
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 May 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00779
 Ignazio Verzicco1
Ignazio Verzicco1 Giuseppe Regolisti2
Giuseppe Regolisti2 Federico Quaini3
Federico Quaini3 Pietro Bocchi1
Pietro Bocchi1 Irene Brusasco1
Irene Brusasco1 Massimiliano Ferrari1
Massimiliano Ferrari1 Giovanni Passeri4
Giovanni Passeri4 Valentina Cannone1Pietro Coghi1
Valentina Cannone1Pietro Coghi1 Enrico Fiaccadori2
Enrico Fiaccadori2 Alessandro Vignali1
Alessandro Vignali1 Riccardo Volpi1,4
Riccardo Volpi1,4 Aderville Cabassi1*
Aderville Cabassi1*The use of antineoplastic drugs has a central role in treatment of patients affected by cancer but is often associated with numerous electrolyte derangements which, in many cases, could represent life-threatening conditions. In fact, while several anti-cancer agents can interfere with kidney function leading to acute kidney injury, proteinuria, and hypertension, in many cases alterations of electrolyte tubular handling and water balance occur. This review summarizes the mechanisms underlying the disturbances of sodium, potassium, magnesium, calcium, and phosphate metabolism during anti-cancer treatment. Platinum compounds are associated with sodium, potassium, and magnesium derangements while alkylating agents and Vinca alkaloids with hyponatremia due to syndrome of inappropriate antidiuretic hormone secretion (SIADH). Novel anti-neoplastic agents, such as targeted therapies (monoclonal antibodies, tyrosine kinase inhibitors, immunomodulators, mammalian target of rapamycin), can induce SIADH-related hyponatremia and, less frequently, urinary sodium loss. The blockade of epidermal growth factor receptor (EGFR) by anti-EGFR antibodies can result in clinically significant magnesium and potassium losses. Finally, the tumor lysis syndrome is associated with hyperphosphatemia, hypocalcemia and hyperkalemia, all of which represent serious complications of chemotherapy. Thus, clinicians should be aware of these side effects of antineoplastic drugs, in order to set out preventive measures and start appropriate treatments.
A series of electrolyte derangements can develop during treatment with anti-cancer drugs. While some of these alterations may be paraneoplastic (1), in many cases specific pharmacodynamic mechanisms can be identified impacting on fluid and electrolyte metabolism. Besides the possible occurrence of acute kidney injury, proteinuria, and hypertension (2), several antineoplastic agents can affect electrolytes tubular handling, as well as urinary water excretion by interfering with antidiuretic hormone (ADH). The aim of this review is to analyze in detail the mechanisms underlying the disorders of the metabolism of sodium, magnesium, potassium, calcium, and phosphate during anti-cancer drug treatment.
An extensive review of English language literature was performed to identify all relevant articles describing the epidemiology, pathogenesis, preventive measures, and outcomes of electrolyte disorders induced by antineoplastic drugs. To this purpose, we searched PubMed, EMBASE™, CINHAL, Web of Science and Cochrane databases for relevant articles. Related search terms were used as follow: (“Hyponatremia”[Mesh]) OR “Diabetes Insipidus, Neurogenic”[Mesh]) OR “Diabetes Insipidus, Nephrogenic”[Mesh]) OR “Hypokalemia”[Mesh]) OR “Magnesium Deficiency”[Mesh]) OR “Hypercalcemia”[Mesh]) OR “Hypocalcemia”[Mesh]) OR “Hypophosphatemia”[Mesh]) AND “Antineoplastic Agents”[Mesh]).
Medical subject heading terms were used to enhance electronic searches. Additional studies of interest were identified by hand searches of references, and at least two reviewers independently reviewed each article for eligibility. Conference proceedings were excluded. The search was last updated on December 12, 2019. Nomenclature of drugs and their molecular targets conforms the recently published IUPHAR/BPS Guide to Pharmacology nomenclature classification (3).
Sodium (Na+) is the main cation of extracellular space, where it is actively extruded from the intracellular space by sodium-potassium ATPase. Total body Na content is pivotal in maintaining extracellular fluid and arterial volume, both related to tubular Na reabsorption and urinary excretion. Serum Na concentration (136-144 mmol/L) depends on the ratio of total body exchangeable Na and potassium to total body water (TBW). Serum Na changes are mainly related to TBW, which is regulated by ADH (4). Hyponatremia (Na<136 mmol/L) is classified as mild, moderate and severe degrees when serum Na level is between 130 and 135, 120 and 129 and lower than 120 mmol/l, respectively (1). In cancer patients, hyponatremia has an overall prevalence up to 47% with mild, moderate and severe degrees accounting for 36, 10, and 1% (5). Hyponatremia is as an independent risk factor for mortality and prolonged hospitalization (6, 7). According to extracellular fluid volume, hypovolemic, hypervolemic, and euvolemic hyponatremia can be distinguished (8). Hypovolemic hyponatremia results from a loss of TBW lower than total body Na and occurs in salt-wasting nephropathy, renal or extrarenal losses or adrenal insufficiency. Hypervolemic hyponatremia results from a rise of TBW greater than the excess of total body Na and occurs in either edematous disorders such as congestive heart failure, decompensated liver cirrhosis, nephrotic syndrome, or in end-stage renal failure. In edematous disorders, when effective arterial volume is reduced and ADH levels are high, the use of non-steroidal anti-inflammatory drugs (both non-selective and cyclooxygenase-2 selective drugs) can affect free water excretion and further increase ADH secretion and activity (9), determining or worsening hyponatremia (10). Euvolemic hyponatremia is the most frequent hypotonic disorder and presents with normal extracellular volume. The syndrome of inappropriate antidiuretic hormone secretion (SIADH) represents the classic euvolemic hyponatremia where ADH secretion, despite the hypo-osmolality state, is not suppressed and is potentiated by several drugs (antineoplastic treatments, antidepressant such as selective serotonin reuptake inhibitors, antipsychotics, anti-epileptics such as carbamazepine, oxcarbazepine, eslicarbazepine, sodium valproate, lamotrigine, levetiracetam, and gabapentin) (11) or alternatively is oversecreted as the expression of a paraneoplastic syndrome. Hyponatremia constitutes a potential complication of thiazide diuretic treatment through mechanisms affecting maximal urinary dilution ability, through stimulation of ADH release and ensuing euvolemic hyponatremia (12). Again, the simultaneous use of non-steroidal anti-inflammatory drugs can facilitate the development of hyponatremia (10).
Conversely, hypernatremia (serum Na > 144 mmol/L) occurs mostly as a result of an excessive loss of TBW relative to Na content, leading to a free water deficit (13). Hypernatremia (prevalence ~3%) is far less frequent than hyponatremia in hospitalized cancer patients (14).
Clinical manifestations of hypo- and hypernatremia are similar, and concern mainly the central nervous system: they are related to the severity and the rate of development of serum Na derangement (13). Both hypo- and hypernatremia can be associated with confusion, behavioral changes, headache, irritability, nausea and vomiting, lethargy, drowsiness/coma, seizures, and respiratory arrest.
Potassium (K+) is the main intracellular cation and is fundamental for resting membrane potential. Altered serum K concentration can modify the electrical activity of excitable cells (cardiac myocytes, skeletal muscle cells and vascular smooth myocytes), leading to serious adverse effects, such as life-threatening arrhythmias. Normal serum K levels range between 3.5 and 5.3 mmol/L. Chemotherapeutic agents may induce serum K derangements mainly through alterations of renal tubular transport. Hypokalemia (K < 3.5 mmol/L) prevalence is around 12% in cancer population patients (15); this figure increases between 43 and 64% in acute leukemia (16). Hyperkalemia (K > 5.3 mmol/L) is often related to tumor lysis syndrome (TLS) or to acute and/or chronic oliguric kidney disease.
Magnesium (Mg++) is the second most abundant divalent cation in the human body (1). It is mainly stored in bone, muscle, and soft tissues, and is important for neurotransmission, protein, and DNA synthesis, hormone-receptor interaction. Its normal serum concentration ranges between 1.6 and 2.6 mg/dL (0.65-1.07 mmol/L), and its homeostasis depends on intestinal absorption and renal excretion. In the kidney, transient receptor potential cation channel, subfamily M, member 6 (TRPM6), a Mg channel located in the apical cellular membrane of the thick ascending limb of Henle's loop and distal convoluted tubule, and exerts the rate-limiting step for Mg tubular transport. The activity of TRPM6 is regulated by the epidermal growth factor (EGF) and its receptor (EGFR) (17, 18). As TRPM6 and EGF/EGFR are mainly expressed in the distal convoluted tubule, this segment represents the main site of regulation of urinary Mg excretion. Mild hypomagnesemia can be pauci-symptomatic, whereas a severe disorder can represent a life-threatening condition. Symptoms involve cardiovascular system, with electrocardiographic alterations (prolonged QT interval), and the neuromuscular system with tremor, paresthesia, tetany, spasms, and seizures (1). Hypomagnesemia is also associated with reduced release and activity of parathyroid hormone (PTH) and reduced synthesis of active vitamin D and its receptors (19). Both hepatic 25-hydroxylation and renal 1α-hydroxylation of vitamin D leading to the active form of 1,25-dihydroxycholecalciferol are magnesium-dependent process (20) In addition, magnesium plays a central role in natural and adaptive immunity by interacting with vitamin D metabolites activity (21). In hospitalized cancer patients, hypomagnesemia (Mg < 1.5 mg/dL) has a prevalence around 17 % although antineoplastic drugs can increase this figure (e.g., up to 90% with cisplatin) (22, 23).
Calcium (Ca++) plays important roles in intracellular signaling, neurotransmission, membrane stability and bone metabolism. Its homeostasis is regulated by PTH via calcium-sensing receptors on parathyroid cells. PTH stimulates tubular Ca reabsorption, mobilizes Ca from bone and enhances the synthesis of 1,25-dihydroxycholecalciferol, which in turn increases Ca intestinal absorption, modulates PTH release and PTH-mediated bone demineralization. Serum total Ca concentration ranges between 8.5 and 10.5 mg/dL, with ionized Ca between 4.7 and 5.2 mg/dL (24). In hospitalized cancer patients, hypocalcemia (Ca < 8.5 mg/dL) has a prevalence around 13% (23). Generally, symptomatic hypocalcemia presents with irritability, tetany, psychosis, and prolonged QT interval (1). Ca levels should be checked in case of hypomagnesemia, because of a concomitant low PTH activity (22). Agents, such as platinum-compounds and anti-EGFR Monoclonal Antibodies (MoAbs), causing hypomagnesemia may also induce hypocalcemia (25). Hypercalcemia is far more common than hypocalcemia, ranging between 20 and 30% in patients with advanced cancer, in particular lung, breast and hematological malignancies (26). Three major mechanisms of hypercalcemia have been identified: (i) PTH-related peptide secretion by cancer cell, (ii) osteolytic lesion, (iii) 1,25-dihydroxycholecalciferol (calcitriol) secretion by the cancer cells.
Phosphate () is predominantly stored as an inorganic salt in bone as hydroxyapatite crystals. In plasma phosphate circulates both as an inorganic anion, or as an organic component of intracellular nucleic acids and cell membranes. Normal phosphate levels range between 2.5 and 4.5 mg/dL, and result from the balance between intestinal absorption, renal excretion, and release from the bone exchangeable fraction which is regulated by PTH, fibroblast growth factor 23 and calcitriol. Fibroblast growth factor 23 and PTH decrease phosphate serum levels by inhibition of tubular reabsorption. Conversely, calcitriol increases the intestinal absorption of phosphate and inhibits PTH secretion. Hypophosphatemia occurs as a result of phosphate redistribution between extra- and intracellular compartments, poor intestinal absorption, increased renal excretion, or prolonged hemodialysis/hemofiltration. Clinical manifestations include weakness, proximal myopathy, rhabdomyolysis, hemolytic anemia, and heart failure. Antineoplastic drugs may induce tubular damage and thus alter phosphate reabsorption, resulting in the development of hypophosphatemia. Proximal convoluted tubule dysfunction determines urinary wasting of phosphate, glucose, urate, and bicarbonate and leads to the acquired Fanconi Syndrome (FS). Phosphate levels lower than 2.5 mg/dL were reported in 49% of patients with advanced cancer whereas a lower fraction (23%) had phosphate levels less than 2.0 mg/dL (27). Hyperphosphatemia (phosphate > 4.5 mg/dL) is related to TLS, particularly in hematologic malignancies, and occurs more frequently as a consequence of chemotherapy than in spontaneous TLS (28).
