RDL1), a p53 receptor required for apoptosis (45), was significantly increased in cells incubated with 8 mM of MCA compared to the control group (Figure 5B and Table 7), as indicated by the significant increase in the values of log2 fragments per kilobase million (FPKM) of UNC5B in BEL-7404 and HepG2.
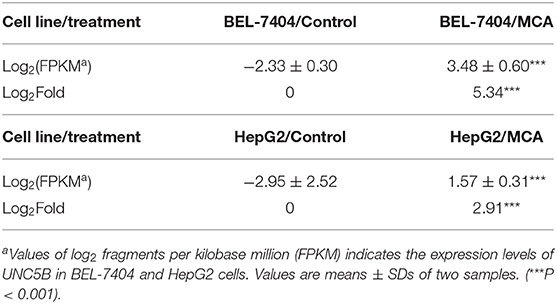
Table 7. The effect of 8 mM of MCA on the expression level of UNC5B in BEL-7404 and HepG2 cell lines.
Discussion
As previously reported, the anticancer drug paclitaxel, a substrate for the ABCB1 transporter (43), inhibited the in vitro growth of the non-drug resistant, parental KB-3-1 cancer cells, whereas paclitaxel was much less potent (920-fold less) in the ABCB1 overexpressing KB-C2 cancer cells that are resistant to paclitaxel. In contrast, the IC50 values of MCA were not significantly different for KB-3-1 (IC50 value of 7.23 mM) and KB-C2 (IC50 value of 8.43 mM) cells. Similarly, paclitaxel was significantly less efficacious in ABCB1 gene transfected HEK293 cells (135-fold less), which overexpress the ABCB1 transporter (46), compared to HEK293 cells transfected with an empty DNA vector that do not overexpress ABCB1 transporter. Furthermore, there was no significant difference in the potency of MCA in the HEK293 cell lines. Our results suggest that the overexpression of the ABCB1 transporter does not confer resistance to MCA and that it has efficacy in MDR KB-C2 cells. Currently, it is unknown as to whether the above-mentioned concentrations of MCA can be obtained in human plasma without producing severe adverse or toxic effects. However, it has been shown that in vitro, MCA is significantly less toxic in normal human liver cells compared to hepatocellular carcinoma cells (42). Additional in vitro, as well as ex-vivo studies, will be required to fully determine the toxicological profile of MCA.
The results of our in vitro study also indicated, as previously reported (47), mitoxantrone, an ABCG2 substrate (44), inhibited the growth of the non-MDR cell line, NCI-H460. However, mitoxantrone was significantly less efficacious in inhibiting the growth of NCI-H460/MX20 cells, which have been shown to be resistant to mitoxantrone caused by ABCG2 transporter overexpression (47), compared with its parental cell line, NCI-H460. Similar to the results we obtained in the ABCB1 overexpressing cell lines, there was no significant difference in the IC50 values for MCA in NCI-H460 (8.53 mM) and NCI-H460/MX30 cells (9.69 mM). Furthermore, the efficacy of mitoxantrone was significantly decreased in HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and HEK293/ABCG2-482-T7 by 9.32-, 14.92-, and 19.49-fold, respectively, compared to HEK-293 cells that express either wild-type and mutant ABCG2. The ABCG2-482-R2 protein is the wild-type protein and it is overexpressed in the transfected cells, whereas the ABCG2 G2 and T7 proteins in the transfected cells overexpress the ABCG2 transporter and have alterations in their substrate and antagonist specificity due to mutations at amino acid 482 (48). Therefore, as previously shown, mitoxantrone was significantly less efficacious in cells transfected with these ABCG2 mutations. In contrast, there was no significant difference in the efficacy of MCA in the ABCG2-R2 (wild-type), ABCG2-G2 and -T7 transfected cells (IC50 values of 8.69, 7.99, and 6.83 mM, respectively). The results suggest that the overexpression of the ABCG2 transporter in cancer cells used in this study does not confer resistance to MCA.
