
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 29 May 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00712
This article is part of the Research Topic Impact of Cancer Plasticity on Drug Resistance and Treatment in Solid Tumors. View all 33 articles
 Caterina F. Donini1,2,3†
Caterina F. Donini1,2,3† Myriam El Helou1,2,4†
Myriam El Helou1,2,4† Anne Wierinckx1,2,5
Anne Wierinckx1,2,5 Balázs Győrffy6
Balázs Győrffy6 Sophie Aires1,2
Sophie Aires1,2 Aurélie Escande7
Aurélie Escande7 Séverine Croze1,5Philippe Clezardin1,8Joël Lachuer1,2,5Mona Diab-Assaf4
Séverine Croze1,5Philippe Clezardin1,8Joël Lachuer1,2,5Mona Diab-Assaf4 Sandra E. Ghayad4
Sandra E. Ghayad4 Béatrice Fervers1,2,3†
Béatrice Fervers1,2,3† Vincent Cavaillès9†
Vincent Cavaillès9† Véronique Maguer-Satta2†
Véronique Maguer-Satta2† Pascale A. Cohen1,2,3,5,8*
Pascale A. Cohen1,2,3,5,8*It is of utmost importance to decipher the role of chronic exposure to low doses of environmental carcinogens on breast cancer progression. The early-transformed triple-negative human mammary MCF10AT1 cells were chronically (60 days) exposed to low doses (10−10 M) of Benzo[a]pyrene (B[a]P), a genotoxic agent, and/or Bisphenol A (BPA), an endocrine disruptor. Our study revealed that exposed MCF10AT1 cells developed, in a time-dependent manner, an acquired phenotype characterized by an increase in cancerous properties (anchorage independent growth and stem-like phenotype). Co-exposure of MCF10AT1 cells to B[a]P and BPA led to a significantly greater aggressive phenotype compared to B[a]P or BPA alone. This study provided new insights into the existence of a functional interplay between the aryl hydrocarbon receptor (AhR) and the G protein-coupled receptor 30 (GPR30) by which chronic and low-dose exposure of B[a]P and/or BPA fosters the progression of MCF10AT1 cells into a more aggressive substage. Experiments using AhR or GPR30 antagonists, siRNA strategies, and RNAseq analysis led us to propose a model in which AhR signaling plays a “driver role” in the AhR/GPR30 cross-talk in mediating long-term and low-dose exposure of B[a]P and/or BPA. Retrospective analysis of two independent breast cancer cohorts revealed that the AhR/GPR30 mRNA expression signature resulted in poor breast cancer prognosis, in particular in the ER-negative and the triple-negative subtypes. Finally, the study identified targeting AhR and/or GPR30 with specific antagonists as a strategy capable of inhibiting carcinogenesis associated with chronic exposure to low doses of B[a]P and BPA in MCF10AT1 cells. Altogether, our results indicate that the engagement of both AhR and GPR30 functions, in particular in an ER-negative/triple-negative context of breast cells, favors tumor progression and leads to poor prognosis.
Progression of human breast epithelial cells from non-cancerous to pre-malignant and of early-transformed mammary cells to malignant stages is a multiyear, multistep, multiscale, and multipath disease process. More than 85% of breast cancers are sporadic and potentially attributable to long-term exposure to environmental factors, such as chemical carcinogens (1–5). Given the increasing evidence that common environmental carcinogens play a significant role in breast cancer, increased attention has been paid to molecular mechanisms through which pollutants affect breast tumor formation, progression, and/or invasion (6–8). The identification of such molecular mechanisms could have several societal and environmental consequences, and may lead to the discovery of human biomarkers of exposure to environmental carcinogens exploitable for breast cancer prevention.
Previous in vitro investigations have mainly been conducted on human mammary epithelial cells or on human breast cancer cells, reflecting the impact of environmental factors on the earlier and later stages of carcinogenesis (9–13). However, little is known on the impact of exposure to pollutants on the breast early-transformed stage. Short-term exposure of cells to carcinogens at micro- to millimolar concentrations was previously typically investigated (1, 2, 14–16) which, while informative, is not optimal in mimicking natural chronic exposure to low doses of environmental carcinogens and to reflect physiologically-achievable levels of environmental mammary carcinogens. Additionally, few studies have attempted to mimic natural environmental exposure by assessing the impact of exposure to a combination of several pollutants with distinct mechanisms of action that may interact or induce a greater adverse effect than the use of individual compounds.
Benzo[a]pyrene (B[a]P), a family member of poly-cyclic aromatic hydrocarbons, is considered to be a tobacco, environmental, and dietary chemical carcinogen classified as Group 1 carcinogen by the IARC (17). B[a]P is a tumor initiator that binds and forms a complex with the aryl hydrocarbon receptor (AhR) (18–20). Upon such activation, the AhR-transcriptional complex activates specific DNA-recognition elements, such as xenobiotic response elements (XREs), and upregulates the expression of genes such as cytochrome P450 isoforms (including CYP1A1). These latter are involved in the metabolic activation of B[a]P in genotoxic metabolites forming DNA adducts relevant for carcinogenesis [for review, (21, 22)]. A growing body of evidence is accumulating implicating the B[a]P/AhR/CYP1A1 pathway in carcinogenesis (23–25). At early stages of carcinogenesis, short-term and millimolar B[a]P doses were shown to induce aggressiveness and transformation of non-cancerous human mammary epithelial cells (26–28). The impact of chronic and low-dose exposure of these cells to B[a]P was scarcely investigated, but seems to foster progression toward the early-transformed stage by favoring increased mesenchymal, stem-like, and anchorage-independent growth properties (9–13, 29).
Bisphenol-A (BPA) is a monomer of polycarbonate plastics and human exposure to BPA mainly occurs through the oral route due to the leaching of BPA in food and beverage containers, but non-dietary sources such as dust, air and cosmetics are also relevant (30). BPA has been the focus of widespread concern due to the fact that it interferes with endocrine signaling pathways even at extremely low doses, and thus belongs to the endocrine-disrupting compounds (EDC) (31, 32). BPA is known to bind to estrogen receptors alpha and beta (ERα and ERβ), to the G protein-coupled receptor 30 (GPR30) but also to the pregnane X receptor (PXR) (31, 33). Although several in vivo studies reported a carcinogenic potential of BPA [reviewed in (32)], the World Health Organization (WHO) indicated that there is currently insufficient evidence on which to base this carcinogenic potential (34). In vitro studies have however revealed that BPA causes adverse effects in non-cancerous mammary epithelial cells or in breast cancer cell lines, including increased cell proliferation, cell stemness, oxidative stress, and alterations of cell signaling pathways involved in carcinogenesis (13, 29, 35–38).
The MCF10 unique model of breast cancer progression comprises a series of isogenic triple-negative cell lines derived from MCF10A cells (MCF10A, MCF10AT1 and MCF10CA1a.cl1 cells). The parental cell line (MCF10A) having been originally isolated from a woman with fibrocystic change (39), the members of the MCF10 series belong to the triple negative/basal-like subtype (ER-negative, progesterone receptor (PR)-negative, HER2-negative) (40–42). These cell lines thus recapitulate the stages of mammary carcinogenesis (43), making this a valuable in vitro model for studying the progression of triple-negative breast cancer (44–46). In the present study, we used MCF10AT1 breast cells which represent the transformed early stage in the MCF10 unique model of breast cancer progression (43, 44) to further characterize the carcinogenic potential of B[a]P and BPA. To our knowledge, these cells have never been used to test the impact of chronic and low-dose exposure to environmental pollutants.
The main objectives of this work were to newly investigate: (i) whether long-term and low-dose exposure to B[a]P and/or BPA triggers the progression of early-transformed mammary cells to a more aggressive stage; (ii) whether their combination enhances the effect of each compound tested individually, in particular whether BPA facilitates the pro-carcinogenic activity of B[a]P; and (iii) to identify candidate strategies capable of inhibiting mammary carcinogenesis linked to chronic exposure to the environmental pollutants B[a]P and/or BPA.
Our data reveal that long-term and low-dose exposure to B[a]P and BPA increases cancerous properties of the MCF10AT1 cell line. Importantly exposure to the two pollutants leads to a greater deleterious impact than the compounds tested individually, and our data highlight the existence of a unique functional cross-talk between AhR and GPR30 in mediating those effects. The clinical relevance of the AhR/GPR30 interplay is validated by the observation of high mRNA expression levels of these two receptors in breast cancer patients as markers of poor prognosis. Finally, this study identified AhR and GPR30 as novel targets for strategies inhibiting the development of cancer-associated properties (AIG and MFE) in early-transformed human mammary cells following long-term and low-dose exposure to B[a]P and BPA.
Early-transformed human mammary MCF10AT1 cells and the MCF10AT1-derived cancerous MCF10CA1a.cl1 cells (Karmanos Institute, Detroit, USA) were purchased from Karmanos Institute (Detroit, USA) and maintained in complete DMEM/Ham's F12 medium with 5% horse serum (Thermo Fisher Scientific, Waltham, USA) and additional supplements: 100 ng/mL cholera enterotoxin, 10 mg/mL insulin, 0.5 mg/mL hydrocortisol, 20 ng/mL epidermal growth factor (Sigma, Saint Louis, USA) 100 units/mL penicillin and 100 mg/mL streptomycin. MCF-7 cells were purchased from ATCC (Teddington, UK) and grown according to the manufacturer's recommendations. HG5LN PXR cells stably expressing PXR (47), were grown in DMEM containing phenol red and 1 g/L glucose with 5% fetal calf serum (FCS) and additional supplements: 1 mg/mL G418 and 0.5 μg/mL puromycin.
B[a]P and BPA were purchased from Sigma (Saint Louis, USA). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), used as a control, was purchased from LGC Standard (Molsheim, France). The GPR30 agonist G1, the GPR30 antagonist G15, and the AhR agonist ITE were from TOCRIS Bioscience (Bristol, UK); the AhR antagonist GNF351 from Calbiochem (Billerica, USA).
MCF10AT1 cells were chronically exposed or not to 10−10 M of B[a]P or to 10−10 M BPA, alone or in combination, during 60 days (≈20 passages) in phenol red-free DMEM/Ham's F12 medium with 5% steroid-depleted, dextran-coated and charcoal-treated horse serum, containing the above-mentioned supplements (further referred to as DCC medium). Unexposed MCF10CA1a.cl1 cells were grown concomitantly in the same medium for 60 days and named MCF10CA1a.cl160d. Media and treatments were changed every 2 days. Cells were frozen every 2 weeks.
Anchorage-independent growth was assessed by soft agar assay as previously described (48). Single-cell suspensions (75 × 103) were seeded onto soft agar, and colonies were counted after 3 weeks of incubation.
Single-cell suspensions were seeded using non-adherent mammosphere culture conditions (49). After 7 days, primary mammospheres (first generation) were counted, collected, trypsinized, and replated for 10 days in non-adherent culture conditions to generate second-generation mammospheres. The culture media were replenished every 2–3 days.
Total RNA extraction, reverse transcription and RT-qPCR measurements were performed as described previously (29, 48). RNA lysates were extracted after exposure to the different molecules tested, untreated cells were used as controls in the presence of the corresponding volume of solvent. One microgram of total RNA from each sample was reverse-transcribed as previously described (48). RT-qPCR measurements were performed using a CFX96 with the SsoAdvanced Universal SYBR green supermix (BioRad, Hercules, USA), according to the manufacturer's recommendations. The primers used to explore the expression of the ERα, ERβ, PXR, AhR, GPR30, CYP1A1, and 28S genes are listed in the Supplementary Table 1.
Western blot experiments were performed as previously described (48) using the following antibodies: AhR (1:1,000, ab2770; Abcam, Paris, France), GPR30 (1:1,000, NBP1-31239; Novus Biologicals, Littleton, USA), α-tubulin (1:10,000, T5168; Sigma), phospho-p42/44 MAPK (1:1,000, 9106; Cell Signaling), p42/44 MAPK (1:1,000, 9102; Cell Signaling, Danvers, USA).
StealthTM siRNAs siRNA-GPR30 and StealthTM siRNAs siRNA-AhR and their corresponding scrambled control RNA (scrambled) were obtained from Ambion (Carlsbad, USA,4390825) and Invitrogen (Carlsbad, USA, AHRHSS100337/336), respectively. Fifty nM of siRNA-GPR30, 5 nM of siRNA-AhR or corresponding scrambled RNA were transfected into MCF10AT1 cells with lipofectamine RNAimax (Invitrogen). Transfections were performed directly at the time of cell seeding. Western blots were performed 48 h post-transfection. Exposure to G1, TCDD, or ITE for RNA collection or luciferase assay was performed 24 h post-transfection.
Cells were plated and then transfected with 150 ng XRE-firefly luciferase reporter plasmid (XRE-luc) (50) and 10 ng Renilla luciferase plasmid (pTK-RL). Twenty-four hours after transfection cells were grown for 8 h in the presence of the indicated treatment, and luciferase activity was then assessed as previously described (48).