Platinum-derived drugs include cisplatin, carboplatin, oxaliplatin and nedaliplatin, and electrolyte disorders associated with these drugs are shown in Table 1. Nephrotoxicity represents the limiting factor of these drugs (2, 22). Compared with cisplatin and nedaliplatin, carboplatin and oxaliplatin appear to be less nephrotoxic and associated with less electrolyte derangements (34). Cisplatin nephrotoxicity results from cell damage in the S3 segment of the proximal tubule, distal convoluted tubules and collecting ducts (22). Electrolyte disorders are also related to cisplatin-induced DNA damage of thiazide-sensitive sodium-chloride co-transporter genes and to the apoptosis of distal tubule cells (34). Cisplatin treatment may cause hyponatremia through SIADH, related to both higher secretion of and sensitivity to ADH (32). Nausea and vomiting, which are common side effects of platinum-derived chemotherapy, are also powerful stimuli for ADH secretion. The incidence of hyponatremia can reach 59% (severe hyponatremia 12%) with cisplatin, whereas 20% is reported with carboplatin (29–31). Rarely, cisplatin-related hyponatremia may result from Renal Salt Wasting Syndrome (33). Hypernatremia can also develop with cisplatin due to acquired nephrogenic diabetes insipidus with ensuing hypotonic polyuria (34).
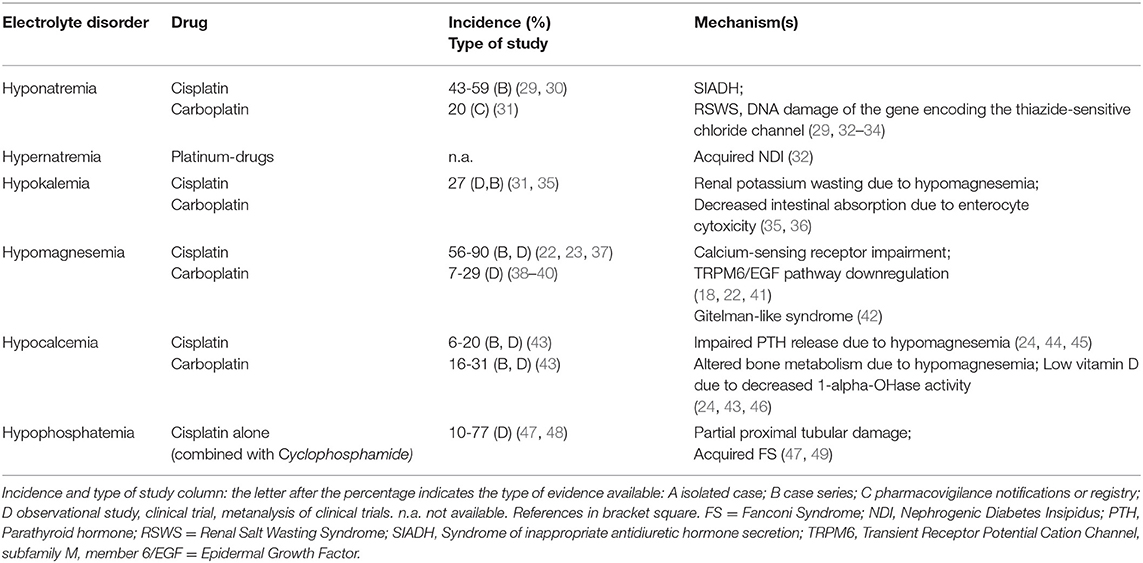
Table 1. Platinum derived drugs.
Platinum-derived agents can induce hypokalemia due to renal K wasting secondary to hypomagnesemia. The incidence of cisplatin-related hypokalemia is around 27% (35). Intracellular magnesium depletion reverts inactivation of voltage-dependent renal outer medulla K channels (ROMK), thus increasing kaliuresis. Increased distal Na delivery or elevated aldosterone levels are also required for exacerbating K wasting (36). Potassium supplementation may fail to correct hypokalemia until hypomagnesemia is corrected.
Hypomagnesemia is the most frequent electrolyte alteration caused by cisplatin and is related to its cumulative dose (50). Hypomagnesemia is associated with shorter survival (35, 50), and its incidence ranges between 56 and 90% of patients receiving cisplatin, being lower with carboplatin (22, 23, 37–39). It is mainly related to an impaired Mg reabsorption in the proximal tubule. However, cisplatin was shown to downregulate the TRPM6/EGF pathway resulting in Mg loss (41), and patients receiving platinum drugs can also develop persistent distal tubular dysfunction with a Gitelman-like syndrome characterized by hypocalciuria, hypomagnesemia and hypokalemic metabolic alkalosis (42).
Cisplatin long-term treatment can cause hypocalcemia in a dose-dependent manner (46); low-dose cisplatin combined with 5-Fluorouracil and interferon-alpha can also induce hypocalcemia (44). Frequently, hypocalcemia is associated with hypomagnesemia (45). Cisplatin can decrease 1-alfa-hydroxylation activity and result in low vitamin D3 levels. In cisplatin-treated patient, hypocalcemia incidence is around 6–20%, and is 16–31% in those treated with carboplatin (43).
Hypophosphatemia may often complicate treatment with platinum agents, with incidence reaching up to 77% (47, 48). Cisplatin causes proximal tubule cell apoptosis and necrosis, resulting in partial or complete acquired FS (49).
Considering the development of the individual ion disturbances, there are also differences related to the cumulative dose and to the cycles of chemotherapy schedule. In particular, among platinum-derived drugs, cisplatin-induced hyponatremia occurred in 50% of the patients after a median critical dose of 195 mg at cycle 2, while higher cumulative doses had to be administered to observe hypokalemia (560 mg at cycle 7). Median critical doses for development of hypomagnesemia and hypocalcemia in 50% of the patients were 160 mg and 240 mg at cycle 2 and 3, respectively, supporting the concept of a lower dose required to induce hypomagnesemia versus others ion disorders (35).
Table 2 shows the electrolyte disorders observed in patients treated with Alkylating agents. These drugs can frequently cause hyponatremia by impairing free water excretion. Hyponatremia usually occurs 12–48 h after the administration of cyclophosphamide (CYC). CYC can induce severe symptomatic hyponatremia through different mechanisms: (i) a tumor lysis-related SIADH due to massive release of ADH or ADH-like peptides from damaged tumor cells or normal pituitary cells. (56), (ii) an ADH-like activity of CYC metabolites on renal collecting tubules (57), (iii) an upregulation of vasopressin V2 receptors and aquaporin-2 channels through the suppression of IL-1 and TNF-α, leading to increased ADH effects (58), (iv) a CYC-induced nephrogenic SIADH with activation of vasopressin V2 receptors in absence of ADH stimulation (59). Although most cases (up to 89%) are related to intravenous treatment with single-pulse high-dose of CYC (51, 69), hypotonic hyponatremia was also reported after low-dose treatment (14% incidence) (51). High doses of CYC (30–40 mg/kg of body weight), moderate doses (20–30 mg/kg) and low doses (<20 mg/kg) can produce hyponatremia with different frequencies (51–53, 69). Indeed, as a half-saline hydration protocol is performed before and after CYC administration in order to force diuresis and minimize the risk of hemorrhagic cystitis, the administration of large volumes of hypotonic fluid can facilitate hypotonic hyponatremia during CYC treatment. Thus, isotonic solutions should be preferred as treatment. Several cases of SIADH-related hyponatremia have also been reported with chlorambucil, melphalan, busulfan, or ifosfamide (54, 60).
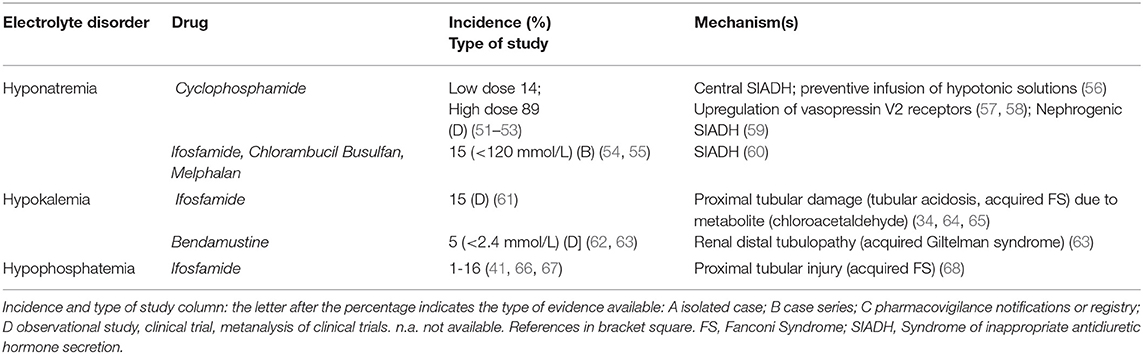
Table 2. Alkylating agents.
Ifosfamide (especially when combined with cisplatin) may induce hypokalemia as a consequence of proximal or distal tubular acidosis, or acquired FS. Ifosfamide enters proximal tubular cells through organic cation transporter 2 and, after metabolization to chloracetaldehyde, induces glutathione depletion and lipid peroxidation (64, 65). Ifosfamide is also associated with proximal tubular injury, phosphaturia, and hypophosphatemia especially at cumulative doses greater than 60 g/m2: its incidence is between 1% (moderate dose, 1–1.5 g/m2 over 1–10 days) and 16 % (high dose, 3.33 g/m2, over 1–4), but can increase if patients are pretreated with cisplatin (47, 55, 68, 70).
Bendamustine, a chemotherapeutic agent designed to have both alkylating and antimetabolite properties, can induce hypokalemia (severe hypokalemia in around 5%) through a distal tubulopathy (acquired Gitelman syndrome) and a mild diuretic effect (62, 63). Special attention should be given to patients with pre-existing hypokalemia and if the cumulative dose of bendamustine exceeds 1,080 mg/m2 in the chemotherapy scheme (63).
Electrolyte disorders reported in target therapies-treated patients are described in Tables 3–5 part I, II, III. Hyponatremia is one of the most frequent derangement. According to a recent meta-analysis, the highest incidence (63.4%) of all-grade hyponatremia was observed in patients treated with Brivanib, a selective type 1 fibroblast growth factor receptor/vascular endothelial growth factor receptor-2 (VEGFR-2) antagonist combined with Cetuximab, a recombinant chimeric monoclonal antibody anti-EGFR (Her1). Hyponatremia was also frequent (31.7%) after monotherapy with Pazopanib, a multikinase inhibitor of VEGFR-2/platelet-derived growth factory receptor (PDGFR) (6). Lower incidence was seen with Afatinib, a protein kinase inhibitor inhibiting EGFR 2 (Her2) (1.7%) (6). Cediranib, a pan-VEGFR tyrosine-kinase inhibitor showed a 65% incidence of overall hyponatremia (72). The highest incidence of severe hyponatraemia (<120 mmol/l) was reported with cetuximab (34.8%), while the lowest incidence with gefitinib, an EGFR inhibitor (1.0%) (6).
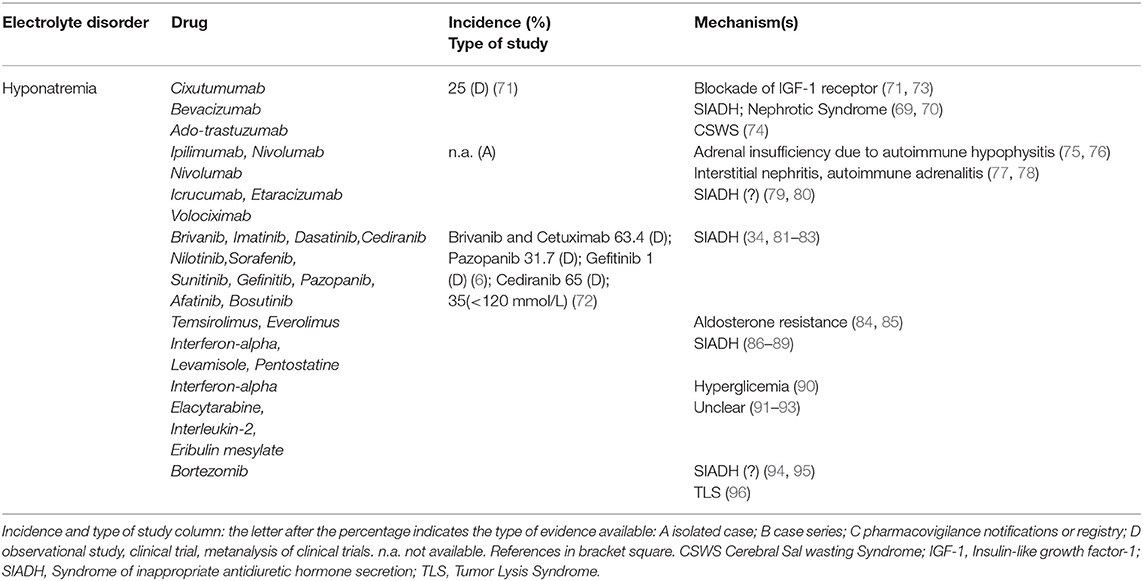
Table 3. Target therapies (part I).
