
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 30 April 2020
Sec. Head and Neck Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00683
This article is part of the Research Topic Head & Neck Cancer and Esophageal Cancer: From Biosignatures to Therapeutics View all 35 articles
 Melvin A. Ambele1,2*
Melvin A. Ambele1,2* Andre van Zyl3
Andre van Zyl3 Michael S. Pepper2
Michael S. Pepper2 Marlene B. van Heerden1Willie F. P. van Heerden1
Marlene B. van Heerden1Willie F. P. van Heerden1The lack of clinical biomarkers for head and neck cancer subtypes limits early diagnosis and monitoring of disease progression. This study investigates genetic alterations in clinically identical tumor, tumor-adjacent dysplastic epithelium (TADE) and normal epithelium (NE) in five oral cancer patients to identify differences and commonalities between oral cancer, TADE and NE. A VELscope®Vx device was used to identify TADE and NE surrounding a clinical tumor for analysis of genetic alterations using the OncoScan® assay. One of the tumor samples examined was an “M” class tumor with a high confidence BRAF:p.G469A:c.1406G>C somatic mutation, which is the first to be reported in oral cancer. Another tumor showed mosaicism in genetic alterations, indicating the presence of multiple clones. Overall, each patient's tumor, TADE and NE showed a distinct genetic profile which indicates intertumoral clonal/genetic diversity. Interestingly, four tumors showed gain of 3q26.2, 5q14.3, 8q24.3, 8q22.3, 14q32.33 and loss/LOH in 9p21.3 while all TADE had LOH on 22q11.23. In addition, some genetic alterations progressed from NE through TADE into tumor in individual patients. Furthermore, no molecular event was identified that is common to all NE and/or TADE that progressed into tumor. This pilot study demonstrates the presence of genetic heterogeneity in oral tumorigenesis, and suggests that there might exist some common genetic alterations between tumors and TADE. However, this observation would need to be further investigated and validated in a larger cohort of oral cancer patients for its potential role in oral tumorigenesis.
The Globocan 2018 statistics reported 887,659 new cases of head and neck cancer (lip & oral cavity; 354,864, salivary gland; 52,799, oropharynx; 92,887, nasopharynx; 129,079, hypopharynx; 80,608 and larynx; 177,422 cases) with 453,307 deaths worldwide (http://gco.iarc.fr/today/fact-sheets-cancers). Significant genomic instability and resultant clonal diversity are hallmark characteristics of head and neck squamous cell carcinoma (HNSCC). Heterogeneity in phenotype, etiology, biology and clinical presentation are common features of HNSCC, and this may account for the dismal 5-year survival rates which are as low as 50% in all patients, despite treatment. Except for tumor human papilloma virus (HPV) status, the molecular risk factors investigated in HNSCC have yielded limited clinical utility. Risk stratification for HNSCC is based largely on the tumor anatomical site, stage and histological characteristics (1). Of interest is oral squamous cell carcinoma (OSCC) that develops through a multistep process involving the accumulation of multiple genetic mutations. This process is influenced both by genetic predisposition and environmental risk factors such as tobacco, alcohol, and HPV infection (2). Differences in tumor evolution and progression as well as resistance to therapy in oral cancer can be attributed to intratumoral clonal heterogeneity and intertumoralal clonal diversity, which are seen in clinical cases of oral cancer and other cancers of the head and neck region (1, 3–6). Understanding the molecular mechanisms of clonal heterogeneity within a tumor and clonal diversity between tumors may lead to better treatment outcomes in affected patients.
Field cancerization is to date the most acceptable molecular progression model for OSCC development. The model was proposed by Slaughter et al. and linked the presence of dysplastic changes in tumor-adjacent epithelium of oral cancer specimens with local recurrence and multifocal areas of cancer development process in many cells as a result of exposure to a carcinogen such as tobacco (7). A previous study in our group has also demonstrated this concept of field cancerization in OSCC with heterozygosity in p16 expression (8). Genetic alterations, mainly loss of heterozygosity (LOH) or deletion in chromosomal regions 9p (CDKN2A) (2, 9), 3p (FHIT & RSSFIA) (10, 11), and 17p (TP53) (2) have been observed in relatively high proportions of dysplastic lesions and are considered early events in oral carcinogenesis. Losses at 13q and 8p are observed more frequently in carcinomas than in dysplasia, and are associated with late stages of oral carcinogenesis (2, 12).
Inactivation of tumor suppressor genes (TSGs) by loss of heterozygosity (LOH) or deletion and/or the activation of oncogenes by gene amplification are the two major types of genetic alterations most often associated with OSCC tumorigenesis (1, 12, 13). In this study, a VELscope®Vx Handpiece (LED Dental Inc, Canada) was used to identify tumor-adjacent dysplastic epithelium (TADE) and normal epithelium (NE) surrounding a clinically detected cancer. The Affymetrix OncoScan® FFPE assay was then used to investigate the genetic alterations in these specimens with the aim of identifying common molecular events in all tumor, TADE and NE samples. Significant findings could be further explored in a larger cohort of oral cancer patients for their potential role in early to late stages of oral tumorigenesis.