In AIG experiments, MCF10AT1 cells were exposed to BPA and/or B[a]P 10−10 M, ITE 10−10 M, or G1 10−10 M for 72 h in the presence or the absence of a 2 h pre-treatment with the AhR antagonist GNF351 10−7 M or the GPR30 antagonist G15 10−8 M. Exposure was maintained throughout the course of the experiments. In MFE assays, MCF10AT1 cells were exposed to BPA and/or B[a]P 10−10 M, ITE 10−10 M, or G1 10−10 M in the presence or the absence of GNF351 10−7 M or G15 10−8 M only during the time-course of the experiments.
RNA isolation, library preparation, and RNA-Seq were performed by the core facility ProfileXpert (Lyon, France) from three independent cell-culture replicates of each tested cell line (unexposed MCF10AT160d cells, B[a]P 10−10 M exposed MCF10AT160d, BPA 10−10 M exposed MCF10AT160d cells, B[a]P+BPA 10−10 M exposed MCF10AT160d). The resulting RNA were isolated using the RNeasy mini kit (Qiagen) according to the manufacturer's protocol and ribosomal depletion was performed with the Ribo-zero gold kit (Epicentre). Libraries were performed from 20 ng ribosomal depleted RNA with the NEXTFLEX® Rapid Directional RNA-Seq Library Prep Kit (BIOO-Scientific). Libraries were sequenced using an Illumina NextSeq 500 platform (flow cell highoutput V2) and a 75 bp paired-end sequencing with ~30–35 million reads per sample. After trimming, reads were aligned to the human genome (hg19) using TopHat-2 v. 2.1.0 and data normalization (FPKM) was performed with Cufflinks software v.2.1.1. Data were logged on the NCBI Gene Expression Omnibus (GEO) website (http://www.ncbi.nlm.nih.gov/geo/) and are available as a GSE142073 dataset. Transcripts were considered as differentially expressed when the p-value of a student non-parametric t-test was ≤ 0.05. The Aryl Hydrocarbon Receptor Signaling Canonical Pathway was evaluated with a functional analysis created with Ingenuity Pathway Analysis software (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). The GPR30 gene expression signature described by Pandey and collaborators (51) was introduced in the Ingenuity Pathway Analysis software to assess the GPR30 signaling pathway.
A total of 4 × 104 cells/well were seeded onto and cultured in 24-well plates. Proliferating cells were analyzed using the Scepter™ 2.0 Cell Counter (Merck Millipore, Billerica, USA).
A total of 104 cells/well were plated onto a 96-well plate and treated for 4 days as indicated. Cell viability was assessed as previously described (52).
Women with primary breast tumors (n = 113) and known clinical follow-up who had not received any therapy before surgery and who relapsed, or not, while receiving endocrine therapy and/or chemotherapy were recruited from the BB-0033-00050, Biological Resources Center (CRB) Centre Léon Bérard, Lyon France (CLB cohort, Supplementary Table 2) (53). This study has been approved by the local ethics committee (CRB Centre Léon Bérard, France). The CRB Centre Léon Bérard is quality certified according NFS96-900 French standard and, ISO 9001 for clinical trials, ensuring scientific rigor for sample conservation, traceability and quality, as well as ethical rules observance and defined rules for transferring samples for research purposes (Ministry of Health for activities authorization n° AC-2019-3426 and DC-2008-99). The material used in the study has been collected in agreement with all applicable laws, rules, and requests of French and European government authorities, including the patients' informed written consents. Extraction of total RNA from frozen tumor samples and RT-qPCR measurements were performed as previously described (52, 53). Univariate analyses were performed using the SPSS™ Software (IBM, USA). The IBM SPSS software (IBM) was used for all statistical analyses in which the prognostic value of AhR and GPR30 mRNA levels was analyzed. The data were divided at the median value of AhR or GPR30 mRNA expression into two groups with either high or low expression levels. The Kaplan-Meier plotter (KMP) cohort was established from a meta-analysis of the gene-expression profiles of 1,877 primary breast cancer samples from patients who had not received any therapy prior to surgery (54). A p < 0.05 was considered statistically significant.
MCF10AT1 cells were chronically (60 days) exposed or not to (B[a]P + BPA) 10−10 M, alone or in combination with GNF351 10−7 M and/or G15 10−8 M. Control experiments were performed in MCF10AT1 cells exposed for 60 days to GNF351 10−7 M and/or G15 10−8 M. The resulting established cells were then tested for AIG and MFE as described above.
The MCF10AT1 cells and the MCF10AT1-derived MCF10CA1a.cl1 respectively represent the early-transformed and cancerous stages in the unique MCF10 model of triple negative breast cancer progression (43, 44). Validation of progression to malignancy of MCF10AT1 cells was investigated by assessing: (i) anchorage-independent growth (AIG), a hallmark of carcinogenesis associated with aggressiveness and metastasis in malignant cells; (ii) cancer stem-like and self-renewing properties by assessing first and second generation mammosphere-forming efficiency (MFE), as a growing body of evidence suggests that cancer stem-like cells are involved in generating and maintaining pre-malignant and malignant lesions (55–57). Consistent with the substage of breast cancer progression displayed by each cell line, MCF10AT1 cells formed significantly fewer and smaller colonies in soft agar (AIG) and significantly fewer mammospheres than the cancerous MCF10CA1a.cl1 cells (Supplementary Figures 1A,B).
In order to identify the impact of B[a]P and BPA on progression to malignancy in conditions mimicking environmental exposure, the MCF10AT1 cells were chronically (60 days) exposed or not to physiologically-relevant concentrations (10−10 M) of B[a]P and/or BPA (58–62) (MCF10AT160d). Unexposed MCF10AT160d and MCF10CA1a.cl160d cells displayed unmodified AIG and MFE phenotypes (Figures 1A,B and Supplementary Figure 1C) compared to the corresponding parental cells (Supplementary Figures 1A,B). MCF10AT160d cells exposed to the carcinogenic B[a]P pollutant at a concentration as low as 10−10 M exhibited significantly increased AIG (Figure 1A), increased colony size (Supplementary Figure 1C) and higher MFE (Figure 1B) compared to the unexposed MCF10AT160d cells. The EDC BPA (10−10 M, 60 days exposure) also enhanced the cancerous properties of the exposed MCF10AT160d cells (significant increase in AIG, in colony size and in MFE), while that impact was always significantly lower than that of 60-days exposure to 10−10 M B[a]P (Figures 1A,B, Supplementary Figure 1C). Of utmost interest, the combination of B[a]P with BPA (10−10 M) had a significantly greater effect than B[a]P (10−10 M) tested individually (Figures 1A,B, Supplementary Figure 1C). The cancerous features displayed by the MCF10AT160d cells exposed to the B[a]P + BPA (10−10 M) combination was at least equivalent (Figure 1A and Supplementary Figure 1C) if not greater (second generation of mammospheres, Figure 1B) than those displayed by the unexposed cancerous MCF10CA1a.cl160d cells. Supplementary Figure 2 eliminated the possibility that the aggressive phenotypes observed in the different exposed MCF10AT160d cells were the consequence of an increase in cell proliferation (compared to unexposed MF10AT160d).
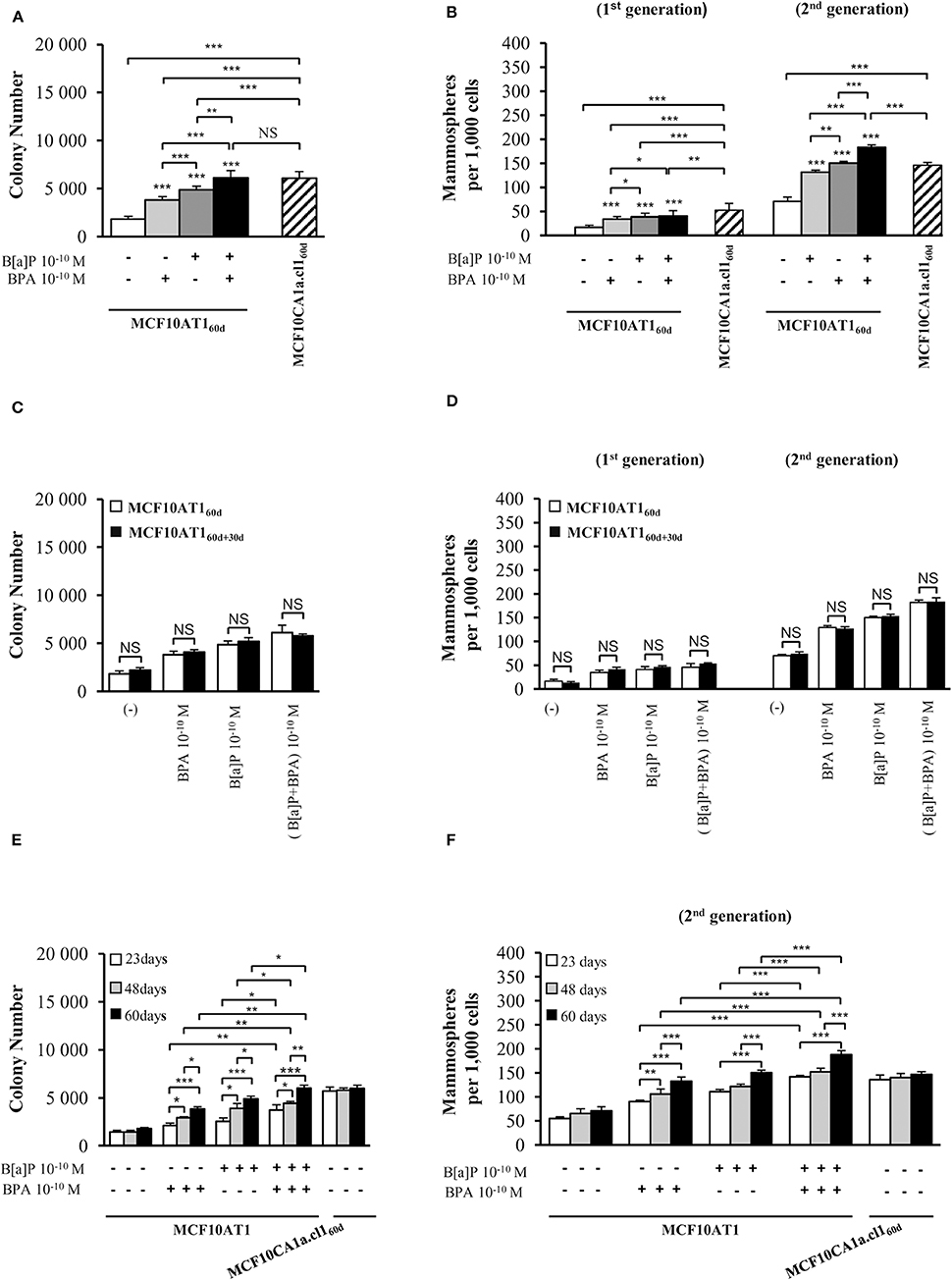
Figure 1. Cumulative and low-dose exposure of BPA and/or B[a]P increases MCF10AT1 cancerous properties (AIG and MFE assays). (A) Average number of MCF10AT160d colonies in soft agar after 60 days of chronic exposure to B[a]P and/or BPA (10−10 M). (B) Average number of primary and secondary MCF10AT160d mammospheres formed after 60 days of chronic exposure to B[a]P and/or BPA (10−10 M). In (A,B), exposure was maintained throughout the course of the experiments. (C,D) After 60 days of chronic exposure to B[a]P and/or BPA (10−10 M), MCF10AT1 cells were grown for a further 30 days without any exposure, and single-cell suspensions of cells were seeded in soft agar (C) or in non-adherent mammosphere culturing conditions (D). (E,F) Time-dependent acquisition of the aggressive phenotype: average number of colonies in soft agar (E) and of secondary mammospheres (F) following 23, 48, or 60 days of chronic exposure to B[a]P and/or BPA (10−10 M). Data illustrated in (A–F) represent mean ± SD of at least three independent experiments, in triplicate. ***p < 0.001, **p < 0.01, *p < 0.05 or NS (not significant) in Student t-test.
Unexposed or exposed MCF10AT160d cells were grown for a further 30 days in the absence of any treatment (MCF10AT160+30d cells), and then tested for AIG and MFE. The number and size of colonies in soft agar (Figure 1C and Supplementary Figure 1D) and the number of mammospheres (Figure 1D) were similar between MCF10AT160d and MCF10AT160+30d cells, thus demonstrating that the aggressive phenotype induced by chronic exposure of MCF10AT1 cells to low-doses of B[a]P and/or BPA is an acquired phenotype.
Taking advantage of the systematic freezing of exposed cells over the experimental time-course, AIG and MFE were retrospectively tested after 23 and 48 days of exposure and compared to 60 days of exposure (Figures 1E,F). The number of colonies in soft agar and mammospheres significantly increased between 23–48 and 48–60 days of chronic exposure, thus demonstrating that the longer MCF10AT1 cells are exposed to low doses of B[a]P and/or BPA, the more aggressive is their resulting phenotype. Once again, the combined exposure to B[a]P + BPA (10−10 M) caused more deleterious effects than those of B[a]P or BPA individually, irrespective of the exposure time.
