 Yuqi Yang1Ning Ji1,2*Qiu-Xu Teng1Chao-Yun Cai1Jing-Quan Wang1
Yuqi Yang1Ning Ji1,2*Qiu-Xu Teng1Chao-Yun Cai1Jing-Quan Wang1 Zhuo-Xun Wu1Zi-Ning Lei1Sabrina Lusvarghi3
Zhuo-Xun Wu1Zi-Ning Lei1Sabrina Lusvarghi3 Suresh V. Ambudkar3
Suresh V. Ambudkar3 Zhe-Sheng Chen1*
Zhe-Sheng Chen1*- 1Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John's University, Queens, NY, United States
- 2State Key Laboratory of Experimental Hematology Institute of Hematology and Hospital of Blood Diseases, Chinese Academy of Medical Science and Peking Union Medical College, Tianjin, China
- 3Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD, United States
Sitravatinib, also called MGCD516 or MG-516, is a broad-spectrum tyrosine kinase inhibitor (TKI) under phase III clinical evaluation. Herein, we explored the activity of sitravatinib toward multidrug resistance (MDR) by emphasizing its inhibitory effect on ATP-binding cassette super-family G member 2 (ABCG2). ABCG2 is a member of ATP-binding cassette (ABC) transporter family and plays a critical role in mediating MDR. Sitravatinb received an outstanding docking score for binding to the human ABCG2 model (PDB code: 6ETI) among thirty screened TKIs. Also, an MTT assay indicated that sitravatinib at 3 μM had the ability to restore the antineoplastic effect of various ABCG2 substrates in both drug-selected and gene-transfected ABCG2-overexpressing cell lines. In further tritium-labeled mitoxantrone transportation study, sitravatinib at 3 μM blocked the efflux function mediated by ABCG2 and as a result, increased the intracellular concentration of anticancer drugs. Interestingly, sitravatinib at 3 μM altered neither protein expression nor subcellular localization of ABCG2. An ATPase assay demonstrated that ATPase activity of ABCG2 was inhibited in a concentration-dependent manner with sitravatinib; thus, the energy source to pump out compounds was interfered. Collectively, the results of this study open new avenues for sitravatinib working as an ABCG2 inhibitor which restores the antineoplastic activity of anticancer drugs known to be ABCG2 substrates.
Introduction
Evidence from clinical studies showed that patients with multidrug resistant (MDR) tumors have a poorer prognosis and decreased likelihood of survival compared to cancer patients with drug sensitive tumors (1). Cancer patients develop cross-resistance to various structurally and functionally unrelated chemotherapeutic agents, resulting in treatment failure (2, 3). Overexpression of ATP-binding cassette (ABC) transporters is a leading cause of MDR (4). ABC transporters can be divided into seven subfamilies (ABCA to ABCG). The overexpression of certain transporters leads to MDR, including, but not limited to, ABCB1 (P-glycoprotein, P-gp), ABCG2 (breast cancer resistance protein, BCRP/MXR) and ABCC1 (multidrug resistance-associated protein 1, MRP1) (4, 5). Functionally, ABCB1 and ABCG2 work as efflux pumps, and are located in the lipid raft of specific cell lines (1, 6), reducing the intracellular level of various antineoplastic agents accumulating in cancer cells.
Currently, there are several approaches to circumvent MDR and to enhance the efficacy of antineoplastic drugs such as chemosensitizers (7), gene therapy (8, 9), immune-oncology (10), nanotechnology (11), or traditional Chinese medicine (12). The chemosensitizers, fumitremorgin C (FTC) and its derivative Ko143 are commonly used reference inhibitors of the ABCG2 transporter (13). However, FTC has neurotoxic effects and was withdrawn from clinical use (13). Furthermore, Ko143 was shown to lack specificity for the ABCG2 transporter at high concentrations and it was metabolically unstable in plasma (14). Therefore, more specific and less toxic inhibitors of ABC transporters are urgently needed for both preclinical and clinical evaluation.
In vitro studies have shown that some, but not all, novel tyrosine kinase inhibitors (TKIs) have ability to inhibit the ABCG2 transporter (15, 16). Clinically, TKIs are used as first- or second- line treatments for certain metastatic cancers (16, 17). However, TKIs have non-specific and “off-target” effects (18), thereby probably explaining why TKIs [1] are used as alternative treatments in the clinical setting and [2] restore the anticancer efficacy of chemotherapeutic drugs in the ABCG2-mediated MDR model.
Sitravatinib, also called MGCD516 or MG-516, is a broad-spectrum TKI targeting MET, TAM (TYRO3, AXL, MerTK), and members of vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and Eph families (17, 19, 20). Notably, it has been reported that sitravatinib has potent antitumor efficacy, that may be due, in part, to altering the tumor microenvironment and restoring the efficacy of immune checkpoint blockade (PD-1) in diverse cancer models (20). Dolan et al. reported that sitravatinib could combat drug resistance caused by sunitinib and axitinib in metastatic human and mouse models (17). Together, all these studies provide us with a clue that sitravatinib has the capability to antagonize MDR in cancer cells. Thus, various studies indicate that sitravatinib is efficacious in reversing or antagonizing MDR in cancer cells. Furthermore, sitravatinib is under nine ongoing clinical trials for various indications, with one being a phase III study (NCT03906071). To date, these studies have proved that intolerable adverse effects or unacceptable toxicity profile are not found under sitravatinib treatment in preclinical or clinical model. In this article, we focus on the antagonizing activity of sitravatinib toward MDR mediated by ABCG2.
Materials and Methods
Chemicals and Reagents
Sitravatinib was purchased from ChemieTek (Indianapolis, IN). Gilteritinib, BMS-777607, merestinib, and LOXO-101 were kindly provided as free samples from Selleckchem (Houston, TX). Topotecan was purchased from Selleckchem (Houstin, TX). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, GA). Dulbecco's modified Eagle medium (DMEM), antibiotics (penicillin/streptomycin [P/S]), and trypsin were obtained from Corning (Corning, NY). Mitoxantrone and SN-38 were purchased from Medkoo Sciences (Chapel Hill, NC). Phosphate buffered saline (PBS) (pH 7.4) was obtained from VWR Chemicals (Solon, OH). Ko143, cisplatin, and G418 were obtained from Enzo Life Sciences (Farmingdale, NY). Dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT) and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). Formaldehyde was obtained from J.T. Baker Chemical (Phillipsburg, NJ). Bovine serum albumin (BSA), 4′,6-diamidino-2-phenylindole (DAPI), PageRulerTM plus pre-stained protein ladder, GAPDH loading control monoclonal antibody (GA1R), PierceTM ECL Western blotting substrate, Alexa Fluor 488 conjugated goat anti-mouse IgG secondary antibody, and liquid scintillation cocktail were purchased from Thermo Fisher Scientific (Rockford, IL). HRP-conjugated rabbit anti-mouse IgG secondary antibody was purchased from Cell Signaling Technology (Dancers, MA). The monoclonal anti-BCRP antibody (BXP-21) was obtained from Millipore (Billerica, MA). [3H]-Mitoxantrone (0.5 Ci·mmol−1) were purchased from Moravek Biochemicals (Brea, CA).
Cell Lines and Cell Culture
The non-small cell lung cancer (NSCLC) cell line, NCI-H460, and the corresponding mitoxantrone-selected NCI-H460/MX20 cells were used. The NCI-H460/MX20 cells were developed and maintained in complete medium containing 20 nM of mitoxantrone and these cells were shown to overexpress the wild-type ABCG2 protein (21). The human colon carcinoma cell line, S1, and its corresponding mitoxantrone-selected S1-M1-80 cells were used. The S1-M1-80 cells were selected and maintained in complete medium containing 80 μM of mitoxantrone and were shown to overexpress a mutant allele (R482G) in the ABCG2 gene (22, 23). In addition, transfected cells were also used in this article. HEK293/pcDNA3.1, HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and HEK293/ABCG2-482-T7 were transfected with either an empty vector pcDNA3.1 or a pcDNA3.1 vector containing a full length ABCG2 encoding arginine (R), glycine (G), or threonine (T) for amino acid at position 482 (24). All transfected cell lines were selected and cultured in complete medium with 2 mg·ml−1 of G418. All cell lines were cultured in DMEM complete medium containing 10% FBS and 1% P/S at 37°C in a humidified incubator supplied with 5% CO2. All drug-resistant cells were grown in drug-free medium for more than 3 weeks and passaged at least 3 generations before experimental use. All drug-selected and gene-transfected cell lines used in this article were kindly provided by Drs. Susan Bates and Robert Robey (NCI, NIH, Bethesda, MD).
