 Shijie Wu1,2†
Shijie Wu1,2† Jiaojiao Zhou1,2†
Jiaojiao Zhou1,2† Kun Zhang1,2Huihui Chen1,2Meng Luo1,2
Kun Zhang1,2Huihui Chen1,2Meng Luo1,2 Yuexin Lu1,2
Yuexin Lu1,2 Yuting Sun1,2Yiding Chen1,2*
Yuting Sun1,2Yiding Chen1,2*- 1Department of Breast Surgery, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2The Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, Zhejiang University School of Medicine, Hangzhou, China
Partner and localizer of BRCA2 (PALB2) is vital for homologous recombination (HR) repair in response to DNA double-strand breaks (DSBs). PALB2 functions as a tumor suppressor and participates in the maintenance of genome integrity. In this review, we summarize the current knowledge of the biological roles of the multifaceted PALB2 protein and of its regulation. Moreover, we describe the link between PALB2 pathogenic variants (PVs) and breast cancer predisposition, aggressive clinicopathological features, and adverse clinical prognosis. We also refer to both the opportunities and challenges that the identification of PALB2 PVs provides in breast cancer genetic counseling and precision medicine.
Introduction
Partner and localizer of BRCA2 (PALB2) is encoded on chromosome 16p12.2 and comprises 1186 residues (1). As a major BRCA2 binding partner, PALB2 licenses the function of BRCA2 and participates in homologous recombination (HR), a faithful DNA double-strand break (DSB) repair pathway in mammalian cells (2–4). Numerous studies have demonstrated that biallelic mutations in PALB2 resulted in a subtype of Fanconi anemia (FA-N), while monoallelic PALB2 mutations predispose carriers to multiple cancers such as breast, pancreatic, and ovarian cancers (5–8).
Breast cancer is the most frequently diagnosed cancer and the major cause of cancer death among women worldwide (9). Approximately 10–15% of breast cancer cases are due to familial and genetic factors, underscoring the great significance of genetic susceptibility in breast cancer development (10). Previous studies have identified a broad range of breast cancer susceptibility genes, including BRCA1, BRCA2, and TP53 (11). However, the high-penetrance BRCA1 and BRCA2 are responsible for only ~20% of the familial aggregation of breast cancer (12, 13), and syndromic breast cancer susceptibility