Collectively, our results suggest that: (i) chronic, low-dose exposure to B[a]P or BPA enhances the cancerous properties of MCF10AT1 cells, with exposure to B[a]P being the most effective; (ii) the resulting aggressive phenotype was acquired and not reversed or softened when exposure was stopped; (iii) the duration of the exposure to pollutants impacted the magnitude of the aggressiveness developed by the exposed cells; (iv) combining BPA to B[a]P had more impact than exposure to B[a]P alone and was at least equivalent or higher than that of cancerous MCF10Ca1a.cl1 cells, supporting progression toward the cancerous substage.
As AhR is the main target of B[a]P and BPA is known to bind to ERα, ERβ, GPR30, and PXR, we investigated the presence of these receptors in MCF10AT1 cells. Supplementary Figure 3 validated that ERα, ERβ, and PXR were not or scarcely expressed in MCF10AT1 and MCF10CA1a.cl1 cells. Conversely, we newly reported that AhR and GPR30 receptors are concomitantly expressed at the mRNA and protein levels in MCF10AT1 and MCF10CA1.cl1 cells (Figures 2A,B). Of interest, AhR expression levels are higher both at the mRNA and protein levels in the MCF10AT1 cells compared to MCF10ACA1a.cl1.
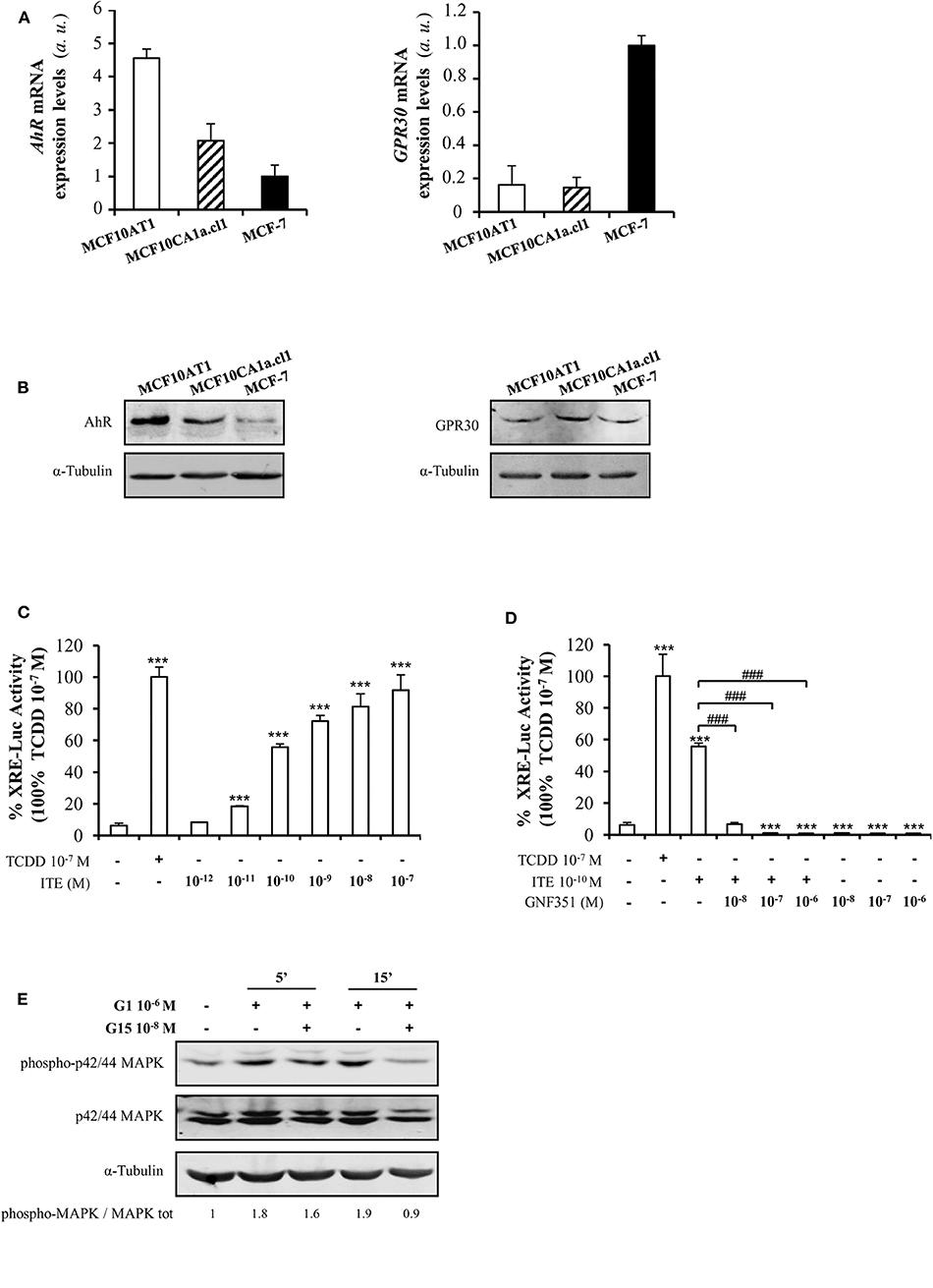
Figure 2. AhR and GPR30 receptors are present and functional in the MCF10AT1 cells. (A) RT-qPCR analysis of AhR and GPR30 mRNA expression levels represented in arbitrary units (a.u.) in the MCF10AT1 and MCF10CA1a.cl1 cells. MCF-7 cells were used as a control. Values represent mean ± SD of three independent experiments conducted in triplicate. (B) Representative Western blot analyses from three independent experiments of AhR and GPR30 protein expression in MCF10AT1 and MCF10CA1a.cl1 cells. MCF-7 cells were used as a control. (C) XRE-luciferase activity following 8 h exposure of MCF10AT1 cells to ITE at the indicated concentrations. TCDD 10−7 M was used as a control and results were expressed as % of TCDD 10−7 M activity. ***p < 0.001 in Student t-test. (D) XRE-luciferase activity upon 8 h of exposure to ITE 10−10 M alone or in combination with GNF351 at the indicated concentrations. TCDD 10−7 M was used as a control, and results were expressed as % of TCDD 10−7 M activity. Student t-tests revealed the statistically significant differences between unexposed and exposed cells: ***p < 0.001; and between ITE and ITE+GNF351: ###p < 0.001. Values in (C,D) represent mean ± SD of three independent experiments. (E) Representative Western blot analyses from three independent experiments of the phospho-MAPK/MAPK ratio upon exposure of MCF10AT1 cells to G1 (GPR30 agonist) for the times indicated, in the presence or absence of a 2 h pre-treatment with G15 (GPR30 antagonist).
Gene reporter experiments performed in MCF10AT1 cells demonstrated that TCDD, a well-known AhR ligand and activator of AhR-direct transcriptional activity (50), led to an increase in XRE-luciferase activity (Figure 2C). The selective AhR agonist ITE (63) also led to a stronger and dose-dependent activation of AhR-driven transcriptional activity in MCF10AT1 cells (Figure 2C). A concentration as low as 10−11 M of ITE was sufficient to give rise to a significant increase in XRE-luc activity (Figure 2C) and the effect of ITE was prevented (Figure 2D) by a 100-fold excess of the AhR antagonist GNF351 (64).
Rapid phosphorylation of MAPK is one of the main downstream effects of GPR30 activation (65). In MCF10AT1 cells, the MAPK pathway is rapidly activated (increase in the phospho-p42/p44 MAPK/MAPK ratio) in the presence of the selective GPR30 agonist G1 (66) and this activation is impaired in the presence of the GPR30 antagonist G15 (67) (Figure 2E), newly revealing the presence of functional GPR30 in MCF10AT1 cells.
We then aimed at investigating the possible involvement of the AhR and/or GPR30 receptors in mediating the tumorigenic effects of exposure to B[a]P and/or BPA 10−10 M in short-term experiments. This was tested in the presence or absence of selective antagonists of AhR or GPR30 (GNF351 and G15, respectively) at non-toxic concentrations (Supplementary Figures 4A,B).
Figure 3A demonstrates that short-term (72 h) exposure to 10−10 M BPA and/or 10−10 M B[a]P were sufficient to generate increased colony numbers in AIG assays. GNF351 and G15 were able to completely inhibit the effects of B[a]P and/or BPA on AIG (Figure 3A). This result was, at least to some extent, expected with the B[a]P/GNF351 and the BPA/G15 combinations, but was totally surprising concerning the BPA/GNF351 and the B[a]P/G15 combinations. In MFE assays conducted with MCF10AT1 cells exposed only during the time-course of the experiments (Figure 3B), the increased number of mammospheres induced by 10−10 M BPA was totally blocked in the presence of G15, as expected, but also in the presence of GNF351. Conversely, B[a]P-induced MFE was completely inhibited by GNF351, while the GPR30 antagonist G15, surprisingly resulted in a partial but significant inhibition. The impact of the G15 or GNF351 on the combination B[a]P+BPA was similar to that observed with B[a]P alone.
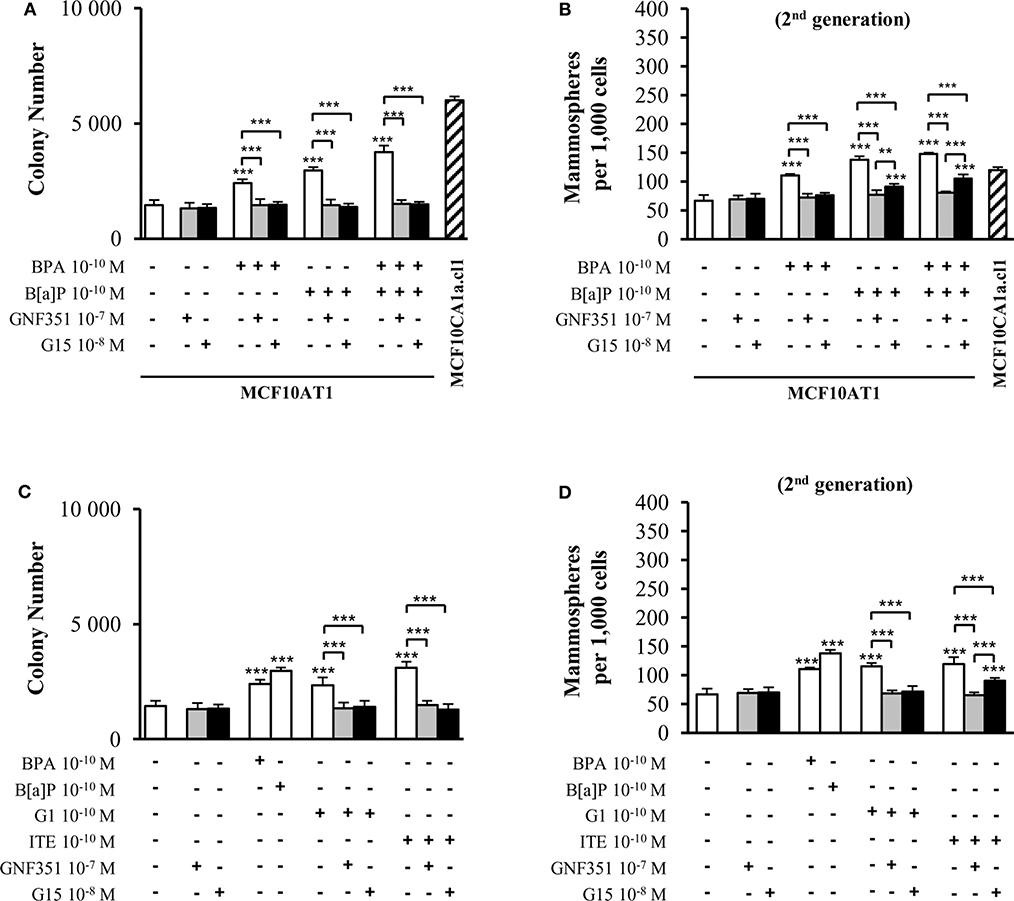
Figure 3. Effects of short-term exposure of MCF10AT1 cells by BPA, B[a]P, ITE, and G1 on AIG and MFE are inhibited by GPR30 and AhR antagonists. Average number of colonies in soft agar (A) or of secondary mammospheres (B) upon exposure to B[a]P and/or BPA 10−10 M for 72 h in the presence or absence of a 2 h pre-treatment with GNF351 10−7 M or G15 10−8 M. Exposure was maintained throughout the course of the experiments. Unexposed MCF10CA1a.cl1 cells were used as a control. (C) Average number of colonies in soft agar or (D) secondary mammospheres formed after 72 h exposure with ITE 10−10 M or G1 10−10 M in the presence or the absence of a 2 h pre-treatment with GNF351 10−7 M or G15 10−8 M. Exposure to BPA 10−10 M or B[a]P 10−10 M was used as controls. Exposure was maintained throughout the course of the experiments. Data illustrated in (A–D) represent mean ± SD of at least three independent experiments, in triplicate. ***p < 0.001, **p < 0.01 in Student t-test.