We also determined the effect of MCA in cancer cells resistant to cisplatin. Similar to previous studies (49–51), the efficacy of cisplatin was significantly lower in the cisplatin-resistant cell lines, BEL-7404/CP20 and KCP-4, compared to their respective, non-cisplatin resistant parental cell lines, BEL-7404 and KB-3-1 cells. Previous in vitro studies have reported that cisplatin resistance in BEL-7404 cells can result from: (1) a significant increase in tolerance to DNA damage (52, 53) and (2) a decrease in the intracellular levels of cisplatin due to the overexpression of efflux proteins and a decrease in the endocytosis of cisplatin (54, 55). Cancer cells can also become resistant to chemotherapeutic drugs by decreasing or abrogating their death by apoptosis (6). Indeed, our in vitro results indicated that the incubation of BEL-7404/CP20 cells, as well as BEL-7404 cells, with 16 mM of MCA for 48 h, significantly increased the percentage of apoptotic cells. Additionally, the incubation of BEL-7404 and BEL-7404/CP20 cells with 5 mM of MCA significantly increased the percentage of cells that were arrested in the G0/G1 phase compared to cells incubated with vehicle. The fact that more cells were arrested in the G0/G1 phase suggests that there will be a lower number of cells that can enter the S phase of the cell cycle, thereby decreasing cellular replication. Thus, MCA inhibits the replication of BEL-7404 and BEL-7404/CP20 cancer cells.
We subsequently conducted experiments to determine the mechanism by which MCA induces the apoptosis in BEL-7404 and HepG2 cells using a differential expression gene (DEG) analysis. This approach can reveal what genes are significantly upregulated or downregulated following exposure to MCA. Our results indicated that the incubation of BEL-7404 and HepG2 cancer cells with 8 mM of MCA significantly increased the expression of the protein, UN5CB, by 5.34- and 2.91- Log2Fold, respectively. UNC5B, also known as the p53-regulated receptor for death and life protein (p53RDL1), is a protein that interacts with the p53 regulated receptor to induce apoptosis (45, 56). It is well-established that the p53 protein plays a key role in tumor suppression and at least 50% of all human cancers have mutations in the p53 gene (57). Furthermore, p53 mediates cell cycle arrest and cell death depending on the context of the cellular environment (58). One of the proteins involved in p53—dependent apoptosis is the netrin receptor family (56). Furthermore, UNC5B is one of netrin-1 receptor family (UNC5A to UNC5D) (59) members that participates in p53-dependent apoptosis as a target gene for p53 via a p53-binding sequence located within its intron 1 gene (45). There is a death domain in UNC5B's intracellular region that serves as a dependence receptor, i.e., the apoptotic effect of UNC5B will be inhibited when it interacts with its biological ligand, netrin-1 (56). A caspase-cleavage sequence, DXXD, which is cleaved by caspase-3 and, further induces cellular apoptosis, is present in UNC5B's intracellular region (45). The genotype of p53 in both BEL-7404 and HepG2 cells are wild-type (60, 61), indicating that the expression of UNC5B in these two cell lines was not affected by p53 mutations. Interestingly, a recent study reported that the incubation of HepG2 cells with MCA significantly upregulated caspase-3 expression, which plays a key role in apoptosis (42). Thus, in our study, the significant in vitro increase in UNC5B expression levels produced by MCA suggests that the increase in the apoptosis of BEL-7404 and HepG2 cells may be due to its activation of the p53-UNC5B apoptosis pathway. In addition, the arrest of the cell cycle of BEL-7404 and BEL-7404/CP20 cells produced by MCA may also be due to its effect on the p53-UNC5B axis. However, additional detailed studies must be conducted to determine how MCA induces the upregulation of UNC5B in cisplatin-resistant BEL-7404 cancer cells.
Materials and Methods
Chemicals
Methyl-Cantharidimide (MCA) was obtained as a gift from Sihuan Bioengineering Co., Ltd. (Jiangsu, China). Dulbecco's modified Eagle's Medium (DMEM), Bovine serum albumin (BSA), penicillin/streptomycin and 0.25% trypsin, fetal bovine serum (FBS) were purchased from Corning Incorporated (Corning, NY). Dimethylsulfoxide (DMSO), 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), paclitaxel, cisplatin, mitoxantrone were purchased from Sigma-Aldrich (St. Louis, MO). Propidium Iodide (PI) and annexin V were purchased from BD biosciences (San Jose, CA). All the rest chemicals were purchased from Sigma Chemical Co (St. Louis, MO).
