 Praveen Vikas
Praveen Vikas Nicholas Borcherding
Nicholas Borcherding Adithya Chennamadhavuni
Adithya Chennamadhavuni Rohan Garje
Rohan Garje
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 28 April 2020
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00570
Immunotherapy has revolutionized the treatment of both hematological malignancies and solid tumors. The use of immunotherapy has improved outcome for patients with cancer across multiple tumor types, including lung, melanoma, ovarian, genitourinary, and more recently breast cancer with durable responses seen even in patients with widespread metastatic disease. Despite the promising results, immunotherapy still helps only a subset of patients due to overall low response rates. Moreover, the response to immunotherapy is highly cancer specific and results have not been as promising in cancers that are considered less immunogenic. The strategies to improve immunotherapy responses have focused on biomarker selection, like PD-L1 status, and usage of combinatorial agents, such as chemotherapy, targeted therapy, and radiotherapy. Of particular interest, DNA-damaging agents have the potential to enhance the response to immunotherapy by promoting neoantigen release, increasing tumor mutational burden, and enhancing PD-L1 expression. Poly-ADP-ribose polymerase (PARP) inhibitors are one such class of drugs that has shown synergy with immunotherapy in preclinical and early clinical studies. PARP-based therapies work through the inhibition of single-strand DNA repair leading to DNA damage, increased tumor mutational burden, making the tumor a more attractive target for immunotherapy. Of the solid tumors reviewed, breast, ovarian, and prostate cancers have demonstrated efficacy in the combination of PARP inhibition and immunotherapy, predominately in BRCA-mutated tumors. However, initial investigations into wildtype BRCA and gastrointestinal tumors have shown moderate overall response or disease control rates, dependent on the tumor type. In contrast, although a number of clinical trials underway, there is a paucity of published results for the use of the combination in lung or urothelial cancers. Overall this article focuses on the promise of combinatorial PARP inhibition and immunotherapy to improve patient outcomes in solid tumors, summarizing both early results and looking toward ongoing trials.
A renaissance of cancer immunotherapy is currently underway for clinicians, researchers, and patients. Broadly speaking, cancer immunotherapy can be thought of as the selected manipulation of the balance between pro-tumor growth inflammation and anti-tumor immune responses (1). A number of different methodologies exist to shift the balance and promote anti-tumor immune response, most notably is immune checkpoint blockade, which act to remove the inhibition of anti-tumor lymphocytes (2). Checkpoint inhibitors (anti-PD-1/PD-L1 and CTLA-4 antibodies) are approved as single agent therapy and in combination with chemotherapy for a variety of cancers (3). Other mechanisms include targeted agents, personalized vaccines, T cell therapies, and dendritic-cell and non-specific agents, likes oncolytic viral therapy and modulators of the tumor microenvironment. Together, these immunotherapies may offer a chance at robust and durable responses for patients, however for the majority of non-immunogenic solid tumors, clear improvements in outcomes are still wanting (4).
PARP inhibitors are more effective in tumors with existing defects in DNA damage repair (DDR), particularly, BRCA1/2 mutations (5). In addition, PARP inhibitors are known to be more effective in tumors carrying somatic mutations in other DNA repair genes, including ATM, ATR, BARD1, BRIP1, CHK1, CHK2, PALB2, RAD51, and FANC, with defects in these genes might exhibit a phenotype similar to BRCAness (6). Functionally, as a DNA damage sensor, PARP enzymes are rapidly recruited to sites of single and double-stranded DNA damage (Figure 1A) (5). Binding to the DNA alters the catalytic domains of the PARP enzymes, leading to the production of ADP-ribose moieties (7, 8). The extension of the poly-ADP-ribose can serve multiple functions, including (1) recruiting effector proteins for DNA repair, (2) interfering with post-translationally modified chromatin proteins, like histones, and (3) can act as an energy sink for NAD+ molecules leading to cell death (5). PARP inhibition is not only thought to block proper DNA repair, but the inhibition can act to trap PARP at the replication fork, preventing transcription or translation (Figure 1A) (9).
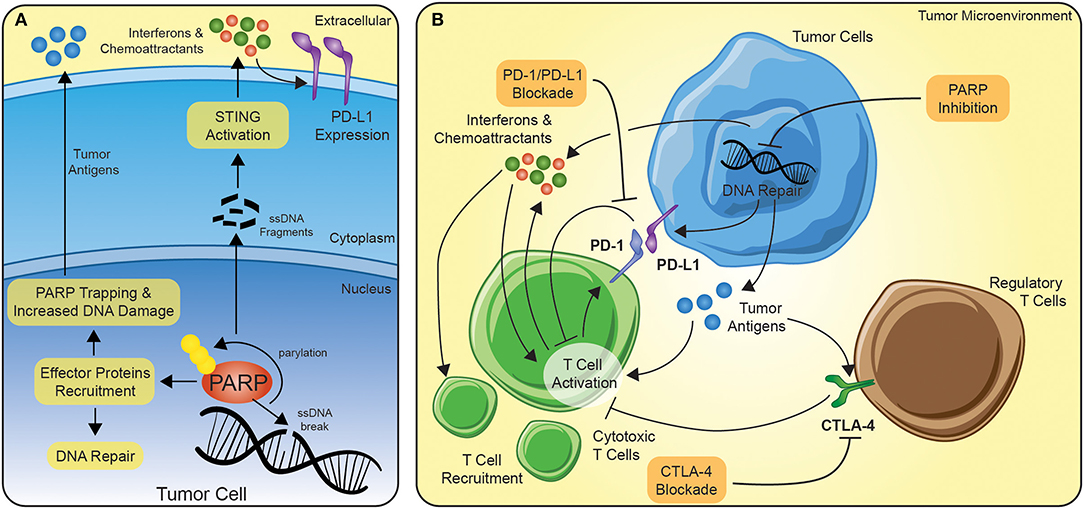
Figure 1. The potential synergy between PARP inhibition and immune checkpoint blockade. (A) In the context of PARP inhibition, tumor cells develop a more immunogenic repertoire of tumor antigens and release interferons/chemoattractants to serve as signals for immune cells. Interferons amplifies the immune activation of T cells and lead to further recruitment of T cells. (B) Immune checkpoint blockade can function on several levels, but principally the FDA-approved agents are directed at PD-1/PD-L1 or CTLA-4. PD-1 is transcriptionally upregulated on activating T cells and serve as a negative regulator of response. By disrupting the connection of PD-1 to PD-L1 expressed on tumor or stromal cells in the tumor microenvironment, the anti-tumor immune response can be sustained. Similarly, CTLA-4 is translocated to the cell surface of activating T cells or constitutively expressed on the cell surface of the suppressive regulatory T cells and dampens activation via the sequestration of co-stimulatory signals.
