 Yena Cho
Yena Cho Yong Kee Kim
Yong Kee Kim- Research Institute of Pharmaceutical Sciences, College of Pharmacy, Sookmyung Women's University, Seoul, South Korea
Multidrug resistance (MDR), which is a significant impediment to the success of cancer chemotherapy, is attributable to various defensive mechanisms in cancer. Initially, overexpression of ATP-binding cassette (ABC) transporters such as P-glycoprotein (P-gp) was considered the most important mechanism for drug resistance; hence, many investigators for a long time focused on the development of specific ABC transporter inhibitors. However, to date their efforts have failed to develop a clinically applicable drug, leaving only a number of problems. The concept of cancer stem cells (CSCs) has provided new directions for both cancer and MDR research. MDR is known to be one of the most important features of CSCs and thus plays a crucial role in cancer recurrence and exacerbation. Therefore, in recent years, research targeting CSCs has been increasing rapidly in search of an effective cancer treatment. Here, we review the drugs that have been studied and developed to overcome MDR and CSCs, and discuss the limitations and future perspectives.
Introduction
Chemotherapy is one of the most effective treatments for cancer; however, its success has been challenged by the acquisition of multidrug resistance (MDR) (1). MDR is caused by sustained (dose-dependent or time-dependent) administration of chemotherapeutic drugs, resulting in cross-resistance to a broad spectrum of structurally- and mechanically-distinct chemotherapeutic drugs (2). There are several mechanisms underlying MDR (3, 4): (i) increased pumping out of the drug through efflux pumps such as P-glycoprotein (P-gp) encoded by ABCB1 (5); (ii) decreased uptake of the drug through transporters; (iii) activation of drug-metabolizing enzymes such as cytochrome P450 and glutathione S-transferase; (iv) activation of DNA repair systems; (v) evasion of apoptosis. The first three of these processes are conducive to the development of resistance by preventing the drug from reaching an effective concentration, while the remaining two mechanisms achieve resistance by detoxifying the action of the drug. Based on the mechanisms described, we have been trying to establish strategies for overcoming MDR. Before discussing them further, we need to understand the existence of cancer stem cells (CSCs) and their roles in cancer biology. CSCs, also known as tumor-initiating cells (TICs), are a small population of cancer cells that have the ability to self-renew and differentiate similar to normal stem cells (NSCs). However, as CSCs are tumorigenic, they can contribute to the aggravation and recurrence of cancer (6). According to a CSC model that explains the relationship between CSC and MDR, increased expression of ATP-binding cassette (ABC) transporters and other genes contributes to the intrinsic resistance of CSCs to chemotherapy (7, 8). CSCs have relatively slow cell-cycle kinetics and are therefore targeted less by chemotherapeutic drugs compared to rapidly dividing cells (9). In addition to the well-known ABC transporters (2, 10), many other drug resistance mechanisms of CSCs have been identified, for example, aldehyde dehydrogenases (ALDHs) (11), epithelial-mesenchymal transition (EMT) (12), epigenetic modifications (13, 14), factors affecting tumor microenvironment, such as hypoxia (15), and signaling pathways (16–18). In this review, we provide a brief outline of MDR mechanisms and focus on the investigated drugs.
Current Strategies to Overcome MDR
Targeting ABC Transporters
ABC transporters including ABCB1, ABCC1, and ABCG2 are expressed in cancer stem/progenitor cells. These transporters have broad drug specificity and pump out a wide range of structurally- and mechanically-unrelated compounds, thereby lowering the intracellular accumulation of these compounds and consequently diminishing their biological efficacies (19). Several chemotherapeutic agents in clinical use are susceptible to ABC transporter-mediated efflux, such as microtubule-targeting taxanes (e.g., docetaxel and paclitaxel) and vinca alkaloids (vinblastine and vincristine), DNA-damaging anthracyclines (daunorubicin and doxorubicin), topoisomerase inhibitors (etoposide and topotecan), and tyrosine kinase inhibitors (dasatinib and gefitinib) (20). Therefore, developing strategies to target ABC transporters is an important area of cancer research, and many studies have been conducted accordingly (21). There are three approaches: (i) regulating the function of ABC transporters using competitive or allosteric inhibitors (Table 1) as well as the antibodies that target ABC transporters, such as UIC2 and MRK16 (86); (ii) regulating gene expression of ABC transporters at the transcriptional or translational level because, as with trabectedin, it is an attractive strategy to control ABC transporters at the transcriptional level by affecting the MDR enhanceosome (87–89); (iii) using anticancer drugs that are poor substrates of P-gp, such as ixabepilone (Table 1). Until ixabepilone was launched, efforts to develop drugs targeting ABC transporters had been a driving force in the development of first-, second-, and third-generation P-gp inhibitors (90, 91). However, it has been reported that first-generation inhibitors have low potency and high toxicity, and second-generation inhibitors have frequent drug-drug interactions (92). With the third-generation inhibitors, there have been many improvements with regard to the drawbacks of the previous generations, but clinical trial data are still insufficient. In effect, most clinical trials have been discontinued. Because NSCs, including hematopoietic stem cells, unrestricted somatic stem cells, and mesenchymal stem cells, also express ABC transporters to protect themselves from cytotoxic agents (93), inhibiting ABC transporters may cause serious side effects such as hematopoietic disorders due to bone marrow dysfunction. Therefore, the emergence of ixabepilone was inevitable and has been well-received. Like taxanes, ixabepilone leads to G2/M phase arrest by stabilizing microtubules and promoting tubulin polymerization. However, ixabepilone has a very important feature (not found in taxanes) effective against cancer cells that acquire MDR following repeated chemotherapy, as this drug is not pumped out through P-gp. Now, developing drugs that are not substrates of P-gp has become a trend for overcoming MDR cancer. In light of ixabepilone, chemical modifications of paclitaxel and vinblastine have also been attempted in succession, producing cabazitaxel and ortataxel, and vinflunine, respectively (Table 1). After these modifications, increased cytotoxic effects were observed in P-gp-overexpressing cell lines (43).