genes such as TP53, PTEN, and CDH1 are estimated to explain just 5% of familial breast cancers (14). Large-scale analyses of multigene panel testing recently confirmed PALB2 as a high-risk breast cancer susceptibility gene (15), and the odds ratio (OR) of PALB2 mutations for breast cancer was comparable to that of BRCA2 mutations (16). Hence, a comprehensive understanding of the biological functions of PALB2 is vital for breast cancer management and precision medicine.
Structures of PALB2 and Its Biological Functions in HR
PALB2, first described by Xia et al. in 2006 (1), has an important role in HR. It mainly serves as a bridging molecule that connects the BRCA complex (BRCA1-PALB2-BRCA2-RAD51) and facilitates the function of RAD51, a protein vital for strand invasion during HR (Figure 1). The role of PALB2 in HR has been shown to involve several protein domains, including a coiled-coil domain, a WD40 domain, and a chromatin-association motif (ChAM) (Figure 2).
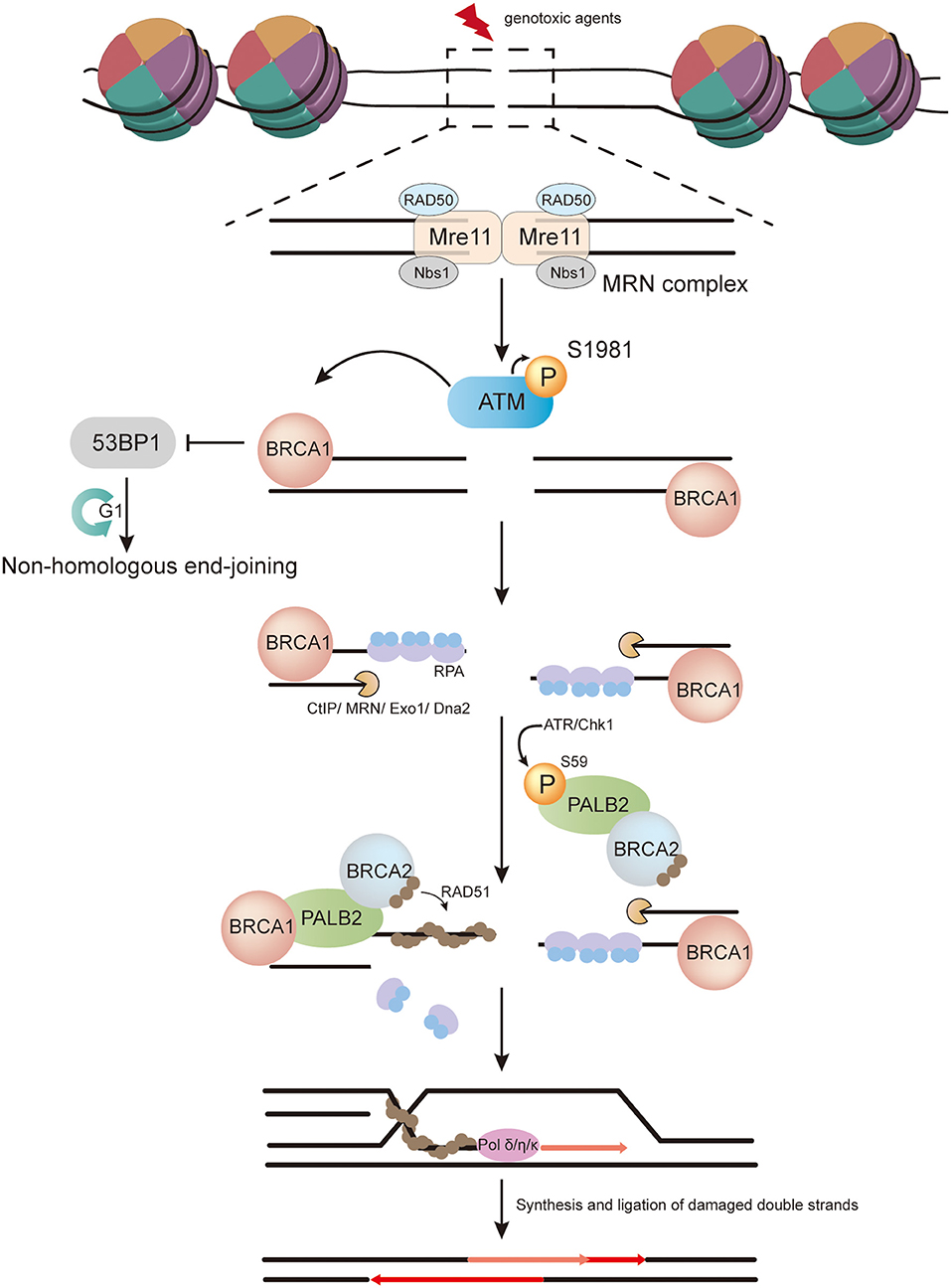
Figure 1. The role of PALB2 in homologous recombination (HR). In response to DNA double-strand breaks (DSBs) induced by genotoxic agents in the S/G2 phase, the Mre11–RAD50–Nbs1 (MRN) complex is recruited to DSBs and promotes ATM recruitment. The inactive ATM dimer then dissociates into active monomers through autophosphorylation at serine 1981. Active ATM monomers phosphorylate H2AX in regions of DSBs and create a platform to recruit BRCA1, which facilitates a shift from non-homologous end-joining to HR. Meanwhile, CtBP-interacting protein (CtIP), in conjunction with the MRN complex, catalyzes 5′-3′ resection at DSBs to generate single-stranded DNA (ssDNA), and further resection is completed by Exo1 exonuclease and Dna2 nuclease/helicase in cooperation with BLM helicase. The resulting ssDNA is then covered by replication protein A (RPA). PALB2 is phosphorylated on S59 by ATR/Chk1, which accelerates its recruitment to sites of damage. Thereafter, BRCA2 is recruited by PALB2. PALB2 and BRCA2 further promote RPA removal and RAD51 loading. The resulting RAD51-ssDNA filament invades the intact sister chromatid and extends the strand with the help of DNA polymerase δ/η/κ. Finally, further restoration and ligation of double strands are carried out.