In cancer cells with defective homologous recombination repair (HRR) and double-stranded DNA repair, PARP inhibition can lead to synthetic lethality, a combinatorial effect caused by deficiencies in multiple pathways (10). Most notably, this approach led to the recent Food and Drug Administration (FDA) approval of PARP inhibition for metastatic breast cancer (11) and as maintenance therapy for ovarian cancers (12), for individuals with germline mutations in BRCA1 or BRCA2. Unlike the implicated role of PARP inhibition in facilitating cell death in cancer cells, new evidence is emerging in which PARP inhibition can enhance the response of immune checkpoint inhibitors (13, 14). PARP inhibition leads to the accumulation of DNA damage and by increasing the amount of DNA in the cytosol, PARP inhibition may trigger the interferon pathways (Figure 1A) (15, 16). Thus, not only can PARP inhibition lead to the accumulation of neoantigens for the anti-tumor immune response, but through the upregulation of interferons, the inhibition may act to prime the tumor microenvironment to facilitate a more profound anti-tumor immune response (Figure 1B). In addition, interferons have been shown to also increase expression of targets of immune blockade, like PD-L1, which suggests a further potential synergy between PARP inhibition and immune checkpoint blockade (17–19). Others have reported PARP-inhibitor-mediated upregulation of PD-L1 via interferon-independent mechanisms (20). This synergy may be the underlying mechanism of the marked preclinical responses seen using CTLA-4 and PARP inhibition in BRCA1-deficient tumors (21).
There exists a strong interplay among host immune system, DNA damage, and inflammation in the tumor microenvironment. Chronic PARP inhibition leads to sustained DNA damage resulting into epigenetic changes on tumor cells, hence making them more vulnerable to T cells and NK cells and finally resulting into increased intrinsic immunogenicity of tumor cells (22). Similarly, combinatorial trials utilizing epigenetic modulators, may increase therapeutic response by altering the expression of DNA repair genes (23). These strategies act to drive the balance of the tumor microenvironment toward anti-tumor immune response by recruiting immune and inflammatory cells while enhancing extrinsic immunogenicity. Combination of DNA damaging agents, like PARP inhibitors, and immune checkpoint blockers, may act as the key in bridging the potential of immunotherapies to a broader number of patients and tumors (Figure 1).
Ovarian cancers represent a heterogenous group of solid tumors, that have had no major change in survival rates since the introduction of platinum agents (24). For example, relatively recent approval of VEGF inhibitor, like bevacizumab, with the standard carboplatin and paclitaxel chemotherapy was based on the demonstrated increased in progression-free survival improvement of 1.5 months (25). Part of the complications for the treatment of ovarian tumors vague constellation of symptoms leading to a late diagnosis (70% are stage III or greater) (26). Single allele hereditary mutations in DNA damage recognition or repair genes account for 24% of ovarian cancers (27). Of the one in five ovarian cancers associated with germline mutations, 65–85% are associated with germline BRCA mutations (28). Since the approval of PARP inhibitors in 2014 as a second line therapy for ovarian cancers (29), PARP inhibition has also been approved for maintenance therapy after response to platinum-based agents (30, 31). Phase III for the use of the combination as a maintenance therapy (NCT02477644) showed improvement in progression-free survival from 22.7 to 24.0 months compared to PARP inhibition alone (32). Additionally, the use of PARP inhibitors with bevacizumab in clinical trials (NCT02354131) have shown an impressive preliminary disease control rate of 92% in 12 patients with BRCA2-mutated ovarian cancers (33).
A number of clinical trials have examined or are currently investigating the synergistic effects of PARP inhibition and immune checkpoint blockade in ovarian cancer (Table 1). For example, the phase Ib basket study of the combination of anti-PD-1 antibody (BGB-A317) with PARP inhibitor (BGB-290) is currently underway for advanced solid tumors without BRCA mutation specifications. In the 34 patients which were treated, seven achieved a partial response (PR) and two women with ovarian cancer achieved a complete response (CR) (34). In the context of patients with known mutations in BRCA1 or BRCA2, the phase II basket study (MEDIOLA) evaluated combination of durvalumab (anti-PD-L1) and olaparib in 32 patients with platinum-sensitive relapsed ovarian cancer (21). The combination of PARP and PD-L1 inhibition showed an overall response rate (ORR) of 63% (6 CR and 14 PR) and a 12-week disease control rate (DCR) of 81%. This combination was well-tolerated with grade 3–4 AEs of anemia (9%), increased pancreatic enzymes (6–9%), and neutropenia (3%). The larger TOPACIO trial showed more modest results in the evaluation of pembrolizumab (anti-PD-1) and niraparib in ovarian and triple-negative breast (TNBC) tumors with BRCA mutations vs. wild-type (wt) BRCA1/2 (35). Across all 60 patients, there was an ORR of 25% and a DCR of 68%, with nearly a third of platinum-resistant ovarian cancers responding. Among the BRCA-mutated tumors, both ORR and DCR were elevated to 45 and 73%, respectively (35). Future trials, such as the ATHENA trial (Table 1), are evaluating the use of nivolumab (anti-PD-1) and rucaparib as maintenance following response to upfront platinum-based therapy in stage III/IV ovarian cancer (NCT03522246). The four arms will include: (1) maintenance nivolumab plus rucaparib, (2) nivolumab plus placebo, (3) rucaparib plus placebo, or (4) placebo alone. Likewise, the combination of PARP inhibition and checkpoint blockade with VEGF inhibition is being evaluated in several phase II trials (Table 1).