Patients with OSSC were referred to the Pretoria Oral and Dental Hospital. Each patient was seen in a specialist clinic where the primary lesion was identified, and a scalpel biopsy was done for histological confirmation of the OSSC. Written informed consent was obtained from all five patients and the study was approved by the Faculty of Health Sciences Research Ethics Committee (Reference number 44/2010).
The VELscope®Vx Handpiece (LED Dental Inc, Canada) is a portable device with a 40–460 nm light source that has been shown to be effective at identifying lesions at risk of developing cancer in clinically normal mucosa (14). This device has a sensitivity of 98% and a specificity of 100% for identification of oral dysplasia and cancer (15). VELscope® is a diagnostic aid that when applied, results in pale green autofluorescence of normal mucosa and shows loss of fluorescence with abnormal tissue. This fluorescence is viewed through the narrow-band filter built into the eyepiece (16). It has also been shown to be effective at identifying lesions at risk of developing cancer in clinically normal mucosa as seen under normal light (14). One study showed that VELscope® is able to identify LOH at 3p and 9p in mucosa surrounding a clinically detected cancer, with margins as wide as 25 mm (17). Hence, each patient received a comprehensive intra-oral clinical examination with the use of the VELscope® to identify TADE and NE sites (Figure 1) for tissue sampling.
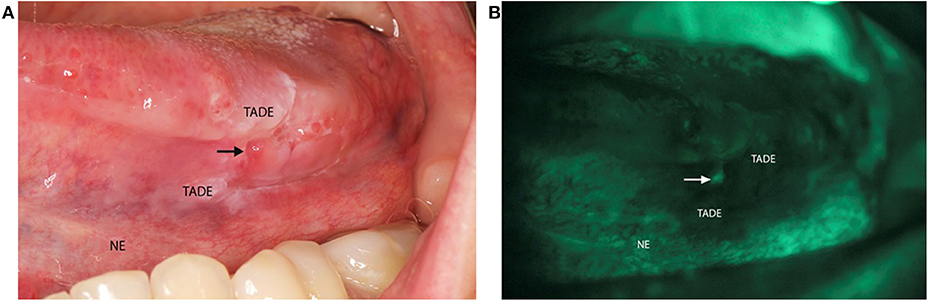
Figure 1. Clinical photo of VELscope® oral cancer screening of a patient. (A) Image of a patient with a squamous cell carcinoma on the left side of the tongue presenting as an ulcer (arrow). The tumor adjacent dysplastic epithelium (TADE) that showed loss of fluorescence with the VELscope® is present around the ulcer with the distant clinical normal epithelium (NE) that did not show loss of fluorescence. (B) Photograph of the VELscope® analysis showing the ulcer (arrow) with surrounding TADE with loss of autofluorescence and NE showing normal autofluorescence. (Number of patients screened = 5).
Formalin fixed paraffin embedded (FFPE) specimens of tumor, TADE and NE were prepared from hemi-glossectomy specimens (Figure 2) with different histological classification (Figure 3). Genomic DNA (gDNA) was extracted from each FFPE sample (tumor, TADE and NE) using the QIAamp DNA FFPE Tissue kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's protocol. DNA was quantified using a Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies) following the manufacturer's recommended protocol. Eighty nanogram gDNA was prepared for each sample and run on the Affymetrix OncoScan® FFPE assay kit (Affymetrix; Thermo Fisher Scientific company) according to the manufacturers' instructions (18). The Affymetrix OncoScan® FFPE assay is a molecular analytical tool that works efficiently on formalin-fixed paraffin-embedded (FFPE) samples. It provides a comprehensive coverage of whole genome copy number alterations, LOH and somatic mutations of genes that have been implicated in cancer and tumor progression by utilizing the molecular inversion Probe (MIP) technology (18). Briefly, to each 80 ng of gDNA sample, copy number variation and somatic mutations MIP mixes was added followed by denaturation for 5 min at 95°C and an overnight annealing for 17 h at 58°C. The product of each sample was split into two wells to which dATP (A) and dTTP (T) (A/T) was added into one well while dGTP (G) and dCTP (C) (G/C) was added to the other well to perform a gap fill reaction according to the manufacture's manual. A cocktail of exonucleases supplied with the OncoScan® kit was used to digest gDNA and uncircularized MIP. Cleavage enzymes supplied with the OncoScan® kit were then used to linearize the circular MIP that had been gap filled by A/T or G/C nucleotides followed by a PCR amplification. A second round of PCR amplification was then performed using Haeii enzyme supplied with the OncoScan® kit to cleave amplicons from the previous PCR reaction. The cleaved fragments were then hybridized onto the OncoScan® assay array overnight for 17 h according the manufacturer's protocol. The arrays were then washed and stained using the Affymetrix GeneChip® Fluidics Station 450 and scanned on the Affymetrix GeneChip® Scanner 3000 7G. Each scanned array generates array images known as a DAT file that were automatically converted into fluorescence intensity (CEL) files by the Affymetrix® GeneChip® Command Console® (AGCC) software version 4. CEL files were imported onto the Affymetrix OncoScanTM Console software version 1.3 and processed to generate OSCHP files which were analyzed using Chromosome Analysis Suite (ChAS) for copy number alterations (CNAs) and LOH in approximately 900 cancer genes as well as for 74 clinically actionable somatic mutations in nine cancer genes (BRAF, EGFR, IDH1, IDH2, KRAS, NRAS, PIK3CA, PTEN, and TP53) (18).