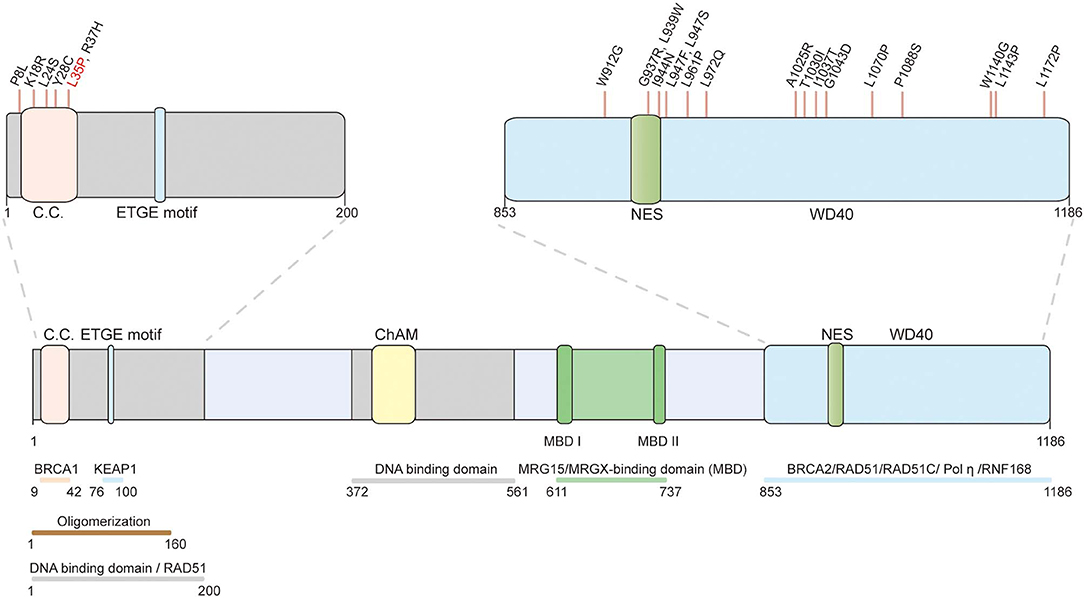
Figure 2. Schematic representation of the PALB2 protein and the position of functionally validated PALB2 pathogenic missense variants. The structural motifs and functional domains of PALB2. C.C.: coiled-coil motif (9–42); ETGE motif (88–94); ChAM: chromatin-association motif (395–446); WD40: WD40-repeats (853–1186); NES: nuclear export sequence (928–945). The validated pathogenic missense variants are marked on top. The only recognized PALB2 pathogenic missense variant (p.L35P) validated by systematic in vitro functional assays is highlighted in red.
The coiled-coil domain is located in the N terminus of PALB2 (residues 9–42) and is responsible for its interaction with BRCA1 (2–4). The L21A, Y28A, and L35A mutations in the PALB2 coiled-coil domain disrupt the BRCA1-PALB2 interaction, impairing the function of PALB2 in HR repair and inducing hypersensitivity to mitomycin C (MMC) treatment (3). In addition to positively regulating HR, the BRCA1-PALB2 interaction is required for preventing single-strand annealing (SSA), which is a deletion-causing DSB repair pathway. Using U2OS/DR-GFP and U2OS/SA-GFP reporter cells, Anantha et al. demonstrated that depletion of either PALB2 or BRCA2 led to impaired HR activity and a substantial increase in SSA, whereas BRCA1 depletion caused a reduction of both HR and SSA activity (17). These results established that BRCA1 is essential for DSB repair, while PALB2 serves to direct the DSB repair toward the HR pathway following resection.
The WD40 domain is located in the PALB2 C-terminus and in the shape of a WD40-type β-propeller with seven blades (18). This domain is involved in the interaction with BRCA2, DNA polymerase η, RAD51, RAD51C, and the ubiquitin ligase RNF168 (5, 19–21). Even a single nucleotide change within the WD40 region (e.g., L939W) can disturb the PALB2-BRCA2 interaction and causes HR deficiency (20). The WD40 domain of PALB2 is also crucial for the interaction with DNA polymerase η, which is vital for the initiation of HR-mediated DNA synthesis and D-loop extension (19). Recently, a hidden nuclear export sequence (NES) was found in the WD40 domain of PALB2. The breast cancer-associated PALB2 truncating mutation, W1038X, exposes this NES, resulting in PALB2 translocation to the cytoplasm and defects in HR (22).
The ChAM is an evolutionarily conserved domain located in the middle region of PALB2 (23). ChAM-deleted PALB2 has a compromised role in supporting MMC-induced RAD51 focus formation, suggesting that ChAM promotes the function of PALB2 through chromatin association (23). The ChAM binds to nucleosomes and participates in the formation of the PALB2-BRCA2-RAD51 complex on chromatin, which rapidly transforms into an active BRCA complex following DSBs (23).
In addition to BRCA complex formation, PALB2 also directly interacts with RAD51 and enhances its strand invasion activity (24, 25). In vitro D-loop assays revealed increased product formation when PALB2 was included in the RAD51 reaction. Moreover, Buisson et al. also identified two DNA-binding domains in PALB2 (24) (Figure 2). More recently, Deveryshetty et al. (26) showed that the main DNA-binding domain (DBD) of PALB2 is located in its N-terminus (N-DBD, residues 1–200). Mutation of just four amino acids in the N-DBD significantly disrupts the HR activity of PALB2. Surprisingly, the authors discovered that the N-DBD of PALB2 enhances RAD51-mediated strand exchange and also promotes a similar reaction in the absence of RAD51. Using strand exchange fluorescent assays, they further demonstrated that PALB2 N-DBD promotes both forward and inverse strand exchange using either DNA or RNA as substrate (26).