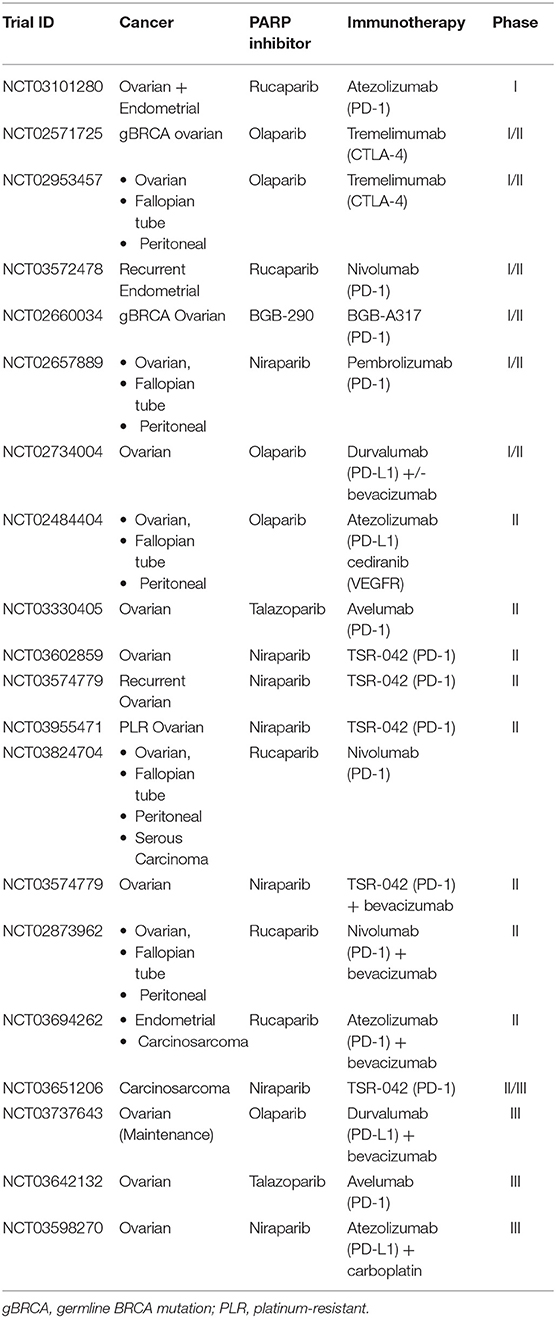
Table 1. Active combinatorial trials in gynecologic cancers.
Breast cancer can be divided clinically by the expression of estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2). These receptors not only inform therapeutic options for a patient, such as selective estrogen modulators, but can act as biomarkers that predict disease course. The absence of these three receptors, a subtype of breast cancer referred to as TNBC, represents a poor prognosis for the respective 15–20% of breast cancers (36). Interestingly, 70–80% of TNBC are referred to as basal-like are defined by expression of basal cell markers and high levels of genomic instability, for example, p53 pathway alterations seen in 85–95% of tumors (36). This genomic instability in basal-like breast cancer (BLBC) is compounded by the characteristic loss in chromosome 5q, consisting of a number of DNA repair genes (36, 37). In addition, 15–20% of BLBC possess mutations in BRCA1 or BRCA2 (36, 38–40). Half of the 15–20% of BLBC tumors with BRCA1 or BRCA2 are a result of germline mutations (41, 42).
Breast cancer with germline mutations in BRCA1 or BRCA2 have been a focus of therapeutic targeting. Monotherapy PARP inhibitors have shown improved responses in germline BRCA-mutated breast cancer compared to conventional chemotherapy. The high ORR of 62.6% with talazoparib vs. control ORR of 27.2% in the phase III EMBARCA trial (NCT01945775) led to the FDA approval of the inhibitor for locally-advanced or metastatic HER2-negative breast cancer with germline mutations in BRCA1/2 (43). Similarly, olaparib was FDA approved based on the results of the OlympiAD trial (NCT02000622) where the response rate was 59.9% in the olaparib group and 28.8% in the standard-therapy group (11). However, germline BRCA mutations only account for 8–10% of BLBC, with questionable efficacy for the majority of breast cancers without said germline mutations (36). Similarly, despite the increase immune infiltration of TNBC/BLBC compared to other breast cancers, the use of single-agent immune checkpoint blockade across all TNBC has found a wide range of objective responses from 5.4 to 33% (44). Thus, the combination of immunotherapy and PARP inhibition is currently being investigated (Table 2) for the promise of synergistic response in wild-type and mutant BRCA breast cancers (44).
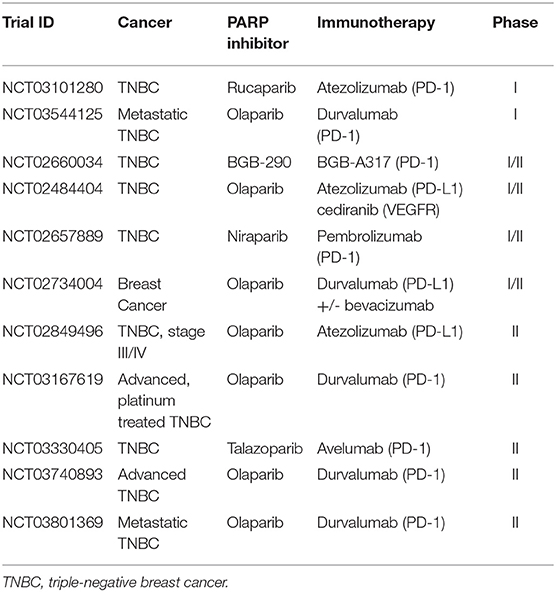
Table 2. Active combinatorial trials in breast cancer.
As theorized, the combination of PARP inhibitor and immunotherapy has yielded promising initial response rates in both BRCA1/2 mutated and BRCA wild-type (BRCAwt) patients. The phase I study evaluating durvalumab (anti-PD-L1 antibody) in combination with olaparib in 12 patients (10 with ovarian cancer and two with TNBC) with 11 of which were BRCAwt, two women achieved PR and eight women had stable disease, achieving an 83% DCR (45). The MEDIOLA phase I/II trial evaluated durvalumab and olaparib combination as a first- or second-line therapy. In 32 patients with germline BRCA mutations, the combinatorial therapy had an ORR of 53% and a 12-weeks DCR of 47% with minimal adverse events (46). These promising results are leading to the expansion of treatment arms in the MEDIOLA trial, especially with the expansion of BRCAwt TNBC patients (47, 48). Similar response was seen in the KEYNOTE-162/TOPACIO trial evaluating pembrolizumab and niraparib with an ORR of 29% patients, eight of the 12 patients with germline BRCA mutations. More intriguingly, ORR was 14.3–19.2% in BRCAwt TNBC, with results still forthcoming (49). This study also found, regardless of BRCA status, an ORR in 33% of PD-L1+ compared to 15% of PD-L1- tumors, suggesting PD-L1 may serve as a proxy marker for the combinatorial therapy, similar to the use in immune checkpoint blockade monotherapy (4, 49).