We then performed the same experiments using G1 and ITE agonists, at 10−10 M non-toxic concentrations (Supplementary Figures 4C,D). Figures 3C,D show similar data as those obtained with B[a]P and/or BPA in Figures 3A,B, namely that: (i) low-dose exposure of MCF10AT1 cells to 10−10 M G1 or 10−10 M ITE gives rise to significantly increased colony number (AIG) or increased MFE in the same range as that observed with 10−10 M BPA or 10−10 M B[a]P, respectively; (ii) G15 and GNF351 were both able to totally inhibit G1-dependent AIG or MFE; (iii) the impact of ITE on MFE was totally blocked in the presence of GNF351, while only partially inhibited by G15.
To confirm the above data supporting the involvement of both GPR30 and AhR receptors in mediating the effects of low dose (10−10 M) BPA, B[a]P, G1, or ITE, we used a siRNA strategy silencing either AhR or GPR30 (Figures 4A,B). We verified that siRNA-AhR or siRNA-GPR30 had, however, no impact on GPR30 or AhR expression, respectively (Figures 4A,B), nor on MCF10AT1 cell viability (Supplementary Figures 4E,F).
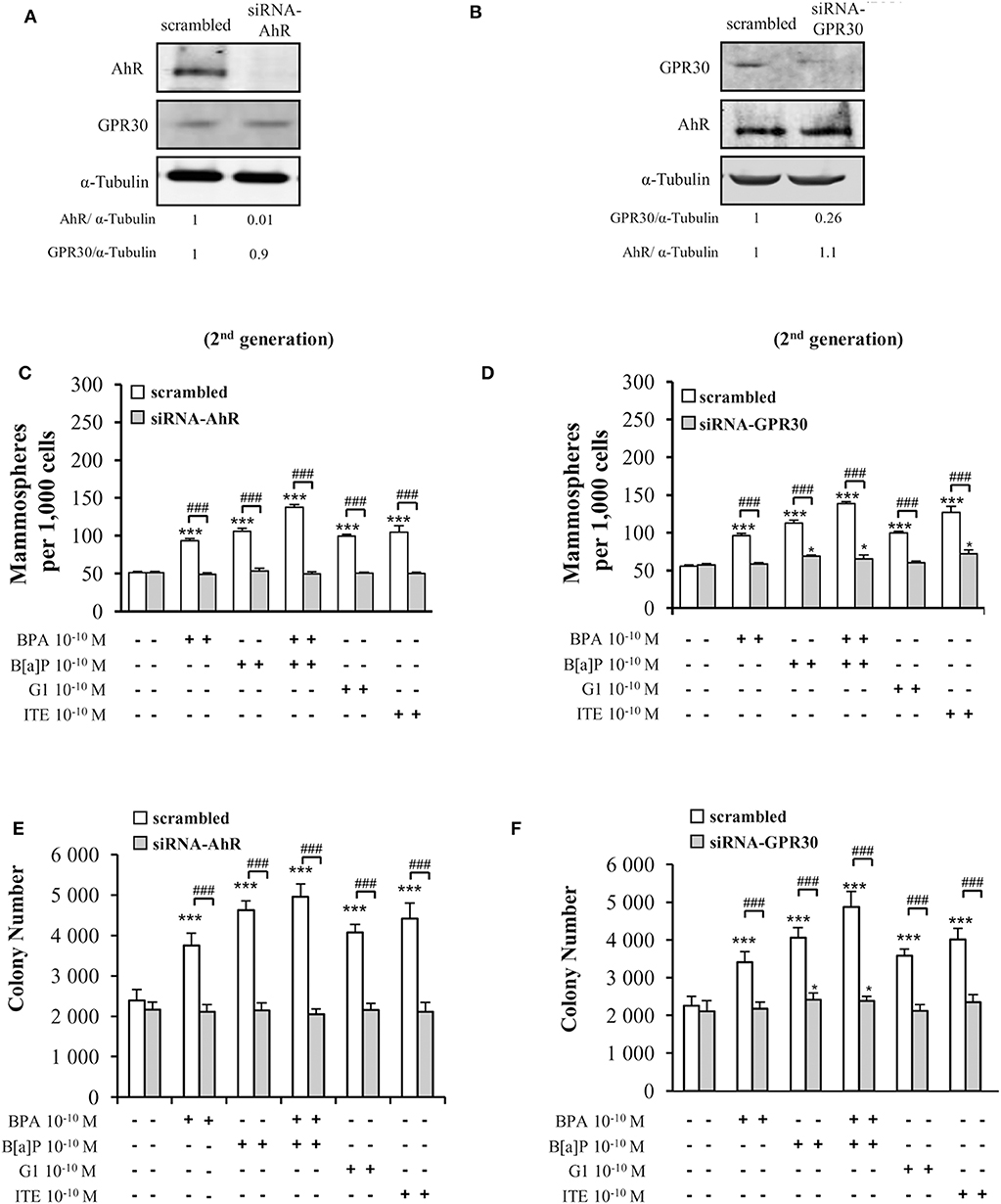
Figure 4. Effects of short-term exposure of BPA, B[a]P, ITE and G1 10−10 M on AIG and MFE are inhibited by siRNA-AhR and siRNA-GPR30. Representative Western blot analysis from three independent experiments of AhR and GPR30 expression in transfected MCF10AT1 cells with (A) siRNA-AhR, (B) siRNA-GPR30 or their scrambled controls. Quantification of protein expression levels was normalized against tubulin expression. (C,D) Secondary mammospheres formation and (E,F) average number of colonies in soft agar, with the following treatments: BPA and/or B[a]P, G1, or ITE, 10−10 M. Cells were transfected with either siRNA-AhR, siRNA-GPR30 or their scrambled controls before being subjected to the treatments. Treatments were maintained throughout the course of experiments. (mean ± SD of 2 independent experiments, in triplicate). ***p < 0.001, *p < 0.05 vs. their respective unexposed; ###p < 0.001 siRNA vs. scrambled in Student t-test.
Findings from AIG and MFE experiments conducted in MCF10AT1 exposed to BPA, B[a]P, G1, or ITE 10−10 M in the presence of siRNA-AhR or of siRNA-GPR30 (Figures 4C–F) corroborated those obtained using the AhR- or GPR30-antagonists (GNF351 and G15, respectively) (Figures 3A–D). Indeed, AhR knock-out resulted in a total inhibition of BPA-, B[a]P-, G1-, or ITE-mediated effects (Figures 4C,E). GPR30 silencing, while totally inhibiting BPA- or G1-mediated effects (Figures 4D,F), was slightly less effective in inhibiting MFE or AIG assays when cells were exposed to B[a]P (Figures 4D,F) or ITE (Figure 4D). The impact of siRNA-AhR or siRNA-GPR30 on the combination B[a]P+BPA was similar to that observed with B[a]P alone (Figures 4C–F).
Altogether these results support the idea that both AhR and GPR30 receptors play a role in mediating the deleterious effects exerted by BPA, G1, B[a]P, or ITE on the MCF10AT1 early-transformed cells. More importantly, our data highlight a functional cross-talk between GPR30 and AhR.
To further decipher the interplay between AhR and GPR30, we performed XRE-luciferase reporter assays. As anticipated, AhR-driven transcriptional activity in MCF10AT1 cells was fostered by TCDD 10−7 M or ITE 10−10 M and lost following AhR silencing (Figure 5A). Very interestingly, the GPR30 agonist G1 was also able to significantly increase XRE-luciferase activity in a dose-dependent manner, as illustrated by its effect at 10−7 M and 10−6 M, which is, to our knowledge, the first such report in the literature (Figure 5B). The G1-induced XRE-luciferase signal was totally inhibited in the presence of GNF351 (Figure 5C) and also in the presence of siRNA-AhR (Figure 5D). Silencing GPR30 by a siRNA-GPR30 strategy gave rise to a partial but significant decrease in G1- (10−6 M) (Figure 5E), TCDD- (10−7 M), and ITE- (10−10 M) (Figure 5F) -dependent activation of XRE-luciferase activity. Altogether, these data recapitulate the cross-talk between AhR and GPR30 on a simplified XRE-luciferase response.
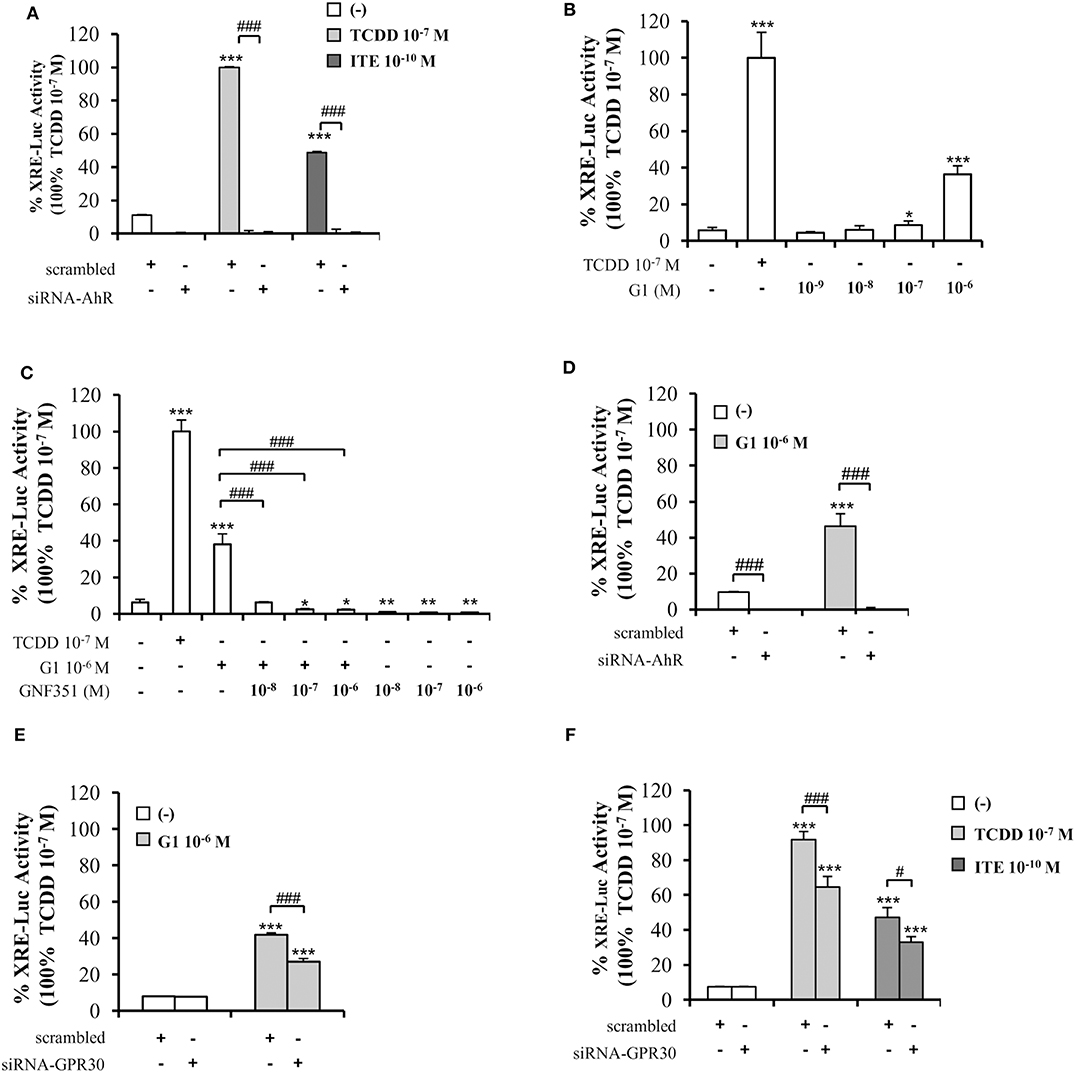
Figure 5. Involvement of GPR30 in AhR transcriptional activity using XRE-luciferase assays. XRE-luciferase activity following 8 h exposure of MCF10AT1 cells to (A) ITE at the indicated concentrations, TCDD 10−7 M was used as a control and results were expressed as % of TCDD 10−7 M activity; in the presence or absence of siRNA-AhR (B) G1 at the indicated concentrations. TCDD 10−7 M was used as a control; (C) G1 10−6 M alone or in combination with GNF351 at the indicated concentrations; TCDD 10−7 M was used as a control. (D,E) XRE-luciferase activity in MCF10AT1 cells transfected with (D) siRNA-AhR, (E) siRNA-GPR30 or their respective scrambled and treated 48 h after siRNA transfection for 8 h with G1 10−6 M. (F) XRE-luciferase activity in MCF10AT1 cells transfected with either scrambled RNA or siRNA-GPR30 and treated for 8 h with TCDD 10−7 M or ITE 10−10 M after 48 h of siRNA transfection. All data represent mean ± SD of three independent experiments conducted in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001; #p < 0.05; ###p < 0.001 in Student t-test.