Cell Lines and Cell Culture
The ABCB1-overexpresing experiments were conducted using the human epidermoid carcinoma cell line, KB-3-1, and its ABCB1-overexpressing, colchicine-selected cells, KB-C2. For the experiments with the ABCG2-overexpresing cells, we used the non-small cell lung cancer (NSCLC) cell line, NCI-H460, and its ABG21-overexpressing, mitoxantrone-selected cells, NCI-H460/MX20. The KB-C2, and NCI-H460/MX20 cells were cultured and maintained as previously reported (47). HEK293/pcDNA3.1, HEK293/ABCB1, HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and HEK293/ABCG2-482-T7 cells were generated by the transfection of an empty vector pcDNA3.1, a pcDNA3.1 vector containing a full length ABCB1 gene or a pcDNA3.1 vector containing a full length ABCG2 gene, with different amino acids at position 482, including threonine, arginine, or glycine (46, 48). G418 (2 mg/ml), with complete culture medium, was used to select the transfected cells. The cisplatin resistant cell line, of KCP-4, a human hepatocellular carcinoma cell line, BEL-7404, and the cisplatin resistant cell line, BEL-7404/CP20, were used in the cisplatin resistance experiments (49–51). KCP-4 cells were maintained in 7 μg/ml cisplatin and BEL-7404/CP20 were maintained in 20 ng/ml of cisplatin with complete DMEM medium. All of the cells were cultured and maintained in DMEM medium containing 1% penicillin/streptomycin, 10% FBS and in a water jacketed CO2 incubator (5% CO2) with thermal protection. All cells were cultured under non-treatment condition for at least 14 days before testing.
MTT Assay
Cell viability and resistance - fold were calculated using the MTT assay as previously reported (62). For the resistance experiments, the cells were digested by 0.25% trypsin-EDTA, resuspended and seeded into a 96-well plate as 5000 cells each well in 180 μL of complete medium. After incubating overnight, MCA and other anticancer drugs were added at different concentrations to produce a final volume of 200 μL. After 72 h incubation, the MTT solution (4 mg/ml) was added to each well and the plates were incubated for another 4 h. After the incubation, the supernatant was removed and 100 μL of DMSO was added and the formazan crystals were dissolved by shaking the plates for 10 min. Finally, the absorbance at 570 nm was measured using a microplate reader (Dynex Technologies, Chantilly, VA). The concentration required to inhibit cell viability by 50% (IC50) was calculated after plotting survival curves using the Bliss method as previously described (63).
Apoptosis Analysis
BEL-7404 and BEL-7404/CP20 cells were incubated with MCA (4, 8, and 16 mM) for 72 h. Cell were harvested and dyed using Propidium Iodide (PI) and annexin V. The apoptosis analysis was conducted using FL-1 and FL-2 of the BD Accuri™ C6 flow cytometer as described previously (64).
Cell Cycle
BEL-7404 and BEL-7404/CP20 cells were incubated with 5 mM of MCA for 48 h. Cell were harvested and fixed using 70% ice cold ethanol, then dyed using Propidium Iodide (PI). The cell cycle analysis was determined using a BD Accuri™ C6 flow cytometer as described previously (65).
Differentially Expressed Genes (DEGs) Analysis
BEL-7404 and HepG2 cells were incubated with 8 mM of MCA 72 h. Cells were digested, harvested, and RNA extraction was performed using a RNA extraction kit provided by QIAGEN. All the expressed genes (DEGs) analysis data were generated and analyzed by Novogene Corporation Inc. (Sacramento, CA).