Recent investigations of urothelial cancers have demonstrated pathogenic or likely pathogenic mutations in DNA repair in up to 15% of urothelial cancers (50, 51). Nearly a third of these mutations were seen in DNA mismatch repair (50). In a phase II clinical trial, the safety and antitumor activity of rucaparib was evaluated in patients with metastatic urothelial cancer irrespective HRR status. However, the study was closed prematurely as there was no adequate clinical response as determined by the independent data monitoring committee. Additional studies with rucaparib and olaparib are planned in a cohort selected for HRR mutations.
The potential synergy and efficacy of combination of PARP inhibitors and checkpoint inhibitors in urothelial cancers are currently being evaluated in multiple clinical trials. One such study is the BISCAY trial, a multi-arm, multi-drug, open-label, phase Ib study with one arm evaluating the safety and tolerability of olaparib as a monotherapy, or in association with durvalumab (anti-PD-L1), for the treatment of metastatic urothelial cancer in patients who have progressed on prior treatment and possess defects in DNA-repair genes (NCT02546661). A similar combination is evaluated in a phase II clinical trial as first-line for platinum-ineligible metastatic urothelial cancer (BAYOU, NCT03459846). Safety and efficacy of these approaches are yet to be reported (Table 3).
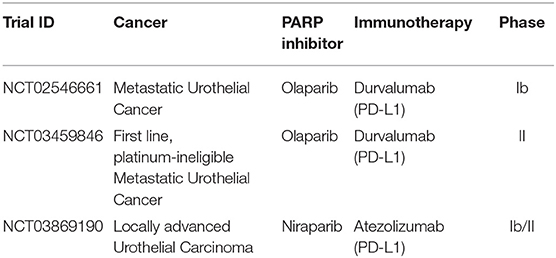
Table 3. PARPi and immunotherapy combination trials in urothelial cancer.
The prevalence of mutations in the DNA repair genes involved in HRR in men with prostate cancer irrespective of age or family history has been estimated at 11–23% (52). In the Trial of PARP Inhibition in Prostate Cancer (TOPARP-A) study, olaparib monotherapy had a composite response (defined as either objective response as per RECIST 1.1 with modified PCWG2 recommendations, a decrease in PSA of ≥ 50%, or a circulating tumor cell count conversion from ≥5 cells per 7·5 mL blood at baseline to <5 cells per 7·5 mL blood) of 88% with a median survival of 13.8 months in a small cohort of heavily pretreated patients with metastatic castrate-resistant prostate cancer (mCRPC) with HRR gene mutation (53). In another study (TOPARP-B), 98 patients with mCRPC with DNA repair gene mutations and prior treatment with chemotherapy and novel antiandrogen therapy were randomized to receive treatment with either olaparib 400 mg BID (Ola400) or olaparib 300 mg BID (Ola 300). The composite response was seen in 54·3 and 39.1% in the Ola400 and Ola300 cohorts, respectively. The radiologic responses were seen predominantly in patients with BRCA 1/2 when compared to ATM and CDK12 gene mutations (54). The preliminary results of the phase III, randomized PROfound study evaluating the efficacy of olaparib in comparison to physician choice novel antiandrogen (pcNHA) in mCRPC with HRR mutations, were presented at the ESMO conference in 2019. In this study, an objective response rate (based on RECIST v1.1 + PCWG3) of 33% was seen in patients with BRCA1, BRCA2, or ATM mutations and treated with olaparib in comparison to pcNHA the ORR was 2.3%. When the response was assessed with the inclusion of other HRR gene mutations, the ORR with olaparib was 21.7%, suggesting differential response with BRCA vs. non-BRCA mutations. The radiographic PFS for those treated with olaparib was 5.82 months, and with pcNHA, it was 3.52 months. At the data cut off, the overall survival was 17.51 and 14.26 months, respectively. The most common adverse events with olaparib monotherapy were anemia, nausea, decreased appetite, and fatigue (55).
The efficacy of niraparib in patients with mCRPC after prior novel hormonal agent and chemotherapy was evaluated in the GALAHAD clinical trial (56). Patients were enrolled based on the presence of HRR gene mutations. In the preliminary results report at the ESMO conference in 2019, 46 patients had BRCA 1/2, and 35 had non-BRCA mutations. The objective response rates were high with BRCA 1/2 when compared to non-BRCA mutations, 41 vs. 9%, respectively. The median PFS in the BRCA 1/2 and non-BRCA cohorts were 8.2 and 5.3 months, respectively. The most common grade 3–4 side effects were myelosuppression, asthenia, and back pain.
The safety and antitumor activity of immunotherapy with olaparib was evaluated in early phase studies. In a phase II, open-label study of 17 patients with mCRPC regardless of HRR mutation status, the combination of durvalumab and olaparib was evaluated (57). Of the 17 patients, 9 had a prostate-specific antigen (PSA) decline of ≥50%, and four had a radiographic response by RECIST v.1.1 criteria. Genomic analysis of the responding patients showed 4 with germline and 2 with bi-allelic somatic alterations in BRCA2 (58). Additionally, one responder had a monoallelic loss of PMS2, a mismatch repair gene, and with a separate responder having a monoallelic loss of BRCA2. The remaining two responding patients had no predisposing gene alterations detected. The most common grade 3 or 4 adverse events were myelosuppression, infection, and nausea (57, 58). In another study, Yu et al. evaluated the efficacy of pembrolizumab along with olaparib in a cohort of 41 patients with mCRPC and HRR wild-type (59). This combination showed a partial response of 7% and disease control rate (DCR) of 29% as per the RECIST v1.1 criteria. The median OS was 14 months with the most common adverse events being anemia, fatigue, and nausea.
PARP inhibitor monotherapy in prostate cancer with HRR gene mutations, especially BRCA1/2, has shown significant antitumor activity. The hypothesis for combining immunotherapy with PARP inhibitors is plausible; however, the data on safety and antitumor activity are still in the early stages. Numerous combination trials to address the safety and efficacy are ongoing (Table 4).
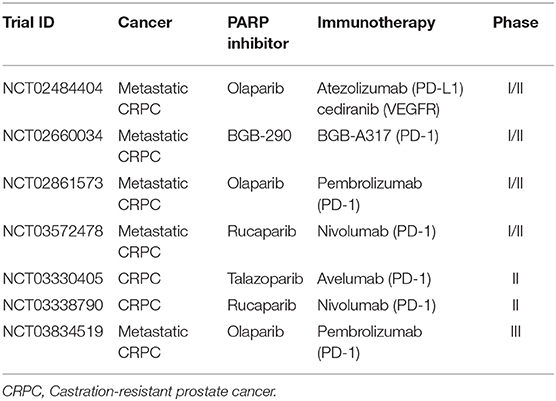
Table 4. Active combinatorial trials in prostate cancer.