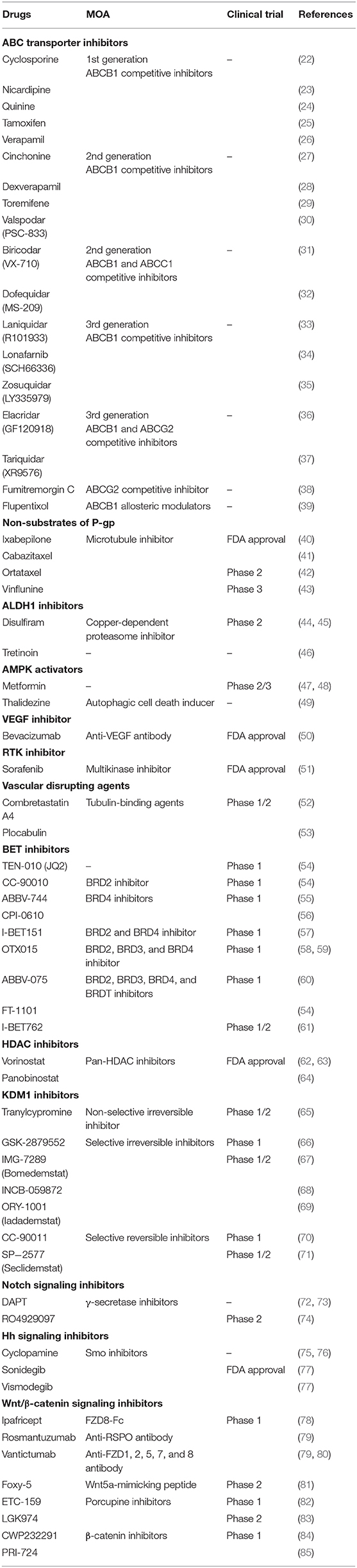
Table 1. Drugs that reverse chemoresistance via various mechanisms.
Targeting Aldehyde Dehydrogenases
ALDH plays an important role in the differentiation of stem cells by converting retinol into retinoic acid, as well as detoxifying the cells by converting aldehyde into carboxylic acid, and thus ALDH is considered a biomarker for stem cells (11, 94). Several studies have shown that ALDH-1 is correlated with CSCs. Gefitinib, an EGFR inhibitor, is used for breast-, lung-, and other cancers. However, it has been confirmed that ALDH1A1-positive CSCs are more resistant to gefitinib than ALDH1A1-negative CSCs (95). ALDH-1 expression is mediated by high expression of Snail, which regulates metastasis as a transcription factor and subsequently causes CSCs to develop resistance to chemotherapy (96). To solve this problem, ALDH inhibitors, such as diethylaminobenzaldehyde, disulfiram, and tretinoin, have been proposed (Table 1). Bromodomain and extra-terminal (BET) inhibitors such as JQ1 have also been proposed due to their ability to suppress ALDH1A1 expression (97).
Targeting Epithelial-Mesenchymal Transition
EMT is a process by which epithelial cells become mesenchymal stem cells. In this process, epithelial cells lose cell polarity and cell-cell adhesion function but gain migratory and invasive functions (98, 99). In other words, EMT causes cancer cells to exhibit stem-like features such as tumorigenicity. EMT also promotes metastasis that occurs mainly due to reduced expression of E-cadherin, which itself is directly repressed by Snail (100). Because EMT plays a crucial role in chemoresistance of CSCs, strategies to inhibit this process can be effective. Some studies have shown that AMP-activated kinase (AMPK) induces apoptosis of cancer cells, inhibits TGF-β-induced EMT and, consequently, can reverse drug resistance (101). AMPK activators that have been proposed include metformin and thalidezine (Table 1). Although the data are still insufficient, these activators seem to have enough potential to overcome chemoresistance. It has also been proposed that histone deacetylase (HDAC) inhibitors suppress EMT and attenuate chemoresistance (102).
Targeting Epigenetic Modifications
Histone acetylation, one of the most common post-translational modifications, is closely associated with CSC chemoresistance. This process, in which lysine residues are acetylated, is tightly regulated by histone acetyltransferases (HATs) and HDACs. Bromodomains (BRD) of the BET family proteins read acetyl-lysine (Kac) residues on histones and regulate gene expression. BET family proteins recruit the positive transcriptional elongation factor (P-TEFb), resulting in a transcriptional cascade of oncogenes (54). Thus, inhibition of BET binding to acetylated histones suppresses cell proliferation and induces apoptosis. JQ1 is a potent, highly specific, and Kac competitive inhibitor for BET family proteins (97). Because JQ1 has a short half-life, its derivatives and structurally similar forms are undergoing clinical trials (Table 1). On the other hand, HDACs, which are epigenetic erasers, induce stem-like features by promoting EMT. HDACs also affect hypoxia-inducible factors (HIFs) and NF-κB related to apoptosis (103, 104), which are components of the tumor microenvironment. HDAC inhibitors, including vorinostat (SAHA) and panobinostat, suppress EGFR expression and reverse EMT (Table 1). In addition, these drugs have been successfully used in combination with BET inhibitors. Such epigenetic combination therapy improves clinical efficacy by reducing Myc expression. Lysine-specific demethylase 1 (KDM1, also known as LSD1) modulates histone methylation, and demethylation of H3K9me2, H3K4me3, and H3K36me3 contributes to KDM1-mediated chemoresistance (105). Inhibition of KDM1 activity was initially observed in monoamine oxidase (MAO) inhibitors such as tranylcypromine, which was accompanied by suppression of the stem cell properties of CSCs in vivo (106). Based on their structure, several selective KDM1 inhibitors have been developed (Table 1). In addition, the activity of KDM1 can be regulated by HDAC inhibitors, as crosstalk exists between KDM1 and HDAC. KDM1 and HDAC1/2 form the CoREST complex, which is associated with silencing gene expression. As a result, combination therapy with KDM1 inhibitors and HDAC inhibitors has been expected to have synergistic effects and has often been evaluated. Recently, KDM1-HDAC dual inhibitors such as corin have been reported (107). BMI1 and EZH2, which induce epigenetic silencing as polycomb group (PcG) members, have been reported to be associated with chemoresistance (108, 109). Besides, tumor suppressor genes are silenced by hypermethylation of promoter regions of DNA, and thus, chemotherapy loses its efficacy against many cancers (110).