Statistical Analysis
All data were collected based on at least three independent experiments. The data were analyzed using a one-way analysis of variance (ANOVA) and post-hoc tests were done a t-test method. The a priori significance level was p < 0.05.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
Y-DL: data curation. Y-DL, X-DD, Z-NL, and YY: formal analysis, investigation, and visualization. Y-DL, YM, CA, D-HY, Y-FF, and Z-SC: methodology. Y-FF and Z-SC: project administration. YM, LL, D-HY, and Z-SC: resources. Z-SC: supervision. Y-DL, YM, X-DD, Z-NL, YY, and LL: validation. Y-DL and YM: writing—original draft. Y-DL, YM, X-DD, Z-NL, YY, CA, D-HY, Y-FF, and Z-SC: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the Department of Pharmaceutical Sciences, at St. John's University for their partial support of this project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Sihuan Bioengineering Co., Ltd. (Jiangsu, China) for providing MCA. We thank Drs. Robert W. Robey and Susan E. Bates (NCI, NIH, Bethesda, MD) for providing the H460, H460/MX20, and ABCG2 gene transfected cell lines. We thank Dr. Gottesman (NCI, NIH, Bethesda, MD) for providing BEL-7404 and BEL-7404/CP20 cell lines. We thank Dr. Shin-Ichi Akiyama (Kagoshima University, Japan) for allowing us to use the KB-3-1, KB-C2, and KCP-4 cell lines.
References
1. Kartal-Yandim M, Adan-Gokbulut A, Baran Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol. (2016) 36:716–26. doi: 10.3109/07388551.2015.1015957
2. Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. (2006) 5:219–34. doi: 10.1038/nrd1984
3. Kaye SB. The multidrug resistance phenotype. Br J Cancer. (1988) 58:691. doi: 10.1038/bjc.1988.291
4. Eckford PD, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem Rev. (2009) 109:2989–3011. doi: 10.1021/cr9000226
5. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat Rev Cancer. (2002) 2:48–58. doi: 10.1038/nrc706
6. Azab F, Vali S, Abraham J, Potter N, Muz B, de la Puente P, et al. PI 3 KCA plays a major role in multiple myeloma and its inhibition with BYL 719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. Br J Haematol. (2014) 165:89–101. doi: 10.1111/bjh.12734
7. Vinod BS, Maliekal TT, Anto RJ. Phytochemicals as chemosensitizers: from molecular mechanism to clinical significance. Antioxidants Redox Signal. (2013) 18:1307–48. doi: 10.1089/ars.2012.4573
8. Casorelli I, Bossa C, Bignami M. DNA damage and repair in human cancer: molecular mechanisms and contribution to therapy-related leukemias. Int J Environ Res Public Health. (2012) 9:2636–57. doi: 10.3390/ijerph9082636
9. Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. (2005) 14(Suppl. 1): 35–48. doi: 10.1159/000086183
10. Saito G, Swanson JA, Lee KD. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Delivery Rev. (2003) 55:199–215. doi: 10.1016/s0169-409x(02)00179-5
11. Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Ann Rev Biochem. (2004) 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723
12. Dassa E, Bouige P. The ABC of ABCs: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. (2001) 152:211–29. doi: 10.1016/S0923-2508(01)01194-9
13. Stavrovskaya AA, Stromskaya TP. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry. (2008) 73:592–604. doi: 10.1134/s0006297908050118
14. Linton KJ, Higgins CF. Structure and function of ABC transporters: the ATP switch provides flexible control. Pflügers Archiv Eur J Physiol. (2007) 453:555–67. doi: 10.1007/s00424-006-0126-x
15. Linton KJ. Structure and function of ABC transporters. Physiology. (2007) 22:122–30. doi: 10.1152/physiol.00046.2006