Cixutumumab, an anti-insulin-like growth factor receptor 1 antibody, induced hyponatremia in 25% of patients in a phase II safety evaluation study (71); the mechanism involves an increase of fractional sodium excretion (73). In patients treated with Icrucumab and Bevacizumab, recombinant human MoAbs against vascular endothelial growth factor receptor-1 and VEGF-A, hyponatremia emerged as a dose-limiting toxicity factor (126). Since Bevacizumab increases the risk of severe proteinuria (127), hypervolemic hyponatremia has also been proposed as a consequence of nephrotic syndrome. Trastuzumab Emtasine, inhibitor of EGFR 2 (Her2) signaling, at a dose of 3.6 mg/kg every 3 weeks, can induce true hyponatremia by a Cerebral Salt Wasting Syndrome as reported in a patient with breast cancer and brain metastasis who needed repeated hospitalizations because of severe hyponatraemia (74). Both Etaracizumab, a humanized monoclonal antibody against αvβ3 integrin receptor, and Volociximab, a chimeric monoclonal antibody directed against human α5β1 integrin, were also associated with severe hyponatremia (79, 80). Ipilimumab, a monoclonal antibody against cytotoxic T lymphocyte antigen-4, may induce hypophysitis leading to hypopituitarism, adrenocortical insufficiency and hyponatremia (75). Loss of the regulatory effects of cortisol on ADH release may induce SIADH. Hyponatremia was also observed after Nivolumab and Pembrolizumab administration, two programmed-death-1 pathway inhibitors. Nivolumab-related hyponatremia can also involve SIADH-independent mechanisms, causing a true Na depletion such as in autoimmune hypophysitis with an isolated ACTH deficiency and secondary adrenal insufficiency (76), or a tubulointerstitial nephritis (77), or an adrenalitis with primary adrenal insufficiency (78). The first case of nivolumab-induced adrenalitis resulting in primary adrenal failure with hyponatraemia was described in a 43-year-old man that started nivolumab (3 mg/kg) at two weekly intervals, whose serum sodium dropped to 127 mmol/L after four cycles (78).
Tyrosine Kinase Inhibitors (TKI) treatment has been associated with SIADH. Hyponatremia-related SIADH was reported in patients affected by Bcr-Abl acute lymphoblastic leukemia treated by Imatinib at a dose of 400 mg/day, and was observed after 11 months of therapy (81), in those affected by refractory chronic myelogenous leukemia treated with Dasatinib (100 mg/day), Nilotinib (400 mg twice/day) and Bosutinib (500 mg/day) (82), as well as in patients with renal carcinoma or with metastatic head and neck cancer treated with Sorafenib (400 mg twice/day for a median duration of therapy of 3.4 months) (83).
Mammalian targets of rapamycin (mTOR) inhibitors. Different grades of hyponatremia have been reported with Temsirolimus and Everolimus, either in monotherapy or in combination, with aldosterone resistance being suggested as the main pathogenetic mechanism (84, 85).
Immunomodulators. Serum Na derangements have been reported with Interferon therapy in the past 2–3 decades (86). Hyponatremia was described after 7 days of Interferon treatment in a patient in therapy with carbamazepine, supporting an additive effect of both drugs on SIADH development (87). A case of translocational hyponatremia due to hyperglycemia was reported in a patient with Interferon-induced diabetes mellitus (90). Levamisole and Pentostatin also appear to be associated with hyponatremia, presumably via SIADH (88, 89). Different degrees of hyponatremia were also observed in patients treated with Eribulin mesilate, Elacytarabine and recombinant Interleukin-2 (91–93), although the mechanisms are not clarified.
Proteasome inhibitor. Hyponatremia secondary to SIADH is not frequent in multiple myeloma even though bortezomib treatment, alone or associated with dexamethasone, was reported to increase ADH secretion and effect (94). In a case report, the finding of the ADH-positivity of monoclonal plasma cells at immunohistochemical analysis, suggested TLS as the pathogenetic mechanism (96). In another case report, a patient treated for multiple myeloma developed SIADH-related hyponatremia 3 cycles after starting 1.3 mg/m2 bortezomib (95).
Treatment with MoAbs and Target Therapies can affect potassium metabolism (Table 4, part II). Cetuximab and other anti-EGFR agents decrease potassium levels through an impairment in magnesium balance (99). Combined grade 3 and 4 hypokalemia (serum K between 2.5–3.0 mmol/L, and <2.5 mmol/L, respectively) had an incidence of 6.2%, and all-grade hypokalemia of 8.0%, in cetuximab-treated patients (97). The anti-HER3 and anti-HER2 antibodies lumretuzumab and pertuzumab combined with paclitaxel induced grade 3 hypokalemia in 40% of patients with breast cancer (98). Bevacizumab can induce hypokalemia due to proximal tubular damage (100). Hypokalemia is also reported with Tremelimumab, Blinatumomab and Eribulin mesylate through an unclear mechanism, possibly via drug-induced diarrhea (102–104). Novel TKI as Volasertib (105) and mTOR inhibitors, particularly Everolimus (101), were associated with hypokalemia.
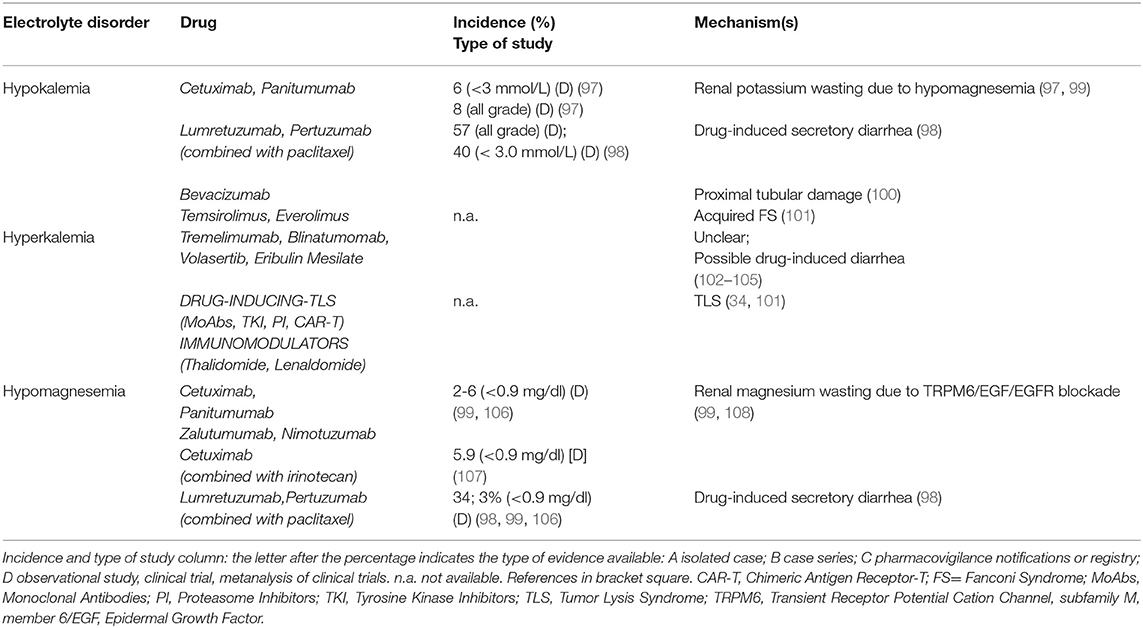
Table 4. Target therapies (part II).
Conversely, hyperkalemia has been observed in cancer patients as a consequence of TLS, sepsis, and adrenal insufficiency due to metastatic disease (Table 4, part II). Other mechanisms include the suppression of insulin release, reducing potassium intracellular uptake, in Octreotide-treated patients, as well as transcellular potassium shift or TLS in Thalidomide-treated patients (at doses ranging between 100 and 400 mg/die) (36). TKI Axitinib induces hyperkalemia through TLS development and distal tubular dysfunction such as in hyperkalemic type 4 renal tubular acidosis (128).
Electrolyte imbalances increased considerably after the introduction of anti-EGFR MoAbs into therapy (129) (Table 4, part II). A recent meta-analysis of 25 randomized controlled trials reported an incidence of hypomagnesemia of 34%, whereas those of hypokalemia and hypocalcemia were 14 and 17%, respectively. Cetuximab increased 6 times the risk of grade 3/4 hypomagnesemia (Mg serum between 0.7-0.9 or <0.7mg/dl, respectively) and grade 3/4 hypokalemia (17, 99, 130). The longer half-life and higher affinity of panitumumab for EGFR, as well as the overexpression of EGFR, are responsible for a high incidence of grade 3/4 hypomagnesemia and hypokalemia in colorectal cancer patients (99). Patients treated with cetuximab 400 mg/m2 at first dose and 250 mg/m2 weekly (or 500 mg/m2 every 2 weeks) or panitumumab 6 mg/kg (or 9 mg/kg according to the tumor types) developed hypomagnesemia and hypokalemia (99). Compared to cetuximab and panitumumab, zalutumumab is associated with less hypomagnesemia and hypokalemia (108). A recent phase 2 trial showed 5.9% incidence of hypomagnesemia when Cetuximab and Irinotecan were co-administrered (107) (Table 4, part II). Hypomagnesemia, by upregulating ROMK activity with ensuing potassium loss, is involved also in anti-EGFR MoAbs-induced hypokalemia (36). Magnesium supplementation should be considered in patients undergoing treatment with Anti-EGFR MoAbs. Some authors suggest empirically prophylactic administration of Mg at the beginning of treatment (1) and oral preparations are preferred in mild hypomagnesemia, while intravenous administration for severe depletion.
Cetuximab and Panitumumab can also induce hypocalcemia related to the underlying hypomagnesemia (99). TKI Imatinib can cause hypocalcemia and muscle cramps in up to 40% of patients. In a young woman with abdominal gastrointestinal stromal tumor, Imatinib (400 mg/day) led to hypocalcemia after 5 months of treatment (109) (Table 5, part III). A direct effect on c-Kit tyrosine kinase receptors of renal tubular cells with ensuing hypocalcemia represents the possible mechanism (110). Osteoclast inhibition and osteoblast activation with bone sequestration of Ca and phosphate and ensuing hypocalcemia may also be involved (111). Sorafenib (112), as well as Axitinib (114) and combined therapy with Erlotinib and Sunitinib (115), can cause hypocalcemia. A case of severe symptomatic hypocalcemia related to Nilotinib was also reported, with an immune-mediated destruction of the parathyroid glands or a drug interference with calcium sensing receptors (CaSRs) and ensuing insufficient PTH secretion being the suggested pathogenic mechanisms (113).
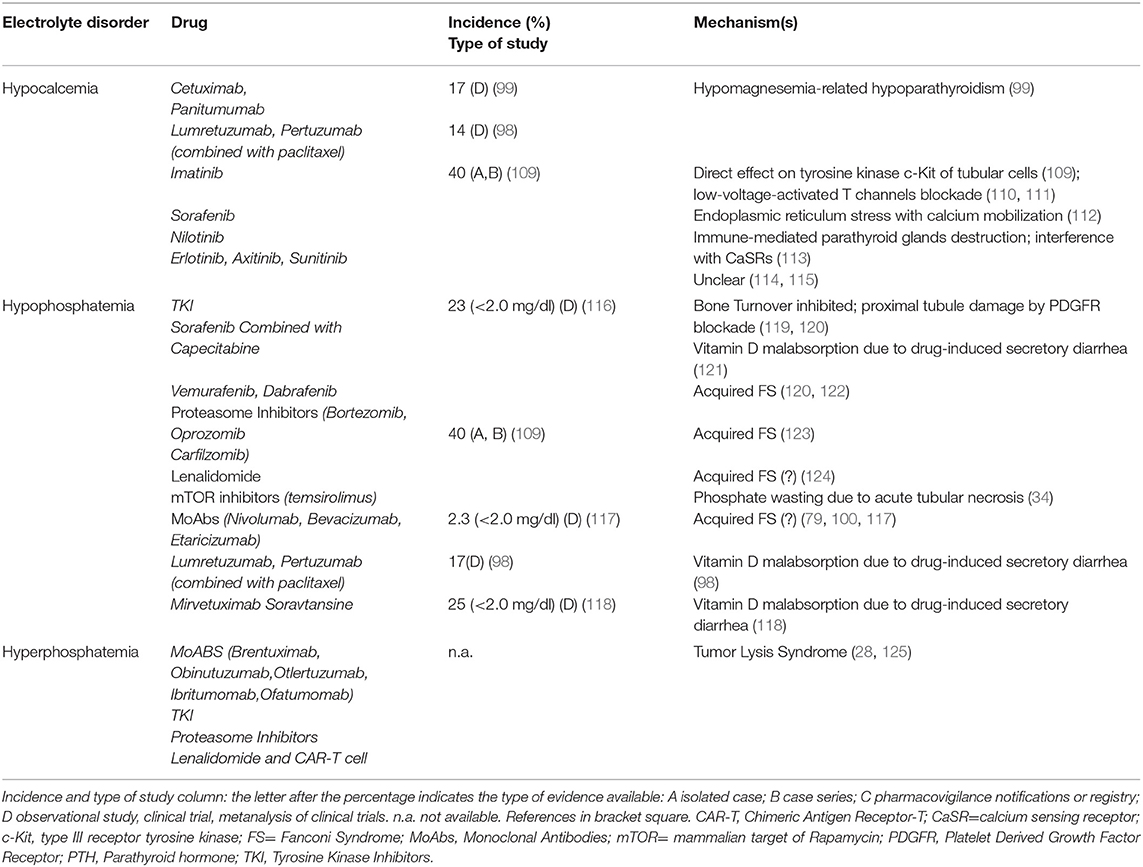
Table 5. Target therapies (part III).