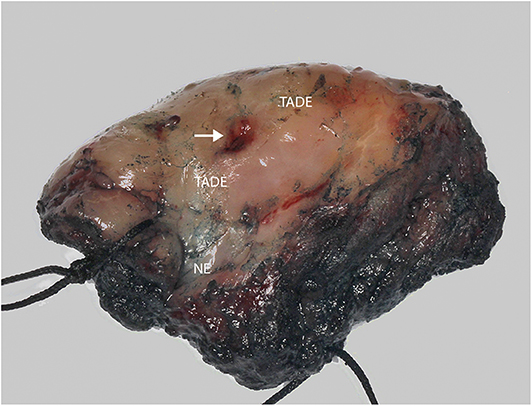
Figure 2. Photograph of hemi-glossectomy specimen. Image showing the carcinoma (arrow) as well as the TADE and NE areas (as determined previously using the VELscope®, number of patients screened = 5).
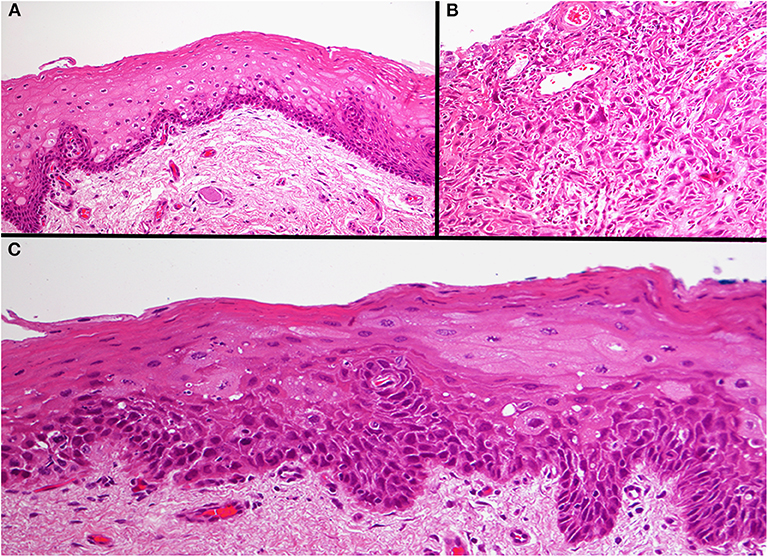
Figure 3. Photomicrograph of the three different areas. Histology of the normal epithelium showing basal cell hyperplasia with no atypia (A). The base of the tumor ulcer consisted of a poorly differentiated squamous cell carcinoma (B). The tumor adjacent dysplastic epithelium (TADE) that showed loss of fluorescence with the VELscope® showed the presence of moderate epithelial dysplasia (C). Original magnification X200.
Fifteen clinical specimens consisting of 5 tumors, 5 TADE and 5 distant NE from five oral cancer patients were examined in this pilot study for genetic changes such as copy number alterations (CNAs), loss of heterozygosity (LOH) and other somatic mutations (SMs). Of the five tumor samples, four were characterized by high CNAs (referred to as C class tumors) and showed a progressive development of genomic instability characterized by LOH and CNAs from NE through TADE to tumor. One patient's tumor was characterized by a high confidence somatic mutation with few CNAs (referred to as an M class tumor) (19) when compared to the TADE and NE in the same patient.
Tumor 1 showed a characteristic HNSCC mutation profile including the amplification of 3q26/28 (TP63, SOX2, PIK3CA), FGFR1, 11q13 (CCND1, FADD, CTTN), E2F1, PTK2, loss of NSD1 and LOH in CDKN2A and TP53 (20). The TADE and NE showed mutations in certain chromosomal regions that do not contain any of the well described HNSCC associated gene mutations. However, some observed molecular events such as the size of a Cn-LOH on 5q14.3 in NE, increased in TADE and changed to a CNL-LOH in the tumor. Also, Cn-LOH on 16q21 in NE and TADE changed to a CNL-LOH in the tumor. Extensive functional studies would be important to validate the significance of these two events in the development and progression of this tumor.
Tumor 2 is an “M” class tumor (tumor driven by mutation rather than CNAs) typified by a high confidence BRAF:p.G469A:c.1406G>C somatic mutation with less genomic instability compared to the TADE and NE. This somatic mutation resulted from the substitution of glycine (G) by alanine (A) at position 469 in BRAF. This mutation has been reported in non-small-cell lung cancer (21–23) and in colorectal cancer (24) but not in oral cancer. Loss in FHIT and RB1 were the only well described HNSCC associated mutations (25) detected in TADE and which were absent from both the tumor and NE, suggesting this TADE is an independent clone and not related to the tumor. Interestingly, the size of a loss on 3p26.3 in NE that increased in this TADE would be important to investigate further for possible role in early genetic event in the NE, since a loss in this region has been suggested to be an independent prognostic factor in OSCC patients (26, 27).