16. Wu CP, Ambudkar SV. The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm Sin B. (2014) 4:105–11. doi: 10.1016/j.apsb.2013.12.001
17. Kim SW, Kwon HY, Chi DW, Shim JH, Park JD, Lee YH, et al. Reversal of P-glycoprotein-mediated multidrug resistance by ginsenoside Rg3. Biochem Pharmacol. (2003) 65:75–82. doi: 10.1016/S0006-2952(02)01446-6
18. Lemos C, Jansen G, Peters GJ. Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. Br J Cancer. (2008) 98:857–62. doi: 10.1038/sj.bjc.6604213
19. Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1−/−/Mdr1a/1b−/−(triple-knockout) and wild-type mice. Mol Cancer Ther. (2008) 7:2280–7. doi: 10.1158/1535-7163.MCT-07-2250
20. Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resistance Updates. (2012) 15:62–9. doi: 10.1016/j.drup.2012.02.001
21. Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ. Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomed Pharmacother. (2013) 67:791–9. doi: 10.1016/j.biopha.2013.08.001
22. Ali MA, Elsalakawy WA. ABCB1 haplotypes but not individual SNPs predict for optimal response/failure in Egyptian patients with chronic-phase chronic myeloid leukemia receiving imatinib mesylate. Med Oncol. (2014) 31:279. doi: 10.1007/s12032-014-0279-y
23. Xie ZY, Lv K, Xiong Y, Guo WH. ABCG2-meditated multidrug resistance and tumor-initiating capacity of side population cells from colon cancer. Oncol Res Treat. (2014) 37:666–72. doi: 10.1159/000368842
24. Liu L, Zuo LF, Guo JW. ABCG2 gene amplification and expression in esophageal cancer cells with acquired adriamycin resistance. Mol Med Rep. (2014) 9:1299–304. doi: 10.3892/mmr.2014.1949
25. Yang B, Ma YF, Liu Y. Elevated expression of Nrf-2 and ABCG2 involved in multi-drug resistance of lung cancer SP cells. Drug Res. (2015) 65:526–31. doi: 10.1055/s-0034-1390458
26. Nakatani T, Konishi T, Miyahara K, Noda N. Three Novel Cantharidin-Related Compounds from the Chinese Blister Beetle, Mylabris phalerata P ALL. Chem Pharm Bull. (2004) 52:807–9. doi: 10.1248/cpb.52.807
27. Liu D, Chen Z. The effects of cantharidin and cantharidin derivates on tumour cells. Anticancer Agents Med Chem. (2009) 9:392–6. doi: 10.2174/1871520610909040392
28. Yeh CB, Su CJ, Hwang JM, Chou MC. Therapeutic effects of cantharidin analogues without bridging ether oxygen on human hepatocellular carcinoma cells. Eur J Med Chem. (2010) 45:3981–5. doi: 10.1016/j.ejmech.2010.05.053
29. Kizaki M, Sagawa M. Mylabris phalerata (Chinese blister beetle) on hematological malignancies. In: Evidence-Based Anticancer Materia Medica. Dordrecht: Springer (2011). p. 375–88. doi: 10.1007/978-94-007-0526-5_17
30. Liang F, Wang MY, Huang WB, Li AJ. Effect of sodium cantharidinate on the angiogenesis of nude mice with human gastric cancer. Zhong Yao Cai. (2011) 34:343–6.
31. Shao H, Hong G, Luo X. Evaluation of sodium cantharidinate/vitamin B6 in the treatment of primary liver cancer. J Cancer Res Ther. (2014) 10:75. doi: 10.4103/0973-1482.139770
32. Deng YY, Zhang W, Li NP, Lei XP, Gong XY, Zhang DM, et al. Cantharidin derivatives from the medicinal insect Mylabris phalerata. Tetrahedron. (2017) 73:5932–9. doi: 10.1016/j.tet.2017.08.034
33. Hsia TC, Lin JH, Hsu SC, Tang NY, Lu HF, Wu SH, et al. Cantharidin induces DNA damage and inhibits DNA repair-associated protein levels in NCI-H460 human lung cancer cells. Environ Toxicol. (2015) 30:1135–43. doi: 10.1002/tox.21986
34. Kuo JH, Shih TY, Lin JP, Lai KC, Lin ML, Yang MD, et al. Cantharidin induces DNA damage and inhibits DNA repair-associated protein expressions in TSGH8301 human bladder cancer cell. Anticancer Res. (2015) 35:795–804.