Targeting Microenvironment
The cellular microenvironment plays an important role in determining cellular behavior. NSCs and CSCs are generated, maintained, and regulated within this microenvironment. The tumor microenvironment (TME) creates a niche for itself that influences not only the proliferation and differentiation of CSCs but also the response to drugs (111). Although cancer-associated fibroblasts (CAFs) as well as inflammation and immune cells are components of the TME, we have to discuss the crucial role of hypoxia in the TME. Hypoxia signaling contributes to chemoresistance of CSCs by increasing the expression of ABC transporters and ALDH (8, 112, 113). In solid tumors, hypoxic regions are necessarily present and lead to angiogenesis through HIF1A and VEGF (114, 115). However, tumor angiogenesis is sloppy; hence, drugs do not reach effective concentrations in hypoxic cells. Thus, VEGF inhibitors such as bevacizumab and receptor tyrosine kinase (RTK) inhibitors such as sorafenib can enhance chemosensitivity. Vascular disrupting agents (VDAs), including tubulin-binding agents such as combretastatin A4 and plocabulin, also enhance chemosensitivity by increasing vessel permeability (Table 1). Of course, we should be careful not to inject these agents prior to chemotherapy because it could impair the delivery of chemotherapeutic drugs by reducing blood supply.
Targeting Signaling Pathways
Signaling pathways control cell responses, particularly the self-renewal, differentiation, and survival of CSCs. Among them, the Notch, Hedgehog (Hh), and Wnt/β-catenin signaling pathways are responsible for drug resistance (116). Notch induces the expression of survivin, which is an anti-apoptotic gene that inhibits apoptosis. This signaling is suppressed by γ-secretase inhibitors (GSIs) such as DAPT and RO4929097, which block the second cleavage of Notch receptors and release of the Notch-IC fragment from the cell membrane (Table 1). Hh signaling is critical in embryogenesis and has been found in many cancers (75, 117). This signaling induces the activation of Smo and Gli1, which are involved in the drug resistance caused by overexpression of P-gp and BCRP (118). Thus, Smo inhibitors such as cyclopamine, vismodegib, and sonidegib can suppress chemoresistance (Table 1). Wnt/β-catenin signaling activated by Frizzleds (FZDs) is also associated with drug resistance due to overexpression of P-gp, BCRP, and MRP (119). Besides, this pathway promotes cell cycling, inhibits apoptosis, and mediates DNA repair processes. Although it has been suggested that the Wnt/β-catenin signaling pathway is undruggable, strategies to target it have been explored because of their various therapeutic potential. Indeed, there has been an effort to develop drugs that inhibit Wnt/β-catenin signaling (Table 1).
Future Perspectives
To date, many chemotherapeutic drugs have been developed and many studies conducted to overcome MDR. The newer strategies that we have covered above have limitations, however, and so more work is needed with different approaches being explored to find effective and lasting treatment. Any strategy for overcoming MDR should not affect NCSs, but only CSCs. There are a few things to pay attention to for selective targeting of CSCs: (i) encapsulation of anticancer drugs in liposomes, micelles, and nanoparticles, i.e., nanotechnology-based drug delivery (120). The improvement of pharmacokinetic properties, tumor-specific delivery due to the enhanced permeability and retention (EPR) effect, and resistance to efflux pumps because of the size-exclusion effect are representative advantages of nanomaterials (NMs) (121–123). Thus, NMs are expected to increase the therapeutic effect and reduce unwanted side effects of anticancer drugs. Moreover, dual drug-loaded NMs can lead to better therapeutic effects (124). For example, pluronics, which are amphiphilic polymers and form nano-sized micellar structures, have been increasingly regarded as CSC modulators (125, 126). Pluronics are not just drug delivery carriers; rather, they inhibit metastasis and activate apoptosis by mediating the release of cytochrome c and apoptosis inducing factor (AIF). In addition, they can alter the microenvironment and suppress CSCs effectively. At present, only SP1049C (doxorubicin with pluronics L61 and F127 micelles) is in phase 3 trials (127), but there are reasons to be optimistic. (ii) Targeting microRNAs (miRNAs), which mediate translational repression and mRNA degradation mainly by binding to the 3′ UTR. Previous studies have shown that miRNAs regulate ABC transporters in CSCs (128, 129); for instance, miR-212, miR-328, miR-451, and so on. Some miRNAs, including miR-21 and miR-222, are oncogenic and up-regulated in cancer cells, while other miRNAs, including miR-15 and miR-181, are tumor suppressive and down-regulated in cancer cells (29, 130). (iii) Targeting MDR mRNA (131, 132), through which antisense oligonucleotides (aODNs) are used to down-regulate the expression of specific genes. Small interfering RNAs (siRNAs) also silence specific genes because they are artificial double-stranded RNA (dsRNA) molecules with functions similar to those of miRNA. If they selectively target tumorigenic genes, cancer cells will become chemosensitive. There are no clinical data yet to suggest that MDR can be completely reversed using this RNA interference (RNAi) technology, but it is likely that drugs related to RNAi will be developed in the near future. (iv) Transferring MDR genes into NSCs, especially bone marrow stem cells (133–135). Most chemotherapeutic drugs suppress the bone marrow and, as a result, blood cells such as erythrocytes, leukocytes, and thrombocytes do not function properly, and the ensuing loss of oxygen transport, immune response, and bleeding control functions causes serious adverse effects. To prevent these adverse effects, gene transfer technology has emerged, making it possible to administer high doses of chemotherapeutic drugs. In the transplantation model of CD34-positive peripheral blood stem cell (PBSC) infection with retroviral vectors containing MDR genes, long-term myeloprotection was achieved, demonstrating the safety of transplantation (136).
Conclusion
Over the past few decades there have been many strategies used to treat cancer, from conventional radiation therapy, chemotherapy to recent targeted therapy, and immunotherapy. It is no exaggeration to say that the flow of this change has been led by the discovery of various factors that cause MDR acquisition in cancer cells, especially ABC transporters. By discovering ABC transporters and identifying their functions, new possibilities have emerged for cancer treatment. Initially, many studies were focused on the direct inhibition of ABC transporters such as P-gp inhibitors. However, because they are less selective and less potent, differ in their in vitro and in vivo data, and often cause severe adverse effects, so far no drugs that directly target or inhibit P-gp have been accepted for clinical use. Although direct inhibition of ABC transporters may not be effective, the transporters remain attractive targets because enhanced drug efflux through the transporters is one of the most important causes of MDR acquisition. Therefore, rather than directly inhibiting the ABC transporters, it would be effective to devise alternative strategies to avoid drug efflux via transporters. The transporter-mediated MDR might be overcome by developing novel anticancer drugs with P-gp non-substrates. In addition, we have introduced alternative approaches for targeting CSCs that focus on ALDHs, EMT, epigenetic modifications, the microenvironment, and signaling pathways (Figure 1). We have also discussed the drugs being developed in each approach: (i) ABC transporter inhibitors and non-substrates; (ii) ALDH inhibitors; (iii) AMPK activators; (iv) BET inhibitors, HDAC inhibitors, and KDM1 inhibitors; (v) VEGF inhibitors, RTK inhibitors, and vascular disrupting agents; (vi) Notch inhibitors, Smo inhibitors, and Wnt/β-catenin inhibitors. One of the big obstacles we are facing now is whether we can selectively influence CSCs only. NSCs and CSCs share the same characteristics; therefore, solving this problem is a complex task. Although promising solutions have been proposed, more research should be conducted to support the arguments relating to nanotechnology-based drug delivery, RNAi technology, and gene transfer technology. It is worth noting that these approaches are directly or indirectly related to ABC transporters. It is hoped that all of the approaches reviewed here will help devise new strategies to overcome MDR and to eradicate MDR cancer.