Tumor 3 showed mosaicism in CNAs which is indicative of a highly heterogenous tumor with multiple clones. This tumor showed mosaicism in the amplification of TP63, PIK3CA, SOX2, FADD, CTTN, CCND1, NOTCH1, E2F1, HRAS, BIRC2, EGFR, MYC, PTK2, loss in FHIT and CDKN2A, and LOH in NSD1, FAT1 and APC. All these mutations constitute a combination of distinct mutation profiles for various molecular sub-types of HNSCC of different etiology (1, 13, 25). Genetic alterations in TADE and NE did not involve any well described HNSCC associated mutations.
Tumor 4 showed amplification of PIKC3A, SOX2, TP63, EGFR, PTK2, NOTCH 1, 2 & 3, AJUBA, TRAF3, ERBB2, MYC, and KMT2D, loss of CDKN2A, FAT1, APC, RB1, KLK12, SMAD4, and PTEN, and LOH of FHIT, CDKN2A, CASP8, TP53 and NFE2L2, which have all been reported in HNSCC (1, 13). TADE and NE had no detectable HNSCC associated mutations. However, the size of a LOH at 3p21.31 in NE and TADE increased in the tumor. In addition, the size of a homozygous deletion of 19p13.2 in NE and TADE that increased in the tumor; this warrants further investigation as it may have played a role in oral cancer development and progression in this patient.
Tumor 5 had a high confidence PIK3CA:p.H1047R:c.3140A>G somatic mutation resulting from the substitution of histidine (H) by arginine (R) at position 1047 of the PIK3CA gene. This somatic mutation has previously been detected in HNSCC patients (28). This tumor also showed additional mutations such as amplification of TP63, SOX2, PIK3CA, FADD, CTTN, CCND1, EGFR, FGFR1, CASP8, SMAD4, TP53, NFE2L2, NOTCH1, 2,& 3, MYC, PTK2, AJUBA, TRAF3, ERBB2, NRAS, KRAS, HRAS, FAT1, KEAP1, E2F1, and SMAD2, loss of RASSF1, FHIT, CDKN2A, KMT2D, RB1, APC, CSMD1, PTPRD, MET, CUL3, and NSD1 and a LOH of TP53, NOTCH1, APC, RB1, CSMD1, PTPRD, CUL3, NSD1, CDKN2A, FHIT, and RASSF1, which have been reported in HNCSS (1, 13, 25). Mutations detected in TADE and NE were not those that have been well described to be associated with HNSCC.
We examined the mutation profile of the three groups of histologically identical sample types (tumor, TADE, and NE) from all five patients in an attempt to identify genetic alterations that are unique to each sample in the different groups (inter-patient heterogeneity) as well as those that are common to all samples in each group, which could potentially be explored in a larger cohort study of oral cancer patients for their clinical significance.
With the exception of tumor 2, all tumor samples showed characteristic mutations of HNSCC including amplification of 3q26.2 (TP63, SOX2, PIK3CA) (1), 5q14.3 (APC) (13, 29, 30), 8q24.3 (PTK2) (25), 8q22.3 (LRP12) (27) as well as loss and/or LOH of 9p21.3 (CDKN2A) (1, 31). Other characteristic HNSCC mutations such as loss of 4q35.1 (FAT1) (1, 31), 5q35.2 (NSD1) (1, 31), LOH of 17p13.2 (TP53) (1, 31), and amplification of 7p11.2 (EGFR) (1, 31), 8q24.21 (MYC) (1, 25, 32) and 11q13.3 (CCND1, FADD, CTTN) (1) were detected in three tumors. Interestingly, we also found genetic alterations not previously described in HNSCC such as amplification of 14q32.33 (LINC00221) and 20q11.22 in 4 tumors, and loss and/or LOH of 5q23.2, 5q35.1 and loss of 3p24.1 (NEK10) in three tumors. There were many other undescribed detectable genetic alterations present in at least 3 tumor samples (Table S1). Despite some common molecular features of HNSCC detected in all tumor samples, the type of alteration (gain, loss or LOH) detected in some of the affected chromosomal locations and in certain HNSCC associated genes, differ from one tumor to another, thereby making each tumor genetically distinct (Table 1).