As CYP1A1 is amongst the canonical genes regulated by AhR with a promoter containing typical XRE, we further investigated endogenous CYP1A1 mRNA levels in MCF10AT1 cells upon ITE and G1 treatment. Consistent with Figures 5, 6 revealed that: (i) CYP1A1 mRNA levels significantly increased upon treatment with TCDD 10−7 M and ITE 10−7 M (Figure 6A); (ii) the impact of ITE 10−7 M was completely impaired in the presence of siRNA-AhR (Figure 6A) or GNF351 10−6 M (Figure 6B), and significantly decreased in the presence of siRNA-GPR30 (Figure 6C). Figures 6D–F highlight that G1 10−6 M resulted in a significant increase in CYP1A1 mRNA levels, and that this was totally impeded in the presence of siRNA-AhR (Figure 6D), GNF351 10−6 M (Figure 6E) and significantly decreased in the presence of siRNA-GPR30 (Figure 6F). Altogether, our XRE-luciferase experiments and RT-qPCR experiments confirmed, at the transcriptional level, that GPR30-dependent mechanisms significantly impact and favor, directly or indirectly, AhR-driven transcriptional-dependent events.
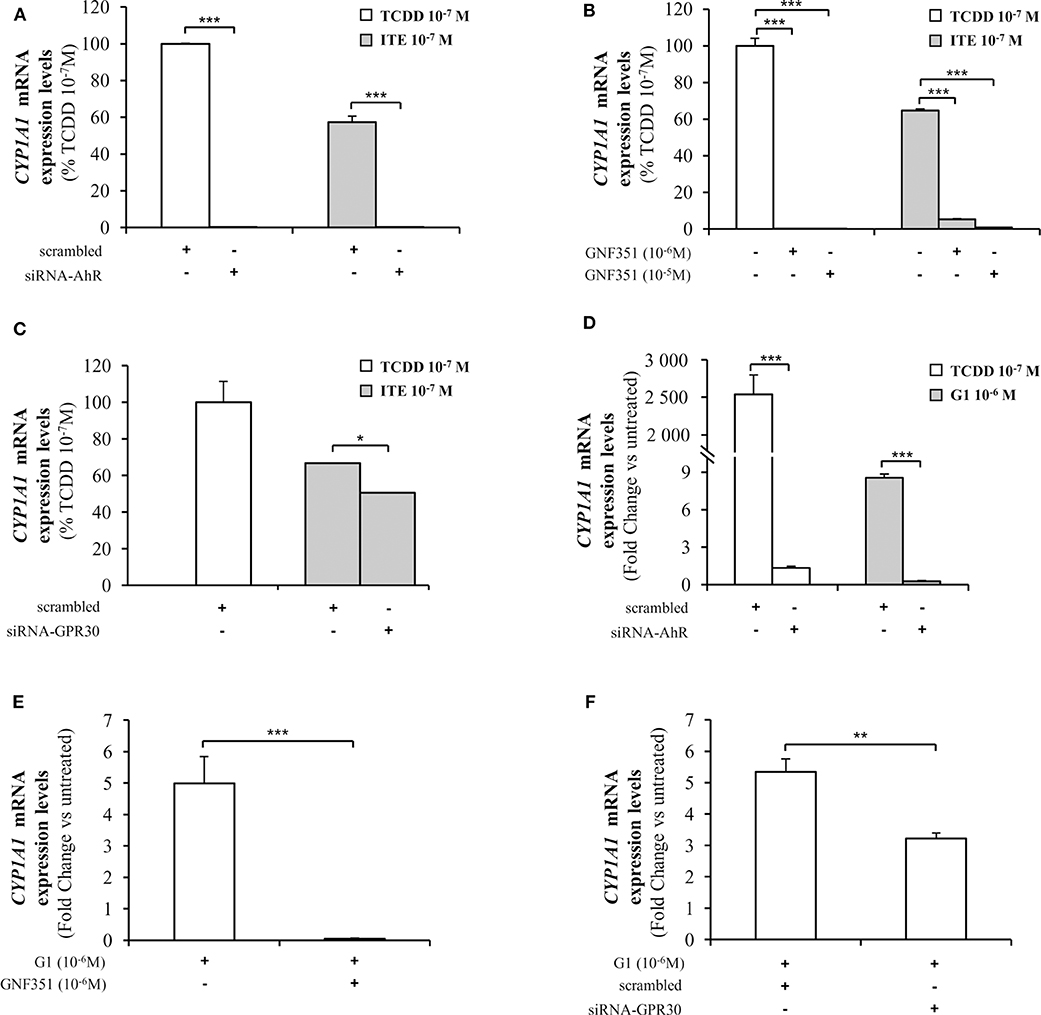
Figure 6. Analysis of CYP1A1 mRNA levels by quantitative RT-qPCR in MCF10AT1 cells. (A) CYP1A1 expression levels in cells transfected with either siRNA-AhR or scrambled RNA after 4 h of exposure to ITE 10−7 M. TCDD 10−7 M was used as a control and results are expressed as % of TCDD 10−7 M activity. (B) CYP1A1 mRNA expression levels in cells treated with GNF351 alone at indicated concentrations or in combination with ITE 10−7 M or TCDD 10−7 M. TCDD 10−7 M was used as a control and results are expressed as % of TCDD 10−7 M activity. (C) CYP1A1 mRNA expression levels in cells transfected with either siRNA-GPR30 or scrambled RNA and exposed to ITE 10−7 M for 4 h. TCDD 10−7 M was used as a control and results are expressed as % of TCDD 10−7 M activity. (D) CYP1A1 mRNA expression levels in cells transfected with either siRNA-AhR or scrambled RNA and exposed to G1 10−6 M or TCDD 10−7 M for 24 h. TCDD 10−7 M was used as a control. (E) CYP1A1 mRNA expression levels in cells treated with G1 10−6 M in the presence or absence of GNF351 10−6 M. (F) CYP1A1 mRNA expression levels in cells transfected with either siRNA-GPR30 or scrambled RNA and exposed to G1 10−6 M for 24 h. Data represent mean ± SD of 3 independent experiments conducted in triplicate. (A–F) The Student t-test was applied to reveal statistically significant differences between treatments: *p < 0.05, **p < 0.01, ***p < 0.001.
Having verified that protein expression levels of AhR and of GPR30 were similar in both the unexposed and the pollutants-exposed MCF10AT160d cells (Supplementary Figure 5A), the RNAseq data of each cell line were analyzed using the Ingenuity Pathway Analysis software. AhR core signaling was identified as significantly dysregulated in the MCF10AT160d exposed cells compared to unexposed control MCF10AT160d cells. Indeed, the significant enrichment of this canonical pathway was observed both in the B[a]P-exposed MCF10AT160d cells (p = 9.39 10−3), in the B[a]P + BPA exposed MCF10AT160d cells (p = 7.56 10−3), but also in the BPA-exposed MCF10AT160d cells (p = 1.26 10−2) (data not shown). Conversely, assessing the GPR30 signature previously described by Pandey and collaborators (51) in the Ingenuity Pathway Analysis software did not reveal any dysregulation in GPR30 signaling when the exposed MCF10AT160d cells were compared with the unexposed control MCF10AT160d cells (data not shown). As rapid phosphorylation of MAPK is one of the main downstream effects of GPR30 activation (65), we assessed the activation status of the MAPK pathway in the unexposed and exposed MCF10AT160d cells. Supplementary Figure 5B demonstrated that chronic and low-dose-exposure of MCF10AT1 cells to B[a]P and/or BPA at 10−10 M did not lead to any activation of p42/p44MAPK (MCF1Ca1.cl160d cells were used as controls). Altogether, our data suggest that AhR signaling is constitutively activated in the MCF10AT160d exposed to pollutants vs. unexposed cells.
To investigate the clinical relevance of AhR and GPR30, we performed RT-qPCR analyses to explore GPR30 and AhR mRNA expression levels in a cohort of 113 human primary breast tumor samples (Table 1) (53). The resulting Kaplan-Meier curves are shown in Figure 7. By univariate analysis, we found that neither GPR30 nor AhR mRNA levels were informative (p = 0.09 and p = 0.41, Figures 7A,B, respectively), while an AhR/GPR30 gene expression signature based on high expression levels of both GPR30 and AhR was significantly associated with shorter overall survival (OS) (p = 0.01, Table 1 and Figure 7C). Regarding breast cancer subclasses, the AhR/GPR30 gene expression signature was more informative in the ER-negative (p = 0.001) than in the ER-positive (p = 0.41) (Table 1, Figures 7D,E) or luminal subclasses and (p = 0.50) (Table 1). The situation was less clear for the HER2-enriched and triple-negative subclasses, considering the limited size of available samples (n =16 and n = 21, respectively). We thus performed retrospective analysis of gene-expression array data using the KMP cohort, which contains a sizeable number of breast cancer patients (1,308 ER-positive, 569 ER-negative, 1,055 luminal, 419 HER2-enriched, and 403 triple-negative/basal-like). The most striking results (Table 2, univariate analysis) indicated that the AhR/GPR30 signature was again more informative than GPR30 mRNA or AhR mRNA levels alone and was associated with shorter OS in the ER-negative subclass (p = 0.005), but not in the ER-positive or the luminal subclasses (Table 2). The “all breast tumor samples” univariate analysis of the KMP cohort did not validate what was observed in the CLB cohort, but this discrepancy might reflect the difference in proportion of the ER-positive and ER-negative subclasses in the two cohorts. Finally, the KMP cohort revealed that the AhR/GPR30 signature was associated with shorter OS in the triple-negative subclass (p = 0.033). Altogether, our data reveal a new/original signature based on a combination of high AhR mRNA expression levels and high GPR30 mRNA expression levels that represents a novel marker for poor prognosis in breast cancer, especially in ER-negative or triple-negative subclasses.
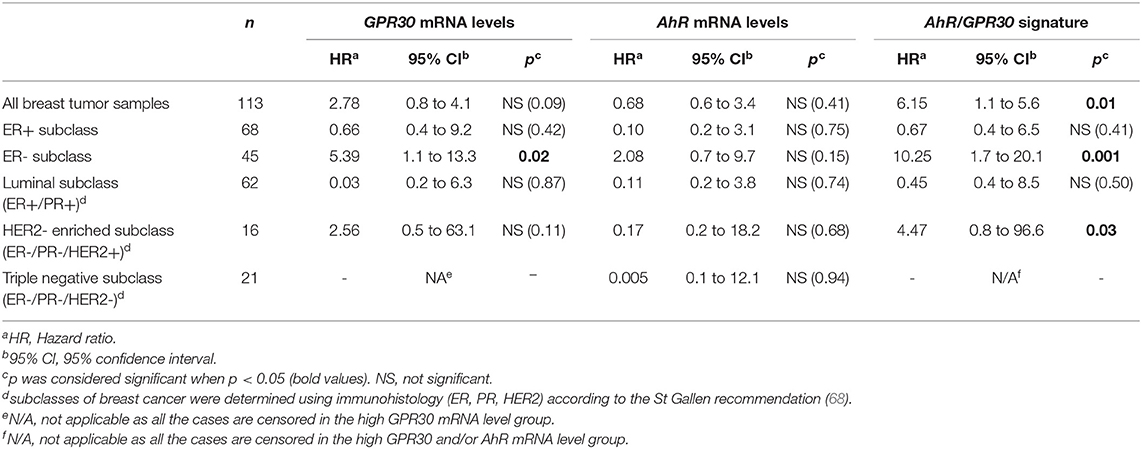
Table 1. Univariate analysis of the GPR30 mRNA expression levels, the AhR mRNA expression levels and the GPR30/AhR mRNA expression signature with regards to overall survival (OS) in different subclasses of the 113 breast cancer samples of the CLB cohort.