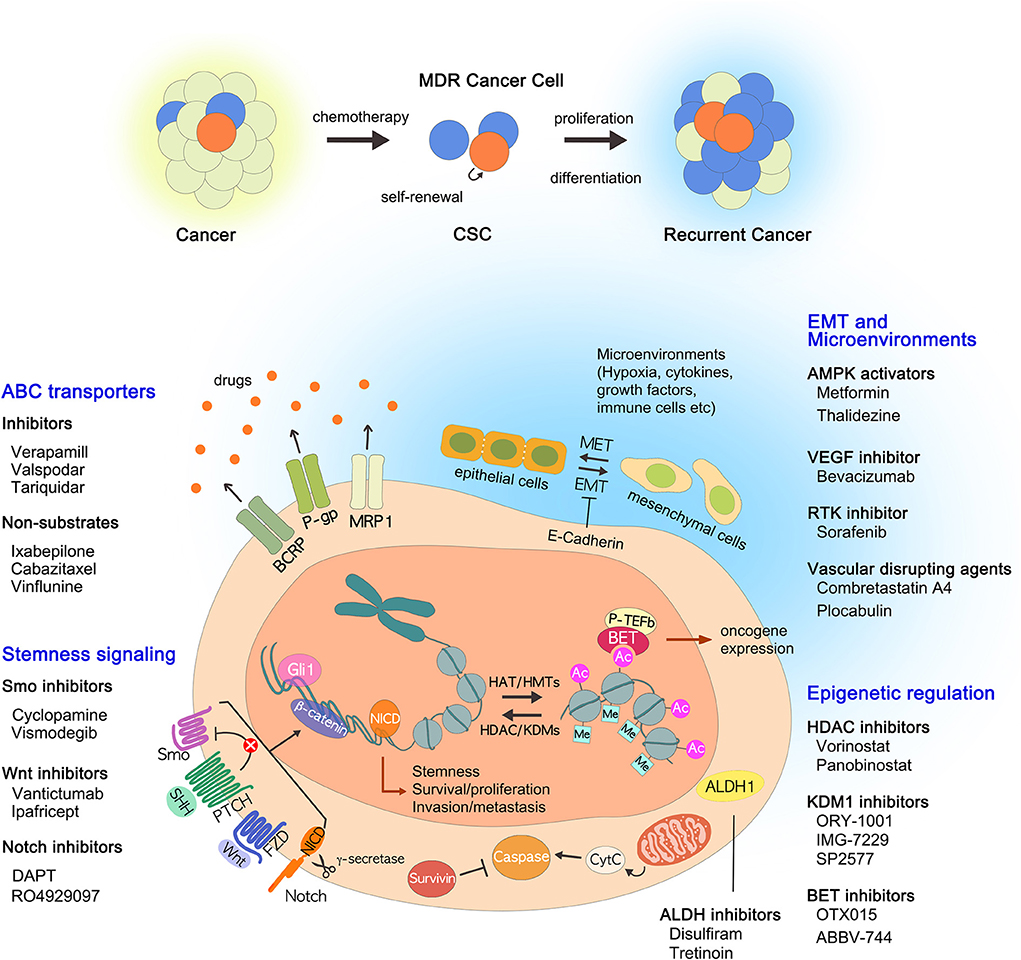
Figure 1. Current strategies to overcome multidrug resistance. In addition to the well-known ABC transporters, many drug resistance mechanisms of CSCs have been identified, including ALDHs, EMT, epigenetic changes, tumor microenvironment, and stemness-related signaling pathways. The drugs that inhibit each pathway are described: (i) ABC transporter inhibitors and non-substrates; (ii) ALDH inhibitors; (iii) AMPK activators; (iv) BET inhibitors, HDAC inhibitors, and KDM1 inhibitors; (v) VEGF inhibitors, RTK inhibitors, and vascular disrupting agents; (VI) Notch inhibitors, Smo inhibitors, and Wnt/β-catenin inhibitors. ABC, ATP binding cassette; ALDHs, aldehyde dehydrogenases; AMPK, AMP-activated kinase; BET, bromodomain and extra-terminal; CSCs, cancer stem cells; EMT, epithelial to mesenchymal transition; HDAC, histone deacetylase; KDM1, lysine-specific demethylase 1; MDR, multidrug resistance; RTK, receptor tyrosine kinase; VEGF, vascular endothelial growth factor.
Author Contributions
YC and YK participated in manuscript drafting and revision of this article.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) [NRF-2018R1A2B2005646 (to YK)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Bokyeong Kim for providing the illustration used herein.