Induced-Fit Docking Analysis and Molecular Dynamic Simulations for ABCG2
Previously reported protocol was used for the molecular docking simulations (16) by using Maestro v11.1 software (Schrödinger, LLC, New York, NY). The structure of sitravatinib after preparation by LigPrep v4.1 to simulate the low-energy pose was subjected to the Glide XP (extra precision) docking default protocol (Schrödinger, LLC, New York, NY) with a pre-prepared cryo-EM structure of the human ABCG2 model (PDB code: 6ETI). The human ABCG2 protein model was confined to the docking grid at the drug-binding cavity by selecting specific amino acids that were deemed to be involved in specific interactions (25). The following induced-fit docking (IFD) was generated based on the best scored results from the Glide XP analysis. The docked sitravatinib-ABCG2 complex simulated based on IFD was then subjected to another 10 ns molecular dynamic (MD) simulation using a previously reported default protocol (26). The docking scores were calculated and reported as kcal·mol−1 and the highest-scoring result was used for further graphical analysis.
Cell Viability Assay
A modified MTT colorimetric assay was conducted to determine the cytotoxic efficacy of the chemotherapeutic agents with or without ABCG2 inhibitors, as described previously (26). In short, each cell line was harvested and seeded evenly into a 96-well plate with a density of 5 × 103 cell·well−1. The next day, cells were pretreated for 2 h with or without sitravatinib, or Ko143, at the indicated concentrations. Following pretreatment, cells were co-cultured with anticancer drugs (mitoxantrone, SN-38, topotecan or cisplatin) at serial concentrations. After a 68 h incubation period, an MTT solution (4 mg·ml−1 in PBS) was added to each well and the cells were further incubated at 37°C for 4 h in the dark. The supernatant was removed, and the resulting formazan crystals were dissolved with DMSO. An accuSkanTM GO UV/Vis Microplate Spectrophotometer (Fisher Sci., Fair Lawn, NJ) was used to measure the absorbance at 570 nm. The half maximal inhibitory concentration (IC50) for each anticancer drug was calculated as previously described (27). DMSO was used as an effective solvent to prepare stock solution (stock concentration is 10 mM) of all compounds. As the highest final concentration in cell viability assay was 100 μM, the final concentration of solvent was 1% in treatment medium (DMEM complete medium containing 10% FBS and 1% P/S). Notably, the control group was treated with solvent only. All experiments were performed independently at least three times performed in triplicate.
[3H]-Mitoxantrone Accumulation and Efflux Assay
The detailed protocol as previously stated was followed (28). Briefly, ABCG2-mediated MDR cells (1 × 106 cell·well−1) were seeded into a 24-well plate and incubated 1 day prior to further study. After 2 h of incubation in a drug-free medium, sitravatinib (0.75 and 3 μM) or a known inhibitor (3 μM), cells were co-incubated with [3H]-mitoxantrone at 37°C for another 2 h. The cells were then washed twice with ice-cold PBS, trypsinized, harvested, and placed in 5 ml of a liquid scintillation cocktail. Radioactivity was measured using a Packard TRI-CARB 1,900 CA liquid scintillation analyzer (Packard Instrument, Downers Grove, IL).
To determine the effect of sitravatinib on the efflux function of the ABCG2 transporter, the tritium-labeled mitoxantrone efflux assay was performed as previously stated (29). As described in the accumulation phase above, cells were pretreated with or without an ABCG2 inhibitor for 2 h followed by 2 h co-treatment with [3H]-mitoxantrone. The cells were then incubated in the presence or absence of an inhibitor at serial time points (0, 30, 60, and 120 min). Subsequently, the cells were washed twice with iced PBS, trypsinized, and transferred into a 5 ml liquid scintillation cocktail. Finally, radioactivity was measured as described above. In this assay, Ko143 served as the reference ABCG2 inhibitor for MDR cell lines. All the experiments were conducted independently three times.
Immunoblotting Analysis
Immunoblotting analysis (i.e., Western blotting) was conducted to detect the protein expression level of ABCG2 after up to 72 h of incubation with or without sitravatinib at 3 μM using a previously reported protocol (26). Briefly, following incubation with the highest non-toxic concentration of sitravatinib for 0, 24, 48, or 72 h, cells were harvested and lysed with lysis buffer (10 mM Tris, 1 mM EDTA, 0.1% SDS, 150 mM NaCl, 1% Triton X-100 and protease inhibitor cocktail) on ice for 20 min, followed by centrifugation at 4°C at 15,000 G for 20 min. The supernatant was collected and a bicinchoninic acid (BCA) Protein Kit (Thermo Scientific, Waltham, MA) was used to quantify the total concentration of protein in each sample. Equal amounts of total protein (20 μg) were loaded and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA). After blocking with 4% non-fat milk for 2 h at room temperature, the membrane was incubated with primary monoclonal antibodies against ABCG2 (at 1:500 dilution) and GAPDH (at 1:2,000 dilution) at 4°C overnight. The next day, after washing three times with Tris buffered saline containing 0.4% Tween 20 (TBST), the membrane was incubated with an HRP-conjugated secondary antibody (at 1:2,000 dilution) for 2 h at room temperature. Finally, the chemiluminescence signal of protein-antibody complex was developed by ECL substrate as per manufacturer's instruction. The relative density of each protein band was analyzed by ImageJ software (NIH, Bethesda, MD). All experiments were repeated three times independently.
Immunofluorescence Assay
Immunofluorescence assay was performed to assess the subcellular localization of membrane protein ABCG2 as previously described (30). In short, each cell line (1 × 104 cell·well−1) was seeded onto a 24-well plate and treated with or without sitravatinib at 3 μM for several time frames (0, 24, 48, and 72 h). After the treatment period, cells were washed with PBS three times, fixed in 4% paraformaldehyde for 15 min, and permeabilized by 0.1% Triton X-100 for 15 min before being blocked with 6% BSA for 2 h. Subsequently, the presence of ABCG2 was detected using anti-BCRP monoclonal antibody (at 1:1,000 dilution) followed by Alexa Fluor 488 conjugated secondary antibody (at 1:1,000 dilution). Finally, 1 μg·ml−1 DAPI was used to counterstain the nuclei. The immunofluorescence images were captured via an EVOS FL Auto fluorescence microscope (Thermo Scientific, Waltham, MA). All the experiments were performed three times independently.
ATPase Assay
The ATPase activity was determined by quantifying the amount of product (inorganic phosphate, Pi) produced after ATP hydrolysis. High Five insect cells expressing ABCG2 were used to prepare membrane vesicles (4). Next, membrane vesicles (10 μg total protein) were combined with assay buffer containing 50 mM MES (pH 6.8), 50 mM KCl, 5 mM sodium azide, 2 mM EGTA, 2 mM DTT, 1 mM ouabain, and 10 mM MgCl2, final volume 100 μL. Then sitravatinib was incubated with the membrane vesicles for 3 min at 37°C. The ATP hydrolysis was initiated by addition of 5 mM of Mg-ATP. After incubation at 37°C for 20 min, the reaction was terminated by addition of 100 μl 5% SDS solution. The amount of Pi was quantified using the method modified from Murphy and Riley (31), which is based on the complex formation of phosphate with potassium-antimonyl-tartarate in acidic ammonium molybdate. After reduction induced by ascorbic acid, the complex displays a stable blue color that was quantified by measuring the absorbance at 880 nm using a spectrophotometer (Bio-Rad, Hercules, CA).
Data and Statistical Analysis
All data are presented as the mean ± SD (standard deviation) obtained from three independent experiments performed in triplicate. Comparisons were made between the control group and the corresponding treatment group. The results were analyzed with one-way or two-way analysis of variance (ANOVA) following Tukey post hoc analysis. The statistical analysis was performed by GraphPad Prism version 6.02 for Windows (GraphPad Software, La Jolla, CA). The a priori significance level was p < 0.05.
Results
Pre-selecting TKIs Using a Molecular Simulation Analysis and a Modified Cell Viability Assay
To predict and compare the possible interaction between thirty TKIs and the human ABCG2 model, an in silico screening was conducted. The glide gscores of all screened TKIs are shown in Table S1. Based on the results from a preliminary study, sitravatinib had a higher docking score for binding to ABCG2 (Figure 1A) and a greater efficacy in reversing the anticancer efficacy of mitoxantrone in ABCG2-overexpressing cell line compared to other TKIs (BMS-777607, LOXO-101, gilteritnib, and merestinib) (Figure 1B). Based on these results, we conducted experiments to determine if sitravatinib was efficacious in attenuating ABCG2-mediated drug resistance in cell lines overexpressing the ABCG2 transporter.