These studies uncovered multiple effects of PALB2 during HR. On the one hand, PALB2 serves as the bridging molecule in the BRCA complex; on the other hand, it potently stimulates strand invasion in HR.
PALB2: A Versatile Player in Biological Regulation
PALB2 and Chromatin Association
Chromatin association is considered indispensable for the biological function of PALB2. In addition to the ChAM, MRG15 is another PALB2-interacting factor involved in PALB2 chromatin association (27) (Figure 3A). In 2009, Sy et al. unveiled MRG15 and another MORF-related gene product, MRGX, as PALB2 cooperators through tandem affinity purification and mass spectrometry analysis (28). MRG15 belongs to the highly conserved MRG protein family (29) and has two functional domains: one is an MRG domain that binds to PALB2 as well as multiple transcriptional regulators (28, 30), the other is N-terminal chromodomain that binds lysine 36-trimethylated histone H3 (H3K36me3) (31), which is mediated by lysine methyltransferase SET domain containing 2 (SETD2) (32). The MRG15-binding region was roughly mapped to the middle region of PALB2 (residues 611–764) and exactly matched two highly conserved regions named MBD-I (residues 611–629) and MBD-II (residues 724–737) (33). Sy et al. found that reconstitution of MRG15-binding domain deleted PALB2 could restore RAD51 foci formation and cell survival after MMC treatment in EUFA1341F PALB2-deficient cells (28). Strikingly, a concurrent study reached a contradictory conclusion, whereby siRNA-mediated MRG15 depletion in cells compromised HR repair efficiency and sensitization to MMC (34). Furthermore, MRG15 knockout murine embryonic fibroblasts exhibited moderate sensitivity to γ-irradiation and decreased capacity for RAD51 nuclear foci formation (35). Considering the studies above, we could propose that the MRG15-PALB2 interaction is involved in HR repair, but may not be critical for this process.
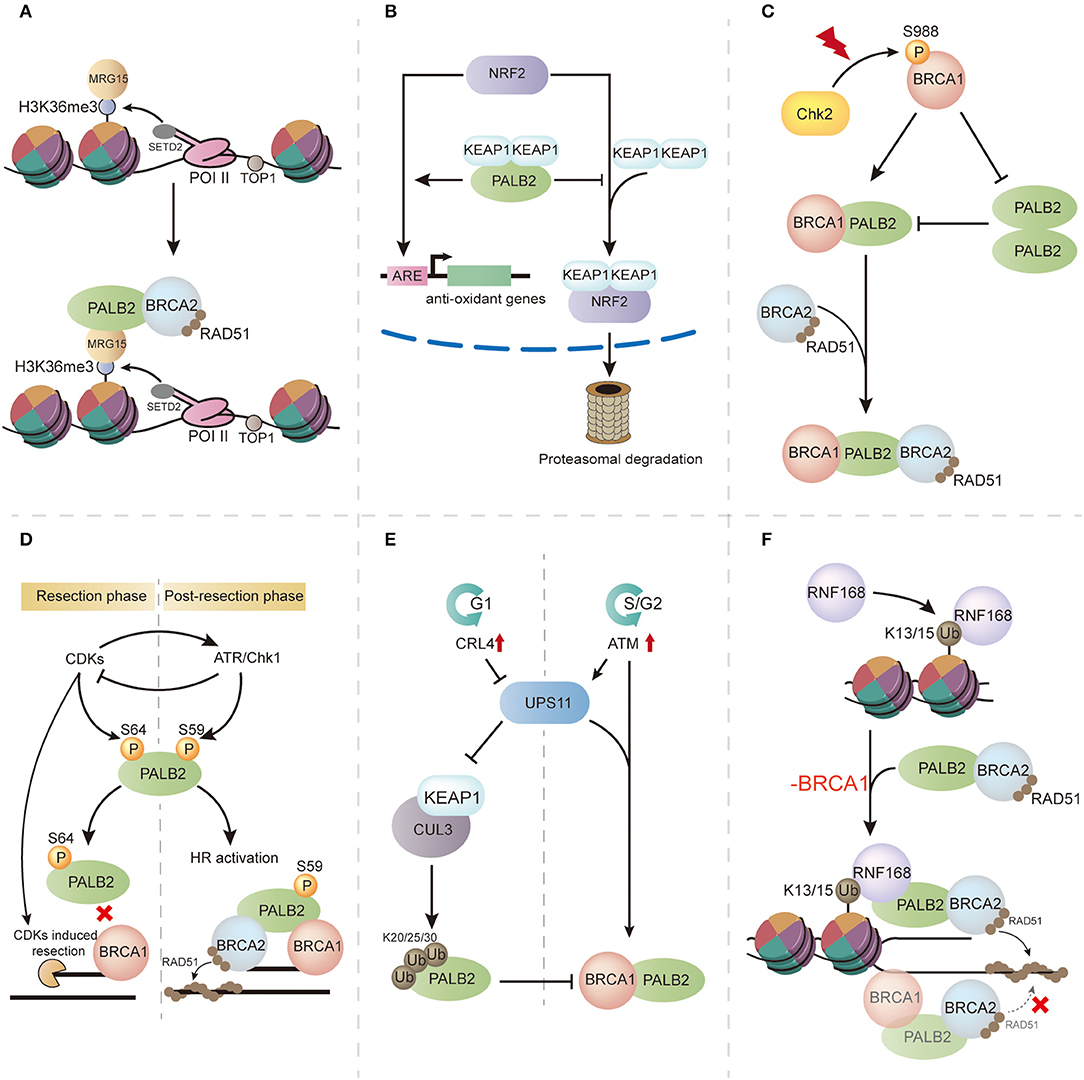
Figure 3. The multifaceted functions of PALB2 and its regulation. (A) PALB2 is recruited through the SETD2/H3K36me3/MRG15 axis and protects transcriptionally active genes from replication stress. (B) PALB2 promotes NRF2 function during oxidative stress by competitively binding KEAP1. (C) Following ionizing radiation (IR), the switch from PALB2 oligomerization to BRCA1-PALB2 interaction is regulated by S988 phosphorylated BRCA1. (D) Phosphorylation events in PALB2 regulation. In the resection phase, high CDKs induce PALB2 phosphorylation at S64, inhibiting its interaction with BRCA1, whereas in post-resection phase, ATR-induced PALB2 phosphorylation at S59 promotes BRCA1-PALB2 binding and enhances HR activity. (E) In the G1 phase of the cell cycle, PALB2 is ubiquitylated by the CUL3–KEAP1 complex, which disrupts BRCA1-PALB2 interaction, whereas in the G2 phase, PALB2 ubiquitylation is neutralized by USP11. (F) RNF168 mediates PALB2 recruitment and RAD51 loading in BRCA1-deficient cells.