References
1. Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. (2003) 10:159–65. doi: 10.1177/107327480301000207
2. Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. (2003) 22:7468–85. doi: 10.1038/sj.onc.1206948
3. Moitra K. Overcoming multidrug resistance in cancer stem cells. BioMed Res Int. (2015) 2015:635745–8. doi: 10.1155/2015/635745
4. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol. (2016) 43:723–37. doi: 10.1111/1440-1681.12581
5. Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. (1999) 39:361–98. doi: 10.1146/annurev.pharmtox.39.1.361
6. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. (2004) 101:14228–33. doi: 10.1073/pnas.0400067101
7. Jung Y, Kim W. Cancer stem cell targeting: are we there yet? Arch Pharm Res. (2015) 38:414–22. doi: 10.1007/s12272-015-0570-2
8. Ma L, Lai D, Liu T, Cheng W, Guo L. Cancer stem-like cells can be isolated with drug selection in human ovarian cancer cell line SKOV3. Acta Biochim Biophys Sin (Shanghai). (2010) 42:593–602. doi: 10.1093/abbs/gmq067
9. Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. (2011) 2011:396076. doi: 10.1155/2011/396076
10. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. (2002) 2:48–58. doi: 10.1038/nrc706
11. Raha D, Wilson TR, Peng J, Peterson D, Yue P, Evangelista M, et al. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. (2014) 74:3579–90. doi: 10.1158/0008-5472.CAN-13-3456
12. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
13. Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. (2017) 3:372–86. doi: 10.1016/j.trecan.2017.04.004
14. Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. (2012) 6:620–36. doi: 10.1016/j.molonc.2012.10.006
15. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. (2012) 1826:272–96. doi: 10.1016/j.bbcan.2012.04.008
16. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell fate determination of human mammary stemprogenitor cells. Breast Cancer Res. (2004) 6:R605–15. doi: 10.1186/bcr920
17. Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. (2010) 29:469–81. doi: 10.1038/onc.2009.392
18. Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. (2008) 68:4287–95. doi: 10.1158/0008-5472.CAN-07-6691
19. Wu CP, Calcagno AM, Ambudkar SV. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr Mol Pharmacol. (2008) 1:93–105. doi: 10.2174/1874467210801020093
20. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. (2000) 11:265–83. doi: 10.1016/s0928-0987(00)00114-7
21. Chung FS, Santiago JS, Jesus MF, Trinidad CV, See MF. Disrupting P-glycoprotein function in clinical settings: what can we learn from the fundamental aspects of this transporter? Am J Cancer Res. (2016) 6:1583–98.
22. Chao NJ, Aihara M, Blume KG, Sikic BI. Modulation of etoposide (VP-16) cytotoxicity by verapamil or cyclosporine in multidrug-resistant human leukemic cell lines and normal bone marrow. Exp Hematol. (1990) 18:1193–8.
23. Jin Y, Bin ZQ, Qiang H, Liang C, Hua C, Jun D, et al. ABCG2 is related with the grade of glioma and resistance to mitoxantone, a chemotherapeutic drug for glioma. J Cancer Res Clin Oncol. (2009) 135:1369–76. doi: 10.1007/s00432-009-0578-4
24. Solary E, Velay I, Chauffert B, Bidan J, Caillot D, Dumas M, et al. Sufficient levels of quinine in the serum circumvent the multidrug resistance of the human leukemic cell line K562/ADM. Cancer. (1991) 68:1714–9. doi: 10.1002/1097-0142(19911015)68:8<1714::AID-CNCR2820680811>3.0.CO;2-2
25. Li C, Kim M, Choi H, Choi J. Effects of baicalein on the pharmacokinetics of tamoxifen and its main metabolite, 4-hydroxytamoxifen, in rats: possible role of cytochrome P450 3A4 and P-glycoprotein inhibition by baicalein. Arch Pharm Res. (2011) 34:1965–72. doi: 10.1007/s12272-011-1117-9
26. Spoelstra EC, Westerhoff HV, Pinedo HM, Dekker H, Lankelma J. The multidrug-resistance-reverser verapamil interferes with cellular P-glycoprotein-mediated pumping of daunorubicin as a non-competing substrate. Eur J Biochem. (1994) 221:363–73. doi: 10.1111/j.1432-1033.1994.tb18748.x
27. Solary E, Mannone L, Moreau D, Caillot D, Casasnovas RO, Guy H, et al. Phase I study of cinchonine, a multidrug resistance reversing agent, combined with the CHVP regimen in relapsed and refractory lymphoproliferative syndromes. Leukemia. (2000) 14:2085–94. doi: 10.1038/sj.leu.2401945
28. Wilson WH, Jamis-Dow C, Bryant G, Balis FM, Klecker RW, Bates SE, et al. Phase I and pharmacokinetic study of the multidrug resistance modulator dexverapamil with EPOCH chemotherapy. J Clin Oncol. (1995) 13:1985–94. doi: 10.1200/JCO.1995.13.8.1985
29. Mubashar M, Harrington KJ, Chaudhary KS, Lalani E, Stamp GW, Peters AM. Differential effects of toremifene on doxorubicin, vinblastine and Tc-99m-sestamibi in P-glycoprotein-expressing breast and head and neck cancer cell lines. Acta Oncol. (2004) 43:443–52. doi: 10.1080/02841860410031048
30. Twentyman PR, Bleehen NM. Resistance modification by PSC-833, a novel non-immunosuppressive cyclosporin [corrected]. Eur J Cancer. (1991) 27:1639–42. doi: 10.1016/0277-5379(91)90435-G
31. Gandhi L, Harding MW, Neubauer M, Langer CJ, Moore M, Ross HJ, et al. A phase II study of the safety and efficacy of the multidrug resistance inhibitor VX-710 combined with doxorubicin and vincristine in patients with recurrent small cell lung cancer. Cancer. (2007) 109:924–32. doi: 10.1002/cncr.22492
32. Nakanishi O, Baba M, Saito A, Yamashita T, Sato W, Abe H, et al. Potentiation of the antitumor activity by a novel quinoline compound, MS-209, in multidrug-resistant solid tumor cell lines. Oncol Res. (1997) 9:61–9.
33. van Zuylen L, Sparreboom A, van der Gaast A, Nooter K, Eskens FA, Brouwer E, et al. Disposition of docetaxel in the presence of P-glycoprotein inhibition by intravenous administration of R101933. Eur J Cancer. (2002) 38:1090–9. doi: 10.1016/S0959-8049(02)00035-7
34. Shi B, Yaremko B, Hajian G, Terracina G, Bishop WR, Liu M, et al. The farnesyl protein transferase inhibitor SCH66336 synergizes with taxanes in vitro and enhances their antitumor activity in vivo. Cancer Chemother Pharmacol. (2000) 46:387–93. doi: 10.1007/s002800000170
35. Green LJ, Marder P, Slapak CA. Modulation by LY335979 of P-glycoprotein function in multidrug-resistant cell lines and human natural killer cells. Biochem Pharmacol. (2001) 61:1393–9. doi: 10.1016/S0006-2952(01)00599-8
36. de Bruin M, Miyake K, Litman T, Robey R, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. (1999) 146:117–26. doi: 10.1016/S0304-3835(99)00182-2
37. Lusvarghi S, Ambudkar SV. ATP-dependent thermostabilization of human P-glycoprotein (ABCB1) is blocked by modulators. Biochem J. (2019) 476:3737–50. doi: 10.1042/BCJ20190736
38. Rabindran SK, He H, Singh M, Brown E, Collins KI, Annable T, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. (1998) 58:5850–8.