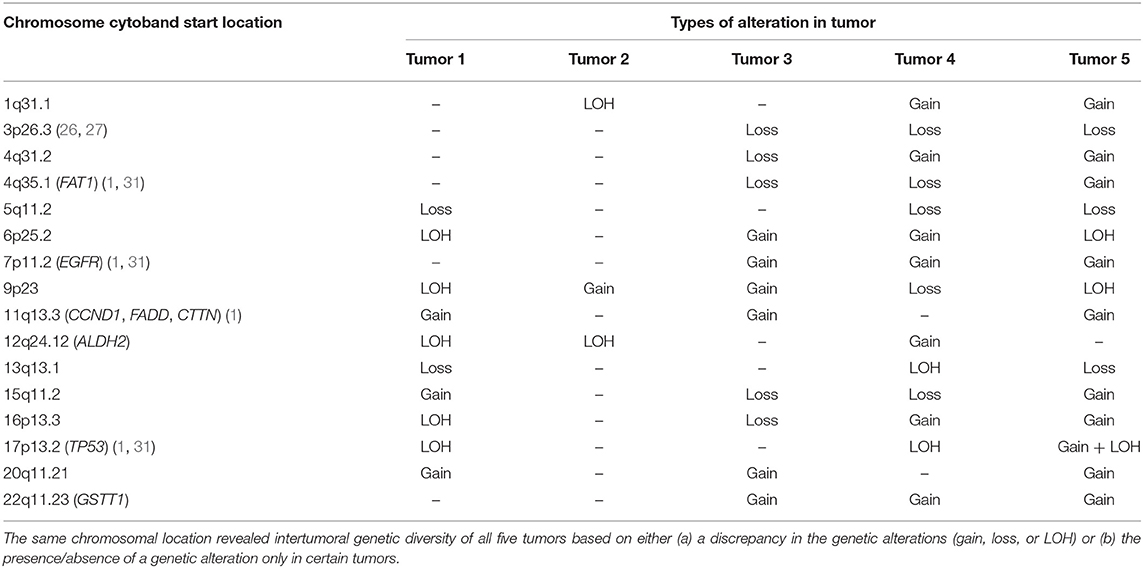
Table 1. Genetic alterations on the same chromosomal location common to at least three tumors.
Except for TADE 2, all TADE showed fewer CNAs compared to their respective tumor samples. The number of genetic alterations that were common to at least 3 TADE were fewer and different from those found among tumor samples (Table 2). Interestingly, a LOH event on 22q11.23 (GSTTP1, LOC391322, GSTT1, GSTTP2) was present in all 5 TADE. Other alterations detected in at least 3 TADE includes amplification of 1p13.3 (GSTM2, GSTM1, GSTM5), 14q32.33 (LINC00221) and LOH of 3p21.1 (NEK4) and 3p21.31. NEK4 has not been reported in HNSCC tumorigenesis and this gene is known to regulate cell entry into replicative senescence as well as the response to double strand DNA damage (33). Thus, LOH of NEK4 in OSCC should be functionally investigated further as it could be suggestive of a putative TSG in the development of dysplastic lesions in the oral mucosal. Genetic alterations of a specific type (gain, loss or LOH) in affected chromosomal locations/genes were consistent among TADE unlike in tumor samples. Notwithstanding the limited number of mutations found to be common among TADE, there were alterations that were present and/or absent in a subset of TADE thereby resulting in each patient's TADE having a unique genetic profile (Table 2).
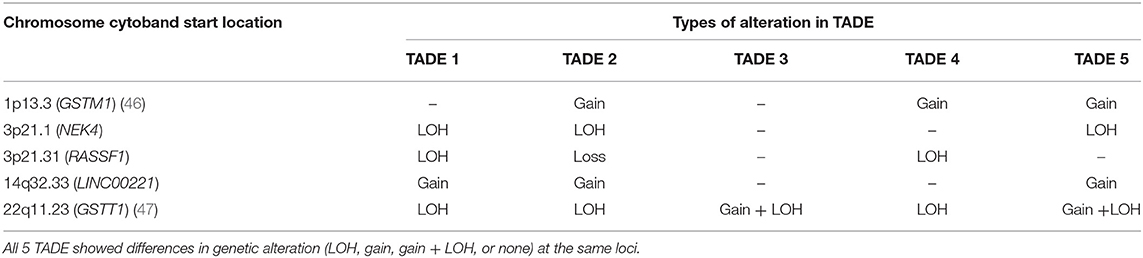
Table 2. Genetic alterations on the same chromosomal location common to at least two TADE.
In all patients, NE had fewer CNAs compared to their respective TADE. Only two genetic alterations were common to at least two out of the five NE (Table 3). Surprisingly, one of the genetic events—loss and/or LOH of 3p21.3—was common in all the five NE. Inactivating mutations in this region have been a consistent finding in cancers, especially HNSCC, and have been associated with early development of dysplastic lesions in HNSCC (13, 27, 34–43). The clinical significance of this genetic event in NE is unknown and warrants further investigation.
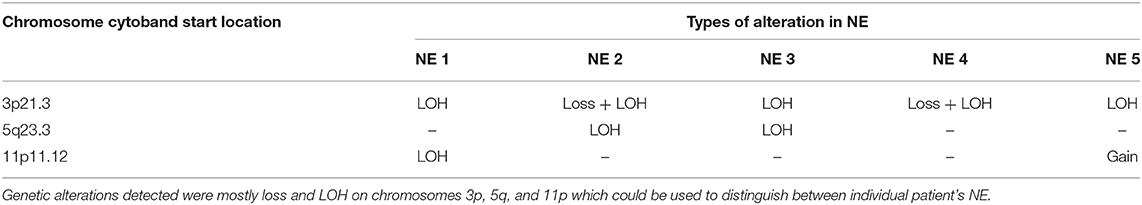
Table 3. Genetic alterations common to at least two NE.