Lung cancers can be subdivided into two major categories by histological examination, small-cell (SCLC) and non-small-cell lung cancers (NSCLC). In the latter, immune checkpoint monotherapy has shown responses ranging from 12 to 45%, dependent on patient selection criteria (4). Several studies are currently aimed at the combination of immune checkpoint blockade and PARP inhibition in NSCLC (Table 5), however, most efforts for improving therapeutic response in NSCLC is the combination of checkpoint blockade with targeted therapies (60) or traditional chemotherapy regimens (61–63). Or similarly, the testing the synergy of PARP inhibition with targeted therapies (64) or chemotherapies (65).
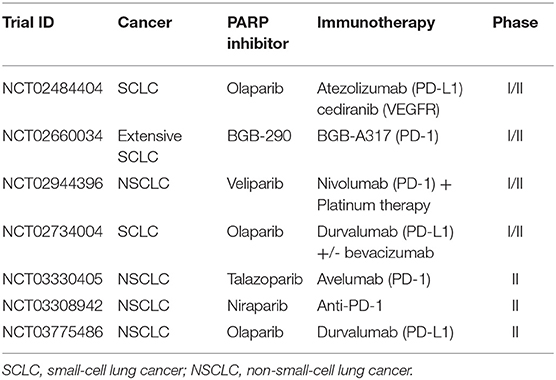
Table 5. Active combinatorial trials in lung cancer.
PARP inhibitors are attractive not only in tumors with underlying homologous recombination deficiencies, but also in tumors associated with high levels of replication stress, such as SCLC. SCLC cell lines have shown upregulation of BCL2 and elevated PARP1 levels (66). Early preclinical data has found PARP inhibition sensitized SCLC to cisplatin therapy (67). The IOLite open label trial (NCT03307785) is testing the safety and tolerability of the combination of anti-PD-1 and niraparib with the proteasome inhibitor bevacizumab in chemotherapy-resistant advance solid tumors, including SCLC (68). This elevated expression of PARP1 in SCLC is even being explored as a dynamic, non-invasive imaging modality to measure the drug-target engagement of PARP inhibitors (69).
The use of the combination immunotherapy and PARP inhibition is particularly interesting the gastrointestinal cancers. Notably, defective DNA mismatch repair and microsatellite instability, seen in Lynch Syndrome and 15% of colon cancer, have phenomenal responses of 25–80% to single-agent immune checkpoint blockade (4). Germline DDR mutations are seen in about 3% in gastric cancer (70), 3–17% in pancreatic cancer (71), and up to 8% in colorectal cancer (72) based on the gene involved and the population group. More relevant to the use of possible combinatorial PARP and checkpoint inhibition, acquired mutations in HRR are seen in about 28.9% in biliary, 20.9% in hepatocellular, 20.8% in gastroesophageal, 15.4% in pancreatic, and 15% in colorectal cancer patients (73). Like previously described cancers, a number of investigations are underway in the context of gastrointestinal tumors with intact and defective DNA repair and have reported responses in gastric and pancreatic cancers (74). The below clinical trials are evaluating the combination of PARP agents with PD1/PD-L1 inhibitors for a synergistic response with trials that are currently underway summarized in Table 6.
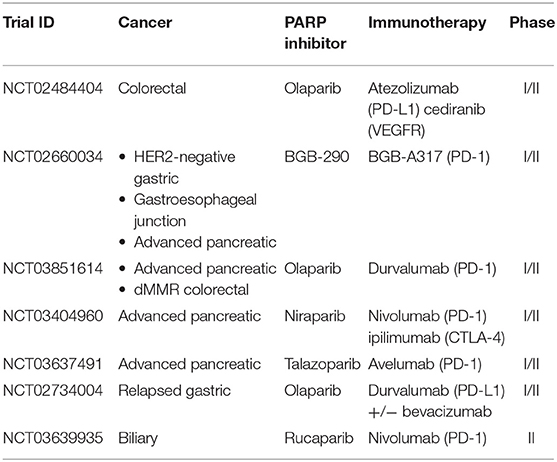
Table 6. Active combinatorial trials in gastrointestinal cancers.
The open-label MEDIOLA phase II basket study evaluating olaparib in combination with durvalumab in patients with relapsing gastric cancer after platinum-based chemotherapy (75). A 4-weeks run-in with olaparib was done to collect biopsies followed by combination therapy with olaparib and durvalumab until disease progression. Among 40 patients included in the study, an ORR of 10% (2 in CR and 2 in PR) was reported with a DCR at 12 weeks of 26% (75). In the patient with controlled disease, the median duration of response about 11.1 months. Grade 3 and grade 4 adverse events were reported for 48 and 8% of treated patients, with 25% of patients developing immune-mediated adverse events. These events principally consisted of anemia (17.5%) and increased lipase (10%). The authors theorized the low 12-weeks disease control rate may have been a result of a high rate of disease progression during the 4-weeks run in treatment with olaparib, which excluded patients from the final evaluation.
The promise of immunotherapy has not become a reality for pancreatic cancers, with low response rates to checkpoint inhibitors (4). With the FDA approval of PARP inhibitors for maintenance therapy in patients with BRCA-mutated pancreatic cancer in late 2019, investigations are proceeding with broadening the therapy using combinatorial approaches (76). For instance, PARPVAX is a phase Ib/II study intended for locally advanced/metastatic pancreatic cancer patients who did not progress after a minimum of 16 weeks of platinum-based chemotherapy. Eligible patients will be in the niraparib with nivolumab (anti-PD-1) arm or niraparib with ipilimumab arm with the primary outcome measure of 6-months progression-free survival in both the arms. However, current trials are underway that move beyond comparing PARP and checkpoint inhibition by adding additional agents. A phase Ib/II study of doublet therapy avelumab with binimetinib (MEK inhibitor); or with the addition of talazoparib (PARP inhibitor) in locally advanced/metastatic RAS-mutant solid tumor patients (including metastatic pancreatic ductal adenocarcinoma) with disease progression after one line of therapy is currently under investigation (Table 6). Another combinatorial approach is seen in the DAPPER phase II basket combination study of durvalumab with olaparib or cediranib (VEGFR inhibitor) in advanced DNA mismatch-repair-proficient colorectal cancer, pancreatic adenocarcinoma, or leiomyosarcoma who failed standard therapy.