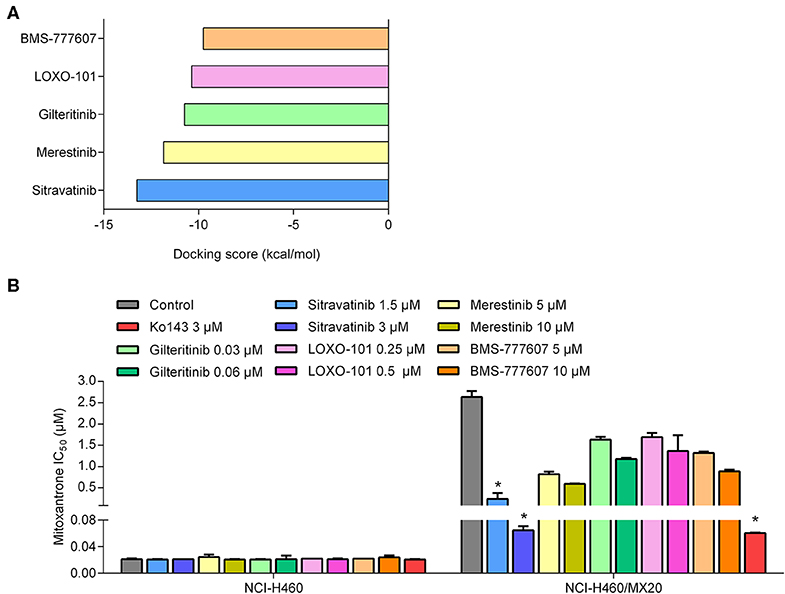
Figure 1. The preliminary study to pre-select possible ABCG2 inhibitor. (A) The docking scores of TKIs were screened by computational simulation analysis. (B) The antagonizing activity of TKIs toward MDR cell lines mediated by ABCG2. Data are represented as mean ± SD from at least three independent assays. *p < 0.05 compared with control group.
Molecular Docking Sitravatinib in the Drug-Binding Pocket of ABCG2
The IFD was carried out to simulate the interactions between sitravatinib and the atomic structure of human ABCG2 homodimer (chain A and B). As shown in Figure 2A, the lowest energy binding pose of sitravatinib was predicted in the drug-binding cavity through hydrophobic interactions with nearby amino acids. Two hydrogen bonds were formed between the ethylamino group and SER440 of ABCG2 chain A, and between the nitrogen in the pyridine ring of sitravatinib and ASN436 of ABCG2 chain A, respectively. Sitravatinib produced the best glide gscore out of all TKIs with a gscore of −13.248 kcal·mol−1, which is indicative of the free binding energy of the ligand. Figure 2B shows that the best-scored pose of sitravatinib occurs in the ABCG2 transmembrane domain with residues GLN 398, VAL 401, THR 402, PHE431, PHE 432, ASN 436, GLN 437, PHE 439, SER 440, SER 441, ARG 482, SER 486, PRO 485, PHE 489, ALA 517, VAL 546, and MET 549 in protein chain A, and PHE 431, PHE 432, THR 435, ASN 436, and MET 549 in protein chain B. Further molecular dynamic simulations of the sitravatinib-ABCG2 complex, as shown in Figures 2C,D, indicated that the position of sitravatinib in binding pocket did not show significant movements or changes after 10 ns. By analyzing the root mean square deviation (RMSD) of protein-drug complex, both the backbone of the protein and the complex were shown to be in a stable conformation after the first 2 ns and maintained this conformation until the end of the simulation (Figure 2E).
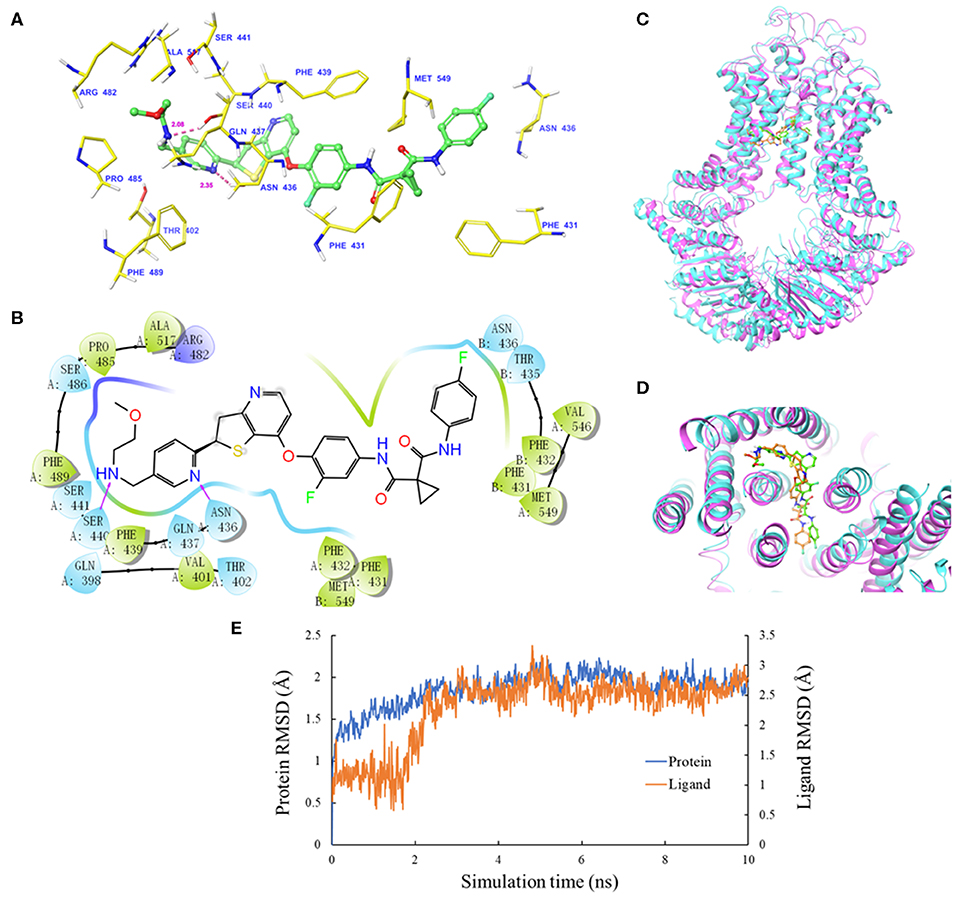
Figure 2. Molecular docking analysis with sitravatinib. (A) The docked conformation of sitravatinib (ball-and-stick model) is shown within the ABCG2 drug-binding cavity, with the atoms colored as follows: carbon, green; hydrogen, white; oxygen, red; nitrogen, blue. Important amino acid residues are described with the same color scheme as above for all atoms, except for carbon atoms in yellow. Dotted pink lines represent hydrogen-bonding interactions and the values of the correlation distances are indicated in Å. (B) The 2D schematic diagram of ligand–receptor interaction between sitravatinib and the human ABCG2 model. Amino acids within 3 Å are indicated as colored bubbles, polar residues are light blue, hydrophobic residues are green, and the positive charged residue is dark blue. Purple arrows denote H-bonds. (C,D) The superimposition of MD pose of sitravatinib within the binding cavity of ABCG2. Sitravatinib molecules are depicted as ball and stick model in faded green or orange for pre- and post-MD, respectively. The ABCG2 structure is in ribbon diagram in faded magenta or faded cyan for pre- and post-MD, respectively. (E) RMSD trajectory of ABCG2 and sitravatinib in sitravatinib-ABCG2 complex over the 10 ns simulation run.
Effect of Sitravatinib on the Efficacy of Antineoplastic Drugs in ABCG2-Mediated MDR Cell Lines
An MTT assay was used to obtain concentration-dependent cell viability curves (concentration range for sitravatinib from 0 to 100 μM) and the non-toxic concentrations for further reversal study. Based on cytotoxicity results, 3 μM concentration was chosen as the highest non-toxic concentration of sitravatinib for further reversal studies (chemical structure of the compound is shown in Figure 3A). At this concentration, the cell survival rate was more than 80% after 72 h treatment period (indicated as dash line in Figures 3B–D).