Intriguingly, a genome-wide analysis evaluating PALB2 chromatin residence revealed a tight relationship between PALB2 chromatin residence and transcriptionally active genes (36). This result confirmed that MRG15-PALB2 interaction is associated with unperturbed chromatin. In 2017, Bleuyard et al. hypothesized the innovative concept that the MRG15-PALB2 interaction within undamaged chromatin maintains chromatin stability during DNA replication (33) (Figure 3A). This idea was supported by genome-wide PALB2 chromatin immunoprecipitation-sequencing analysis, which indicated gathering of PALB2 at H3K36me3-modified genes through the SETD2/H3K36me3/MRG15 axis. Moreover, expression of MRG15 binding-defective PALB2 leads to compromised proliferation, DNA stress, and genome instability when compared with wild-type PALB2 expression in EUFA1341 cells (33). These findings indicate that the MRG15-PALB2 complex may be a genomic stabilizer within active genes, which renders PALB2 immediately available following DNA damage and guarantees a rapid response to replication stress, thereby maintaining genome stability. In addition to MRG15-PALB2 interaction, PALB2 is also recruited by phosphorylated replication protein A (RPA) during replication stress. Murphy et al. revealed that phosphorylation of RPA during replication stress stimulates the recruitment of PALB2 and increases the stability of PALB2 chromatin binding, making PALB2 available to alleviate replication stress and facilitating the recovery of stalled replication forks (37).
PALB2 and Oxidative Stress
KEAP1, an oxidative stress mediator that negatively regulates the function of the antioxidant transcription factor NRF2, was revealed to bind PALB2 by coimmunoprecipitation (38). Surprisingly, a highly conserved 7-aa motif (LDEETGE) within the KEAP1 binding domain of PALB2 (residues 76–100) was identical to the ETGE motif of NRF2 that binds KEAP1, implying that PALB2 may promote the role of NRF2 by competitively binding KEAP1 (Figure 3B). This was supported by increased levels of reactive oxygen species (ROS) and reduced expression of NRF2-regulated genes after PALB2 depletion. Thus, this study unveiled a unique function of PALB2 during oxidative stress and provided a possible link between the oxidative stress response and PALB2-associated cancer formation.