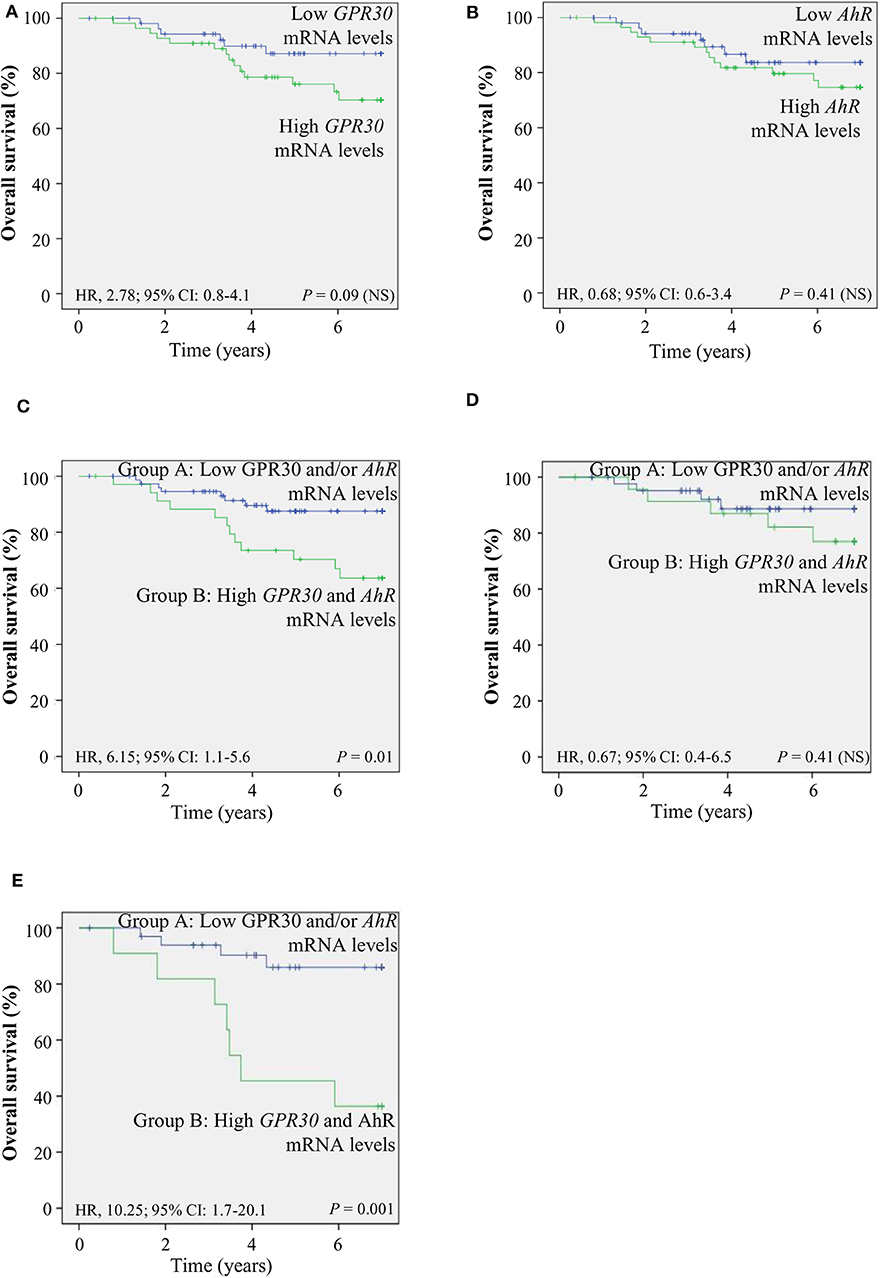
Figure 7. The AhR/GPR30 mRNA expression signature is of poor prognosis and is associated with shorter overall survival (OS). Kaplan-Meier analysis (univariate analysis) of OS in the CLB breast cancer cohort of: (A) GPR30 mRNA expression levels; (B) AhR mRNA expression levels; (C–E) Patients were divided into two groups: patients expressing low GPR30 mRNA levels and/or low levels of AhR mRNA (group A) and patients expressing high mRNA levels of GPR30 and AhR (group B). (C) AhR/GPR30 gene expression signature in all breast tumor samples. (D) GPR30/AhR gene expression signature in the ER-positive subclass. (E) GPR30/AhR mRNA expression signature in the ER-negative subclass. NS, not significant.
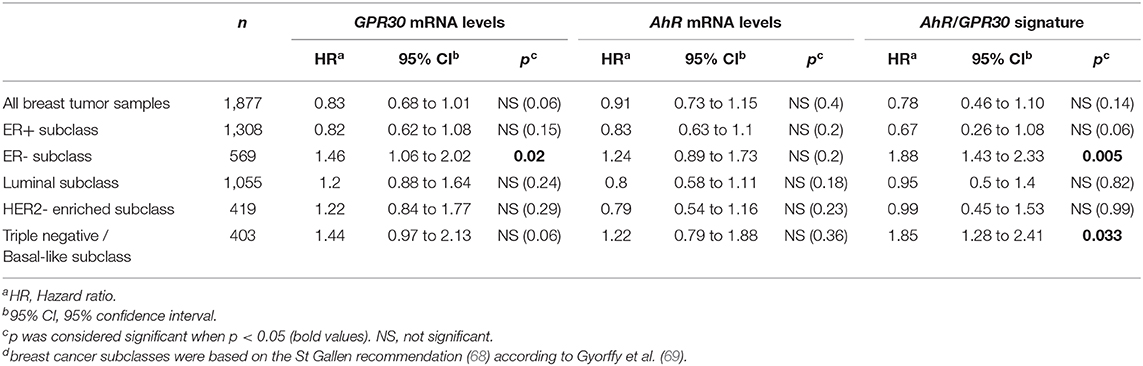
Table 2. Univariate analysis of the GPR30 mRNA expression levels, the AhR mRNA expression levels and the GPR30/AhR mRNA expression signature with regards to overall survival (OS) in different subclasses of the 1,877 breast cancer samples of the Kaplan-Meier plotter (KMP) cohort.
To identify candidate strategies capable of blocking mammary carcinogenesis associated with chronic exposure to low doses (10−10M) of the environmental pollutants B[a]P and BPA, MCF10AT1 cells were exposed for 60 days, in the presence or the absence of the AhR antagonist GNF351 10−7 M and/or the GPR30 antagonist G15 10−8 M (Figures 8A,B). Exposure for 60 days to the two antagonists, alone or in combination, had no impact on MFE and AIG. Strikingly, co-exposure for 60 days to B[a]P + BPA 10−10 M with GNF351 10−7 M and/or G15 10−8 M was sufficient to inhibit the development of pollutants-driven enhancement of cancerous properties (AIG and MFE) in MCF10AT1 cells (Figures 8A,B).
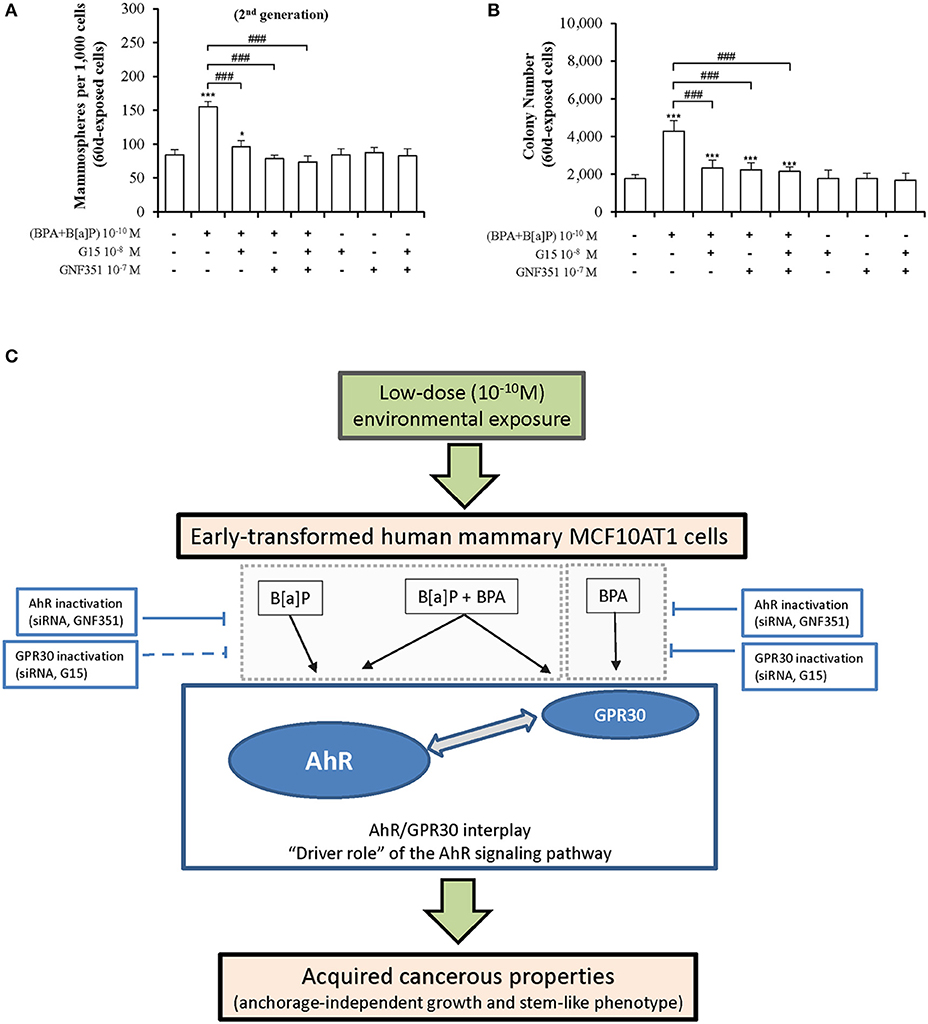
Figure 8. Long-term and low-dose exposure to B[a]P and/or BPA in promoting cancerous properties of early-transformed human mammary cells is driven by AhR/GPR30 cross-talk. Inhibition of the effects due to chronic and low-dose exposure of MCF10AT1 cells to BPA+B[a]P (10−10 M), using AhR and/or GPR30 antagonists: (A) formation of secondary mammospheres, (B) AIG. The MCF10AT1 cells were chronically (60 days) unexposed or exposed to B[a]P + BPA 10−10 M, alone or in combination with GNF351 10−7 M and/or G15 10−8 M. Control experiments were performed in MCF10AT1 cells exposed for 60 days to GNF351 10−7 M and/or G15 10−8 M (mean ± SD of 3 independent experiments, in triplicate). *p < 0.05, ***p < 0.001 vs. unexposed; ###p < 0.001 vs. BPA+B[a]P (10−10 M); (C) Diagram summarizing our findings.
A growing body of in vitro and in vivo experimental evidence suggests the implication of environmental factors in the development and progression of breast cancer. People are chronically exposed to a mixture of environmental factors, usually present at low-dose concentrations, constituting a complex exposome. B[a]P is detected at picomolar concentrations in body fluids and tissues of cancer patients (58–60). BPA is detected at nanomolar concentrations in human samples such as serum, urine and maternal milk (61, 62). The major obstacles in studying the impact of these two pollutants in vitro on breast tumorigenesis are thus the choice of a relevant cellular model and the use of relevant exposure conditions (long-term and low-dose exposure) mimicking natural exposure. The few studies having investigated the impact of chronic and low-dose exposure of B[a]P or BAP on tumor progression have focused on the early stage of carcinogenesis (non-transformed epithelial cells) (9–13, 29) MCF10AT1 breast cells (representing the early-transformed stage of the unique MCF10 model of mammary progression from normal epithelium to triple negative breast cancer (43, 44) have, to our knowledge, never been used to test the impact of chronic and low-dose exposure to environmental pollutants. While the genotoxic and pro-carcinogenic B[a]P and the EDC BPA are two of the most studied pollutants, no previous studies, to our knowledge, have investigated whether the combination of BPA and B[a]P, each compound possessing distinct mechanisms of action, induces potentiating effects on tumor progression.
The present study aimed at addressing: (i) the effects of long-term, cumulative exposure to low doses of B[a]P or BPA on the mammary early-transformed substage; (ii) whether the combination of BPA with B[a]P, each compound possessing distinct mechanisms of action, impacts breast tumor progression differently from what might be observed with each compound tested alone. Our study reveals that long term and low dose (10−10 M) exposure to B[a]P and/or BPA increases the cancerous properties (AIG and cancer stem-like properties) of the early-transformed MCF10AT1 cells, and the longer the cells were exposed, the greater was the impact. Co-exposure of MCF10AT1 cells with the B[a]P and BPA led to a significantly greater aggressive phenotype compared to B[a]P alone, suggesting that BPA facilitates the pro-carcinogenic activity of B[a]P and supporting the potentiating effects of distinct pollutants present in the exposome. Importantly, the aggressiveness developed by the exposed MCF10AT160d cells was acquired and not softened or reverted after stopping B[a]P and/or BPA exposure. Altogether, our data reveal that long-term and low dose exposure to B[a]P and/or BPA irreversibly favors the evolution of early-transformed human mammary cells toward breast tumor progression.
Mechanistically, while B[a]P and BPA are well-known activators of AhR and GPR30, respectively, our short-term exposure experiments highlight that the MFE and AIG response triggered by exposure of MCF10AT1 cells to B[a]P and/or BPA occurs through a functional cross-talk between AhR and GPR30. Regarding the impact of inactivating AhR or GPR30 by two different strategies (using an antagonist molecule or a siRNA strategy), the BPA, BPA+B[a]P, or G1 impact on AIG and MFE was totally reversed when AhR was inhibited, while the GPR30 inactivation globally seemed to lead to a significant but not total, reversion of B[a]P, BPA+B[a]P, or ITE impact (in particular in MFE experiments). Finally, this study provided evidence that long-term (60 days) co-exposure to B[a]P + BPA 10−10 M with the AhR antagonist GNF351 10−7 M and/or the GPR30 antagonist G15 10−8 M inhibited the development of pollutants-driven enhancement of cancerous properties in MCF10AT1 cells.
The AhR/CYP1A1 pathway participates in carcinogenesis by mediating stem properties, the formation of mammospheres, expansion of breast cancer stem cells, and the transcriptional activity of AhR is mainly implicated in such effects (24). Our study also reveals that GPR30 participates and favors AhR-dependent transcriptional activity in early-transformed breast cells. Indeed, the GPR30 agonist G1 was able to stimulate AhR-dependent driven activity and CYP1A1 gene transcription. GPR30 silencing by a siRNA strategy led to a significant, yet moderate, decrease in ITE-, G1-, or TCDD-mediated activation of AhR-driven transcriptional activity or CYP1A1 transcription. This suggests that GPR30-dependent mechanisms other than those influencing the AhR-driven transcriptional mechanisms might be involved in the AhR/GPR30 cross-talk impacting AIG and MFE. Altogether, our in vitro data support a model in which GPR30 is involved in an AhR-dependent network leading to increased cancerous properties of early-transformed mammary cells.