We examined genetic alterations present in NE and/or TADE that were also found in tumor and as such could denote progression through clonal expansion from a single progenitor clone. Such progressive genetic alterations from NE and/or TADE to tumor could be suggestive of an early marker for oral cancer development and/or progression. We could not find a single genetic event that was common to all five NE and/or TADE which progressed to tumor. This demonstrates a high level of genetic diversity among all five patients' samples. Nevertheless, the activating mutation of the novel putative oncogene LINC00221 (14q32.33) and the inactivating mutation of the novel putative TSG NEK4 (3p21.1) that were detected in TADE1 & 5, were retained as they progressed into their respective tumors.
Genetic heterogeneity in HNSCC has been described in tumors from various anatomical sites analyzed using different techniques (3, 5, 44). This pilot study reports analysis of the genome of tumor, TADE and NE from five patients with OSCC with the aim of identifying molecular events in tumor, TADE and NE samples, which could be explored further in a larger cohort study of oral cancer patients for their significance in oral tumorigenesis. Tumor from each patient showed a unique interpatient mutation profile. Differences in the types of genetic alterations (gain, loss and/or LOH) at the same chromosomal location and in some HNSCC associated genes observed in tumor samples are indicative of intertumoral genetic diversity. Tumor 2 showed a high confidence BRAF:p.G469A:c.1406G>C somatic mutation (classified as “M” class tumor), which is the first to be reported in oral cancer. This tumor sample showed less genomic instability compared to TADE and NE. “M” class tumors have previously been observed in oral cancer (1, 19). Except for tumor 2, all tumors were characterized by high CNAs classified as “C” class tumors. Tumor 3 showed mosaicism in CNAs, which is indicative of intratumoral clonal heterogeneity. Lack of uniformity in the mutational landscape of all 5 tumors suggests intertumoral clonal/genetic diversity. Except for tumor 2, all tumors showed focal deletion of 3p and amplification of 5p and 8q which contain genes not previously described to be associated with HNSCC. Furthermore, amplification of 14q32.33 (LINC00221) and 20q11.22 in all 4 tumors and a loss and/or LOH of 5q23.2, 5q35.1 and 3p24.1 (NEK10) in three tumors are interesting findings which would require more detailed functional studies to evaluate their possible role as putative oncogenes and TSGs, respectively, in these regions. NEK10, not previously described in HNSCC tumorigenesis, has been reported to mediate G2/M cell cycle arrest (45) and could be a potential TSG in oral cancer.
Interestingly, TADE from all 5 patients showed inactivating mutations of 22q11.23 (GSTTP1, LOC391322, GSTT1, GSTTP2). The clinical significance of this genetic alteration in all five patients is unknown. However, given the presence of a traditional risk factor (tobacco consumption) in all patients, further studies are warranted.
Surprisingly, all five NE showed inactivating mutations of 3p21.3, known to contain TSGs or resident cancer genes. Alterations in this region are one of the most consistent genetic events reported not only in HNSCC but also in other cancer types (13, 27, 34–43). The presence of this genetic alteration in NE of all patients is therefore very surprising; could it possibly be that this particular region of the human genome is naturally very unstable? Further investigation of NE in both healthy individuals and oral cancer patients would be beneficial in understanding this genomic region and its associated genetic alterations.
Amplification of 3q26.2, 5q14.3, 8q24.3, 8q22.3, 14q32.33, 20q11.22 together with a loss/LOH on 9p21.3 was detected in four out of the five oral cancer samples, and a LOH on 22q11.23 detected in all five TADE. Furthermore, no molecular event was identified that is common to all NE and/or TADE that progressed into tumor. The small sample size limits the clinical significance of these findings. We therefore recommend further studies in a larger cohort of oral cancer patients to determine their significance in oral cancer biology.
The datasets generated for this study are available on request to the corresponding author.
The studies involving human participants were reviewed and approved by University of Pretoria Faculty of Health Sciences Research Ethics Committee (Reference number 44/2010). The patients/participants provided their written informed consent to participate in this study.
MA performed the OncoScan® assay, data analysis, interpretation, and wrote the first draft of the manuscript. AZ conceptualized the study, performed clinical examination of patients, the Velscope® screening, data interpretation, and edited the manuscript, MP contributed to the study design, data interpretation, edited the manuscript, and provided funding for the study. MH prepared FFPE specimen of all tissues, performed DNA isolation, and edited the manuscript. WH conceptualized the study, performed clinical examination of patients, data interpretation, edited the manuscript, and provided funding for the study.