PALB2 and Cell-Cycle Checkpoint Control
Cell-cycle checkpoints are essential for DNA damage repair following genotoxic exposure because of their role in constraining cell-cycle progression and providing time for accurate DSB repair by HR, thereby guaranteeing genome stability (39). Menzel et al. performed a high-throughput siRNA screen to explore potential G2 checkpoint modulators, and identified PALB2 as a main G2 checkpoint maintainer (40). Depletion of PALB2 led to G2 checkpoint dysregulation and premature checkpoint recovery. In the same study, the role of PALB2 in the maintenance of the G2 checkpoint was seemly independent of the HR pathway, as RAD51 depletion did not compromise G2 checkpoint control. More recently, Simhadri et al. proposed a novel model in which PALB2 serves as a nexus that connects BRCA1 and BRCA2 in G2/M checkpoint control (41). Consistent with this view, disturbing the interactions of BRCA1-PALB2 or BRCA2-PALB2, using the L35P or A1025R mutant of PALB2, respectively, severely impaired the checkpoint response. Notably, BRCA1-PALB2 interaction seems to be critical for checkpoint initiation, whereas BRCA2-PALB2 interaction plays a more significant role in checkpoint maintenance. Although these studies have unveiled the role of PALB2 in checkpoint control, it remains unclear how exactly PALB2 participates in the pathway.
Regulation of PALB2
The biological functions of PALB2 are strictly regulated. To date, many mechanisms of its regulation have been elucidated, including PALB2 oligomerization, phosphorylation, ubiquitylation, and interaction with RNF168.
PALB2 Oligomerization
PALB2 oligomerization negatively regulates HR through its coiled-coil domain (42, 43) (Figure 3C). Overexpression of the PALB2 coiled-coil domain markedly impairs RAD51 filament formation, suggestive of competition between PALB2 oligomerization and BRCA1-PALB2 interaction. Meanwhile, immunoprecipitation analyses showed that the presence of BRCA1 completely abrogated PALB2 self-interaction, indicating that PALB2 self-interaction can be inhibited by its interaction with BRCA1 (43). As the Chk2-induced BRCA1 phosphorylation of S988 is important for HR activity (44), Buisson et al. proposed that BRCA1 phosphorylation may lead to a molecular switch from PALB2 homodimerization to BRCA1-PALB2 interaction, thereby promoting HR (43) (Figure 3C). Song et al. recently reported that PALB2 homodimerization is mediated by an antiparallel coiled-coil leucine zipper (45). Mutation of residue Leu24, a key stabilizer at the dimer interface, greatly reduces PALB2 homodimer stability and results in genomic instability in mutated cells, suggesting an important role of PALB2 oligomerization in HR regulation.
PALB2 Phosphorylation
PALB2 phosphorylation is also critical for its modulation. Three N-terminal S/Q sites of PALB2 (S59, S157, and S376) were found to be phosphorylated following ionizing radiation, and the phosphorylation events were mediated by ataxia telangiectasia mutated protein (ATM) and ATM and Rad3-related kinase (ATR) (46, 47). Phosphorylation-deficient PALB2 failed to promote RAD51 foci formation, leading to impaired HR and genome instability, highlighting the role of phosphorylation in PALB2 regulation (47). Strikingly, Buisson et al. (48) demonstrated a phosphorylation conversion at S59 and S64 on PALB2 during the phosphorylation process (Figure 3D). In this model, PALB2 is first phosphorylated at S64, a cyclin-dependent kinase (CDK) site, and high CDK activity actuates DNA end resection and ATR activation. Activated ATR then induces S59 phosphorylation and suppresses CDK activity, followed by hypo-phosphorylation of S64 and a strengthened BRCA1-PALB2 interaction (48). This CDK-ATR switch is crucial for attaining optimal levels of PALB2 at DSBs.
PALB2 Ubiquitylation
Ubiquitylation has also been reported to regulate PALB2 function via cell-cycle control (Figure 3E). In the G1 phase, the CUL3–KEAP1 complex ubiquitylates PALB2 on its N terminus, which is the BRCA1-binding region, to suppress BRCA1-PALB2 interaction, ultimately inhibiting HR. As cells enter the S/G2 phase, PALB2 ubiquitylation is neutralized by USP11, a deubiquitylase that is antagonized by CRL4 in the G1 phase. Restoration of BRCA1-PALB2 interaction facilitates BRCA complex formation and induces HR repair (49).