We thus propose a model (Figure 8C) in which AhR signaling plays a “driving role” in the AhR/GPR30 cross-talk in mediating the effects of long-term and low dose exposure of B[a]P and/or BPA on AIG and MFE in MCF10AT1 cells. The relevance of our model was supported by our RNAseq data demonstrating that the canonical AhR signaling pathway was significantly enriched in the B[a]P-exposed MCF10AT160d cells, in the B[a]P + BPA exposed MCF10AT160d cells, but also in the BPA-exposed MCF10AT160d cells. Strengthening our findings, previous studies support a role for AhR and breast tumor progression: (i) AhR expression levels were significantly up-regulated in human breast ductal carcinoma in situ and breast cancer tissues compared to normal/benign breast tissues (70, 71); (ii) an in vivo model of breast tumorigenesis suggests that AhR is constitutively activated at early stages of mammary tumorigenesis (72); (iii) the prognostic value of AhR seems to be dependent on the activation/inactivation of metastatic processes (73).
While this research was ongoing, a study was published supporting our data by demonstrating in the ERα-negative SKBR3 breast cancer cells that the environmental pollutant 3-methylcholantrene, mainly known to exert its carcinogenic effects through AhR, stimulates cell growth response through a functional interaction between AhR and GPR30 (74). However, in the ERα-positive MCF-7 breast cancer cell line, possessing the well-identified close cross-talk between AhR and ERα [for review (75)], 10−5 M G1 was demonstrated to increase transcription of CYP1A1 mRNA in AhR-dependent, but GPR30-independent, mechanisms (76). As MCF10AT1 cells used in this study are triple-negative cells and the SKBR3 cells are ERα-negative, one cannot exclude that the detrimental functional cross-talk between AhR and GPR30 might be exacerbated in such a specific cellular breast context. Supporting previous data highlighted that in the ERα-negative/triple negative context, an AhR-signaling reinforces cell aggressiveness and induces breast cancer stem cells (77–80).
Altogether, our in vitro data thus demonstrated the role of the AhR/GPR30 cross-talk in favoring tumor progression, at least in a triple-negative breast context. Previous controversial studies emerged regarding the prognostic value of AhR or GPR30 expression levels in breast cancers (71, 73, 80–84), and these discrepancies have been suggested as possibly related to the breast cancer subgroup or the substage considered. In the present study, the clinical relevance of our in vitro findings was further reinforced by retrospective analysis of two independent breast cancer cohorts, showing that in only ER-negative or triple-negative breast cancer subclasses, the gene signature involving both AhR and GPR30 mRNA levels were of poor prognosis.
Hence, we have provided novel insights into the progression of early-transformed human mammary cells upon long-term and low-dose exposure to B[a]P and/or BPA and deciphered the involvement of the functional crosstalk occurring between AhR and GPR30 leading to an exacerbated AhR-driven network. Our in vitro and retrospective data analyses further support the idea that the deleterious impact of this cross-talk might be of utmost importance in the progression of ER-negative or triple-negative breast cancers. More importantly, strategies targeting AhR and/or GPR30 were demonstrated to be efficient in inhibiting the deleterious impact of cumulative and low-dose exposure to B[a]P and/or BPA in early-transformed MCF10AT1 cells. The identification of such molecular mechanisms may help in the discovery of human biomarkers of environmental carcinogen exposure and the development of preventive strategies.
The datasets generated from this study can be found in the NCBI Gene Expression Omnibus (GEO) [http://www.ncbi.nlm.nih.gov/geo/ (GSE142073)].
This study has been approved by the local ethics committee (CRB Centre Léon Bérard, France). All subjects gave written informed consent.
PAC, VM-S, VC, and BF participated in the design of the study. CD, ME, SG, and SA performed the in vitro data. PAC and CD performed the biostatistical analysis on the CLB cohort. BG performed the biostatistical analysis on the KMP cohort. AW, SC, and JL performed and analyzed the RNAseq data. SG, MD-A, AE, and PC participated in scientific discussions. PAC, CD, and ME wrote the manuscript. BG, AW, PC, JL, VC, VM-S, BF, CD, SG, and ME participated in the scientific revisions of the manuscript. PAC supervised the study.
CD was funded by the ANR (2011 ANR-CESA-018-01, Agence Nationale de la Recherche, France) and the University of Lyon 1. ME was funded by a PhD grant from the Lebanese University. This work was funded by the ANR (2011 ANR-CESA-018-01), Lyon Biopôle, and supported by grants from the Ligue nationale contre le cancer LNCC (committees 42 and 71).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the Centre de Ressources Biologiques (CRB) of the Centre Léon Bérard (BB-0033-00050). We are grateful to B. Manship for editing. We thank Dr. Balaguer for providing the HG5LN PXR cell line and the XRE-luc plasmid. Some of the work performed by ME during her thesis has been partly included in this article (85).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00712/full#supplementary-material
B[a]P, benzo[a]pyrene; AhR, aryl hydrocarbon receptor; XRE, xenobiotic response element; BPA, bisphenol-A; EDC, endocrine-disrupting compound; ERα, estrogen receptor alpha; ERβ, estrogen receptor beta; GPR30, G protein-coupled receptor 30; PXR, pregnane X receptor; PR, progesterone receptor; AIG, anchorage-independent growth; MFE, mammosphere formation efficiency; RT-qPCR, real-time quantitative polymerase chain reaction; TCDD, 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin; OS, overall survival.
1. Guengerich FP. Metabolism of chemical carcinogens. Carcinogenesis. (2000) 21:345–51. doi: 10.1093/carcin/21.3.345
2. DeBruin LS, Josephy PD. Perspectives on the chemical etiology of breast cancer. Environ Health Perspect. (2002) 110(Suppl.1):119–28. doi: 10.1002/sc271_5_1835
3. Hecht SS. Tobacco smoke carcinogens and breast cancer. Environ Mol Mutagen. (2002) 39:119–26. doi: 10.1002/em.10071
4. Roukos DH, Murray S, Briasoulis E. Molecular genetic tools shape a roadmap towards a more accurate prognostic prediction and personalized management of cancer. Cancer Biol Ther. (2007) 6:308–12. doi: 10.4161/cbt.6.3.3994
5. Kelloff GJ. Cancer Chemoprevention Totowa, NJ: Humana Press Inc. (2005). doi: 10.1007/978-1-59259-768-0
6. Benigni R, Bossa C. Mechanisms of chemical carcinogenicity and mutagenicity: a review with implications for predictive toxicology. Chem Rev. (2011) 111:2507–36. doi: 10.1021/cr100222q
7. Rudel RA, Fenton SE, Ackerman JM, Euling SY, Makris SL. Environmental exposures and mammary gland development: state of the science, public health implications, and research recommendations. Environ Health Perspect. (2011) 119:1053–61. doi: 10.1289/ehp.1002864
8. Koual M, Cano-Sancho G, Bats AS, Tomkiewicz C, Kaddouch-Amar Y, Douay-Hauser N, et al. Associations between persistent organic pollutants and risk of breast cancer metastasis. Environ Int. (2019) 132:105028. doi: 10.1016/j.envint.2019.105028
9. Siriwardhana N, Wang HC. Precancerous carcinogenesis of human breast epithelial cells by chronic exposure to benzo[a]pyrene. Mol Carcinog. (2008) 47:338–48. doi: 10.1002/mc.20392
10. Rathore K, Wang HC. Green tea catechin extract in intervention of chronic breast cell carcinogenesis induced by environmental carcinogens. Mol Carcinog. (2012) 51:280–9. doi: 10.1002/mc.20844
11. Rathore K, Wang HC. Mesenchymal and stem-like cell properties targeted in suppression of chronically-induced breast cell carcinogenesis. Cancer Lett. (2013) 333:113–23. doi: 10.1016/j.canlet.2013.01.030S0304-383500062-1
12. Pluchino LA, Wang HC. Chronic exposure to combined carcinogens enhances breast cell carcinogenesis with mesenchymal and stem-like cell properties. PLoS ONE. (2014) 9:e108698. doi: 10.1371/journal.pone.0108698
13. Clement F, Xu X, Donini CF, Clement A, Omarjee S, Delay E, et al. Long-term exposure to bisphenol A or benzo(a)pyrene alters the fate of human mammary epithelial stem cells in response to BMP2 and BMP4, by pre-activating BMP signaling. Cell Death Differ. (2017) 24:155–66. doi: 10.1038/cdd.2016.107
14. Mehta RG. Experimental basis for the prevention of breast cancer. Eur J Cancer. (2000) 36:1275–82. doi: 10.1002/S0959-804900100-3
15. Caruso JA, Reiners JJJr, Emond J, Shultz T, Tainsky MA, Alaoui-Jamali M, et al. Genetic alteration of chromosome 8 is a common feature of human mammary epithelial cell lines transformed in vitro with benzo[a]pyrene. Mutat Res. (2001) 473:85–99. doi: 10.1016/s0027-5107(00)00140-8
16. Jaiswal AS, Panda H, Pampo CA, Siemann DW, Gairola CG, Hromas R, et al. Adenomatous polyposis coli-mediated accumulation of abasic DNA lesions lead to cigarette smoke condensate-induced neoplastic transformation of normal breast epithelial cells. Neoplasia. (2013) 15:454–60. doi: 10.1593/neo.13176
17. IARC. Chemical agents and related occupations. IARC Monogr Eval Carcinog Risks Hum. (2012) 100:9–562.
18. Rundle A, Tang D, Hibshoosh H, Estabrook A, Schnabel F, Cao W, et al. The relationship between genetic damage from polycyclic aromatic hydrocarbons in breast tissue and breast cancer. Carcinogenesis. (2000) 21:1281–9.
19. Shi S, Yoon DY, Hodge-Bell KC, Bebenek IG, Whitekus MJ, Zhang R, et al. The aryl hydrocarbon receptor nuclear translocator (Arnt) is required for tumor initiation by benzo[a]pyrene. Carcinogenesis. (2009) 30:1957–61. doi: 10.1093/carcin/bgp201
20. Kasala ER, Bodduluru LN, Barua CC, Sriram CS, Gogoi R. Benzo(a)pyrene induced lung cancer: role of dietary phytochemicals in chemoprevention. Pharmacol Rep. (2015) 67:996–1009. doi: 10.1016/j.pharep.2015.03.004
21. Keshava C, Divi RL, Whipkey DL, Frye BL, McCanlies E, Kuo M, et al. Induction of CYP1A1 and CYP1B1 and formation of carcinogen-DNA adducts in normal human mammary epithelial cells treated with benzo[a]pyrene. Cancer Lett. (2005) 221:213–24. doi: 10.1016/j.canlet.2004.08.038
22. Shi Q, Godschalk RWL, van Schooten FJ. Inflammation and the chemical carcinogen benzo[a]pyrene: partners in crime. Mutat Res. (2017) 774:12–24. doi: 10.1016/j.mrrev.2017.08.003
23. Divi RL, Lindeman TL, Shockley ME, Keshava C, Weston A, Poirier MC. Correlation between CYP1A1 transcript, protein level, enzyme activity and DNA adduct formation in normal human mammary epithelial cell strains exposed to benzo[a]pyrene. Mutagenesis. (2014) 29:409–17. doi: 10.1093/mutage/geu049
24. Al-Dhfyan A, Alhoshani A, Korashy HM. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol Cancer. (2017) 16:14. doi: 10.1186/s12943-016-0570-y
25. Vazquez-Gomez G, Rocha-Zavaleta L, Rodriguez-Sosa M, Petrosyan P, Rubio-Lightbourn J. Benzo[a]pyrene activates an AhR/Src/ERK axis that contributes to CYP1A1 induction and stable DNA adducts formation in lung cells. Toxicol Lett. (2018) 289:54–62. doi: 10.1016/j.toxlet.2018.03.012
26. Calaf G, Russo J. Transformation of human breast epithelial cells by chemical carcinogens. Carcinogenesis. (1993) 14:483–92.