This work was funded by the Cancer Association of South Africa (WFPvH) and the South African Medical Research Council (MSP—Flagship and Extramural Stem Cell Unit).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the five oral cancer subjects that consented and participated in this study and Izak Storm at BIOCOM Africa and Jo Mcbride at the Centre for Proteomic & Genomic Research, Cape Town for their technical support with the OncoScan® assay.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00683/full#supplementary-material
1. The Cancer Genome Atlas N, Lawrence MS, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. (2015) 517:576. doi: 10.1038/nature14129
2. Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. (1996) 56:2488–92. doi: 10.1016/S0194-5998(96)80631-0
3. Mroz EA, Rocco JW. Intra-tumor heterogeneity in head and neck cancer and its clinical implications. World J Otorhinolaryngol Head Neck Surg. (2016) 2:60–7. doi: 10.1016/j.wjorl.2016.05.007
4. Zhang XC, Xu C, Mitchell RM, Zhang B, Zhao D, Li Y, et al. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia. (2013) 15:1371–8. doi: 10.1593/neo.131400
5. Sarode G, Sarode SC, Tupkari J, Patil S. Is oral squamous cell carcinoma unique in terms of intra- and inter-tumoral heterogeneity? Transl Res Oral Oncol. (2017) 2:2057178X17703578. doi: 10.1177/2057178X17703578
6. Klussmann JP. Head and neck cancer - new insights into a heterogeneous disease. Oncol Res Treat. (2017) 40:318–9. doi: 10.1159/000477255
7. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. (1953) 6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q
8. Ambele MA, Pepper MS, van Heerden MB, van Heerden WFP. Heterozygosity of p16 expression in an oral squamous cell carcinoma with associated loss of heterozygosity and copy number alterations. Head Neck. (2019) 41:E62–5. doi: 10.1002/hed.25566
9. Mao L, Lee JS, Fan YH, Ro JY, Batsakis JG, Lippman S, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med. (1996) 2:682–5. doi: 10.1038/nm0696-682
10. Masayesva BG, Ha P, Garrett-Mayer E, Pilkington T, Mao R, Pevsner J, et al. Gene expression alterations over large chromosomal regions in cancers include multiple genes unrelated to malignant progression. Proc Natl Acad Sci USA. (2004) 101:8715–20. doi: 10.1073/pnas.0400027101
11. Garnis C, Baldwin C, Zhang L, Rosin MP, Lam WL. Use of complete coverage array comparative genomic hybridization to define copy number alterations on chromosome 3p in oral squamous cell carcinomas. Cancer Res. (2003) 63:8582−5.
12. Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dental Res. (2008) 87:14–32. doi: 10.1177/154405910808700104
13. Kasamatsu A, Uzawa K, Usukura K, Koike K, Nakashima D, Ishigami T, et al. Loss of heterozygosity in oral cancer. Oral Sci Int. (2011) 8:37–43. doi: 10.1016/S1348-8643(11)00027-9
14. Poh CF, Ng SP, Williams PM, Zhang L, Laronde DM, Lane P, et al. Direct fluorescence visualization of clinically occult high-risk oral premalignant disease using a simple hand-held device. Head Neck. (2007) 29:71–6. doi: 10.1002/hed.20468
15. Lane PM, Gilhuly T, Whitehead P, Zeng H, Poh CF, Ng S, et al. Simple device for the direct visualization of oral-cavity tissue fluorescence. J Biomed Opt. (2006) 11:024006. doi: 10.1117/1.2193157
16. Lingen MW, Kalmar JR, Karrison T, Speight PM. Critical evaluation of diagnostic aids for the detection of oral cancer. Oral Oncol. (2008) 44:10–22. doi: 10.1016/j.oraloncology.2007.06.011
17. Poh CF, Zhang L, Anderson DW, Durham JS, Williams PM, Priddy RW, et al. Fluorescence visualization detection of field alterations in tumor margins of oral cancer patients. Clin Cancer Res. (2006) 12:6716–22. doi: 10.1158/1078-0432.CCR-06-1317
18. Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, et al. Cross-laboratory validation of the OncoScan(R) FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genom. (2015) 8:5. doi: 10.1186/s12920-015-0079-z
19. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. (2013) 45:1127–33. doi: 10.1038/ng.2762
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
21. Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. (2013) 19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657
22. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
23. Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. (2011) 29:2046–51. doi: 10.1200/JCO.2010.33.1280
24. Rizzo S, Bronte G, Fanale D, Corsini L, Silvestris N, Santini D, et al. Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer Treat Rev. (2010) 36 (Suppl. 3):S56–61. doi: 10.1016/S0305-7372(10)70021-9
25. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. (2011) 11:9–22. doi: 10.1038/nrc2982
26. Uchida K, Oga A, Nakao M, Mano T, Mihara M, Kawauchi S, et al. Loss of 3p26.3 is an independent prognostic factor in patients with oral squamous cell carcinoma. Oncol Rep. (2011) 26:463–9. doi: 10.3892/or.2011.1327