PALB2 and RNF168
The E3 ubiquitin ligase RNF168 was recently found to promote PALB2 accumulation in the S/G2 phase and facilitate DNA repair. It was supported by the restoration of PALB2 foci in endogenous RNF168-depleted S/G2 cells after re-expression of RNF168. The intrinsic mechanism is a physical interaction between the WD40 domain of PALB2 and the newly uncovered PALB2-interacting domain of RNF168 (21). Recently, Zong et al. revealed that RNF168-driven PALB2 recruitment, a BRCA1-independent pathway, serves as a backup to maintain DNA repair by HR (50) (Figure 3F). In this model, PALB2 recruitment is mainly orchestrated by BRCA1 in BRCA1-proficient cells, and an RNF168-driven pattern is applied as an auxiliary. Nevertheless, in BRCA1 mutated cells, RNF168-mediated PALB2 recruitment plays a vital alternative role for RAD51 loading and genome stability. Considering the unambiguous association between RNF168 and PALB2, inhibiting RNF168 signaling in BRCA1-insufficient cancers may be an effective therapeutic strategy (51).
PALB2 and Diseases
PALB2 and Fanconi Anemia
Fanconi anemia (FA) is a rare human genetic instability syndrome associated with diverse developmental defects, early-onset bone marrow failure, and cancer predisposition, mainly to acute myeloid leukemia and head and neck squamous cell carcinoma. Cells derived from FA patients are hypersensitive to DNA crosslinking agents such as MMC and cisplatin, and this hallmark is commonly used for the clinical diagnosis of FA (52). To date, 22 FA-related proteins have been identified in the FA-BRCA pathway for DNA interstrand cross-link repair (53–55), and PALB2 serves as a mediator in the BRCA pathway (56). In 2007, Xia et al. (5) reported a new subtype of Fanconi anemia (FA-N) resulted from biallelic mutations in PALB2 (also known as FANCN). A PALB2-deficient Fanconi anemia cell line showed impaired RAD51 foci formation and hypersensitivity to MMC treatment (5). Notably, FA-N patients are at a high risk of developing embryonal cancer, similar to that seen in patients with biallelic BRCA2 mutations, but differing from that observed for patients of other FA subtypes (57). These findings emphasize the important role of PALB2 in maintaining genomic stability and tumor suppression.
PALB2 and Breast Cancer
PALB2 is tightly correlated with breast cancer and has been associated with breast cancer predisposition, clinicopathological features, and prognosis.
In 2007, Rahman et al. (6) provided a profile of PALB2 mutations in breast cancer predisposition. The authors determined the frequency of PALB2 monoallelic truncating variants in a familial breast cancer cohort negative for BRCA mutations (10/923, 1.1%), which was far more common than in controls (0/1,084, 0%; p = 0.0004). They also revealed that individuals with monoallelic PALB2-mutations had a 2.3-fold increased risk of breast cancer compared with controls (95% confidence interval [CI], 1.4–3.9; p = 0.0025) (6), hinting that monoallelic PALB2 mutations may have a more moderate role in breast cancer predisposition than monoallelic BRCA2 variants (58, 59). At the same time, research in Finland identified a recurrent mutation, c.1592delT, in 1% of unselected breast cancer patients (60). This frameshift mutation resulted in a 40% cumulative risk of developing breast cancer by age 70 (95% CI, 17–77) (61), similar to that for BRCA2 mutation carriers (~45%; 95% CI, 31–56) (62), implying a striking role of PALB2 in predisposition to breast cancer. Subsequently, multiple population-based screenings of PALB2-truncating mutations reported 2–30-fold increases in breast cancer risk for PALB2-truncating mutations carriers (6, 63–67). In 2014, Antoniou et al. (64) estimated the age-specific relative risk for PALB2 mutation carriers, which was highest among women before age 40 years (relative risk, 8–9) then gradually declined with age, with the lowest risk after the age of 60 years (relative risk, ~5). Meanwhile, female PALB2 mutation carriers showed an estimated cumulative breast cancer risk of 35% (95% CI, 26–46) by age 70 (64). A recent international study from 21 countries that comprised 524 families with PALB2 pathogenic variants (PVs) revealed that the estimated relative risk associated with PALB2 PVs for breast cancer in females was 7.18 (95% CI, 5.82–8.85; p = 6.5 × 10−76). The authors also showed that the estimated relative risk for female breast cancer declined with age, varying from 13.1 at young ages to 4.69 for older ages, and the estimated female breast cancer risk was 53% (95% CI, 44–63) to age 80 years (68).