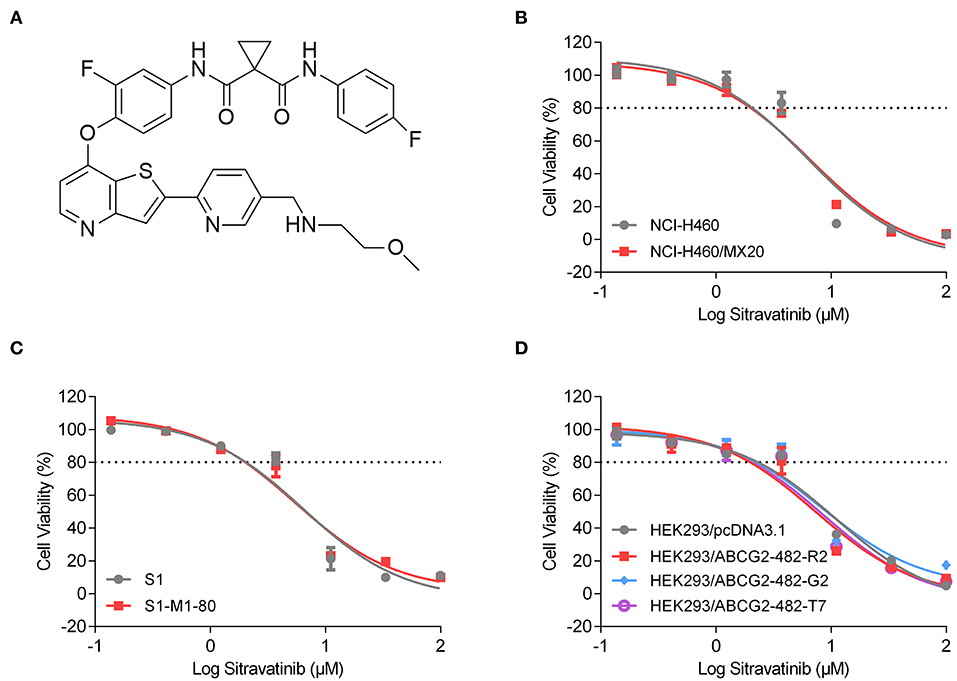
Figure 3. Chemical structure and the cytotoxic activity of sitravatinib in ABCG2-overexpressing cell lines and their corresponding sensitive cell lines. (A) Chemical structure of sitravatinib. The cell viability-concentration curves for NCI-H460/MX20 and NCI-H460 (B), S1-M1-80 and S1 (C), HEK293 cell lines transfected with full length ABCG2 or empty vector (D) after treatment with serial concentrations of sitravatinib for 72 h. Data are shown as mean ± SD, obtained from at least three experiments performed independently.
The reversal study showed that sitravatinib made the NCI-H460/MX20 and S1-M1-80 cells became more sensitive to mitoxantrone, SN-38 and topotecan in a concentration-dependent manner (Figures 4A–F). Additionally, it increased the efficacy of these antineoplastic agents in ACBG2-transfected HEK293 cell lines, including HEK293/ABCG2-482-R2, HEK293/ABCG2-482-G2, and HEK293/ABCG2-482-T7 (Figures 5A–C). Importantly, sitravatinib had a significant reversal effect on both wild-type and mutant ABCG2 overexpressing cells, whereas this effect could not be found in corresponding parental cells NCI-H460, S1, or HEK293/pcDNA3.1. It is notable that the reversal activity in ABCG2-overexpressing cell lines is comparable to its counterpart positive control treatment Ko143 at the same concentration level, which served as a reference inhibitor of ABCG2. In addition, sitravatinib did not affect the anticancer effect of cisplatin, a drug that is not the substrate of ABCG2, in neither drug-sensitive nor ABCG2-overexpressing cell lines (Figures 4G,H, 5D). The fold-reduction of IC50 values for chemotherapeutic drugs with or without inhibitors are shown in Tables S2, S3.
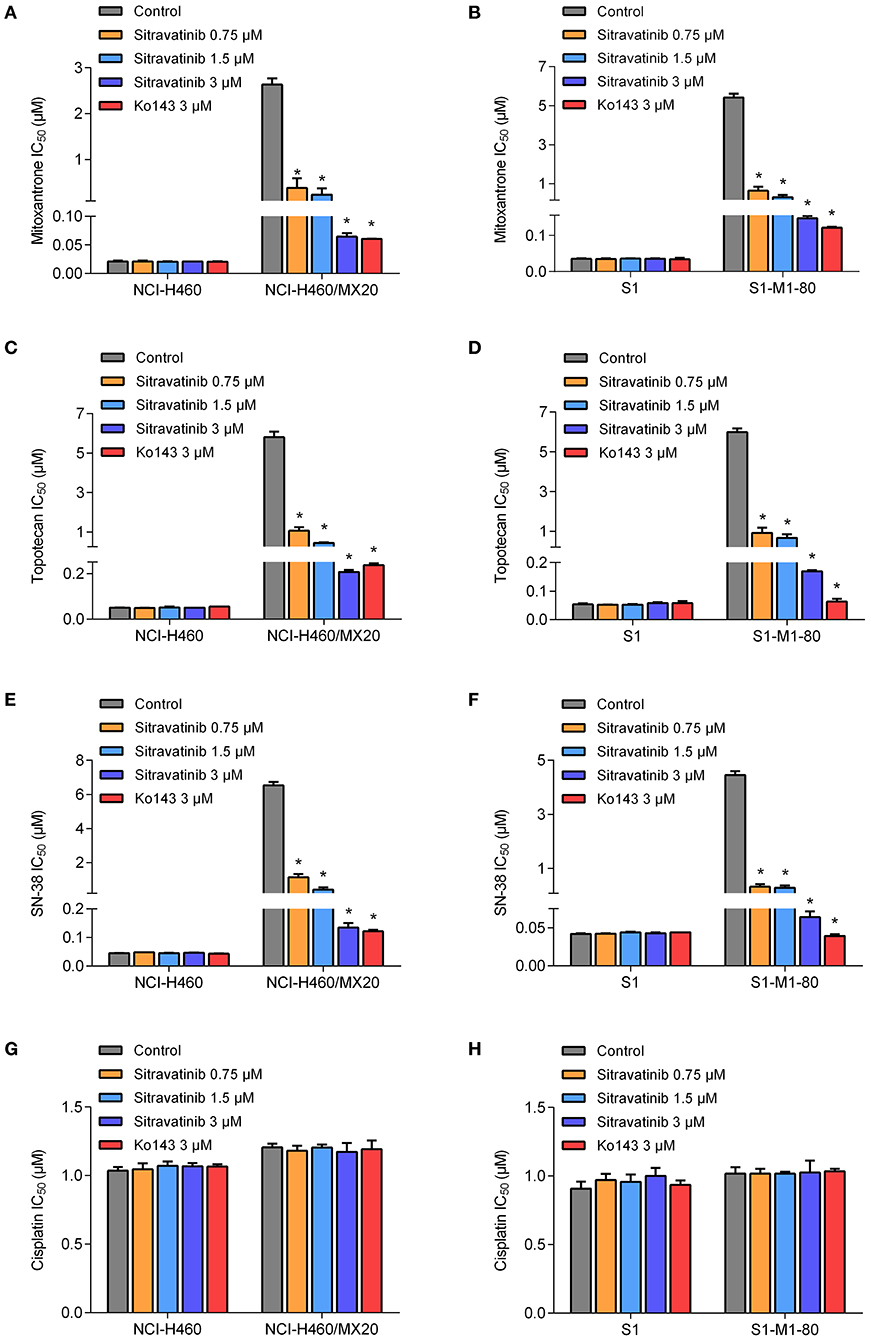
Figure 4. Drug sensitivity of drug-selected ABCG2-overexpressing cell lines to various chemotherapeutic agents in the absence or presence of sitravatinib. The IC50 values of mitoxantrone (A,B), topotecan (C,D), SN-38 (E,F), and cisplatin (G,H) in NCI-H460 and its mitoxantrone-selected resistance cell line (NCI-H460/MX20), S1 and its mitoxantrone-selected resistance cell line (S1-M1-80). Data were collected from independent experiments repeated at least three times and are shown as mean ± SD. *p < 0.05 compared with control group.