TKIs (Imatinib, Sunitinib, Ceritinib) affect also phosphate balance via inhibition of bone turnover (Table 5, part III) (119, 131). TKI may affect bone turnover by inhibiting PDGFR in proximal tubular cells and induce, as in the case of imatinib, an acquired FS (120). Hypophosphatemia can be worsened by concomitant conditions, as in the case of sorafenib-induced diarrhea with consequent vitamin D malabsorption (121). Novel target therapies may also induce different grades of hypophosphatemia. Vemurafenib and rarely, Dabrafenib, both competitive inhibitors of mutated BRAF kinase, may cause acute (1–2 weeks) or subacute (1–2 months) tubular toxicity. Acute toxicity is associated with acquired FS while subacute toxicity to an immuno-allergic interstitial nephritis (122). Diarrhea-induced vitamin D malabsorption can be also responsible for hypophosphatemia in 25% of patients treated with Mirvetuximab Soravtansine, a folate receptor α-targeting antibody-drug conjugate (118). Finally, acquired FS was also reported in patients treated with Perifosine, a protein kinase B inhibitor (132), Lenalidomide (after three weeks at a dose of 15 mg/day) (124), proteasome inhibitors (123), mTOR inhibitors (34), and MoAbs (Nivolumab, Etaracizumab, Bevacizumab, with nivolumab showing a 2.3% incidence of grade 3 [<2.0 mg/dl] hypopophatemia) (79, 100, 117) (Table 5, part III). Among proteasome inhibitors, hypophosphatemia development was related to subcutaneous administration of 1.3 mg/m2 bortezomib during a 21-day cycle for 16 cycles, combined with thalidomide, dexamethasone, and panobinostat (133).
Finally, a special emphasis must be given to various target therapy drugs (Table 4, part II and Table 5, part III) causing diarrhea, a mechanism leading to a combination of electrolyte derangements including hypokalemia, hypomagnesemia, hypocalcemia, hypophosphatemia, normal anion gap (hyperchloremic) metabolic acidosis due to bicarbonate loss, and severe hypovolemia (102–104, 118, 121). Target therapy, and in general chemotherapy, can cause nausea and vomiting. Excessive vomiting, especially over a prolonged period of time, leads to hypovolemia and hypochloremic metabolic alkalosis due to loss of chloride and hydrogen ions that can be associated with hypokalemia and hypomagnesemia (134).
Vinca Alkaloids, including vincristine, vinblastin, and vindesine, may induce hyponatremia via SIADH, that commonly occurs 1–3 weeks after drug administration (range 2–21 days). The incidence of moderate-severe hyponatremia is around 11% (135) with vincristine, but a higher incidence was reported with vinblastine after 7 days of treatment (136) or even earlier (after 36 hours) after a variable vinblastine dose between 0.2 and 6 mg/m2/day (137).
SIADH can result from a direct neurotoxic effect on the hypothalamus with altered control of ADH secretion (138). Interestingly, antifungal azole therapy, that inhibits Vinca Alkaloids metabolism, leads to severe neurotoxicity and SIADH-related hyponatremia (139). Hypocalcemia has also been reported in Vinka alkaloyds-treated patients but its incidence is not known; the involved mechanism is the impairment of microtubule polymerization (140). Table 6 reports the electrolyte disorders associated with Vinka alkaloid agents.
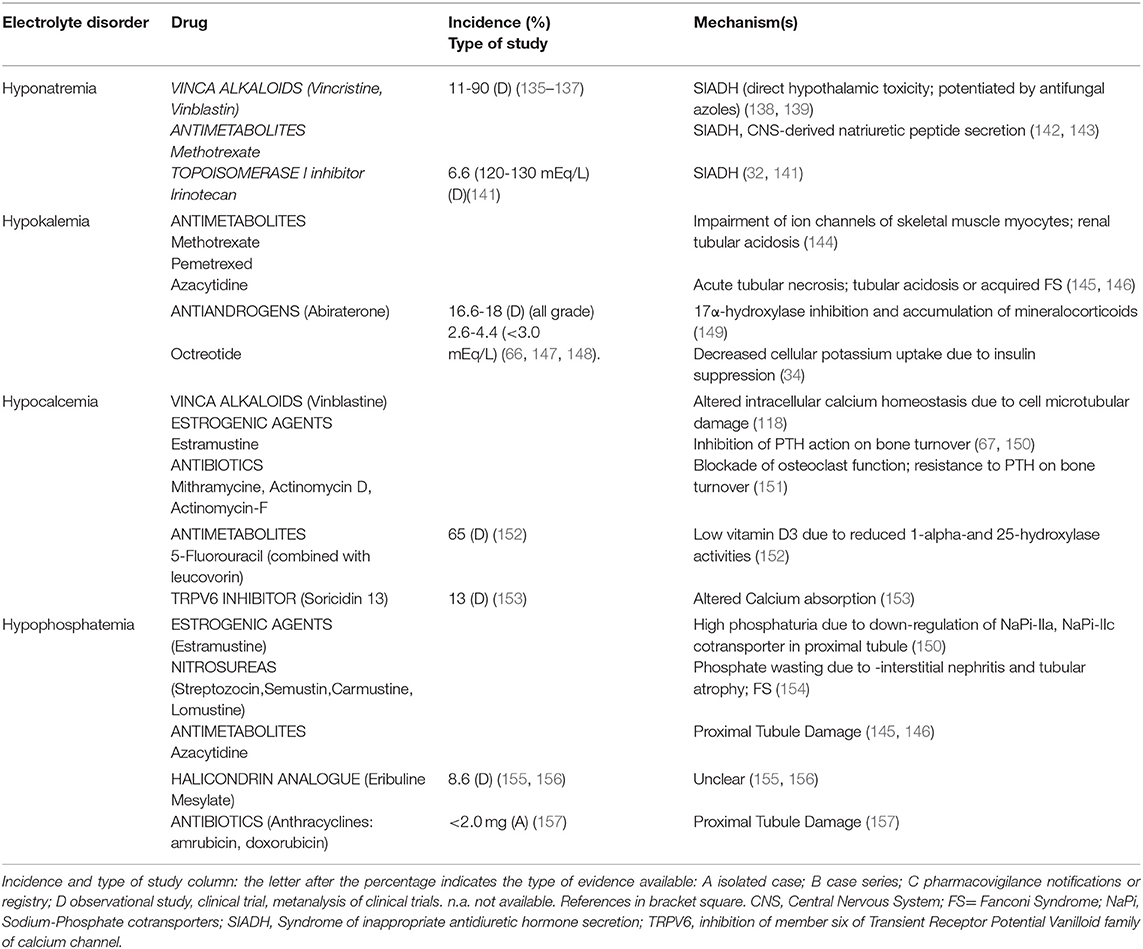
Table 6. Miscellaneous.
Both increased secretion of ADH (142) or a central nervous system-derived natriuretic peptide (143) are described as potential mechanisms of Methotrexate-induced hyponatremia (Table 6). Methotrexate (at a dose of 12 g/m2) can induce severe hypokalemia, as observed in a patient with transient hypokalemic tetraparesis occurring after intravenous high-dose administration (144). Azacytidine (75 mg/m2/day administered subcutaneously or intravenously, days 1–7 of each 28-day cycle for 6 cycles) may also induce hypokalemia and hypophosphatemia; potassium depletion can persist for weeks after stopping the drug, and necessitates prolonged parenteral supplementation (145, 146). The combination of 5-Fluorouracil (at a dose of 425–600 mg/m2/day for 5 consecutive days) with 5-formyl tetrahydrofolic acid (leucovorin, 20 mg/m2/day) can induce hypocalcemia due to inhibition of vitamin D 1- and 25-hydroxylation, leading to low levels of calcitriol (Table 6) (152).
Antiandrogens. Abiraterone inhibits both testicular and extra-testicular androgen synthesis by inhibiting 17α- hydroxylase and 17–20 liase resulting in decreased testosterone levels. The inhibition of 17α-hydroxylase leads to the accumulation of upstream mineralocorticoids that increase epithelial Na channel and the voltage-dependent ROMK activities in the distal nephron, resulting in increased cortical collecting duct potassium secretion and ensuing hypokalemia (149). The incidence of all-grade hypokalemia related to abiraterone (at a standard dose of 1,000 mg/day) ranges between 16.6 and 18% and between 2.6 and 4.4% when grade 3/4 hypokalemia (less than 3.0 mmol/L) is considered, and occurs after 2–4 weeks from the beginning of therapy (Table 6) (66, 149). Symptoms associated with mineralocorticoid excess and hypokalemia are managed by co-administration of low-dose prednisone, potassium chloride supplementation and/or a mineralcorticoid antagonist.
Estrogenic agents. Oral and intravenous administration of Estramustine (two 2,000 mg/m2 doses administered weekly) is associated with hypocalcemia and vitamin D3 deficiency after 4–5 weeks (67) (Table 6). Hypocalcemia may be due to inhibition of PTH action and impaired calcium mobilization from bone (67). Several studies reported phosphate wasting during high-dose estrogen treatments, hypothesizing a down-regulation of sodium phosphate co-transporters (NaPi-IIa and NaPi-IIc) in the proximal convoluted tubule through the activation of estrogen receptor-alpha, independently from the Klotho/FGF- 23 and PTH pathways (150) (Table 6).
Antibiotics. Mithramycin associates with hypocalcemia by blocking osteoclast function and PTH action on gut and bone directly, or by causing vitamin D resistance. Actinomicyn-D and Actinomycin-F also induce hypocalcemia by interfering with bone mineralization (151) (Table 6).
Nitrosureas. Streptozocin has a greater nephrotoxicity profile than other molecules, such as carmustine and lomustine. These drugs may induce renal toxicity through interstitial nephritis and tubular atrophy, resulting in FS (154).
Halichondrin B Analogue. Eribuline mesylate, a nontaxane inhibitor of microtubule dynamics has been associated with hypophosphatemia (around 8.6% in a phase I trial, at doses of 2.0 mg/m2/week, and with the liposomal formulation of the drug at doses between 1.0 and 3.5 mg/m2/week) (155, 156), but also with hyponatremia, hypomagnesemia, and hypokalemia (3–10%) (Table 6); the exact mechanisms of ion derangements have not yet been clarified.
TRPV6 Inhibitor. SOR-C13 reduces calcium intestinal absorption by the inhibition of member six of Transient Receptor Potential Vanilloid family of calcium channel (153).
Tumor Lysis Syndrome often occurs as a consequence of cytotoxic therapies, mostly in patients affected by hematological malignancies, leading to rapid release of cell constituents. It can be complicated by life-threatening conditions such as cardiac arrhythmias, acute kidney injury and neurologic impairment (158). The electrolyte derangements observed in TLS include hyperphosphatemia, hyperkalemia, and hypocalcemia, with the latter being the result of calcium precipitation in soft tissues secondary to acute hyperphosphatemia. The highest incidence of TLS is observed after Dinaciclib and Alvocidib treatment (between 15 and 53 % in acute leukemia trials) whereas the incidence is 8–10% for Venetoclax, CAR-T cell, Obinutuzumab, and <5% with Brentuximab, Carfilzomib, Lenalidomide, Dasatinib and Oprozomib. Idelalisib, and Ofatumumab have no reported cases of TLS (125).
First-line therapy represents the regimen(s) that is(are) generally accepted for the initial treatment of a specific type and stage of cancer, and is intended to cure the tumor when possible. The strategy of treatment should consider several factors, such as histology and molecular pathology of cancer, and age and comorbidities of the patient in order to set adequate therapeutic decisions (159).