27. Vincent-Chong VK, Salahshourifar I, Woo KM, Anwar A, Razali R, Gudimella R, et al. Genome wide profiling in oral squamous cell carcinoma identifies a four genetic marker signature of prognostic significance. PLoS ONE. (2017) 12:e0174865. doi: 10.1371/journal.pone.0174865
28. Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. (2013) 73:276–84. doi: 10.1158/0008-5472.CAN-12-1726
29. Largey JS, Meltzer SJ, Sauk JJ, Hebert CA, Archibald DW. Loss of heterozygosity involving the APC gene in oral squamous cell carcinomas. Oral Surg Oral Med Oral Pathol. (1994) 77:260–3. doi: 10.1016/0030-4220(94)90295-X
30. Rivero ER, Horta MC, Silva Guerra EN, Ferraz AR, Nunes FD. Loss of heterozygosity of the APC gene in oral squamous cell carcinoma. Pathol Res Pract. (2008) 204:793–7. doi: 10.1016/j.prp.2008.05.003
31. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. (2018) 18:269–82. doi: 10.1038/nrc.2018.11
32. Rodrigo JP, Lazo PS, Ramos S, Alvarez I, Suarez C. MYC amplification in squamous cell carcinomas of the head and neck. Arch Otolaryngol Head Neck Surg. (1996) 122:504–7. doi: 10.1001/archotol.1996.01890170038008
33. Nguyen CL, Possemato R, Bauerlein EL, Xie A, Scully R, Hahn WC. Nek4 regulates entry into replicative senescence and the response to DNA damage in human fibroblasts. Mol Cell Biol. (2012) 32:3963–77. doi: 10.1128/MCB.00436-12
34. Ghosh S, Ghosh A, Maiti GP, Alam N, Roy A, Roy B, et al. Alterations of 3p21.31 tumor suppressor genes in head and neck squamous cell carcinoma: Correlation with progression and prognosis. Int J Cancer. (2008) 123:2594–604. doi: 10.1002/ijc.23834
35. Angeloni D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief Funct Genom Proteom. (2007) 6:19–39. doi: 10.1093/bfgp/elm007
36. Marsit CJ, Hasegawa M, Hirao T, Kim DH, Aldape K, Hinds PW, et al. Loss of heterozygosity of chromosome 3p21 is associated with mutant TP53 and better patient survival in non-small-cell lung cancer. Cancer Res. (2004) 64:8702–7. doi: 10.1158/0008-5472.CAN-04-2558
37. Wang K, Ling T, Wu H, Zhang J. Screening of candidate tumor-suppressor genes in 3p21.3 and investigation of the methylation of gene promoters in oral squamous cell carcinoma. Oncol Rep. (2013) 29:1175–82. doi: 10.3892/or.2012.2213
38. Dasgupta S, Chakraborty SB, Roy A, Roychowdhury S, Panda CK. Differential deletions of chromosome 3p are associated with the development of uterine cervical carcinoma in Indian patients. Mol Pathol. (2003) 56:263–9. doi: 10.1136/mp.56.5.263
39. Martin CL, Reshmi SC, Ried T, Gottberg W, Wilson JW, Reddy JK, et al. Chromosomal imbalances in oral squamous cell carcinoma: examination of 31 cell lines and review of the literature. Oral Oncol. (2008) 44:369–82. doi: 10.1016/j.oraloncology.2007.05.003
40. Hogg RP, Honorio S, Martinez A, Agathanggelou A, Dallol A, Fullwood P, et al. Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer. (2002) 38:1585–92. doi: 10.1016/S0959-8049(01)00422-1
41. Maestro R, Gasparotto D, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Three discrete regions of deletion at 3p in head and neck cancers. Cancer Res. (1993) 53:5775–9.
42. Chakraborty SB, Sabbir MG, Roy A, Sengupta A, Panda CK. Multiple deletions in chromosome 3p are associated with the development of head and neck squamous cell carcinoma. Int J Human Genet. (2003) 3:79–87. doi: 10.1080/09723757.2003.11885832
43. Chakraborty SB, Dasgupta S, Roy A, Sengupta A, Ray B, Roychoudhury S, et al. Differential deletions in 3p are associated with the development of head and neck squamous cell carcinoma in Indian patients. Cancer Genet Cytogenet. (2003) 146:130–8. doi: 10.1016/S0165-4608(03)00127-4
44. Mroz EA, Tward AD, Hammon RJ, Ren Y, Rocco JW. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. (2015) 12:e1001786. doi: 10.1371/journal.pmed.1001786
45. Moniz LS, Stambolic V. Nek10 mediates G2/M cell cycle arrest and MEK autoactivation in response to UV irradiation. Mol Cell Biol. (2011) 31:30–42. doi: 10.1128/MCB.00648-10
46. Zhang X, Huang M, Wu X, Kadlubar S, Lin J, Yu X, et al. GSTM1 copy number and promoter haplotype as predictors for risk of recurrence and/or second primary tumor in patients with head and neck cancer. Pharmacogenom Personal Med. (2013) 6:9–17. doi: 10.2147/PGPM.S35949
Keywords: oral squamous cell carcinomas, head and neck cancer, genomic heterogeneity, intratumor clonal heterogeneity, intertumoral clonal diversity, OncoScan® FFPE assay, VELscope®Vx device
Citation: Ambele MA, van Zyl A, Pepper MS, van Heerden MB and van Heerden WFP (2020) Amplification of 3q26.2, 5q14.3, 8q24.3, 8q22.3, and 14q32.33 Are Possible Common Genetic Alterations in Oral Cancer Patients. Front. Oncol. 10:683. doi: 10.3389/fonc.2020.00683
Received: 04 October 2019; Accepted: 14 April 2020;
Published: 30 April 2020.
Edited by:
Victor C. Kok, Asia University, TaiwanReviewed by:
Anu R. I., MVR Cancer Centre and Research Institute, IndiaCopyright © 2020 Ambele, van Zyl, Pepper, van Heerden and van Heerden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melvin A. Ambele, bWVsdmluLmFtYmVsZUB1cC5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.