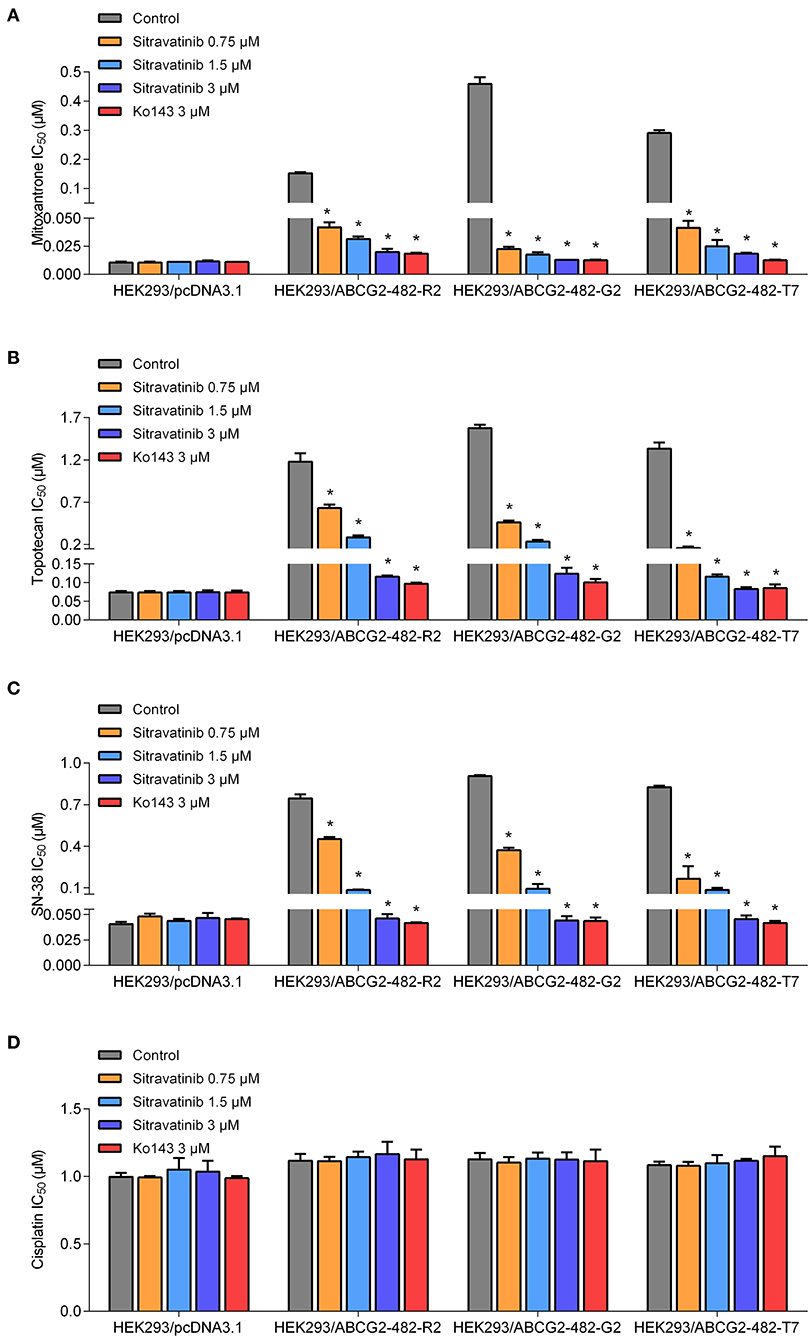
Figure 5. The antagonizing effect of sitravatinib in ABCG2-transfected HEK293 cell lines and the parental cell line HEK293/pcDNA3.1. The IC50 values of mitoxantrone (A), topotecan (B), SN-38 (C), and cisplatin (D) in transfected cell lines. Data are presented as mean ± SD. All data were obtained from at least three independent assays. *p < 0.05 compared with control group.
Overall, these results denoted that sitravatinib can improve the efficacy of multiple chemotherapeutic agents in ABCG2-mediated MDR cell lines in a concentration-dependent manner.
Modulation of ABCG2-Mediated Efflux of [3H]-Mitoxantrone in MDR Cell Lines
In order to understand the mechanism of action of sitravatinib in reversing MDR, several mechanism-based assays were performed. To evaluate the function of ABCG2, the intracellular concentration of tritium-labeled chemotherapeutic drug was quantified in parental cells and cells overexpressing ABCG2 with or without an inhibitor.
Sitravatinib at 3 μM concentration significantly increased the intracellular [3H]-mitoxantrone accumulation level from 38.7 to 129.2% or from 31.6 to 91.0% in NCI-H460/MX20 or S1-M1-80 cells, respectively, but not in the corresponding parental cell line counterparts (NCI-H460 and S1), as shown in Figures 6A,B. In this study, Ko143 served as reference ABCG2 inhibitor. Hence, these results suggested that sitravatinib increases the intracellular accumulation of [3H]-mitoxantrone in ABCG2-mediated MDR cells.
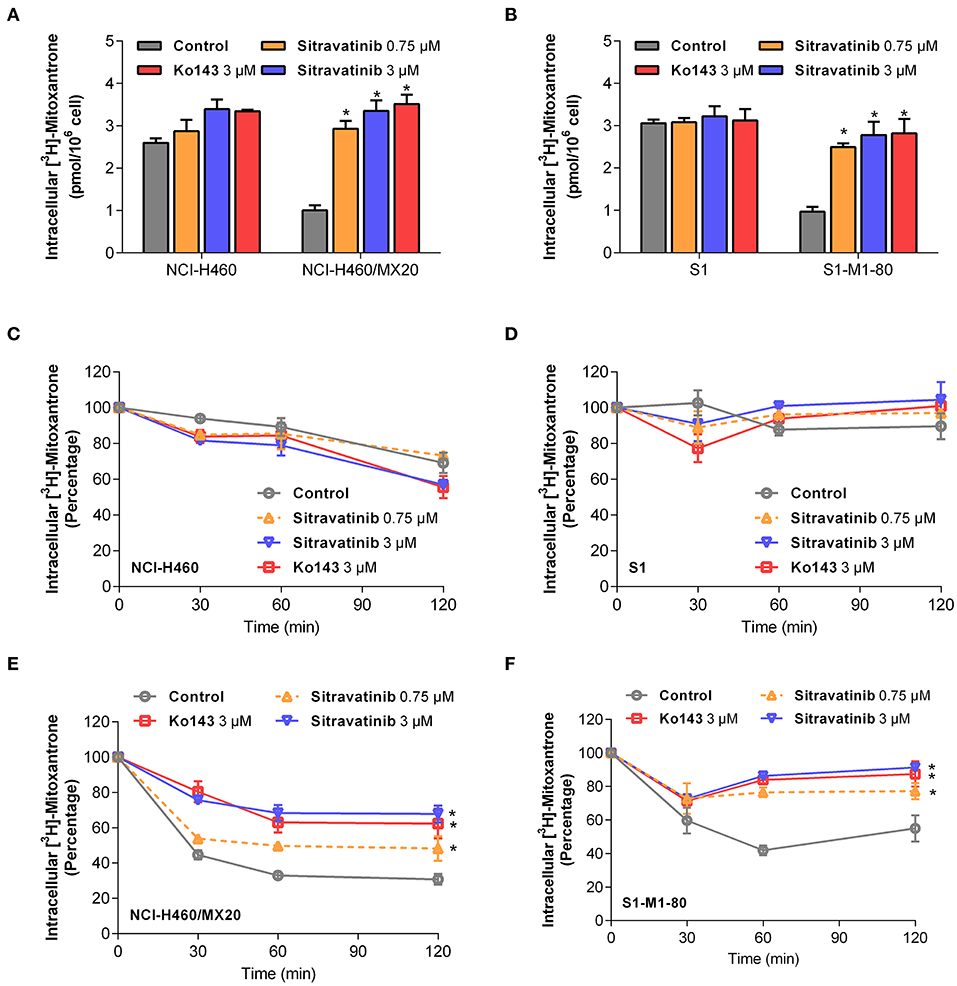
Figure 6. Sitravatinib inhibits ABCG2-mediated mitoxantrone transport. The accumulation of tritium-labeled mitoxantrone in NCI-H460 and NCI-H460/MX20 (A), S1 and S1-M1-80 (B). The ABCG2-mediated efflux activity in NCI-H460 (C), S1 (E) and their corresponding mitoxantrone-selected cell line NCI-H460/MX20 (D), S1-M1-80 (F) at a series of time points (0, 30, 60, 120 min). Data from at least three independent experiments are shown as mean ± SD. *p < 0.05 compared with control group.
Furthermore, in order to further understand the enhanced intracellular accumulation of [3H]-mitoxantrone in MDR cells, a time-course study was conducted to assess the efflux function of ABCG2 by quantifying the level of intercellular [3H]-substrate at serial time points in the presence or absence of inhibitors. As shown in Figures 6E,F, after 120 min, NCI-H460/MX20 and S1-M1-80 cells without ABCG2 inhibitor treatment maintained only 30 and 55% of [3H]-mitoxantrone, respectively. On the contrary, when treated with 3 μM sitravatinib, 68 and 91% [3H]-mitoxantrone was accumulated in NCI-H460/MX20 and S1-M1-80 cells, respectively. In contrast, sitravatinib did not significantly change the efflux function in parental cells (NCI-H460 or S1) at different time points (Figures 6C,D).