In 2018, according to World Cancer Research Fund International, breast, colorectal, lung, and cervical cancers are the most common cancers in the female population whereas lung, prostate, and colorectal cancers are the most common malignancies in the male population (160). Chemotherapy with platinum-based drugs (preferably cisplatin) is recommended in the majority of lung cancers, as a first-line treatment for locally advanced non-small cell lung cancer (NSCLC), small-cell lung cancer (SCLC), unresectable malignant pleural mesothelioma and in association with immunotherapy (checkpoint inhibitors such as Pembrolizumab, an inhibitor of programmed cell death protein 1 pathway) in metastatic NSCLC and in those positive for programmed cell death protein 1. In patients with NSCLC and with a sensitizing EGFR mutation, first-line therapy considers TKI (erlotinib, afatinib). The most frequent electrolyte disorders in patients treated with platinum-derived drugs are mainly hypomagnesemia and hyponatremia, with the latter being related to cisplatin, pembrolizumab or TKI combined effect. Less frequently patients can also show hypophosphatemia, hypocalcemia, and hypokalemia (161–165) (Tables 1, 3–5). Androgen deprivation therapy (Goserelin, a luteinizing hormone releasing hormone agonist) and Docetaxel, a semisynthetic taxane, are used as first-line treatments for advanced metastatic prostate cancer; in castration-resistant prostate cancer patients the treatment includes abiraterone, which can lead to resistant hypokalemia (Table 6) (166). Early colon cancer first-line treatment includes adjuvant therapy based on several regimens responsible for electrolyte derangements. Leucovorin in combination with 5-Fluorouracil can often induce hypocalcemia; the FOLFOX (Leucovorin, 5-Fluorouracil, Oxaliplatin) or CAPOX (Capecitabin, the prodrug of 5-FU + Oxaliplatin) regimens can induce hypomagnesemia, hyponatremia and hypocalcemia, but with lower frequency compared to Cisplatin (34, 167). First-line therapy in metastatic colorectal cancer consists in chemotherapy doublet by combining the above regimens with the anti-EGFR antibodies (in patients with wild-type RAS), or bevacizumab (in the case of mutated RAS), or in chemotherapy triplet (FOLFOXIRI: 5-Fluorouracil, leucovorin, irinotecan, and oxaliplatin) plus bevacizumab in case of BRAF mutation: hyponatremia, hypomagnesemia, hypocalcemia, hypokalemia, and hypophosphatemia are the ion disorders that these patients can experience (Tables 1, 6) (168). The most frequently chemotherapeutic regimens used in early estrogen receptor-negative breast cancer are represented by anthracyclines and/or taxanes, although in selected patients a CYC/Methotrexate/5-Fluorouracil combination can also be used. In Her2-positive patients, first-line therapy includes chemotherapy combined with anti-Her2 drugs as trastuzumab and pertuzumab. Triple-negative breast cancer are treated with chemotherapy alone. In early estrogen receptor-positive breast cancer, endocrine therapy should be added to the above-mentioned treatments (169). In advanced breast cancer anthracycline or taxane-based regimens, preferably as single agents, would usually be considered as first-line treatment for Her2-negative tumors. Capecitabine can also be a valuable option. In patients affected by Her2-positive advanced breast cancer first-line regimens include trastuzumab combined with vinorelbine or a taxane (170). Patients treated with a CYC/Methotrexate/5-Fluorouracil regimen can often experience hyponatremia because of the combined effects of Methotrexate and CYC, particularly when CYC is administered at higher doses. This disorder can be further amplified by combination with trastuzumab. The treatment with pertuzumab is complicated by secretory diarrhea leading to hypokalemia, hypomagnesemia and, due to vitamin D malabsorption, to hypocalcemia and hypophosphatemia. Hypocalcemia can also occur because of decreased calcitriol generation induced by 5-Fluorouracil or its prodrug Capecitabine; hypophosphatemia may develop, due to proximal tubule damage caused by anthracyclines (Tables 2–6) (152). Special attention should also be given to the presence of a concurrent hypokalemia and hypomagnesemia in patients following an anthracycline-based regimen; the cardiotoxic effects of these drugs can be amplified by the presence of these ion disturbances and be a harbinger of threatening arrhythmic events.
Sodium: dysnatremias (both hypo- and hypernatremia), should be carefully evaluated for the cause (or causes) and treated with special attention to the timing of disturbance onset and the rate of correction. It is mandatory to know if the disturbance is acute (<48 h) or chronic (>48 h); in the case of acute hyponatremia with neurologic alterations, a rapid infusion of hypertonic saline is required to increase serum Na concentration by 1–2 mmol/L per hour. In chronic hyponatremia the correction rate should be 4–8 mmol/L per day, or even less (4–6-mmol/L per day) if there is a particularly high risk of osmotic demyelination syndrome. While the Adrogue-Madias equation can be used, when starting the infusion of a hypertonic solution, to predict serum Na concentration with therapy, several limitations have been described using this formula, and a close monitoring of the serum Na concentration is mandatory during treatment of patients with severe hyponatremia (171). In patients affected by SIADH and mild to moderate hyponatremia, fluid restriction is traditionally the first-line therapy; if fluid restriction is unsuccessful, pharmacological treatment with loop diuretics, urea, or vaptans should be considered.
In case of hypernatremia, sodium alterations should be corrected based on water deficit equation bearing in mind the importance of cause and the time of development of the electrolyte derangement and the rate of correction (13). Moreover, ongoing losses due to perspiration and urine output should be taken into account. The objective in patients with chronic hypernatremia is to lower sodium levels by a maximum of 10 mEq/L per day (less than 0.5 mEq/L/h is considered safe), whereas in those with acute hypernatremia the objective is to lower sodium levels by 1–2 mEq/L/h to restore normal levels in less than 24 h (172).
Other electrolyte disorders (K, Ca, Mg, P): the electrolytes depletions should be corrected with oral or intravenous supplementation. Treatment of K disorders in patients affected by malignancies is similar to that in the normal population. In case of hypokalemia and hypocalcemia secondary to hypomagnesemia, magnesium depletion should be primarily corrected (21). In case of acute hypercalcemia, in order to reduce calcium serum levels, an intravenous 0.9% saline infusion should be initially administered (usually 200 to 500 mL/h) since most patients are volume depleted. In the absence of renal or heart failure, loop diuretic therapy to increase urinary calcium excretion is not recommended; only if an impairment in the excretion of the fluid load is anticipated a loop diuretic should be considered. Bone calcium mobilization blockade with intravenous bisphophonates as zoledronic acid (a dose-adjustment in presence of renal failure is required) are frequently used in the treatment of acute hypercalcemia. Recently, treatment with denosumab, another antiresorptive drug with a long term effect, is increasingly being used. Patients affected by hypophosphatemia should receive medical treatment with vitamin D (calcitriol is preferred) and phosphate supplementation (1 to 3 g/day) (173).
A vast array of traditional and novel antineoplastic drugs, currently available for cancer treatments, may induce serious and potentially life-threatening derangements in serum electrolyte concentrations, via mechanisms such as nephrotoxic tubular damage, diarrhea induction, and/or TLS. Oncologists and clinicians should be aware of these crucial side effects of antineoplastic therapies, in order to set out preventive measures and start promptly appropriate treatments when needed.
IV, GR, and AC: conceived and designed the review. IV, GR, FQ, PB, IB, MF, GP, VC, PC, EF, AV, RV, and AC: collected the data and contributed to the analysis of literature data. IV, GR, EF, and AC: performed the analysis of all data. IV, GR, FQ, and AC: discussion of the results. IV, GR, and AC: wrote the paper.
This study was funded by University of Parma FIL 2017-2018, Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. De las Penas R, Escobar Y, Henao F, Blasco A, Rodrìguez C.A. SEOM guidelines on hydroelectrolytic disorders. Clin Transl Oncol. (2014) 16:1051–9. doi: 10.1007/s12094-014-1234-2
2. Rosner MH, Perazella MA. Acute kidney injury in patients with cancer. N Engl J Med. (2017) 376:1770–81. doi: 10.1056/NEJMra1613984
3. Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al. The coincise guide to pharmacology 2017/18: overview. Br J Pharmacol. (2017) 174(Suppl 1):S1–S6. doi: 10.1111/bph.13882
4. Antunes-Rodrigues J, de Castro M, Elias LL, Valença MM, McCann SM. Neuroendocrine control of body fluid metabolism. Physiol Rev. (2004) 84:169–208. doi: 10.1152/physrev.00017.2003
5. Doshi SM, Shah P, Lei X, Lahoti A, Salahudeen AK. Hyponatremia in hospitalized cancer patients and its impact on clinical outcomes. Am J Kidney Dis. (2012) 59:222–8. doi: 10.1053/j.ajkd.2011.08.029
6. Berardi R, Santoni M, Rinaldi S, Nunzi E, Smerilli A, Caramanti M, et al. Risk of hyponatraemia in cancer patients treated with targeted therapies: a Systematic review and meta-Analysis of clinical trials. PLoS ONE. (2016) 11:e0152079. doi: 10.1371/journal.pone.0152079
7. Berardi R, Caramanti M, Castagnani M, Guglielmi S, Marcucci F, Savini A, et al. Hyponatremia is a predictor of hospital length and cost of stay and outcome in cancer patients. Support Care Cancer. (2015) 23:3095–101. doi: 10.1007/s00520-015-2683-z
8. Verbalis JG, Goldsmith SR, Greenberg A, Korzelius C, Schrier RW, Sterns RH, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. (2013) 126:S1–42. doi: 10.1016/j.amjmed.2013.07.006
9. Bolignano D, Cabassi A, Fiaccadori E, Ghigo E, Pasquali R, Peracino A, et al. Copeptin (CTproAVP), a new tool for understanding the role of vasopressin in pathophysiology. Clin Chem Lab Med. (2014) 52:1447–56. doi: 10.1515/cclm-2014-0379
10. Cabassi A, Tedeschi S, Perlini S, Verzicco I, Volpi R, Gonzi G, et al. Non-steroidal anti-inflammatory drug effects on renal and cardiovascular function: from physiology to clinical practice. Eur J Prev Cardiol. (2019) 2019:2047487319848105. doi: 10.1177/2047487319848105
11. Lu X, Wang X. Hyponatremia induced by antiepileptic drugs in patients with epilepsy. Expert Opin Drug Saf. (2017) 16:77–87. doi: 10.1080/14740338.2017.1248399
12. Clark BA, Shannon RP, Rosa RM, Epstein FH. Increased susceptibility to thiazide-induced hyponatremia in the elderly. J Am Soc Nephrol. (1994) 5:1106–11.