35. Su CC, Liu SH, Lee KI, Huang KT, Lu TH, Fang KM, et al. Cantharidin induces apoptosis through the calcium/PKC-regulated endoplasmic reticulum stress pathway in human bladder cancer cells. Am J Chin Med. (2015) 43:581–600. doi: 10.1142/S0192415X15500366
36. Tian X, Zeng G, Li X, Wu Z, Wang L. Cantharidin inhibits cell proliferation and promotes apoptosis in tongue squamous cell carcinoma through suppression of miR-214 and regulation of p53 and Bcl-2/Bax. Oncol Rep. (2015) 33:3061–8. doi: 10.3892/or.2015.3942
37. Kim A, Im M, Ma JY. Sosiho-tang ameliorates cachexia-related symptoms in mice bearing colon 26 adenocarcinoma by reducing systemic inflammation and muscle loss. Oncol Rep. (2016) 35:1841–50. doi: 10.3892/or.2015.4527
38. Ren Y, Zhang SW, Xie ZH, Xu XM, Chen LL, Lou ZG, et al. Cantharidin induces G2/M arrest and triggers apoptosis in renal cell carcinoma. Mol Med Rep. (2016) 14:5614–8. doi: 10.3892/mmr.2016.5963
39. Li CC, Yu FS, Fan MJ, Chen YY, Lien JC, Chou YC, et al. Anticancer effects of cantharidin in A431 human skin cancer (Epidermoid carcinoma) cells in vitro and in vivo. Environ Toxicol. (2017) 32:723–38. doi: 10.1002/tox.22273
40. Wang G, Dong J, Deng L. Overview of cantharidin and its analogues. Curr Med Chem. (2018) 25:2034–44. doi: 10.2174/0929867324666170414165253
41. Wei FC, Du J, Wei NN, Wang Y, Li SW. Progress and application of cantharidin and its derivatives. Prog Modern Biomed. (2012) 12:1586–9.
42. Huang X, Xie W, Yu X, Fan C, Wang J, Cao Y, et al. Methyl-cantharidimide inhibits growth of human hepatocellular carcinoma cells by inducing cell cycle arrest and promoting apoptosis. Front Oncol. (2019) 9:1234. doi: 10.3389/fonc.2019.01234
43. Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Investig. (1996) 97:2517–24. doi: 10.1172/JCI118699
44. Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene. (2003) 22:7340–58. doi: 10.1038/sj.onc.1206938
45. Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H. p53RDL1 regulates p53-dependent apoptosis. Nat Cell Biol. (2003) 5:216–23. doi: 10.1038/ncb943
46. Newman MJ, Rodarte JC, Benbatoul KD, Romano SJ, Zhang C, Krane S, et al. Discovery and characterization of OC144-093, a novel inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Res. (2000) 60:2964–72.
47. Fan YF, Zhang W, Zeng L, Lei ZN, Cai CY, Gupta P, et al. Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett. (2018) 421:186–98. doi: 10.1016/j.canlet.2018.01.021
48. Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, et al. Mutations at amino-acid 482 in the ABCG 2 gene affect substrate and antagonist specificity. Br J Cancer. (2003) 89:1971–8. doi: 10.1038/sj.bjc.6601370
49. Fujii R, Mutoh M, Sumizawa T, Chen ZS, Yoshimura A, Akiyama SI. Adenosine triphosphate-dependent transport of leukotriene C4 by membrane vesicles prepared from cisplatin-resistant human epidermoid carcinoma tumor cells. JNCI J Natl Cancer Institute. (1994) 86:1781–4. doi: 10.1093/jnci/86.23.1781
50. Fujii RI, Mutoh M, Niwa K, Yamada K, Aikou T, Nakagawa M, et al. Active efflux system for cisplatin in cisplatin-resistant human KB cells. Jap J Cancer Res. (1994) 85:426–33. doi: 10.1111/j.1349-7006.1994.tb02376.x
51. Shen DW, Akiyama SI, Schoenlein P, Pastan I, Gottesman MM. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: cross-resistance and protein changes. Br J Cancer. (1995) 71:676–83. doi: 10.1038/bjc.1995.134