Taken together, these results demonstrated that sitravatinib could enhance the intracellular accumulation of a tritium-labeled chemotherapeutic drug by blocking the efflux function mediated by ABCG2.
Effect of Sitravatinib on Expression and Localization of ABCG2 in ABCG2-Mediated MDR Cell Lines
It is known that the reversal mechanism of action could involve either downregulation of protein expression level and/or alternation of subcellular localization of the transporter (4). Therefore, immunoblotting analysis and immunofluorescence assay were conducted to detect the expression and localization of ABCG2 protein, respectively.
The expression level of ABCG2 in NCI-H460/MX20 and S1-M1-80 was not significantly altered even after 72 h treatment with 3 μM sitravatinib, Figures 7A,B. In addition, ABCG2 was detected on the cell membrane surface after the cells treated with 3 μM sitravatinib for 0, 24, 48, or 72 h, Figures 7C,D.
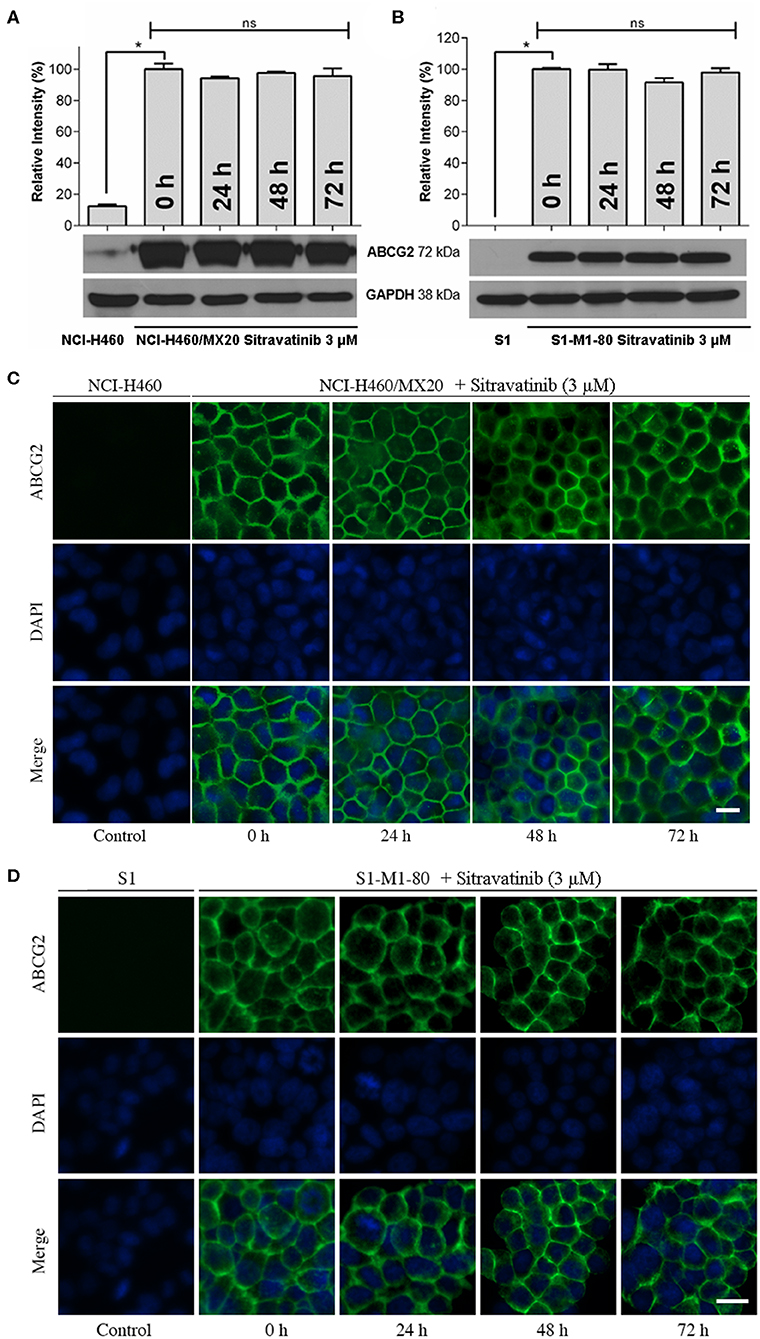
Figure 7. Sitravatinib does not affect protein expression level nor the subcellular localization of ABCG2. Immunoblotting analysis showing the expression level of ABCG2 in MDR cell lines overexpressing ABCG2 NCI-H460/MX20 (A) and S1-M1-80 (B) compared to the corresponding parental cell lines. The relative density of each band is shown as mean ± SD, collected from at least three independent assays. *p < 0.05 compared with control group. Immunofluorescence assay indicating the subcellular localization of membrane protein ABCG2 of NCI-H460/MX20 (C) and S1-M1-80 (D) and the corresponding parental cell lines. Color: ABCG2 (Green), DAPI (Blue). Scale bar:10 μm.
Therefore, long-term treatment at the highest non-toxic concentration of sitravatinib (3 μM) neither downregulates the expression level nor affects the subcellular localization of ABCG2 in ABCG2-mediated MDR cell lines.
Effect of Sitravatinib on ABCG2 ATPase Activity
It is documented that ATP hydrolysis is the energy source for ABC transporters to pump out endogenous and exogenous toxicants (32, 33). Hence, the effect of sitravatinib on ABCG2 ATPase activity was evaluated. Herein, the ABCG2-mediated ATP hydrolysis was measured in membrane vesicles after incubation with serial concentrations of sitravatinib (0–20 μM). Sitravatinib inhibited 56.2% of the basal ATPase activity, and the inhibitory effect reached 50% at 0.9 μM, see Figure 8. These results indicate that sitravatinib inhibits ABCG2 ATPase activity in a concentration-dependent manner. ATPase data combined with molecular docking suggest that sitravatinib interacts with the drug-binding pocket of the ABCG2.
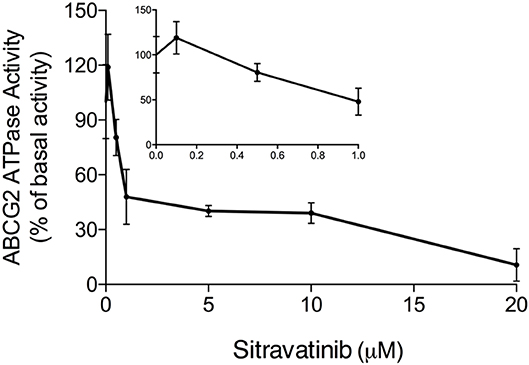
Figure 8. Sitravatinib inhibits ABCG2 ATPase activity in a concentration-dependent manner. The effect of sitravatinib on ATPase activity of ABCG2 in insect cell membrane vesicles was determined as described in the Materials and Methods Section. Data were obtained from independent assays repeated at least three times and are presented as mean ± SD.
Discussion and Conclusion
It is well-known that ABC transporters contribute to MDR and as a result, limit the anticancer efficacy of numerous chemotherapeutic agents in the clinical setting. Due to their efflux function, ABC transporters can extrude many structurally and functionally unrelated anticancer drugs (34), resulting in poor prognosis and low survival rates in cancer patients. In the past decades, many researchers have attempted to synthesize or screen potential inhibitors of ABC transporters to reverse MDR (16, 35–38). However, high toxicity and drug-drug interactions remain to be a challenge (39). To date, many researchers using in vivo and ex vivo models have demonstrated that TKIs have ability to restore the sensitivity of substrate antineoplastic drugs of ABC transporters for effective chemotherapy (40–43). Most recently, Chen et al. conducted a phase I clinical evaluation in a population of patients to reveal that cyclosporine A (CsA), a competitive ABCB1 inhibitor, could combat drug resistance caused by brentuximab vedotin in relapsed/refractory Hodgkin lymphoma with tolerable and feasible profile (44). These studies provided growing evidence that inhibitors of ABC transporters have promising possibility in the future preclinical and clinical use. Thus, it is meaningful to find potential inhibitors of ABC transporters even though many obstacles exist. It has been documented that some TKIs could behave as substrates or inhibitors of ABC transporters depending on different settings (45). Collectively, this suggests that TKIs with inhibitory activity toward ABCG2 in combination with conventional chemotherapeutic agents can be a promising strategy to circumvent MDR.