13. Cabassi A, Tedeschi S. Severity of community acquired hypernatremia is an independent predictor of mortality: a matter of water balance and rate of correction. Intern Emerg Med. (2017) 12:909–11. doi: 10.1007/s11739-017-1693-x
14. Salahudeen AK, Doshi SM, Shah P. The frequency, cost, and clinical outcomes of hypernatremia in patients hospitalized to a comprehensive cancer center. Support Care Cancer. (2013) 21:1871–8. doi: 10.1007/s00520-013-1734-6
15. Bowman BT. Electrolyte disorders associated with cancer. J Onco-Nephrology. (2017) 1:30–5. doi: 10.5301/jo-n.5000004
16. Milionis HJ, Bourantas CL, Siamopoulos KC, Elisaf MS. Acid-base and electrolyte abnormalities in patients with acute leukemia. Am J Hematol. (1999) 62:201–7. doi: 10.1002/(SICI)1096-8652(199912)62:4<201::AID-AJH1>3.0.CO;2-1
17. Izzedine H, Perazella MA. Adverse kidney effects of epidermal growth factor receptor inhibitors. Nephrol Dial Transplant. (2017) 32:1089–97. doi: 10.1093/ndt/gfw467
18. Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, et al. TRPM6 forms the mg2+ influx channel involved in intestinal and renal mg2+ absorption. J Biol Chem. (2004) 279:19–25. doi: 10.1074/jbc.M311201200
19. Rude RK, Singer FR, Gruber HE. Skeletal and hormonal effects of magnesium deficiency. J Am Coll Nutr. (2009) 28:131–41. doi: 10.1080/07315724.2009.10719764
20. Rodriguez-Ortiz ME, Canalejo A, Herencia C, Martínez-Moreno JM, Peralta-Ramírez A, Perez-Martinez P, et al. Magnesium modulates parathyroid hormone secretion and upregulates parathyroid receptor expression at moderately low calcium concentration. Nephrol Dial Transplant. (2014) 29:282–9. doi: 10.1093/ndt/gft400
22. Lajer H, Daugaard G. Cisplatin and hypomagnesemia. Cancer Treat Rev. (1999) 25:47–58. doi: 10.1053/ctrv.1999.0097
23. D'Erasmo E, Celi FS, Acca M, Minisola S, Aliberti G, Mazzuoli GF. Hypocalcemia and hypomagnesemia in cancer patients. Biomed Pharmacother. (1991) 45:315–7. doi: 10.1016/0753-3322(91)90087-A
24. Liamis G, Milionis HJ, Elisaf M. A review of drug-induced hypocalcemia. J Bone Miner Metab. (2009) 27:635–42. doi: 10.1007/s00774-009-0119-x
25. Mune T, Yasuda K, Ishii M, Matsunaga T, Miura K. Tetany due to hypomagnesemia induced by cisplatin and doxorubicin treatment for synovial sarcoma. Int Med. (1993) 32:434–7. doi: 10.2169/internalmedicine.32.434
26. Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. (2005) 352:373–9. doi: 10.1056/NEJMcp042806
27. Yoshida T, Taguchi D, Fukuda K, Shimazu K, Inoue M, Murata K, et al. Incidence of hypophosphatemia in advanced cancer patients: a recent report from a single institution. Int J Clin Oncol. (2017) 22:244–49. doi: 10.1007/s10147-016-1063-0
28. Stephanos K, Picard L. Pediatric oncologic emergencies. Emerg Med Clin North Am. (2018) 36:527–35. doi: 10.1016/j.emc.2018.04.007
29. Liamis G, Milionis H, Elisaf M. A review of drug-induced hyponatremia. Am J Kidney Dis. (2008) 52:144–53. doi: 10.1053/j.ajkd.2008.03.004
30. Shimada A, Takeuchi H, Fukuda K, Suda K, Nakamura R, Wada N, et al. Hyponatremia in patients with esophageal cancer treated with chemotherapy including cisplatin. Esophagus. (2018) 15:209–16. doi: 10.1007/s10388-018-0615-y
31. Ma Y, Hou L, Yu F, Lu G, Qin S, Xie R, et al. Incidence and physiological mechanism of carboplatin-induced electrolyte abnormality among patients with non-small cell lung cancer. Oncotarget. (2017) 8:18417–423. doi: 10.18632/oncotarget.12813
32. Kwon DY, Han GH, Ulak R, Ki KD, Lee JM, Lee S.K. Syndrome of inappropriate antidiuretic hormone secretion following irinotecan-cisplatin administration as a treatment for recurrent ovarian clear cell carcinoma. Obstet Gynecol Sci. (2017) 60:115–7. doi: 10.5468/ogs.2017.60.1.115
33. Hamdi T, Latta S, Jallad B, Kheir F, Alhosaini MN, Patel A. Cisplatin-Induced renal salt-wasting syndrome. Southern Medical Journal. (2010) 103:793–9. doi: 10.1097/SMJ.0b013e3181e63682
34. Liamis G, Filippatos GD, Elisaf MS. Electrolyte disorders associated with the use of anticancer drugs. Eur J Pharmacol. (2016) 777:78–87. doi: 10.1016/j.ejphar.2016.02.064
35. Anand AS, Nikhil S. Incidence and spectrum of electrolyte disturbances in cisplatin based chemotherapy. Int J Res Med Sci. (2015) 3:3824–9. doi: 10.18203/2320-6012.ijrms20151450
36. Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. (2007) 18:2649–52. doi: 10.1681/ASN.2007070792
37. Yamamoto Y, Watanabe K, Matsushita H, Tsukiyama I, Matsuura K, Wakatsuki A. The incidence of cisplatin-induced hypomagnesemia in cervical cancer patients receiving cisplatin alone. Yakugaku Zasshi. (2017) 137:79–82. doi: 10.1248/yakushi.16-00185
38. Foster BJ, Clagett-Carr K, Leyland-Jones B, Hoth D. Results of nCI-sponsored phase i trials with carboplatin. Cancer Treat Rev. (1985) 12 (suppl A):43–9. doi: 10.1016/0305-7372(85)90017-9
39. O'Sullivan D. Late effects of chemotherapeutic agents on renal function in childhood cancer survivors: a review of the literature. Ir J Med Sci. (2017) 186:49–55. doi: 10.1007/s11845-016-1473-z
40. Skinner R, Cotterill SJ, Stevens MC. Risk factors for nephrotoxicity after ifosfamide treatment in children: a uKCCSG late effects group study. United Kingdom Children's Cancer Study Group. Br J Cancer. (2000) 82:1636–45. doi: 10.1054/bjoc.2000.1214
41. Ledeganck KJ, Boulet GA, Bogers JJ, Verpooten GA, De Winter BY. The tRPM6/EGF pathway is downregulated in a rat model of cisplatin nephrotoxicity. PLOS ONE. (2013) 8:e57016 doi: 10.1371/journal.pone.0057016
42. Panichpisal K, Angulo-Pernett F, Selhi S, Nugent KM. Gitelman-like syndrome after cisplatin therapy: a case report and literature review. BMC Nephrol. (2006) 7:10. doi: 10.1186/1471-2369-7-10
43. Yeung SCJ, Escalante C. Oncologic Emergencies. Hamilton, ON: BC Decker (2002). doi: 10.1002/9781119000822.hfcm141
44. Hoffmann K, Marten A, Lindel K, Fritz S, Jager D, Buchler MW, et al. Major combined electrolyte deficiency during therapy with low-dose cisplatin, 5-fluorouracil and interpheron alpha: report on several cases and review of literature. BMC Cancer. (2006) 6:128. doi: 10.1186/1471-2407-6-128
45. Yamamoto M, Yamaguchi T, Yamauchi M, Yano S, Sugimoto T. Acute-onset hypomagnesemia-induced hypocalcemia caused by the refractoriness of bones and renal tubules to parathyroid hormone. J Bone Miner Metab. (2011) 29:752–7. doi: 10.1007/s00774-011-0275-7
46. Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. (2003) 23:460–4. doi: 10.1016/S0270-9295(03)00089-5
47. Liamis G., Milionis HJ, Elisaf M. Medication induced hypophosphatemia: a review. QJM. (2010) 103:449–59. doi: 10.1093/qjmed/hcq039
48. Tsimberidou AM, O'Brien SM, Cortes JE, Faderl S, Andreeff M, Kantarjian HM, et al. Phase iI study of fludarabine, cytarabine (Ara-C), cyclophosphamide, cisplatin and gM-CSF (FACPGM) in patients with richter's syndrome or refractory lymphoproliferative disorders. Leuk Lymphoma. (2002) 43:767–2. doi: 10.1080/10428190290016872
49. Perazella MA. Onco-nephrology: renal toxicities of chemotherapeutic agents. Clin J Am Soc Nephrol. (2012) 7:1713–21. doi: 10.2215/CJN.02780312
50. Ariceta G, Rodriguez-Soriano J, Vallo A, Navajas A. Acute and chronic effect of cisplatin therapy on renal magnesium homeostasis. Med Pediatr Oncol. (1997) 28:35–40. doi: 10.1002/(SICI)1096-911X(199701)28:1<35::AID-MPO7>3.0.CO;2-U
51. Lee YC, Park JS, Lee CH, Bae SC, Kim IS, Kang CM, et al. Hyponatraemia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transplant. (2010) 25:1520–4. doi: 10.1093/ndt/gfp657
52. DeFronzo RA, Braine H, Colvin M, Davis PJ. Water intoxication in man after cyclophosphamide therapy. Time course and relation to drug activation. Ann Intern Med. (1973) 78:861–869. doi: 10.7326/0003-4819-78-6-861
53. Bressler RB, Huston DP. Water intoxication following moderate-dose intravenous cyclophosphamide. Arch Intern Med. (1985) 145:548–9. doi: 10.1001/archinte.145.3.548
54. Pratt CB, Douglass EC, Kovnar EH, Heideman R, Kun L, Avery L, et al. A phase i study of ifosfamide given on alternate days to treat children with brain tumors. Cancer. (1993) 71:3666–9. doi: 10.1002/1097-0142(19930601)71:11<3666::AID-CNCR2820711132>3.0.CO;2-E
55. Church DN, Hassan AB, Harper SJ, Wakeley CJ, Price CG. Osteomalacia as a late metabolic complication of ifosfamide chemotherapy in young adults: illustrative cases and review of the literature. Sarcoma. (2007) 2007:91586. doi: 10.1155/2007/91586
56. Munro AH, Crompton GH. Inappropriate antidiuretic hormone secretion in oat-cell carcinoma of bronchus. Aggravation of hyponatraemia by intravenous cyclophosphamide. Thorax. (1972) 27:640–2. doi: 10.1136/thx.27.5.640
57. Bode U, Self SM, Levine AS. Studies on the antidiuretic effect of cyclophosphamide: vasopressin release and sodium excretion. Med Pediatr Oncol. (1980) 8:295–303. doi: 10.1002/mpo.2950080312
58. Park SJ, Kim JH, Shin JI. Insight on mechanism of hyponatremia induced by low-dose intravenous pulse cyclophosphamide. Nephrol Dial Transpl. (2010) 25:3453–59. doi: 10.1093/ndt/gfq429
59. Kim S, Choi HJ, Jo CH, Park JS, Kwon TH, Kim GH. Cyclophosphamide-induced vasopressin-independent activation of aquaporin-2 in the rat kidney. Am J Physiol Renal Physiol. (2015) 309:F474–F83. doi: 10.1152/ajprenal.00477.2014
60. Culine S, Ghosn M, Droz JP. Inappropriate antidiuretic hormone secretion induced by ifosfamide. Eur J Cancer. (1990) 26:922. doi: 10.1016/0277-5379(90)90205-8
61. Oberlin O, Fawaz O, Rey A, Niaudet P, Ridola V, Orbach D, et al. Long-term evaluation of ifosfamide-related nephrotoxicity in children. J Clin Oncol. (2009) 27:5350–5. doi: 10.1200/JCO.2008.17.5257
62. FDA. Approval for Bendamustine Hydrochloride. (2009). Available online at: http://www.cancer.gov/cancertopics/druginfo/fda-bendamustine-hydrochloride
63. Rogers A, Gandhi P, Bautista J, Tang J. Acquired gitelman syndrome secondary to bendamustine use. R I Med J. (2013) 101:36–38.