52. Johnson SW, Shen DW, Pastan I, Gottesman MM, Hamilton TC. Cross-resistance, cisplatin accumulation, and platinum–DNA adduct formation and removal in cisplatin-sensitive and-resistant human hepatoma cell lines. Exp Cell Res. (1996) 226:133–9. doi: 10.1006/excr.1996.0211
53. Liang XJ, Finkel T, Shen DW, Yin JJ, Aszalos A, Gottesman MM. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol Cancer Res. (2008) 6:1499–506. doi: 10.1158/1541-7786.MCR-07-2130
54. Zhang Y, Maurizi MR. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochimica et Biophysica Acta. (2016) 1862:252–64. doi: 10.1016/j.bbadis.2015.12.005
55. Shen DW, Liang XJ, Gawinowicz MA, Gottesman MM. Identification of cytoskeletal [14C] carboplatin-binding proteins reveals reduced expression and disorganization of actin and filamin in cisplatin-resistant cell lines. Mol Pharmacol. (2004) 66:789–93. doi: 10.1124/mol.66.4.789
56. Llambi F, Causeret F, Bloch-Gallego E, Mehlen P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. (2001) 20:2715–22. doi: 10.1093/emboj/20.11.2715
57. Levine AJ. The~ 53 proto-oncogene can act as a suppressor of transformation. Cell. (1989) 57:1083–93. doi: 10.1016/0092-8674(89)90045-7
59. Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M. Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature. (1997) 386:833–8. doi: 10.1038/386833a0
60. Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. (1995) 92:8493–7. doi: 10.1073/pnas.92.18.8493
61. Zhou X, Yang G, Huang R, Chen X, Hu G. SVH-B interacts directly with p53 and suppresses the transcriptional activity of p53. FEBS Lett. (2007) 581:4943–8. doi: 10.1016/j.febslet.2007.09.025
62. Wang B, Ma LY, Wang JQ, Lei ZN, Gupta P, Zhao YD, et al. Discovery of 5-cyano-6-phenylpyrimidin derivatives containing an acylurea moiety as orally bioavailable reversal agents against P-glycoprotein-mediated mutidrug resistance. J Med Chem. (2018) 61:5988–6001. doi: 10.1021/acs.jmedchem.8b00335
63. Zhang W, Fan YF, Cai CY, Wang JQ, Teng QX, Lei ZN, et al. Olmutinib (BI1482694/HM61713), a novel epidermal growth factor receptor tyrosine kinase inhibitor, reverses ABCG2-mediated multidrug resistance in cancer cells. Front Pharmacol. (2018) 9:1097. doi: 10.3389/fphar.2018.01097
64. Ciardiello F, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. (2000) 6:2053–63.
Keywords: methyl-cantharidimide (MCA), multidrug resistance (MDR), ABCB1, ABCG2, cisplatin resistance, cell apoptosis, unc-5 netrin receptor B (UNC5B)
Citation: Li Y-D, Mao Y, Dong X-D, Lei Z-N, Yang Y, Lin L, Ashby CR Jr, Yang D-H, Fan Y-F and Chen Z-S (2020) Methyl-Cantharidimide (MCA) Has Anticancer Efficacy in ABCB1- and ABCG2-Overexpressing and Cisplatin Resistant Cancer Cells. Front. Oncol. 10:932. doi: 10.3389/fonc.2020.00932
Received: 21 April 2020; Accepted: 12 May 2020;
Published: 26 June 2020.
Edited by:
Jin-Ming Yang, University of Kentucky, United StatesReviewed by:
Tatsuhiko Furukawa, Kagoshima University, JapanJun Lin, Stony Brook Medicine, United States
Copyright © 2020 Li, Mao, Dong, Lei, Yang, Lin, Ashby, Yang, Fan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying-Fang Fan, ZmFueWYwNjg3MDAmI3gwMDA0MDtzaW5hLmNvbQ==; Zhe-Sheng Chen, Y2hlbnomI3gwMDA0MDtzdGpvaG5zLmVkdQ==
†These authors have contributed equally to this work