Using molecular docking and cell viability assay we first screened thirty TKIs that share active pharmacophoric features of ABC transporter inhibitors, such as the aromatic system, benzamido groups, or methoxyphenyl groups (46). The five compounds with the highest glide gscores were chosen to further examine their MDR reversal activity in cell lines expressing ABCG2. These screening assays showed that sitravatinib had an outstanding docking score within the drug-binding pocket in the transmembrane domain of the homodimer of human ABCG2, and also demonstrated excellent inhibitory activity in the ABCG2-mediated MDR cell lines.
In silico analysis of the simulated molecular docking showed specific interactions between sitravatinib and the human ABCG2 drug-binding pocket. The high glide gscore (−13.248 kcal·mol−1) suggested strong affinity of sitravatinib. The best-scored pose of other known ABCG2 inhibitors, such as selonsertib, ulixertinib and NVP-TAE684, received glide gscores of −12.278, −11.501, and −12.929 kcal·mol−1, respectively (4, 28, 47), which indicates that the affinity of sitravatinib and ABCG2 protein model may be comparable to other known ABCG2 inhibitors. To verify whether this docking ligand could be removed from the binding pocket, further MD simulations were performed. The MD simulations indicated that the binding complex did not change significantly in 10 ns, with the maximal RMSD of sitravatinib being approximately 2 Å. These results suggested that sitravatinib could bind stably to the substrate-binding cavity of human ABCG2 with high affinity.
Cell viability assay demonstrated that the highest non-toxic concentration of sitravatinib was 3 μM. Furthermore, the modified MTT colorimetric assay on ABCG2-mediated MDR cell lines supported the conclusion that the inhibitory effect of sitravatinib was strictly associated with the expression of ABCG2. This was further corroborated using ABCG2-transfected HEK293 cell lines, in which ABCG2 is the solo contributor to MDR. By contrast, the reversal effect of sitravatinib was not found in any of the sensitive cell lines. Notably, sitravatinib did not change the IC50 values of cisplatin, which is not a substrate of ABCG2.
A mechanism-based assay was conducted to examine the underlying mechanism of action of the antagonizing activity of sitravatinib in ABCG2-overexpressing cell lines. ABCG2 was shown as an efflux pump (48). Hence, tritium-labeled mitoxantrone-mediated accumulation and efflux assays were conducted to evaluate the pump function of ABCG2. Pre-treatment of the ABCG2 overexpressing cell lines with sitravatinib significantly improved the intracellular accumulation of mitoxantrone, which is a well-established substrate of ABCG2, but not in the corresponding sensitive cell lines. In addition, the ABCG2-expressing cell lines effluxed less amount of the chemotherapeutic drug after incubation with sitravatinib at non-toxic concentrations, compared with their counterparts in parental cell lines. This demonstrated that sitravatinib could impede the ABCG2 efflux function and in turn increase the intracellular accumulation of anticancer drug; thus, sitravatinib at non-toxic concentrations could remarkably antagonize ABCG2-mediated MDR and improve the antineoplastic efficacy of chemotherapeutic agents.
Mechanistically, it is well-documented that several MDR inhibitors downregulate the level of ABCG2 in the plasma membrane to hinder its pump function (4, 15, 49). To evaluate if this is the case with sitravatinib, Western blotting and immunofluorescence assays were conducted. Our results show that sitravatinib did not alter the expression level or the subcellular localization of ABCG2 in ABCG2-expressing cell lines. Collectively, we summarized that sitravatinib directly inhibits the pump function of ABCG2 without changing the expression level or its subcellular localization. Furthermore, the hydrolysis of ATP is the energy source for ABC transporter-mediated efflux of endogenous and exogenous toxicants (32, 33). Therefore, an ATPase assay was performed to determine the effect of sitravatinib on the ATPase activity of ABCG2. The results demonstrated that sitravatinib could partially inhibit ABCG2 ATPase activity suggesting that sitravatinib has the ability to interfere with the transport function of ABCG2 by interfering with its ATPase activity.
In conclusion, our study revealed that sitravatinib is a potential ABCG2 inhibitor with an acceptable toxicity profile. The sensitizing effect of sitravatinib toward different MDR cell lines is due to inhibition of ABCG2 function. In the future, we will focus on long-term exposure to sitravatinib in vivo and ex vivo to further evaluate the antagonizing effect and its toxicity profile as an ABCG2 inhibitor.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
YY and Z-SC designed the study. YY, NJ, Q-XT, and J-QW gave contribution to perform experiments. C-YC, Z-NL, Z-XW, SL, and SA provided technical and material support. YY wrote the first draft. All authors discussed the results and implications and developed the manuscript at all stages.
Funding
This work was supported by NIH (No. 1R15GM116043-01) to Z-SC, SL, and SA were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are grateful to Drs. Susan Bates and Robert Robey (NCI, NIH, Bethesda, MD) for providing drug-selected and transfected cell lines we used in this article. We also thank Dr. Tanaji T. Talele (St. John's University, New York, NY) for providing the computing resources for the docking analysis. We thank Dr. Charles R. Ashby Jr. (St. John's University, New York, NY) for editing the article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00700/full#supplementary-material
References
1. Kartal-Yandim M, Adan-Gokbulut A, Baran Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol. (2016) 36:716–26. doi: 10.3109/07388551.2015.1015957
2. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. (2010) 10:147–56. doi: 10.1038/nrc2789
3. Amiri-Kordestani L, Basseville A, Kurdziel K, Fojo AT, Bates SE. Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist Updat. (2012) 15:50–61. doi: 10.1016/j.drup.2012.02.002
4. Ji N, Yang Y, Cai CY, Lei ZN, Wang JQ, Gupta P, et al. Selonsertib. (GS-4997), an ASK1 inhibitor, antagonizes multidrug resistance in ABCB1- and ABCG2-overexpressing cancer cells. Cancer Lett. (2019) 440–1:82–93. doi: 10.1016/j.canlet.2018.10.007
5. Dean M, Allikmets R. Complete characterization of the human ABC gene family. J Bioenerg Biomembr. (2001) 33:475–9. doi: 10.1023/A:1012823120935
6. Klappe K, Hummel I, Hoekstra D, Kok JW. Lipid dependence of ABC transporter localization and function. Chem Phys Lipids. (2009) 161:57–64. doi: 10.1016/j.chemphyslip.2009.07.004
7. Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW. Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett. (2017) 405:100–10. doi: 10.1016/j.canlet.2017.07.023
8. Shi R, Peng H, Yuan X, Zhang X, Zhang Y, Fan D, et al. Down-regulation of c-fos by shRNA sensitizes adriamycin-resistant MCF-7/ADR cells to chemotherapeutic agents via P-glycoprotein inhibition and apoptosis augmentation. J Cell Biochem. (2013) 114:1890–900. doi: 10.1002/jcb.24533
9. Lage H. Gene therapeutic approaches to overcome ABCB1-mediated drug resistance. Recent Results Cancer Res. (2016) 209:87–94. doi: 10.1007/978-3-319-42934-2_6
10. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer. (2017) 74:55–72. doi: 10.1016/j.ejca.2017.01.001
11. Yin J, Lang T, Cun D, Zheng Z, Huang Y, Yin Q, et al. pH-sensitive nano-complexes overcome drug resistance and inhibit metastasis of breast cancer by silencing akt expression. Theranostics. (2017) 7:4204–16. doi: 10.7150/thno.21516
12. Xu WL, Shen HL, Ao ZF, Chen BA, Xia W, Gao F, et al. Combination of tetrandrine as a potential-reversing agent with daunorubicin, etoposide and cytarabine for the treatment of refractory and relapsed acute myelogenous leukemia. Leuk Res. (2006) 30:407–13. doi: 10.1016/j.leukres.2005.08.005
13. Toyoda Y, Takada T, Suzuki H. Inhibitors of human ABCG2: from technical background to recent updates with clinical implications. Front Pharmacol. (2019) 10:208. doi: 10.3389/fphar.2019.00208
14. Weidner LD, Zoghbi SS, Lu S, Shukla S, Ambudkar SV, Pike VW, et al. The inhibitor Ko143 is not specific for ABCG2. J Pharmacol Exp Ther. (2015) 354:384–93. doi: 10.1124/jpet.115.225482
15. Zhang GN, Zhang YK, Wang YJ, Gupta P, Ashby CR Jr, Alqahtani S, et al. Epidermal growth factor receptor (EGFR) inhibitor PD153035 reverses ABCG2-mediated multidrug resistance in non-small cell lung cancer: in vitro and in vivo. Cancer Lett. (2018) 424:19–29. doi: 10.1016/j.canlet.2018.02.040
16. Zhang YK, Wang YJ, Lei ZN, Zhang GN, Zhang XY, Wang DS, et al. Regorafenib antagonizes BCRP-mediated multidrug resistance in colon cancer. Cancer Lett. (2019) 442:104–12. doi: 10.1016/j.canlet.2018.10.032
17. Dolan M, Mastri M, Tracz A, Christensen JG, Chatta G, Ebos JML. Enhanced efficacy of sitravatinib in metastatic models of antiangiogenic therapy resistance. PLoS ONE. (2019) 14:e0220101. doi: 10.1371/journal.pone.0220101
18. Mastri M, Lee CR, Tracz A, Kerbel RS, Dolan M, Shi Y, et al. Tumor-independent host secretomes induced by angiogenesis and immune-checkpoint inhibitors. Mol Cancer Ther. (2018) 17:1602–12. doi: 10.1158/1535-7163.MCT-17-1066
19. Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. (2016) 7:4093–109. doi: 10.18632/oncotarget.6547
20. Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.124184
21. Henrich CJ, Bokesch HR, Dean M, Bates SE, Robey RW, Goncharova EI, et al. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J Biomol Screen. (2006) 11:176–83. doi: 10.1177/1087057105284576
22. Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. (1999) 59:8–13.
23. Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. (2001) 61:6635–9.
24. Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. (2003) 89:1971–8. doi: 10.1038/sj.bjc.6601370
25. Jackson SM, Manolaridis I, Kowal J, Zechner M, Taylor NMI, Bause M, et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol. (2018) 25:333–40. doi: 10.1038/s41594-018-0049-1
26. Zhang YK, Zhang XY, Zhang GN, Wang YJ, Xu H, Zhang D, et al. Selective reversal of BCRP-mediated MDR by VEGFR-2 inhibitor ZM323881. Biochem Pharmacol. (2017) 132:29–37. doi: 10.1016/j.bcp.2017.02.019
27. Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. (1987) 47:936–42.
28. Ji N, Yang Y, Lei ZN, Cai CY, Wang JQ, Gupta P, et al. Ulixertinib. (BVD-523) antagonizes ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Biochem Pharmacol. (2018) 158:274–85. doi: 10.1016/j.bcp.2018.10.028
29. Ji N, Yang Y, Cai CY, Wang JQ, Lei ZN, Wu ZX, et al. Midostaurin reverses ABCB1-mediated multidrug resistance, an in vitro study. Front Oncol. (2019) 9:514. doi: 10.3389/fonc.2019.00514
30. Ji N, Yang Y, Cai CY, Lei ZN, Wang JQ, Gupta P, et al. VS-4718 antagonizes multidrug resistance in ABCB1- and ABCG2-overexpressing cancer cells by inhibiting the efflux function of ABC transporters. Front Pharmacol. (2018) 9:1236. doi: 10.3389/fphar.2018.01236
31. Ojida A, Mito-oka Y, Sada K, Hamachi I. Molecular recognition and fluorescence sensing of monophosphorylated peptides in aqueous solution by bis(zinc(II)-dipicolylamine)-based artificial receptors. J Am Chem Soc. (2004) 126:2454–63. doi: 10.1021/ja038277x
32. Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F. The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance. (MDR). Curr Drug Targets. (2006) 7:893–909. doi: 10.2174/138945006777709520
33. Han LW, Gao C, Mao Q. An update on expression and function of P-gp/ABCB1 and BCRP/ABCG2 in the placenta and fetus. Expert Opin Drug Metab Toxicol. (2018) 14:817–29. doi: 10.1080/17425255.2018.1499726
34. Theodoulou FL, Kerr ID. ABC transporter research: going strong 40 years on. Biochem Soc Trans. (2015) 43:1033–40. doi: 10.1042/BST20150139
35. Cui H, Zhang AJ, Chen M, Liu JJ. ABC transporter inhibitors in reversing multidrug resistance to chemotherapy. Curr Drug Targets. (2015) 16:1356–71. doi: 10.2174/1389450116666150330113506
36. Hsiao SH, Murakami M, Yeh N, Li YQ, Hung TH, Wu YS, et al. The positive inotropic agent DPI-201106 selectively reverses ABCB1-mediated multidrug resistance in cancer cell lines. Cancer Lett. (2018) 434:81–90. doi: 10.1016/j.canlet.2018.07.022
37. Cui Q, Cai CY, Gao HL, Ren L, Ji N, Gupta P, et al. Glesatinib, a c-MET/SMO dual inhibitor, antagonizes P-glycoprotein mediated multidrug resistance in cancer cells. Front Oncol. (2019) 9:313. doi: 10.3389/fonc.2019.00313
38. Wang JQ, Wang B, Lei ZN, Teng QX, Li JY, Zhang W, et al. Derivative of 5-cyano-6-phenylpyrimidin antagonizes ABCB1- and ABCG2-mediated multidrug resistance. Eur J Pharmacol. (2019) 863:172611. doi: 10.1016/j.ejphar.2019.172611
39. Wu CP, Murakami M, Hsiao SH, Liu TC, Yeh N, Li YQ, et al. SIS3, a specific inhibitor of Smad3 reverses ABCB1- and ABCG2-mediated multidrug resistance in cancer cell lines. Cancer Lett. (2018) 433:259–72. doi: 10.1016/j.canlet.2018.07.004
40. Zhao XQ, Xie JD, Chen XG, Sim HM, Zhang X, Liang YJ, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol. (2012) 82:47–58. doi: 10.1124/mol.111.076299
41. Zhang H, Patel A, Ma SL, Li XJ, Zhang YK, Yang PQ, et al. In vitro, in vivo and ex vivo characterization of ibrutinib: a potent inhibitor of the efflux function of the transporter MRP1. Br J Pharmacol. (2014) 171:5845–57. doi: 10.1111/bph.12889
42. Yang K, Chen Y, To KK, Wang F, Li D, Chen L, et al. Alectinib (CH5424802) antagonizes ABCB1- and ABCG2-mediated multidrug resistance in vitro, in vivo and ex vivo. Exp Mol Med. (2017) 49:e303. doi: 10.1038/emm.2016.168
43. Yang L, Li M, Wang F, Zhen C, Luo M, Fang X, et al. Ceritinib enhances the efficacy of substrate chemotherapeutic agent in human abcb1-overexpressing leukemia cells in vitro, in vivo and ex-vivo. Cell Physiol Biochem. (2018) 46:2487–99. doi: 10.1159/000489655
44. Chen R, Herrera AF, Hou J, Chen L, Wu J, Guo Y, et al. Inhibition of MDR1 overcomes resistance to brentuximab vedotin in hodgkin lymphoma. Clin Cancer Res. (2020) 26:1034–44. doi: 10.1158/1078-0432.CCR-19-1768
45. Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L. Overcoming ABC transporter-mediated multidrug resistance: the dual role of tyrosine kinase inhibitors as multitargeting agents. Eur J Med Chem. (2017) 142:271–89. doi: 10.1016/j.ejmech.2017.07.062
46. Nicolle E, Boumendjel A, Macalou S, Genoux E, Ahmed-Belkacem A, Carrupt PA, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev. (2009) 61:34–46. doi: 10.1016/j.addr.2008.10.004
47. Wang J, Wang JQ, Cai CY, Cui Q, Yang Y, Wu ZX, et al. Reversal effect of ALK inhibitor NVP-TAE684 on ABCG2-overexpressing cancer cells. Front Oncol. (2020) 10:228. doi: 10.3389/fonc.2020.00228
48. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. (2018) 18:452–64. doi: 10.1038/s41568-018-0005-8
Keywords: sitravatinib, tyrosine kinase inhibitor, multidrug resistance, ATP-binding cassette (ABC) transporters, ATP-binding cassette super-family G member 2 (ABCG2)
Citation: Yang Y, Ji N, Teng Q-X, Cai C-Y, Wang J-Q, Wu Z-X, Lei Z-N, Lusvarghi S, Ambudkar SV and Chen Z-S (2020) Sitravatinib, a Tyrosine Kinase Inhibitor, Inhibits the Transport Function of ABCG2 and Restores Sensitivity to Chemotherapy-Resistant Cancer Cells in vitro. Front. Oncol. 10:700. doi: 10.3389/fonc.2020.00700
Received: 19 January 2020; Accepted: 14 April 2020;
Published: 12 May 2020.
Edited by:
Nehad M. Ayoub, Jordan University of Science and Technology, JordanReviewed by:
Xiang Chen, Central South University, ChinaVishwa Khare, Eurofins Viracor, United States
Hua Zhu, The Ohio State University, United States
Copyright © 2020 Yang, Ji, Teng, Cai, Wang, Wu, Lei, Lusvarghi, Ambudkar and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Ji, jining@ihcams.ac.cn; Zhe-Sheng Chen, chenz@stjohns.edu