64. Leem AY, Kim HS, Yoo BW, Kang BD, Kim MH, Rha SY, et al. Ifosfamide-induced fanconi syndrome with diabetes insipidus. Korean J Intern Med. (2014) 29:246–9. doi: 10.3904/kjim.2014.29.2.246
65. Ingemi AI, Bota VM, Peguero A, Charpentier M. Fanconi's syndrome and nephrogenic diabetes insipidus in an adult treated with ifosfamide. Pharmacotherapy. (2012) 32:e12–6. doi: 10.1002/PHAR.1013
66. Yamamoto Y, Akashi Y, Minami T, Nozawa M, Kiba K, Yoshikawa M, et al. Serious hypokalemia associated with abiraterone acetate in patients with castration-Resistant prostate cancer. Case Rep Urol. (2018) 2018:1414395. doi: 10.1155/2018/1414395
67. Madison DL, Beer TM, Bliziotes MM. Acute estramustine induced hypocalcemia unmasking severe vitamin d deficiency. Am J Med. (2002) 112:680–1. doi: 10.1016/S0002-9343(02)01101-4
68. Stava CJ, Jimenez C, Hu MI, Vassilopoulou-Sellin R. Skeletal sequelae of cancer and cancer treatment. J Cancer Surviv. (2009) 3:75–88. doi: 10.1007/s11764-009-0083-4
69. Gilbar PJ, Richmond J, Wood J, Sullivan A. Syndrome of inappropriate antidiuretic hormone secretion induced by a single dose of oral cyclophosphamide. Ann Pharmacotherapy. (2012) 46:e23. doi: 10.1345/aph.1R296
70. Lee BS, Lee JH, Kang HG, Hahn H, Lee JH, Shin HY, et al. Ifosfamide nephrotoxicity in pediatric cancer patients. Pediatr Nephrol. (2001) 16:796–9. doi: 10.1007/s004670100658
71. Abou-Alfa GK, Capanu M, O'Reilly EM, Ma J, Chou JF, Gansukh B, et al. A phase iI study of cixutumumab (IMC-A12, nSC742460) in advanced hepatocellular carcinoma. J Hepatol. (2014) 60:319–42. doi: 10.1016/j.jhep.2013.09.008
72. Zhu AX, Ancukiewicz M, Supko JG, Sahani DV, Blaszkowsky LS, Meyerhardt JA, et al. Efficacy, safety, pharmacokinetics and biomarkers of cediranib monotherapy in advanced hepatocellular carcinoma: a phase iI study. Clin Cancer Res. (2013) 19:1557–66. doi: 10.1158/1078-0432.CCR-12-3041
73. Fernández-Rodriguez CM, Prada I, Andrade A, Moreiras M, Guitián R, Aller R, et al. Disturbed synthesis of insulin-like growth factor i and its binding proteins may influence renal function changes in liver cirrhosis. Dig Dis Sci. (2001) 46:1313–20. doi: 10.1023/A:1010631800505
74. Kolarich AR, Reynolds BA, Heldermon CD. Ado-trastuzumab emtasine associated hyponatremia and intracranial hemorrhage. Acta Oncologica. (2014) 53:1434–6. doi: 10.3109/0284186X.2014.920959
75. Barnard ZR, Walcott BP, Kahle KT, Nahed BV, Coumans JV. Hyponatremia associated with ipilimumab-induced hypophysitis. Med Oncol. (2012) 29:374–7. doi: 10.1007/s12032-010-9794-7
76. Okano Y, Satoh T, Horiguchi K, Toyoda M, Osaki A, Matsumoto S, et al. Nivolumab-induced hypophysitis in a patient with advanced malignant melanoma. Endocrine J. (2016) 63:905–12. doi: 10.1507/endocrj.EJ16-0161
77. Vandiver JW, Singer Z, Harshberger C. Severe hyponatremia and immune nephritis following an initial infusion of nivolumab. Target Oncol. (2016) 11:553–6. doi: 10.1007/s11523-016-0426-9
78. Trainer H, Hulse P, Higham CE, Trainer P, Lorigan P. Hyponatremia secondary to nivolumab-induced primary adrenal failure. Endocrinol Diabetes Metab Case Rep. (2016) 2016:108. doi: 10.1530/EDM-16-0108
79. Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S, et al. Phase i and pharmacokinetic study of etaracizumab (Abegrin™), a humanized monoclonal antibody against αvβ3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. (2008) 26:35–43. doi: 10.1007/s10637-007-9077-0
80. Besse B, Tsao LC, Chao DT, Fang Y, Soria JC, Almokadem S, et al. Phase ib safety and pharmacokinetic study of volociximab, an anti-α5β1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann Oncol. (2013) 24:90–6. doi: 10.1093/annonc/mds281
81. Liapis K, Apostolidis J, Charitaki E, Panitsas F, Harhalakis N, Nikiforakis E. Syndrome of inappropriate secretion of andiuretic hormone associated with imatinib. Ann Pharmacother. (2008) 42:1882–6. doi: 10.1345/aph.1L410
82. Hill J, Shields J, Passero V. Tyrosine kinase inhibitors-associated syndrome of inappropriate secretion of anti-diuretic hormone. J Oncol Pharm Practice. (2016) 22:729–32. doi: 10.1177/1078155215592023
83. Lalami Y, Garcia C, Flamen P, Ameye L, Paesmans M, Awada A. Phase iI trial evaluating the efficacy of sorafenib (BAY 43-9006) and correlating early fluorodeoxyglucose positron emission tomography-CT response to outcome in patients with recurrent and/or metastatic head and neck cancer. Head Neck. (2016) 38:347–54. doi: 10.1002/hed.23898
84. Yeo W, Chan SL, Mo FK, Chu CM, Hui JW, Tong JH, et al. PhaseI/II study of temsirolimus for patients with unresectable hepatocellular carcinoma (HCC)- a correlative study to explore potential biomarkers for response. BMC Cancer. (2015) 15:395. doi: 10.1186/s12885-015-1334-6
85. Saba NF, Hurwitz SJ, Magliocca K, Kim S, Owonikoko TK, Harvey D, et al. Phase1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer. (2014) 120:3940–51. doi: 10.1002/cncr.28965
86. Lei KI, Wickham NW, Johnson PJ. Severe hyponatremia due to syndrome of inappropriate secretion of antidiuretic hormone in a patient receiving interferon-alpha for chronic myeloid leukemia. Am J Hematol. (1995) 49:100. doi: 10.1002/ajh.2830490123
87. Tanaka M, Kamoi K, Takahashi T. Interferon-alpha is a predisponing risk factor for carbamazepine-induced hyponatremia: a case of syndrome of inappropriate antidiuresis caused by interferon-alpha therapy. Int J Gen Med. (2008) 1:21–5. doi: 10.2147/IJGM.S2595
88. Tweedy C, Silverberg DA, Scott L. Levamisole-Induced syndrome of inappropriate antidiuretic hormone. N Engl J Med. (1992) 326:1164. doi: 10.1056/NEJM199204233261716
89. Bruno JJ, Canada TW. Possible pentostatin-Induced symptomatic hyponatremia. Pharmacotherapy. (2007) 27:164–9. doi: 10.1592/phco.27.1.164
90. Soetekouw PM, Koopman M, Burger D, Tjan-Heijnen VC, De Mulder PH. Severe hyponatremia caused by hyperglycaemia due to interferon alpha therapy in advanced renal cell carcinoma. Acta Oncol. (2009) 48:154–6. doi: 10.1080/02841860802047403
91. Arnold SM, Moon J, Williamson SK, Atkins JN, Ou SH, LeBlanc M, et al. Phase iI evaluation of eribulin mesylate (E7389, nSC 707389) in patients with metastatic or recurrent squamous cell carcinoma of the head and neck: southwest oncology group trial S0618. Invest New Drugs. (2011) 29:352–9. doi: 10.1007/s10637-009-9348-z
92. O'Brien S, Rizzieri DA, Vey N, Ravandi F, Krug UO, Sekeres MA, et al. Elacytarabine has single-agent activity in patients with advanced acute myeloid leukaemia. Br J Haematol. (2012) 158:581–8. doi: 10.1111/j.1365-2141.2012.09186.x
93. Ribeiro RC, Rill D, Roberson PK, Furman WL, Pratt CB, Brenner M, et al. Continous infusion of interleukin-2 in children with refractory malignancies. Cancer. (1993) 72:623–8. doi: 10.1002/1097-0142(19930715)72:2<623::AID-CNCR2820720248>3.0.CO;2-S
94. Peng B, Chen H, Lou X. Bortezomib-induced syndrome of inappropriate antidiuresis in a patient with multiple myeloma: a case report and literature review. Int J Clin Pharmacol Ther. (2017) 55:910–14. doi: 10.5414/CP203109
95. Lv CL, Li J. Bortezomib as a probable cause of the syndrome of inappropriate antidiuretic hormone secretion: a case report and review of the literature. Mol Clin Oncol. (2017) 7:667–72. doi: 10.3892/mco.2017.1366
96. Paydas A. Hyponatremia is not a rare adverse event in bortezomib-treated patients. Chemotherapy. (2011) 57:434–6. doi: 10.1159/000334088
97. Cao Y, Liu L, Liao C, Tan A, Gao F. Meta-analysis of incidence and risk of hypokalemia with cetuximab-based therapy for advanced cancer. Cancer Chemother Pharmacol. (2010) 66:37–42. doi: 10.1007/s00280-009-1131-5
98. Schneeweiss A, Park-Simon TW, Albanell J, Ulrik Lassen, Javier Corts, Veronique Dieras, et al. Phase ib study evaluating safety and clinical activity of the anti-HER3 antibody lumretuzumab combined with the anti-HER2 antibody pertuzumab and paclitaxel in hER3-positive, hER2-low metastatic breast cancer. Invest New Drugs. (2018) 36:848–59. doi: 10.1007/s10637-018-0562-4
99. Wang Q, Qi Y, Zhang D, Gong C, Yao A, Xiao Y, et al. Electrolyte disorders assessment in solid tumor patients treated with anti-EGFR monoclonal antibodies: a pooled analysis of 25 randomized clinical trials. Tumour Biol. (2015) 36:3471–82. doi: 10.1007/s13277-014-2983-9
100. Shaikh A, Wiisanene ME, Gunderson HD, Leung N. Acquired fanconi syndrome after treatment with capecitabine, irinotecan, and bevacizumab. Ann Pharmacother. (2009) 43:1370–3. doi: 10.1345/aph.1M120
101. Johnston PB, LaPlant B, McPhail E, Habermann TM, Inwards DJ, Micallef IN, et al. Everolimus combined with r-CHOP-21 for new, untreated, diffuse large b-cell lymphoma (NCCTG (1085). [Alliance]): safety and efficacy results of a phase 1 and feasibility trial. Lancet Haematol. (2016) 3:e309–16. doi: 10.1016/S2352-3026(16)30040-0
102. Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase iI study of the anti-cytotoxic t-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. (2010) 28:3485–90. doi: 10.1200/JCO.2010.28.3994
103. von Stackelberg A, Locatelli F, Zugmaier G, Rupert Handgretinger, Tanya M., Trippett C, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol. (2016) 34:4381–89. doi: 10.1200/JCO.2016.67.3301
104. Sakiyama T, Tsurutani J, Iwasa T, H Kawakami, Y Nonagase, T Yoshida, et al. A phase i dose-escalation study of eribulin and s-1 for metastatic breast cancer. Br J Cancer. (2015) 112:819–24. doi: 10.1038/bjc.2015.10
105. Kobayashi Y, Yamauchi T, Kiyoi H, Sakura T, Hata T, Ando K, et al. Phase i trial of volasertib, a polo-like kinase inhibitor, in japanese patients with acute myeloid leukemia. Cancer Sci. (2015) 106:1590–5 doi: 10.1111/cas.12814
106. Patt Y, Rojas-Hernandez C, Fekrazad HM, Bansal P, Lee FC. Phase iI trial of sorafenib in combination with capecitabine in patients with hepatocellular carcinoma: iNST 08-20. Oncologist. (2017) 22:1158–e16. doi: 10.1634/theoncologist.2017-0168
107. Terazawa T, Nishitani H, Kato K, Hashimoto H, Akiyoshi K, Ito Y„, et al. Phase iI study of cetuximab with irinotecan for kRAS wild-type colorectal cancer in japanese patients. Asia Pac J Clin Oncol. (2017) 13:e132–e7. doi: 10.1111/ajco.12405
108. Saloura V, Cohen EE, Licitra L, Billan S, Dinis J, Lisby S, et al. An open label single-arm, phase iI trial of zalutumumab, a human monoclonal anti-EGFR antibody, in patients with platinum-refractory squamous cell carcinoma of the head and neck. Cancer Chemother Pharmacol. (2014) 73:1227–39. doi: 10.1007/s00280-014-2459-z
109. Zekri JM, Robinson MH, Woll PJ. Relative hypocalcaemia and muscle cramps in patients receiving imatinib for gastrointestinal stromal tumour. Sarcoma. (2006) 2006:1–3. doi: 10.1155/SRCM/2006/48948
110. Cataldi M, Gaudino A, Lariccia V, Russo M, Amoroso S, di Renzo G, et al. Imatinib-mesylate blocks recombinant t-type calcium channels expressed in human embrionic kidney-293 cells by protein tyrosine-kinase independent mechanisms. J Pharmacol Exp Ther. (2004) 309:208–15. doi: 10.1124/jpet.103.061184
111. Vandyke K, Fitter S, Dewar AL, Hughes TP, Zannettino AC. Dysregulation of bone remodeling by imatinib mesylate. Blood. (2010) 115:766–4. doi: 10.1182/blood-2009-08-237404
112. Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P, et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol. (2007) 27:5499–13. doi: 10.1128/MCB.01080-06
113. Petric M, Miljak A, Milicevic T, Radman M. A rare case of hypocalcemia induced by nilotinib. Endoc Oncol Metab. (2017) 3:32–5. doi: 10.21040/eom/2017.3.1.5
114. Locati LD, Licitra L, Agate L, Ou SH, Boucher A, Jarzab B, et al. Treatment of advanced thyroid cancer with axitinib: phase 2 study with pharmacokinetic/Pharmadynamic and quality-of-Life assessments. Cancer. (2014) 120:2694–703. doi: 10.1002/cncr.28766
115. Ruinemans GM, Balemans C, Mattijssen V, Wiersma-van Tilburg AJ, Smit HJ. Fatal necrotizing pancreatitis during combined treatment with erlotinib and sunitinib. Lung Cancer. (2010) 70:364–5. doi: 10.1016/j.lungcan.2010.08.004
116. Kondo R, Watanabe S, Shoji S, Ichikawa K, Abe T, Baba J, et al. A phase iI study of irinotecan for patients with previously treated small-Cell lung cancer. Oncology. (2018) 94:223–32. doi: 10.1159/000486622
117. Tie Y, Ma X, Zhu C, Mao Y, Shen K, Wei X, et al. Safety and efficacy of nivolumab in the treatment of cancers: a meta-analysis of 27 prospective clinical trials. Int J Cancer. (2017) 140:948–58. doi: 10.1002/ijc.30501
118. Moore KN, Borghaei H, O'Malley DM, Jeong W, Seward SM, Bauer TM, et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer. (2017) 123:3080–87. doi: 10.1002/cncr.30736
119. Berman E, Nicolaides M, Maki RG. Altered bone and mineral metabolism in patients receiving imatinib mesylate. N Engl J Med. (2006) 354:2006–13. doi: 10.1056/NEJMoa051140
120. François H, Coppo P, Hayman JP, Fouqueray B, Mougenot B, Ronco P. Partial fanconi syndrome induced by imatinib therapy: a novel cause of urinary phosphate loss. Am J Kidney Dis. (2008) 51:298–301. doi: 10.1053/j.ajkd.2007.10.039
121. Mir O, Coriat R, Boudou-Rouquette P, Durand JP, Goldwasser F. Sorafenib-induced diarrhea and hypophosphatemia: mechanisms and therapeutic implications. Ann Oncol. (2012) 23:280–1. doi: 10.1093/annonc/mdr525
122. Wanchoo R, Jhaveri KD, Deray G, Launay-Vacher V. Renal effects of bRAF inhibitors: a systematic review by the cancer and the kidney international network. Clin Kidney J. (2016) 9:245–51. doi: 10.1093/ckj/sfv149
123. Popat R, Brown SR, Flanagan L, Hall A, Gregory W, Kishore B, et al. Bortezomib, thalidomide, dexamethasone and panobinostat for patients with relapsed multiple myeloma (MUK-six): a multicentre, open-label, phase 1/2 trial. Lancet Haematol. (2016) 3:e572–e80. doi: 10.1016/S2352-3026(16)30165-X
124. Glezerman IG, Kewalramani T, Jhaveri K. Reversible fanconi syndrome due to lenalidomide. NDT Plus. (2008) 1:215–7. doi: 10.1093/ndtplus/sfn044
125. Howard SC, Trifilio S, Gregory TK, Baxter N, McBride A. Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review. Ann Hematol. (2016) 95:563–73. doi: 10.1007/s00277-015-2585-7
126. Wu PF, Lin CH, Kuo CH, Chen WW, Yeh DC, Liao HW, et al. A pilot study of bevacizumab combined with etoposide and cisplatin in breast cancer patients with leptomeningeal carcinomatosis. BMC Cancer. (2015) 15:299. doi: 10.1186/s12885-015-1290-1
127. Wu S, Kim C, Baer L, Zhu X. Bevacizumab increases risk for severe proteinuria in cancer patients. J Am Soc Nephrol. (2010) 21:1381–9. doi: 10.1681/ASN.2010020167
128. Godo S, Yoshida Y, Kawamorita N, Mitsuzuka K, Kawazoe Y, Fujita M, et al. Life-threatening hyperkalemia associated with axitinib treatment in patients with recurrent renal carcinoma. Intern Med. (2018) 57:2895–900. doi: 10.2169/internalmedicine.0262-17
129. Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. (2008) 358:1160–74. doi: 10.1056/NEJMra0707704