27. Russo J, Tahin Q, Lareef MH, Hu YF, Russo IH. Neoplastic transformation of human breast epithelial cells by estrogens and chemical carcinogens. Environ Mol Mutagen. (2002) 39:254–63. doi: 10.1002/em.10052
28. Guo J, Xu Y, Ji W, Song L, Dai C, Zhan L. Effects of exposure to benzo[a]pyrene on metastasis of breast cancer are mediated through ROS-ERK-MMP9 axis signaling. Toxicol Lett. (2015) 234:201–10. doi: 10.1016/j.toxlet.2015.02.016S0378-427400083-1
29. Chapellier M, Bachelard-Cascales E, Schmidt X, Clement F, Treilleux I, Delay E, et al. Disequilibrium of BMP2 levels in the breast stem cell niche launches epithelial transformation by overamplifying BMPR1B cell response. Stem Cell Reports. (2015) 4:239–54. doi: 10.1016/j.stemcr.2014.12.007
30. Giulivo M, Lopez de Alda M, Capri E, Barcelo D. Human exposure to endocrine disrupting compounds: their role in reproductive systems, metabolic syndrome and breast cancer. A review. Environ Res. (2016) 151:251–64. doi: 10.1016/j.envres.2016.07.011
31. Delfosse V, Grimaldi M, le Maire A, Bourguet W, Balaguer P. (Nuclear receptor profiling of bisphenol-A and its halogenated analogues. Vitam Horm. 2014) 94:229–51. doi: 10.1016/B978-0-12-800095-3
32. Seachrist DD, Bonk KW, Ho SM, Prins GS, Soto AM, Keri RA. A review of the carcinogenic potential of bisphenol A. Reprod Toxicol. (2016) 59:167–82. doi: 10.1016/j.reprotox.2015.09.006
33. Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. (2006) 102:175–9. doi: 10.1016/j.jsbmb.2006.09.017
34. FAO/WHO J. F. W. E. M., t.R.T.a.H.A.o,.B. A. O. O., Organization, W. H., Nations, F.a.A.O.o.t.U., and, F. A. O./W. H. O., et al. Toxicological and health aspects of Bisphenol A : report of Joint FAO/WHO Expert Meeting 2-5 November 2010 and report of Stakeholder Meeting on Bisphenol A, 1 November 2010 Ottawa, Canada / Food and Agriculture Organization of the United Nations, World Health Organization. Geneva: World Health Organization (2011).
35. Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C, et al. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol. (2007) 24:178–98. doi: 10.1016/j.reprotox.2007.05.010
36. Fernandez SV, Russo J. Estrogen and xenoestrogens in breast cancer. Toxicol Pathol. (2010) 38:110–22. doi: 10.1177/0192623309354108
37. Jenkins S, Betancourt AM, Wang J, Lamartiniere CA. Endocrine-active chemicals in mammary cancer causation and prevention. J Steroid Biochem Mol Biol. (2012) 129:191–200. doi: 10.1016/j.jsbmb.2011.06.003
38. Marchese S, Silva E. Disruption of 3D MCF-12A breast cell cultures by estrogens–an in vitro model for ER-mediated changes indicative of hormonal carcinogenesis. PLoS ONE. (2012) 7:e45767. doi: 10.1371/journal.pone.0045767
39. Soule HD, Maloney TM, Wolman SR, Peterson WDJr, Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. (1990) 50:6075–86.
40. Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. (2006) 10:515–27. doi: 10.1016/j.ccr.2006.10.008
41. Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. (2008) 68:989–97. doi: 10.1158/0008-5472.CAN-07-2017
42. Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D, et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS ONE. (2010) 5:e9201. doi: 10.1371/journal.pone.0009201
43. Miller FR. Xenograft models of premalignant breast disease. J Mammary Gland Biol Neoplasia. (2000) 5:379–91. doi: 10.1023/a:1009577811584
44. Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. (1996) 148:313–9.
45. Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. (2008) 10:314–29. doi: 10.1593/neo.07970
46. Banda M, Speyer CL, Semma SN, Osuala KO, Kounalakis N, Torres Torres KE, et al. Metabotropic glutamate receptor-1 contributes to progression in triple negative breast cancer. PLoS ONE. (2014) 9:e81126. doi: 10.1371/journal.pone.0081126
47. Lemaire G, Benod C, Nahoum V, Pillon A, Boussioux AM, Guichou JF, et al. Discovery of a highly active ligand of human pregnane x receptor: a case study from pharmacophore modeling and virtual screening to “in vivo” biological activity. Mol Pharmacol. (2007) 72:572–81. doi: 10.1124/mol.106.033415
48. Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bieche I, et al. ZNF217 is a marker of poor prognosis in breast cancer that drives epithelial-mesenchymal transition and invasion. Cancer Res. (2012) 72:3593–606. doi: 10.1158/0008-5472.CAN-11-3095
49. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. (2003) 17:1253–70. doi: 10.1101/gad.1061803
50. Rasmussen MK, Balaguer P, Ekstrand B, Daujat-Chavanieu M, Gerbal-Chaloin S. Skatole (3-Methylindole) is a partial aryl hydrocarbon receptor agonist and induces CYP1A1/2 and CYP1B1 expression in primary human hepatocytes. PLoS ONE. (2016) 11:e0154629. doi: 10.1371/journal.pone.0154629
51. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. (2009) 28:523–32. doi: 10.1038/emboj.2008.304
52. Thollet A, Vendrell JA, Payen L, Ghayad SE, Ben Larbi S, Grisard E, et al. ZNF217 confers resistance to the pro-apoptotic signals of paclitaxel and aberrant expression of Aurora-A in breast cancer cells. Mol Cancer. (2010) 9:291. doi: 10.1186/1476-4598-9-291
53. Nguyen NT, Vendrell JA, Poulard C, Gyorffy B, Goddard-Leon S, Bieche I, et al. A functional interplay between ZNF217 and estrogen receptor alpha exists in luminal breast cancers. Mol Oncol. (2014) 8:1441–57. doi: 10.1016/j.molonc.2014.05.013
54. Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, et al. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. (2013) 140:219–32. doi: 10.1007/s10549-013-2622-y
55. Farnie G, Clarke RB. Mammary stem cells and breast cancer–role of Notch signalling. Stem Cell Rev. (2007) 3:169–75. doi: 10.1007/s12015-007-0023-5
56. Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. (2008) 75:75–84. doi: 10.1159/000123845000123845
57. Kakarala M, Wicha MS. Implications of the cancer stem-cell hypothesis for breast cancer prevention and therapy. J Clin Oncol. (2008) 26:2813–20. doi: 10.1200/JCO.2008.16.3931
58. Obana H, Hori S, Kashimoto T, Kunita N. Polycyclic aromatic hydrocarbons in human fat and liver. Bull Environ Contam Toxicol. (1981) 27:23–7.
59. Hecht SS, Carmella SG, Chen M, Dor Koch JF, Miller AT, Murphy SE, et al. Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. (1999) 59:590–6.
60. Besaratinia A, Maas LM, Brouwer EMC, Moonen EJC, De Kok TMCM, Wesseling GJ, et al. A molecular dosimetry approach to assess human exposure to environmental tobacco smoke in pubs. Carcinogenesis. (2002) 23:1171–6. doi: 10.1093/carcin/23.7.1171
61. Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect. (2010) 118:1055–70. doi: 10.1289/ehp.0901716
62. Teeguarden JG, Twaddle NC, Churchwell MI, Yang X, Fisher JW, Seryak LM, et al. 24-hour human urine and serum profiles of bisphenol A: evidence against sublingual absorption following ingestion in soup. Toxicol Appl Pharmacol. (2015) 288:131–42. doi: 10.1016/j.taap.2015.01.009
63. Song Z, Pollenz RS. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Mol Pharmacol. (2002) 62:806–16. doi: 10.1124/mol.62.4.806
64. Smith KJ, Murray IA, Tanos R, Tellew J, Boitano AE, Bisson WH, et al. Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther. (2011) 338:318–27. doi: 10.1124/jpet.110.178392
65. Filardo EJ, Quinn JA, Bland KI, Frackelton ARJr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. (2000) 14:1649–60. doi: 10.1210/mend.14.10.0532
66. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. (2006) 2:207–12. doi: 10.1038/nchembio775
67. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. (2009) 5:421–7. doi: 10.1038/nchembio.168
68. Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thurlimann B, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. (2013) 24:2206–23. doi: 10.1093/annonc/mdt303
69. Gyorffy B, Benke Z, Lanczky A, Balazs B, Szallasi Z, Timar J, et al. RecurrenceOnline: an online analysis tool to determine breast cancer recurrence and hormone receptor status using microarray data. Breast Cancer Res Treat. (2012) 132:1025–34. doi: 10.1007/s10549-011-1676-y
70. Li ZD, Wang K, Yang XW, Zhuang ZG, Wang JJ, Tong XW. Expression of aryl hydrocarbon receptor in relation to p53 status and clinicopathological parameters in breast cancer. Int J Clin Exp Pathol. (2014) 7:7931–7.
71. Saito R, Miki Y, Hata S, Takagi K, Iida S, Oba Y, et al. Aryl hydrocarbon receptor in breast cancer-a newly defined prognostic marker. Horm Cancer. (2014) 5:11–21. doi: 10.1007/s12672-013-0160-z
72. Trombino AF, Near RI, Matulka RA, Yang S, Hafer LJ, Toselli PA, et al. Expression of the aryl hydrocarbon receptor/transcription factor (AhR) and AhR-regulated CYP1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res Treat. (2000) 63:117–31. doi: 10.1023/a:1006443104670
73. Jeschke U, Zhang X, Kuhn C, Jalaguier S, Colinge J, Pfender K, et al. The prognostic impact of the Aryl Hydrocarbon Receptor (AhR) in primary breast cancer depends on the lymph node status. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20051016
74. Cirillo F, Lappano R, Bruno L, Rizzuti B, Grande F, Guzzi R, et al. AHR and GPER mediate the stimulatory effects induced by 3-methylcholanthrene in breast cancer cells and cancer-associated fibroblasts (CAFs). J Exp Clin Cancer Res. (2019) 38:335. doi: 10.1186/s13046-019-1337-2
75. Tarnow P, Tralau T, Luch A. Chemical activation of estrogen and aryl hydrocarbon receptor signaling pathways and their interaction in toxicology and metabolism. Expert Opin Drug Metab Toxicol. (2019) 15:219–29. doi: 10.1080/17425255.2019.1569627
76. Tarnow P, Tralau T, Luch A. G protein-coupled receptor 30 ligand G-1 increases aryl hydrocarbon receptor signalling by inhibition of tubulin assembly and cell cycle arrest in human MCF-7 cells. Arch Toxicol. (2016) 90:1939–48. doi: 10.1007/s00204-015-1615-5
77. Romagnolo DF, Papoutsis AJ, Laukaitis C, Selmin OI. Constitutive expression of AhR and BRCA-1 promoter CpG hypermethylation as biomarkers of ERalpha-negative breast tumorigenesis. BMC Cancer. (2015) 15:1026. doi: 10.1186/s12885-015-2044-9
78. Novikov O, Wang Z, Stanford EA, Parks AJ, Ramirez-Cardenas A, Landesman E, et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol Pharmacol. (2016) 90:674–88. doi: 10.1124/mol.116.105361
79. Stanford EA, Wang Z, Novikov O, Mulas F, Landesman-Bollag E, Monti S, et al. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. (2016) 14:20. doi: 10.1186/s12915-016-0240-y
80. Mohamed HT, Gadalla R, El-Husseiny N, Hassan H, Wang Z, Ibrahim SA, et al. Inflammatory breast cancer: Activation of the aryl hydrocarbon receptor and its target CYP1B1 correlates closely with Wnt5a/b-beta-catenin signalling, the stem cell phenotype and disease progression. J Adv Res. (2019) 16:75–86. doi: 10.1016/j.jare.2018.11.006
81. Lappano R, Pisano A, Maggiolini M. GPER function in breast cancer: an overview. Front Endocrinol. (2014) 5:66. doi: 10.3389/fendo.2014.00066
82. O'Donnell EF, Koch DC, Bisson WH, Jang HS, Kolluri SK. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. (2014) 5:e1038. doi: 10.1038/cddis.2013.549
83. Vacher S, Castagnet P, Chemlali W, Lallemand F, Meseure D, Pocard M, et al. High AHR expression in breast tumors correlates with expression of genes from several signaling pathways namely inflammation and endogenous tryptophan metabolism. PLoS ONE. (2018) 13:e0190619. doi: 10.1371/journal.pone.0190619
84. Hsu LH, Chu NM, Lin YF, Kao SH. G-protein coupled estrogen receptor in breast cancer. Int J Mol Sci. (2019) 20:e20306. doi: 10.3390/ijms20020306
Keywords: environmental factors, Benzo[a]pyrene, Bisphenol-A, breast cancer, tumor progression, AhR, GPR30
Citation: Donini CF, El Helou M, Wierinckx A, Győrffy B, Aires S, Escande A, Croze S, Clezardin P, Lachuer J, Diab-Assaf M, Ghayad SE, Fervers B, Cavaillès V, Maguer-Satta V and Cohen PA (2020) Long-Term Exposure of Early-Transformed Human Mammary Cells to Low Doses of Benzo[a]pyrene and/or Bisphenol A Enhances Their Cancerous Phenotype via an AhR/GPR30 Interplay. Front. Oncol. 10:712. doi: 10.3389/fonc.2020.00712
Received: 29 November 2019; Accepted: 15 April 2020;
Published: 29 May 2020.
Edited by:
Hamid Morjani, Université de Reims Champagne-Ardenne, FranceReviewed by:
Christos Roussakis, Université de Nantes, FranceCopyright © 2020 Donini, El Helou, Wierinckx, Győrffy, Aires, Escande, Croze, Clezardin, Lachuer, Diab-Assaf, Ghayad, Fervers, Cavaillès, Maguer-Satta and Cohen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascale A. Cohen, cGFzY2FsZS5jb2hlbkB1bml2LWx5b24xLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.