39. Ghosh P, Moitra K, Maki N, Dey S. Allosteric modulation of the human P-glycoprotein involves conformational changes mimicking catalytic transition intermediates. Arch Biochem Biophys. (2006) 450:100–12. doi: 10.1016/j.abb.2006.02.025
40. Fornier MN. Ixabepilone, first in a new class of antineoplastic agents: the natural epothilones and their analogues. Clin Breast Cancer. (2007) 7:9–15. doi: 10.3816/CBC.2007.n.036
41. Di Nunno V, Mollica V, Massari F. Cabazitaxel in metastatic prostate cancer. N Engl J Med. (2020) 382:1286. doi: 10.1056/NEJMc2000990
42. Brooks TA, Minderman H, O'Loughlin KL, Pera P, Ojima I, Baer MR, et al. Taxane-based reversal agents modulate drug resistance mediated by P-glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol Cancer Ther. (2003) 2:1195–205.
43. Waghray D, Zhang Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment. J Med Chem. (2018) 61:5108–21. doi: 10.1021/acs.jmedchem.7b01457
44. MacDonagh L, Gallagher MF, Ffrench B, Gasch C, Breen E, Gray SG, et al. Targeting the cancer stem cell marker, aldehyde dehydrogenase 1, to circumvent cisplatin resistance in NSCLC. Oncotarget. (2017) 8:72544–63. doi: 10.18632/oncotarget.19881
45. Toledo-Guzman ME, Hernandez MI, Gomez-Gallegos AA, Ortiz-Sanchez E. ALDH as a stem cell marker in solid tumors. Curr Stem Cell Res Ther. (2019) 14:375–88. doi: 10.2174/1574888X13666180810120012
46. Moreb JS, Ucar-Bilyeu DA, Khan A. Use of retinoic acid/aldehyde dehydrogenase pathway as potential targeted therapy against cancer stem cells. Cancer Chemother Pharmacol. (2017) 79:295–301. doi: 10.1007/s00280-016-3213-5
47. Choi YK, Park KG. Metabolic roles of AMPK and metformin in cancer cells. Mol Cells. (2013) 36:279–87. doi: 10.1007/s10059-013-0169-8
48. Qu C, Qu C, Zhang W, Zhang W, Zheng G, Zheng G, et al. Metformin reverses multidrug resistance and epithelial–mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol Cell Biochem. (2014) 386:63–71. doi: 10.1007/s11010-013-1845-x
49. Law BYK, Gordillo-Martínez F, Qu YQ, Zhang N, Xu SW, Coghi PS, et al. Thalidezine, a novel AMPK activator, eliminates apoptosis-resistant cancer cells through energy-mediated autophagic cell death. Oncotarget. (2017) 8:30077–91. doi: 10.18632/oncotarget.15616
50. Heijmen L, Ter Voert EG, Punt CJ, Heerschap A, Oyen WJ, Bussink J, et al. Monitoring hypoxia and vasculature during bevacizumab treatment in a murine colorectal cancer model. Contrast Media Mol Imaging. (2014) 9:237–45. doi: 10.1002/cmmi.1564
51. Qiu Y, Shan W, Yang Y, Jin M, Dai Y, Yang H, et al. Reversal of sorafenib resistance in hepatocellular carcinoma: epigenetically regulated disruption of 14-3-3η/hypoxia-inducible factor-1α. Cell Death Discov. (2019) 5:120. doi: 10.1038/s41420-019-0200-8
52. Close A. Antiangiogenesis and vascular disrupting agents in cancer: circumventing resistance and augmenting their therapeutic utility. Future Med Chem. (2016) 8:443–62. doi: 10.4155/fmc.16.6
53. Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. (2006) 66:11520–39. doi: 10.1158/0008-5472.CAN-06-2848
54. Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T, et al. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA. (2019) 5:FSO372. doi: 10.4155/fsoa-2018-0115
55. Bolden JE, Tasdemir N, Dow LE, van Es JH, Wilkinson JE, Zhao Z, et al. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. (2014) 8:1919–29. doi: 10.1016/j.celrep.2014.08.025
56. Siu KT, Ramachandran J, Yee AJ, Eda H, Santo L, Panaroni C, et al. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia. (2017) 31:1760–9. doi: 10.1038/leu.2016.355
57. Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar C, Wahlestedt C, et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. (2014) 289:35494–502. doi: 10.1074/jbc.M114.595348
58. Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. (2015) 6:17698–712. doi: 10.18632/oncotarget.4131
59. Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res. (2015) 21:1628–38. doi: 10.1158/1078-0432.CCR-14-1561
60. Bui MH, Lin X, Albert DH, Li L, Lam LT, Faivre EJ, et al. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. (2017) 77:2976–89. doi: 10.1158/0008-5472.CAN-16-1793
61. Zhang D, Leal AS, Carapellucci S, Zydeck K, Sporn MB, Liby KT. Chemoprevention of preclinical breast and lung cancer with the bromodomain inhibitor I-BET 762. Cancer Prev Res. (2018) 11:143–56. doi: 10.1158/1940-6207.CAPR-17-0264
62. Bruzzese F, Leone A, Rocco M, Carbone C, Piro G, Caraglia M, et al. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J Cell Physiol. (2011) 226:2378–90. doi: 10.1002/jcp.22574
63. Di Costanzo A, Del Gaudio N, Migliaccio A, Altucci L. Epigenetic drugs against cancer: an evolving landscape. Arch Toxicol. (2014) 88:1651–68. doi: 10.1007/s00204-014-1315-6
64. Ling Y, Liu J, Qian J, Meng C, Guo J, Gao W, et al. Recent advances in multi-target drugs targeting protein kinases and histone deacetylases in cancer therapy. Curr Med Chem. (2020). doi: 10.2174/0929867327666200102115720. [Epub ahead of print].
65. Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc. (2010) 132:6827–33. doi: 10.1021/ja101557k
66. Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. (2015) 28:57–69. doi: 10.1016/j.ccell.2015.06.002
67. Fu X, Zhang P, Yu B. Advances toward LSD1 inhibitors for cancer therapy. Future Med Chem. (2017) 9:1227–42. doi: 10.4155/fmc-2017-0068
68. Shaw HV, Koval A, Katanaev VL. Targeting the Wnt signalling pathway in cancer: prospects and perils. Swiss Med Wkly. (2019) 149:w20129. doi: 10.4414/smw.2019.20129
69. Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. (2018) 33:495–511.e12. doi: 10.1016/j.ccell.2018.02.002
70. Hollebecque A, de Bono JS, Plummer R, Isambert N, Martin-Romano P, Baudin E, et al. Phase I study of CC-90011 in patients with advanced solid tumors and relapsed/refractory non-Hodgkin lymphoma (R/R NHL). Ann Oncol. (2019) 29(Suppl. 8):viii649–69. doi: 10.1093/annonc/mdy303
71. Raffaella Soldi, Tithi, Ghosh Halder, Alexis Weston, Trason Thode, Kevin Drenner, Rhonda Lewis, et al. The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. bioRxiv. doi: 10.1101/2020.01.10.902528
72. Young MJ, Wu YH, Chiu WT, Weng TY, Huang YF, Chou CY. All-trans retinoic acid downregulates ALDH1-mediated stemness and inhibits tumour formation in ovarian cancer cells. Carcinogenesis. (2015) 36:498–507. doi: 10.1093/carcin/bgv018
73. Meng RD, Shelton CC, Li Y, Qin L, Notterman D, Paty PB, et al. gamma-secretase inhibitors abrogate oxaliplatin-induced activation of the notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. (2009) 69:573–82. doi: 10.1158/0008-5472.CAN-08-2088
74. Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. (2015) 121:432–40. doi: 10.1002/cncr.29055
75. Song Z, Yue W, Wei B, Wang N, Li T, Guan L, et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE. (2011) 6:e17687. doi: 10.1371/journal.pone.0017687
76. Dirix L. Discovery and exploitation of novel targets by approved drugs. J Clin Oncol. (2014) 32:720–1. doi: 10.1200/JCO.2013.53.7118
77. Dummer R, Ascierto PA, Basset-Seguin N, Dreno B, Garbe C, Gutzmer R, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. (2020). doi: 10.1111/jdv.16230. [Epub ahead of print].
78. Jimeno A, Gordon M, Chugh R, Messersmith W, Mendelson D, Dupont J, et al. A first-in-human phase I study of the anticancer stem cell agent Ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin Cancer Res. (2017) 23:7490–7. doi: 10.1158/1078-0432.CCR-17-2157
79. Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int J Mol Med. (2017) 40:587–606. doi: 10.3892/ijmm.2017.3071
80. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. (2017) 51:1357–69. doi: 10.3892/ijo.2017.4129
81. Safholm A, Leandersson K, Dejmek J, Nielsen CK, Villoutreix BO, Andersson T. A formylated hexapeptide ligand mimics the ability of Wnt-5a to impair migration of human breast epithelial cells. J Biol Chem. (2006) 281:2740–9. doi: 10.1074/jbc.M508386200
82. Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. (2016) 35:2197–207. doi: 10.1038/onc.2015.280
83. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci USA. (2013) 110:20224–9. doi: 10.1073/pnas.1314239110
84. Pak S, Park S, Kim Y, Park JH, Park CH, Lee KJ, et al. Correction to: the small molecule WNT/beta-catenin inhibitor CWP232291 blocks the growth of castration-resistant prostate cancer by activating the endoplasmic reticulum stress pathway. J Exp Clin Cancer Res. (2019) 38:440–1. doi: 10.1186/s13046-019-1451-1
85. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. (2014) 33:2169–78. doi: 10.1038/onc.2013.169
86. Ritchie TK, Kwon H, Atkins WM. Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem. (2011) 286:39489–96. doi: 10.1074/jbc.M111.284554
87. Scotto KW, Johnson RA. Transcription of the multidrug resistance gene MDR1: a therapeutic target. Mol Interv. (2001) 1:117–25.
88. Scotto KW. Transcriptional regulation of ABC drug transporters. Oncogene. (2003) 22:7496–511. doi: 10.1038/sj.onc.1206950
89. Souid S, Aissaoui D, Srairi-Abid N, Essafi-Benkhadir K. Trabectedin (yondelis®) as a therapeutic option in gynecological cancers: a focus on its mechanisms of action, clinical activity, and genomic predictors of drug response. Curr Drug Targets. (2020) 21. doi: 10.2174/1389450121666200128161733. [Epub ahead of print].
90. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. (2006) 5:219–34. doi: 10.1038/nrd1984
91. Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem. (2012) 19:1946–2025. doi: 10.2174/092986712800167392
92. Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. (2013) 7:27–34. doi: 10.4137/DTI.S12519
93. Shackleton M. Normal stem cells and cancer stem cells: similar and different. Semin Cancer Biol. (2010) 20:85–92. doi: 10.1016/j.semcancer.2010.04.002
94. Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, Chao NJ, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA. (2006) 103:11707–12. doi: 10.1073/pnas.0603806103
95. Huang CP, Tsai MF, Chang TH, Tang WC, Chen SY, Lai HH, et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. (2013) 328:144–51. doi: 10.1016/j.canlet.2012.08.021
96. Zhou W, Lv R, Qi W, Wu D, Xu Y, Liu W. Snail contributes to the maintenance of stem cell like phenotype cells in human pancreatic cancer. PLoS ONE. (2014) 9:e87409. doi: 10.1371/journal.pone.0087409
97. Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. (2016) 76:6320–30. doi: 10.1158/0008-5472.CAN-16-0854
98. Celia-Terrassa T, Jolly MK. Cancer stem cells and epithelial-to-mesenchymal transition in cancer metastasis. Cold Spring Harb Perspect Med. (2019) a036905. doi: 10.1101/cshperspect.a036905. [Epub ahead of print].
99. Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. (2014) 14:306–21. doi: 10.1016/j.stem.2014.02.002
100. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. (2014) 16:488–94. doi: 10.1038/ncb2976
101. Lin H, Li N, He H, Ying Y, Sunkara S, Luo L, et al. AMPK inhibits the stimulatory effects of TGF-beta on Smad2/3 activity, cell migration, and epithelial-to-mesenchymal transition. Mol Pharmacol. (2015) 88:1062–71. doi: 10.1124/mol.115.099549
102. Sakamoto T, Kobayashi S, Yamada D, Nagano H, Tomokuni A, Tomimaru Y, et al. A histone deacetylase inhibitor suppresses epithelial-mesenchymal transition and attenuates chemoresistance in biliary tract cancer. PLoS ONE. (2016) 11:e0145985. doi: 10.1371/journal.pone.0145985
103. Hwang JW, Cho H, Lee JY, Jeon Y, Kim SN, Lee SJ, et al. The synthetic ajoene analog SPA3015 induces apoptotic cell death through crosstalk between NF-kappaB and PPARgamma in multidrug-resistant cancer cells. Food Chem Toxicol. (2016) 96:35–42. doi: 10.1016/j.fct.2016.07.020
104. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. (2009) 8:33–40. doi: 10.1038/nrd2781
105. McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. (2011) 18:867–74. doi: 10.1038/nsmb.2084
106. Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim Biophys Acta. (2014) 1839:1416–32. doi: 10.1016/j.bbagrm.2014.05.009
107. Kalin JH, Wu M, Gomez AV, Song Y, Das J, Hayward D, et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. (2018) 9:53–4. doi: 10.1038/s41467-017-02242-4
108. Crea F, Danesi R, Farrar WL. Cancer stem cell epigenetics and chemoresistance. Epigenomics. (2009) 1:63–79. doi: 10.2217/epi.09.4
109. Pietersen AM, Horlings HM, Hauptmann M, Langerod A, Ajouaou A, Cornelissen-Steijger P, et al. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. (2008) 10:R109. doi: 10.1186/bcr2214
110. Ibanez de Caceres I, Cortes-Sempere M, Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, Manguan-Garcia C, et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene. (2010) 29:1681–90. doi: 10.1038/onc.2009.454
111. Lau EY, Ho NP, Lee TK. Cancer stem cells and their microenvironment: biology and therapeutic implications. Stem Cells Int. (2017) 2017:3714190. doi: 10.1155/2017/3714190
112. Seo EJ, Kim DK, Jang IH, Choi EJ, Shin SH, Lee SI, et al. Hypoxia-NOTCH1-SOX2 signaling is important for maintaining cancer stem cells in ovarian cancer. Oncotarget. (2016) 7:55624–38. doi: 10.18632/oncotarget.10954
113. Garson K, Vanderhyden BC. Epithelial ovarian cancer stem cells: underlying complexity of a simple paradigm. Reproduction. (2015) 149:59. doi: 10.1530/REP-14-0234
114. Hajizadeh F, Okoye I, Esmaily M, Ghasemi Chaleshtari M, Masjedi A, Azizi G, et al. Hypoxia inducible factors in the tumor microenvironment as therapeutic targets of cancer stem cells. Life Sci. (2019) 237:116952. doi: 10.1016/j.lfs.2019.116952
115. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. (2019) 18:157–9. doi: 10.1186/s12943-019-1089-9
116. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting notch, hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
117. Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer. (2012) 131:30–40. doi: 10.1002/ijc.26323
118. Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. (2007) 26:5674–9. doi: 10.1038/sj.onc.1210356
119. Flahaut M, Meier R, Coulon A, Nardou KA, Niggli FK, Martinet D, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene. (2009) 28:2245–56. doi: 10.1038/onc.2009.80
120. Zhang M, Liu E, Cui Y, Huang Y. Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer. Cancer Biol Med. (2017) 14:212–27. doi: 10.20892/j.issn.2095-3941.2017.0054
121. Gao Z, Zhang L, Sun Y. Nanotechnology applied to overcome tumor drug resistance. J Control Release. (2012) 162:45–55. doi: 10.1016/j.jconrel.2012.05.051
122. Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. (2013) 48:416–27. doi: 10.1016/j.ejps.2012.12.006
123. Rempe R, Cramer S, Qiao R, Galla HJ. Strategies to overcome the barrier: use of nanoparticles as carriers and modulators of barrier properties. Cell Tissue Res. (2014) 355:717–26. doi: 10.1007/s00441-014-1819-7
124. Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS Nano. (2013) 7:9518–25. doi: 10.1021/nn405674m
125. Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv Drug Deliv Rev. (2002) 54:759–79. doi: 10.1016/S0169-409X(02)00047-9
126. Dehghankelishadi P, Dorkoosh F. Pluronic based nano-delivery systems; Prospective warrior in war against cancer. Nanomed Res J. (2016) 1:1–7. doi: 10.7508/nmrj.2016.01.001
127. Alakhova DY, Zhao Y, Li S, Kabanov AV. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS ONE. (2013) 8:e72238. doi: 10.1371/journal.pone.0072238
128. DeSano JT, Xu L. MicroRNA regulation of cancer stem cells and therapeutic implications. AAPS J. (2009) 11:682–92. doi: 10.1208/s12248-009-9147-7
129. Xing F, Wu K, Watabe K. MicroRNAs in cancer stem cells: new regulators of stemness. Curr Pharm Des. (2014) 20:5319–27. doi: 10.2174/1381612820666140128210912
130. Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol. (2010) 79:817–24. doi: 10.1016/j.bcp.2009.10.017
131. Zou S, Cao N, Cheng D, Zheng R, Wang J, Zhu K, et al. Enhanced apoptosis of ovarian cancer cells via nanocarrier-mediated codelivery of siRNA and doxorubicin. Int J Nanomed. (2012) 7:3823–35. doi: 10.2147/IJN.S29328
132. Xiong X, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. (2011) 5:5202–13. doi: 10.1021/nn2013707
133. Budak-Alpdogan T, Banerjee D, Bertino JR. Hematopoietic stem cell gene therapy with drug resistance genes: an update. Cancer Gene Ther. (2005) 12:849–63. doi: 10.1038/sj.cgt.7700866
134. Zaboikin M, Srinivasakumar N, Schuening F. Gene therapy with drug resistance genes. Cancer Gene Ther. (2006) 13:335–45. doi: 10.1038/sj.cgt.7700912
135. Bertino JR. Transfer of drug resistance genes into hematopoietic stem cells for marrow protection. Oncologist. (2008) 13:1036–42. doi: 10.1634/theoncologist.2008-0173
136. O'Shaughnessy JA, Cowan KH, Nienhuis AW, McDonagh KT, Sorrentino BP, Dunbar CE, et al. Retroviral mediated transfer of the human multidrug resistance gene (MDR-1) into hematopoietic stem cells during autologous transplantation after intensive chemotherapy for metastatic breast cancer. Hum Gene Ther. (1994) 5:891–911. doi: 10.1089/hum.1994.5.7-891
Keywords: multidrug resistance, ABC transporters, P-glycoprotein, cancer stem cells, epigenetics
Citation: Cho Y and Kim YK (2020) Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 10:764. doi: 10.3389/fonc.2020.00764
Received: 10 March 2020; Accepted: 21 April 2020;
Published: 02 June 2020.
Edited by:
Sungpil Yoon, Sungkyunkwan University, South KoreaReviewed by:
Hyung Sik Kim, Sungkyunkwan University, South KoreaXiaoju Wang, University of Michigan, United States
Copyright © 2020 Cho and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Kee Kim, eWtzbmJrQHNvb2tteXVuZy5hYy5rcg==