130. Saif MW. Management of hypomagnesemia in cancer patients receiving chemotherapy. J Support Oncol. (2008) 6:243–8.
131. Shaw AT, Engelman JA. Ceritinib in aLK-rearranged non-small-cell lung cancer. N Engl J Med. (2014) 370:2537–9. doi: 10.1056/NEJMc1404894
132. Jakubowiak AJ, Richardson PG, Zimmerman T, Alsina M, Kaufman JL, Kandarpa M, et al. Perifosine plus lenalidomide and dexamethasone in relapsed and relapsed/refractory multiple myeloma: a phase i Multiple myeloma research consortium study. Br J Haematol. (2012) 158:472–80. doi: 10.1111/j.1365-2141.2012.09173.x
133. Popat R, Brown SR, Flanagan L, Hall A, Gregory W, Kishore B, et al. Extended follow-up and the feasibility of panobinostat maintenance for patients with relapsed multiple myeloma treated with bortezomib, thalidomide, dexamethasone plus panobinostat (MUK six open label, multi-centre phase i/II clinical trial). Br J Haematol. (2019) 185:573–8. doi: 10.1111/bjh.15551
134. Hesketh PJ. Chemotherapy-induced nausea and vomiting. N Engl J Med. (2008) 358:2482–94. doi: 10.1056/NEJMra0706547
135. Janczar S, Zalewska-Szewczyk B, Mlynarski W. Severe hyponatremia in a single-Center series of 84 homogenously treated children with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. (2017) 39:e54–e8. doi: 10.1097/MPH.0000000000000758
136. Castillo JJ, Vincent M, Justice E. Diagnosis and management of hyponatremia in cancer patients. Oncologist. (2012) 17:756–65. doi: 10.1634/theoncologist.2011-0400
137. Salcedo I, Aguayo A, Zuñiga J. Syndrome of inappropriate antidiuretic hormone secretion in a patient with a nodular lymphocyte-Predominant hodgkin lymphoma. A case report and review of literature. J Hematol Oncol Res. (2015) 2:1–6. doi: 10.14302/issn.2372-6601.jhor-15-657
138. Robertson GL, Bhoopalam N, Zelkowitz LJ. Vincristine neurotoxicity and abnormal secretion of antidiuretic hormone. Arch Intern Med. (1973) 132:717–20. doi: 10.1001/archinte.132.5.717
139. Zhou H, Li L, Zhou Y, Han Y. Syndrome of inappropriate antidiuretic hormone secretion from concomitant use of itraconazole and vindesine. J Clin Pharm Ther. (2018) 43:137–40. doi: 10.1111/jcpt.12598
140. Ohya K, Ogura H. The effect of colchicine or vinblastine on the blood calcium levels in rats. Eur J Pharmacol. (1993) 248:111–9. doi: 10.1016/0926-6917(93)90032-L
141. Liu W, Qdaisat A, Soliman PT, Ramondetta L, Lopez G, Narayanan S, et al. Hypomagnesemia and survival in patients with ovarian cancer who received chemotherapy with carboplatin. Oncologist. (2019) 24:e312–e7. doi: 10.1634/theoncologist.2018-0465
142. Frahm H, Von Hulst M. Increased secretion of vasopressin and edema formation in high dosage methotrexate therapy. Z Gesamte Inn Med. (1988) 43:411–4.
143. Diskin C, Dansby LM, Radcliff L, Carter TB, Graves E, Graves A. Recurrent hyponatremia after intrathecal methotrexate not related to antidiuretic hormone: is a natriuretic peptide activated? Am J Med Sci. (2006) 331:37–9. doi: 10.1097/00000441-200601000-00011
144. Thuss-Patience PC, Peters U, Jurkat-Rott K, Pink D, Kretzschmar A, Dörken B, et al. Acute hypokalemic tetraparesis induced by intravenous methotrexate. J Clin Oncol. (2003) 21:1896–7. doi: 10.1200/JCO.2003.99.008
145. Peterson BA, Collins AJ, Vogelzang NJ, Bloomfield CD. 5-Azacytidine and renal tubular dysfunction. Blood. (1981) 57:182–5. doi: 10.1182/blood.V57.1.182.182
146. Abdelall W, Kantarjian HM, Borthakur G, Garcia-Manero G, Patel KP, Jabbour EJ, et al. The combination of quizartinib with azacitidine or low dose cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: interim report of a phase I/II trial. Blood (2006) 128:1642.
147. Sternberg CN, Castellano D, Daugaard G, Géczi L, Hotte SJ, Mainwaring PN, et al. Abiraterone acetate for patients with metastatic castration-resistant prostate cancer progressing after chemotherapy: final analysis of a multicentre, open-label, early-access protocol trial. Lancet Oncol. (2014) 15:1263–8. doi: 10.1016/S1470-2045(14)70417-6
148. Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2015) 16:152–60. doi: 10.1016/S1470-2045(14)71205-7
149. Stein MN, Patel N, Bershadskiy A, Sokoloff A, Singer EA. Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer. Asian J Androl. (2014) 16:387–400. doi: 10.4103/1008-682X.129133
150. Webster R, Sheriff S, Faroqui R, Siddiqui F, Hawse JR, Amlal H. Klotho/fibroblast growth facter 23- and pTH-independent estrogen receptor-alpha-mediated direct downreguation of naPi-IIa by estrogen in the mouse kidney. Am J Physiol Renal Physiol. (2016) 311:F249–59. doi: 10.1152/ajprenal.00542.2015
151. Reyes EL, Talley RW. The hypocalcemic effects of actinomycin f and mithramycin. Henry Ford Medical J. (1970) 18:115–20.
152. Kido Y, Okamura T, Tomikawa M, Yamamoto M, Shiraishi M, Okada Y, et al. Hypocalcemia associated with 5-fluorouracil and low dose leucovorin in patients with advanced colorectal or gastric carcinomas. Cancer. (1996) 78:1794–7. doi: 10.1002/(SICI)1097-0142(19961015)78:8<1794::AID-CNCR21>3.0.CO;2-S
153. Fu S, Hirte H, Welch S, Ilenchuk TT, Lutes T, Rice C, et al. First-in-human phase i study of sOR-C13, a tRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Invest New Drugs. (2017) 35:324–33. doi: 10.1007/s10637-017-0438-z
154. Schacht RG, Feiner HD, Gallo GR, Lieberman A, Baldwin DS. Nephrotoxicity of nitrosoureas. Cancer. (1981) 48:1328–34. doi: 10.1002/1097-0142(19810915)48:6<1328::AID-CNCR2820480613>3.0.CO;2-N
155. Morgan RJ, Synold TW, Longmate JA, Quinn DI, Gandara D, Lenz HJ, et al. Pharmacodynamics (PD) and pharmacokinetics(PK) of e7389(eribulin, halicondrin b analog) during a phase i trial in patients with advanced solid tumors: a california cancer consortium trial. Cancer Chemother Pharmacol. (2015) 76:897–907. doi: 10.1007/s00280-015-2868-7
156. Evans TRJ, Dean E, Molife LR, Lopez J, Malcom R, Fatima EK, et al. Phase 1 dose-finding and pharmacokinetic study of eribulin-liposomal formulation in patients with solid tumours. Br J Cancer. (2019) 120:379–86. doi: 10.1038/s41416-019-0377-x
157. Sakurada H, Kawase Y, Mizuno H, Naito K, Yamamura M. A case of hypophosphatemia induced by administration of amrubicin in a patient with small cell lung cancer. Gan To Kagaku Ryoho. (2018) 45:1369–71.
158. Kitchlu A, McArthur E, Amir E, Booth CM, Sutradhar R, Majeed H, et al. Acute kidney injury in patients receiving systemic treatment for cancer: a Population-Based cohort study. J Natl Cancer Inst. (2018) 7:727–36. doi: 10.1093/jnci/djy167
159. Falco M, Palma G, Rea D, De Biase D, Scala S, D'Aiuto M, et al. Tumour biomarkers: homeostasis as a novel prognostic indicator. Open Biol. (2016) 6:60254. doi: 10.1098/rsob.160254
160. World Cancer Research Fund International. Available online at: https://www.wcrf.org/dietandcancer/about
161. Postmus PE, Kerr KM, Oudkerk M, Senan S, Waller DA, Vansteenkiste J, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2017) 28(Suppl. 4):iv1–21. doi: 10.1093/annonc/mdx222
162. Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2018) 29(Suppl. 4):iv192–237. doi: 10.1093/annonc/mdy275
163. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csoszi T, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
164. Früh M, De Ruysscher D, Popat S, Crinò L, Peters S, Felip E, et al. Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2013) 24(Suppl. 6):vi99–105. doi: 10.1093/annonc/mdt178
165. Baas P, Fennell, Kerr K, Van Schil PE, Haas RL, Peters S. Malignant pleural mesothelioma: eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2015) 26(Suppl. 5):v31–9. doi: 10.1093/annonc/mdv199
166. Parker C, Gillessen S, Heidenreich A, Horwich A. on behalf of the eSMO guidelines committee cancer of the prostate: eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2015) 26(Suppl. 5):v69–77. doi: 10.1093/annonc/mdv222
167. Labianca R, Nordlinger B, Beretta GD, Mosconi S, Mandalà M, Cervantes A, et al. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2013) 24(Suppl. 6):vi64-72. doi: 10.1093/annonc/mdt354
168. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. (2016) 27:1386–422. doi: 10.1093/annonc/mdw235
169. Cardoso F, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rubio IT, et al. Early breast cancer: eSMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2019) 30:1194–220. doi: 10.1093/annonc/mdz173
170. Cardoso F, Senkus E, Costa A, Papadopoulos E, Aapro M, André F, et al. 4th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 4). Ann Oncol. (2018) 29:1634–57. doi: 10.1093/annonc/mdy192
171. Berl T. The adrogue-Madias formula revisited. Clin J Am Soc Nephrol. (2007) 2:1098–9. doi: 10.2215/CJN.03300807
172. Sterns RH. Disorders of plasma sodium–causes, consequences, and correction. N Engl J Med. (2015) 372:55–65. doi: 10.1056/NEJMra1404489
Keywords: electrolytes abnormalities, antineoplastic drug exposure, antidiuretic hormone (ADH), renal tubulopathies, tumor lysis syndrome
Citation: Verzicco I, Regolisti G, Quaini F, Bocchi P, Brusasco I, Ferrari M, Passeri G, Cannone V, Coghi P, Fiaccadori E, Vignali A, Volpi R and Cabassi A (2020) Electrolyte Disorders Induced by Antineoplastic Drugs. Front. Oncol. 10:779. doi: 10.3389/fonc.2020.00779
Received: 28 December 2019; Accepted: 22 April 2020;
Published: 19 May 2020.
Edited by:
Olivier Feron, Université Catholique de Louvain, BelgiumReviewed by:
Antonio Barbieri, Istituto Nazionale Tumori Fondazione G. Pascale (IRCCS), ItalyCopyright © 2020 Verzicco, Regolisti, Quaini, Bocchi, Brusasco, Ferrari, Passeri, Cannone, Coghi, Fiaccadori, Vignali, Volpi and Cabassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aderville Cabassi, YWRlcnZpbGxlLmNhYmFzc2lAdW5pcHIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.