The combination of PARP inhibitor and immune checkpoint agents targeting PD/PD-L1, appears to be safe, potentially synergistic and may represent a compelling strategy in treatment for a variety of tumors. There is difficulty in the concluding of synergy between the use of PARP inhibitors and immunotherapy, as a number of clinical trials summarized are in (1) early phases safety trials, (2) utilize patients in advanced clinical stages or after failure of multiple therapies, and (3) compare response to standard chemotherapy regimens. Moreover, these combinatorial trials do not have multiple arms comparing the treatment with one agent and often differ in inclusion criteria from the single-agent PARP or immune checkpoint trials. In the context of HRR-deficient tumors, remarkable responses have been reported in ovarian (ORR 45–63%, DCR 73–81%) and breast cancer (ORR 53% DCR: 47%) (21, 34, 46). Underlying these observations is the assumption that PARP inhibitors may act to prime the tumor microenvironment and increase anti-tumor immune cells, acting like an adjuvant for immunotherapy. For tumors that harbor a known alteration in HRR genes, such as BRCA1 or BRCA2, higher tumor mutational burden and a greater number of tumor-infiltrating lymphocytes has been seen in breast and ovarian cancer (77, 78). In these instances, the combination therapy seems to act by tipping the balance in favor of an anti-tumor immune response in the setting of a primed tumor microenvironment. This theory is supported by the observations of the CTLA-4 inhibition working in synergy with PARP inhibition in BRCA1 deficient tumors (21).
As summarized in this article the open question remains on translating the efficacy of the combination of PARP inhibition with immune checkpoint blockade in cancers intact DNA repair machinery. The majority of the clinical trials currently underway are investigating that very question. Early reports have shown more modest ORR of 14–19% in breast (49), 10% in gastric (68), and 7% in prostate cancers (59). Despite the low ORR for prostate cancer, there was a 46% DCR for advanced, therapy-resistant, metastatic disease (59). Mechanistic investigations are hopeful, with findings that include PARP inhibition induces PD-L1 expression in homologous-proficient breast cancer (17, 19). PARP inhibition can also promote the accumulation of cytosolic DNA fragments due to unresolved DNA lesions, which in turn activates the DNA sensing cGMP-synthase-stimulator-of-interferon genes (cGAS-STING) pathway (79). This stimulates the production of type I interferons, and in turn, induces antitumor immunity that may be further enhanced by checkpoint inhibitor blockade, independent of BRCAness (80). Other strategies that act to further prime the tumor microenvironment before or concomitantly with immunotherapy are being evaluated, such as the use of radiation or chemotherapy (4).
Combining PARP inhibitors with immunotherapy has the potential to improve outcomes in variety of solid tumors but several challenges remain. Most notably, lack of predictable responses in most tumor types and delineation of patients that would benefit from combination vs. sequential therapies. Identifying biomarkers of DNA damage repair and immune responsiveness would have to be streamlined. Various factors such as tumor vs. liquid biopsy, somatic vs. germline mutations, PDL-1 positivity on tumor cells vs. immune cells, nature and degree of DNA damage can potentially impact on clinical outcomes. Additionally, challenges remain in identifying meaningful endpoints of such combinations therapy like overall survival vs. response rates. Duration and frequency of such combination therapy and associated impact on health care costs should be considered. Research efforts should be focused not only on evaluating safety and efficacy, but also on biomarkers that can accurately predict benefit from such combination. The preliminary results of various studies evaluating the efficacy of PARP inhibitors and immunotherapy are promising in various solid tumors. The final results of the ongoing phase III clinical trials will eventually determine their overall efficacy and clinical benefit for the patients.
PV conceptualized the review, PV and NB contributed equally. AC and RJ contributed to the significant portions of the manuscript. All authors were involved in writing and editing and approval of final manuscript.
The authors are supported by the National Cancer Institute of the National Institutes of Health under award number CA206255 (NB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature. (2018) 562:20–1. doi: 10.1038/d41586-018-06751-0
3. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
4. Borcherding N, Kolb R, Gullicksrud J, Vikas P, Zhu Y, Zhang W. Keeping tumors in check: a mechanistic review of clinical response and resistance to immune checkpoint blockade in cancer. J Mol Biol. (2018) 430:2014–29. doi: 10.1016/j.jmb.2018.05.030
5. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. (2010) 10:293–301. doi: 10.1038/nrc2812
6. Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. (2004) 4:814–9. doi: 10.1038/nrc1457
7. Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. (2009) 37:3723–38. doi: 10.1093/nar/gkp229
8. Tao Z, Gao P, Liu HW. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J Am Chem Soc. (2009) 131:14258–60. doi: 10.1021/ja906135d
9. Pommier Y, O'Connor MJ, De Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. (2016) 8:362ps17. doi: 10.1126/scitranslmed.aaf9246
10. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. (2012) 84:137–46. doi: 10.1016/j.bcp.2012.03.018
11. Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
12. Moore K, Colombo N, Scambia G, Kim B-G, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. (2018) 379:2495–505. doi: 10.1056/NEJMoa1810858
13. Nesic K, Wakefield M, Kondrashova O, Scott CL, McNeish IA. Targeting DNA repair: the genome as a potential biomarker. J Pathol. (2018) 244:586–97. doi: 10.1002/path.5025
14. Mouw KW, Goldberg MS, Konstantinopoulos PA, D'Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. (2017) 7:675–93. doi: 10.1158/2159-8290.CD-17-0226
15. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
16. Ho SSW, Zhang WYL, Tan NYJ, Khatoo M, Suter MA, Tripathi S, et al. The DNA Structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity. (2016) 44:1177–89. doi: 10.1016/j.immuni.2016.04.010
17. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) 19:1189–201. doi: 10.1016/j.celrep.2017.04.031
18. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. (2017) 8:1–11. doi: 10.1038/s41467-017-01883-9
19. Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. (2018) 109:43–53. doi: 10.1111/cas.13424
20. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JMJL, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. (2017) 23:3711–20. doi: 10.1158/1078-0432.CCR-16-3215
21. Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA -mutated (gBRCA m) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. (2018) 149:246–47. doi: 10.1016/j.ygyno.2018.04.555
22. Maio M, Covre A, Fratta E, Di Giacomo AM, Taverna P, Natali PG, et al. Molecular pathways: at the crossroads of cancer epigenetics and immunotherapy. Clin Cancer Res. (2015) 21:4040–7. doi: 10.1158/1078-0432.CCR-14-2914
23. Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. (2017) 87:227–39. doi: 10.1016/j.molimm.2017.04.012
24. Vaughan S, Coward JI, Bast RC, Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. (2011) 11:719–25. doi: 10.1038/nrc3144
25. Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. (2011) 365:2484–96. doi: 10.1056/NEJMoa1103799
26. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. (2018) 68:284–96. doi: 10.3322/caac.21456
27. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci. (2011) 108:18032–7. doi: 10.1073/pnas.1115052108
28. Toss A, Tomasello C, Razzaboni E, Contu G, Grandi G, Cagnacci A, et al. Hereditary ovarian cancer: not only BRCA 1 and 2 genes. Biomed Res Int. (2015). 2015:341723. doi: 10.1155/2015/341723
29. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. (2015) 33:244. doi: 10.1200/JCO.2014.56.2728
30. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. (2017) 18:1274–84. doi: 10.1016/S1470-2045(17)30469-2
31. Friedlander M, Gebski V, Gibbs E, Davies L, Bloomfield R, Hilpert F, et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised tria. Lancet Oncol. (2018) 19:1126–1134. doi: 10.1016/S1470-2045(18)30343-7
32. Ray-Coquard IL, Pautier P, Pignata S, Pérol D, González-Martín A, Sevelda P, et al. LBA2_PR Phase III PAOLA-1/ENGOT-ov25 trial: olaparib plus bevacizumab (bev) as maintenance therapy in patients (pts) with newly diagnosed, advanced ovarian cancer (OC) treated with platinum-based chemotherapy (PCh) plus bev. Ann Oncol. (2019) 30:mdz394–053. doi: 10.1093/annonc/mdz394.053
33. Mirza MR, Bergmann TK, Mau-Sørensen M, Christensen R dePont, Åvall-Lundqvist E, Birrer MJ, et al. A phase I study of the PARP inhibitor niraparib in combination with bevacizumab in platinum-sensitive epithelial ovarian cancer: NSGO AVANOVA1/ENGOT-OV24. Cancer Chemother Pharmacol. (2019) 84:791–8. doi: 10.1007/s00280-019-03917-z
34. Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. (2019) 20:1306–15. doi: 10.1016/S1470-2045(19)30396-1
35. Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): a phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J Clin Oncol. (2018) 36:106. doi: 10.1200/JCO.2018.36.15_suppl.106
36. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61. doi: 10.1038/nature11412
37. Weigman VJ, Chao H-H, Shabalin AA, He X, Parker JS, Nordgard SH, et al. Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res Treat. (2012) 133:865–80. doi: 10.1007/s10549-011-1846-y
38. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. (2016) 13:674–90. doi: 10.1038/nrclinonc.2016.66
39. Foulkes WD. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. CancerSpectrum Knowl Environ. (2003) 95:1482–5. doi: 10.1093/jnci/djg050
40. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. (2012) 486:395–9. doi: 10.1038/nature10933
41. Miki Y, Swensen J, Shattuck-eidens D, Futreal PA, Harshman K, Tavtigian S, et al. Strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. (1994) 266:66–71. doi: 10.1126/science.7545954
42. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for brca1 and brca2 mutation carriers. JAMA. (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
43. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
44. Vikas P, Borcherding N, Zhang W. The clinical promise of immunotherapy in triple-negative breast cancer. Cancer Manag Res. (2018) 10:6823. doi: 10.2147/CMAR.S185176
45. Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety & clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with Poly (ADP-Ribose) polymerase inhibitor olaparib or vascular endothelial growth factor Receptor 1-3 inhibitor cediranib in women's cancers: a dose-escalati. J Clin Oncol. (2017) 35:2193. doi: 10.1200/JCO.2016.72.1340
46. Domchek S, Postel-Vinay S, Im S-A, Hee Park Y, Delord J-P, Italiano A, et al. Abstract PD5-04: an open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): updated results in patients with germline BRCA -mutated (g BRCA m) metastatic breast cancer (MBC). Cancer Res. (2019) 79. doi: 10.1158/1538-7445.SABCS18-PD5-04
47. Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Abstract OT3-05-03: MEDIOLA: an open-label, phase I/II basket study of olaparib (PARP inhibitor) and durvalumab (anti-PD-L1 antibody)–Additional breast cancer cohorts. Cancer Res. (2019) 79. doi: 10.1158/1538-7445.SABCS18-OT3-05-03
48. Mitri ZI, Vuky J, Kemmer KA, Savin MA, Parmar S, Kolodzie AK, et al. A phase II trial of olaparib and durvalumab in metastatic BRCA wild type triple-negative breast cancer. J Clin Oncol. (2019) 37:TPS1111. doi: 10.1200/JCO.2019.37.15_suppl.TPS1111
49. Vinayak S, Tolaney SM, Schwartzberg LS, Mita MM, McCann GA-L, Tan AR, et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial. J Clin Oncol. (2018) 36:1011. doi: 10.1200/JCO.2018.36.15_suppl.1011
50. Abou Alaiwi S, Nassar A, Mouw KW, Kwiatkowski DJ, Choueiri TK, Curran C, et al. Germline variants in urothelial carcinoma: analysis of pathogenic and likely pathogenic variants in 645 subjects. J Clin Oncol. (2019) 37:1528. doi: 10.1200/JCO.2019.37.15_suppl.1528
51. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. (2017) 171:540–56. doi: 10.1016/j.cell.2017.09.007
52. Robinson D, Allen EM Van, Sawyers CL, Chinnaiyan AM, Robinson D, Allen EM Van, et al. Integrative clinical genomics of advanced prostate resource integrative clinical genomics of advanced prostate cancer. Cell. (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
53. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
54. Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. (2020) 21:162–74. doi: 10.1016/S1470-2045(19)30684-9
55. Hussain M, Mateo J, Fizazi K, Saad F, Shore ND, Sandhu S, et al. LBA12_PR PROfound: phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations. Ann Oncol. (2019) 30:mdz394–039. doi: 10.1093/annonc/mdz394.039
56. Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara PN, et al. LBA50 Pre-specified interim analysis of GALAHAD: a phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann Oncol. (2019) 30:mdz394–043. doi: 10.1093/annonc/mdz394.043
57. Karzai F, Madan RA, Owens H, Hankin A, Couvillon A, Cordes LM, et al. Combination of PDL-1 and PARP inhibition in an unselected population with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. (2018) 35:5026. doi: 10.1200/JCO.2017.35.15_suppl.5026
58. Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. (2018) 6:141. doi: 10.1186/s40425-018-0463-2
59. Yu EY, Massard C, Retz M, Tafreshi A, Carles Galceran J, Hammerer P, et al. Keynote-365 cohort a: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. (2019) 37:145. doi: 10.1200/JCO.2019.37.7_suppl.145
60. Grohé C, Blau W, Gleiber W, Haas S, Mueller-Huesmann H, Schulze M, et al. Nintedanib plus docetaxel in lung adenocarcinoma patients (pts) following treatment with immune checkpoint inhibitors (ICIs): preliminary efficacy and safety results of the non-interventional study VARGADO. J Clin Oncol. (2019) 37:9074. doi: 10.1200/JCO.2019.37.15_suppl.9074
61. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
62. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
63. Offin M, Xu C, Jain H, Makhnin A, Hayes SA, Plodkowski AJ, et al. Efficacy of ramucirumab and docetaxel given either before or after immune checkpoint inhibitors in patients with lung cancers. J Clin Oncol. (2019) 37:9078. doi: 10.1200/JCO.2019.37.15_suppl.9078
64. Campelo RG, Arrieta Rodriguez OG, Massuti B, Rodriguez-Abreu D, Ortega Granados AL, Majem M, et al. Combination of gefitinib and olaparib versus gefitinib alone in EGFR mutant non-small-cell lung cancer (NSCLC): a randomized phase 2 study (GOAL, Spanish lung cancer group). J Clin Oncol. (2018) 36:9012. doi: 10.1200/JCO.2018.36.15_suppl.9012
65. Argiris A, Miao J, Cristea MC, Chen AM, Sands J, Decker RH, et al. S1206: a dose-finding study followed by a phase II randomized placebo-controlled trial of chemoradiotherapy (CRT) with or without veliparib in stage III non-small cell lung cancer (NSCLC). J Clin Oncol. (2019) 37:8523. doi: 10.1200/JCO.2019.37.15_suppl.8523
66. Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. (2012) 2:798–811. doi: 10.1158/2159-8290.CD-12-0112
67. Cheng H, Zhang Z, Borczuk A, Powell CA, Balajee AS, Lieberman HB, et al. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1-low non-small cell lung cancer cells. Carcinogenesis. (2013) 34:739–49. doi: 10.1093/carcin/bgs393
68. Gabrail NY, Bessudo A, Hamilton EP, Sachdev JC, Patel MR, Rodon Ahnert J, et al. IOLite: multipart, phase 1b, dose-finding study of the PD-1 inhibitor dostarlimab in combination with the PARP inhibitor niraparib±bevacizumab (bev), or with platinum-based chemotherapy±bev for advanced cancer. J Clin Oncol. (2019) 37:2560. doi: 10.1200/JCO.2019.37.15_suppl.2560
69. Carney B, Kossatz S, Lok BH, Schneeberger V, Gangangari KK, Pillarsetty NVK, et al. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat Commun. (2018) 9:1–13. doi: 10.1038/s41467-017-02096-w
70. Young K, Starling N, Cunningham D. Targeting deficient DNA damage repair in gastric cancer. Expert Opin Pharmacother. (2016) 244:586–97. doi: 10.1080/14656566.2016.1217992
71. Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. (2012) 51:14–24. doi: 10.1002/mc.20855
72. Cercek A, Kemel Y, Mandelker D, Stewart C, Arnold AG, Sheehan M, et al. Prevalence of germline genetic alterations in colorectal cancer patients. J Clin Oncol. (2018) 36:1577. doi: 10.1200/JCO.2018.36.15_suppl.1577
73. Heeke AL, Pishvaian MJ, Lynce F, Xiu J, Brody JR, Chen W-J, et al. Prevalence of homologous recombination–related gene mutations across multiple cancer types. JCO Precis Oncol. (2018) 2:1–3. doi: 10.1200/PO.17.00286
74. Friedlander M, Meniawy T, Markman B, Mileshkin LR, Harnett PR, Millward M, et al. A phase 1b study of the anti-PD-1 monoclonal antibody BGB-A317 (A317) in combination with the PARP inhibitor BGB-290 (290) in advanced solid tumors. J Clin Oncol. (2017) 35:3013. doi: 10.1200/JCO.2017.35.15_suppl.3013
75. Bang Y-J, Kaufman B, Geva R, Stemmer SM, Hong S-H, Lee J-S, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in patients with relapsed gastric cancer. J Clin Oncol. (2019) 37:140. doi: 10.1200/JCO.2019.37.4_suppl.140
76. Kindler HL, Hammel P, Reni M, Van Cutsem E, Mercade TM, Hall MJ, et al. Olaparib as maintenance treatment following first-line platinum-based chemotherapy (PBC) in patients (pts) with a germline BRCA mutation and metastatic pancreatic cancer (mPC): phase III POLO trial. J Clin Oncol. (2019) 37:LBA4. doi: 10.1200/JCO.2019.37.18_suppl.LBA4
77. Vinayak S, Gray RJ, Adams S, Jensen KC, Manola J, Afghahi A, et al. Association of increased tumor-infiltrating lymphocytes (TILs) with immunomodulatory (IM) triple-negative breast cancer (TNBC) subtype and response to neoadjuvant platinum-based therapy in PrECOG0105. ASCO. (2014) 14:1000. doi: 10.1200/jco.2014.32.15_suppl.1000
78. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. (2016) 7:13587. doi: 10.18632/oncotarget.7277
79. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. (2019) 9:722–37. doi: 10.1158/2159-8290.CD-18-1218
Keywords: immunotherapy, PARP inhibitor, combination therapy, breast cancer, prostate cancer, gynecologic cancer, gastrointestinal cancers, solid tumors
Citation: Vikas P, Borcherding N, Chennamadhavuni A and Garje R (2020) Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 10:570. doi: 10.3389/fonc.2020.00570
Received: 29 October 2019; Accepted: 30 March 2020;
Published: 28 April 2020.
Edited by:
Catherine Sautes-Fridman, INSERM U1138 Centre de Recherche des Cordeliers, FranceReviewed by:
Rodabe N. Amaria, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2020 Vikas, Borcherding, Chennamadhavuni and Garje. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Praveen Vikas, cHJhdmVlbi12aWthc0B1aW93YS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.