Male breast cancer (MBC) is a rare disease that accounts for <1% of all breast cancer cases (69). However, ~20% of MBC patients have a family history of breast cancer (70), highlighting a strong correlation between genetic susceptibility genes and MBC. Two high-penetrance breast cancer genes, BRCA1 and BRCA2, are thought to be responsible for only 13% of MBC (71), and multiple genetic factors remain unknown. To date, many PVs of PALB2 have been reported in MBC patients (72–76). In 2017, Pritzlaff et al. uncovered that PALB2 variants significantly increased the risk of MBC (OR, 6.6; p = 0.01) (77). This viewpoint was further supported by an Italian population-based multicenter study in 2019 (78). Rizzolo et al. found that PALB2 was the most frequently mutated gene (1.2%) among non-BRCA1/2 altered MBC patients, and deleterious PALB2 variants conferred a 9.63- to 17.30-fold increased risk of MBC (78). More recently, Yang et al. further showed an estimated MBC relative risk of 7.34 (95% CI, 1.28–42.18; p = 0.026) for PALB2 PVs carriers by analyzing data from 524 families with PALB2 PVs from 21 countries (68).
Several studies have also found that PALB2-mutated breast cancer is associated with aggressive clinicopathological features. In 2009, Heikkinen et al. reported that breast cancer patients harboring the PALB2 c.1592delT mutation were more likely to present the triple-negative phenotype (54.5%, p < 0.0001), characterized by the absent expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (79), than other familial (12.2%) or sporadic (9.4%) breast cancer patients (80). This finding was further supported by other population-based screening studies (10, 81–84). Heikkinen et al. also showed that PALB2-mutated breast cancer patients were more likely to present at an advanced disease stage (p = 0.0027 and p = 0.0017, respectively) and have a higher Ki67 level (p = 0.0004 and p = 0.0490, respectively) compared with other familial or sporadic patients (80).
In 2015, Cybulski et al. first evaluated the prognostic effects of two PALB2 deleterious mutations (509_510delGA and 172_175delTTGT) in Poland (83). In this study, the 10-year survival rate for female breast cancer patients with a PALB2 mutation was 48.0% (95% CI, 36.5–63.2), significantly lower than that for PALB2 mutation-negative female breast cancer patients (74.7%; 95% CI, 73.5–75.8). The 10-year adjusted hazard ratio for all-cause mortality was 2.27 (95% CI, 1.64–3.15; p < 0.0001), indicating an adverse prognostic effect of PALB2 in breast cancer (83). More recently, a population-based screening of breast cancer susceptibility genes in China further confirmed the prognostic value of PALB2 in breast cancer; patients with a PALB2 mutation presented shorter overall survival compared with noncarriers (adjusted hazard ratio, 8.38; 95% CI, 2.19–32.11; p = 0.002) (85).
PALB2 and Other Cancers
In addition to breast cancer, PALB2 has also been identified as a susceptibility gene for pancreatic cancer. Jones et al. (86) first discovered a germline PALB2-truncating mutation (c.172_175delTTGT) in a familial pancreatic cancer (FPC) patient, and three PALB2-truncating mutations were further identified in 96 additional FPC patients (3.1%). In contrast, no PALB2-truncating mutation was found in 1,084 normal individuals (86). Subsequent studies also revealed the prevalence of PALB2 deleterious mutations in patients with FPC (~3–4%) (87, 88), validating the role of PALB2 mutations in pancreatic cancer predisposition. To date, several studies have indicated that PALB2 mutations are associated with ovarian cancer (8, 89, 90). Although the mutation frequency was low, Norquist et al. demonstrated that PALB2 mutation carriers had a significantly higher risk of ovarian cancer compared with the NHLBI Exome Sequencing Project (OR, 10.2; 95% CI, 2.2–47.0; p < 0.001) or the Exome Aggregation Consortium database (OR, 4.4; 95% CI, 2.1–9.1; p < 0.001) (8). Pathogenic PALB2 mutations have also been identified in patients with other cancers, such as gastric and prostate cancer; however, whether these mutations confer an increased cancer risk for these cancer types requires further research (91–94).
PALB2 and Precision Medicine
Excluding BRCA1/2, PVs in PALB2 contribute most significantly to the mutation detection rate in multigene testing panels for hereditary breast cancer (95). Thus, germline PALB2 status is crucial for breast cancer risk assessment in individuals with an apparent family history of breast cancer. To date, several ethnic-specific PALB2 recurrent mutations have been reported, and related cancer predisposition risks have been established in distinct territories (60, 63, 82, 83, 96, 97) (Table 1). In these regions, genotyping for specific PALB2 PVs can be applied as a cost-effective tactic in high-risk individuals. For most high-risk people who are not in these specific regions, multigene panel testing that includes PALB2 is an advisable choice for genetic counseling (98–100). According to National Comprehensive Cancer Network (NCCN) guidelines, annual mammogram with consideration of tomosynthesis and breast magnetic resonance imaging with contrast are recommended for people with PALB2 PVs/likely PVs from the age of 30 to detect cancer at an early stage (101).
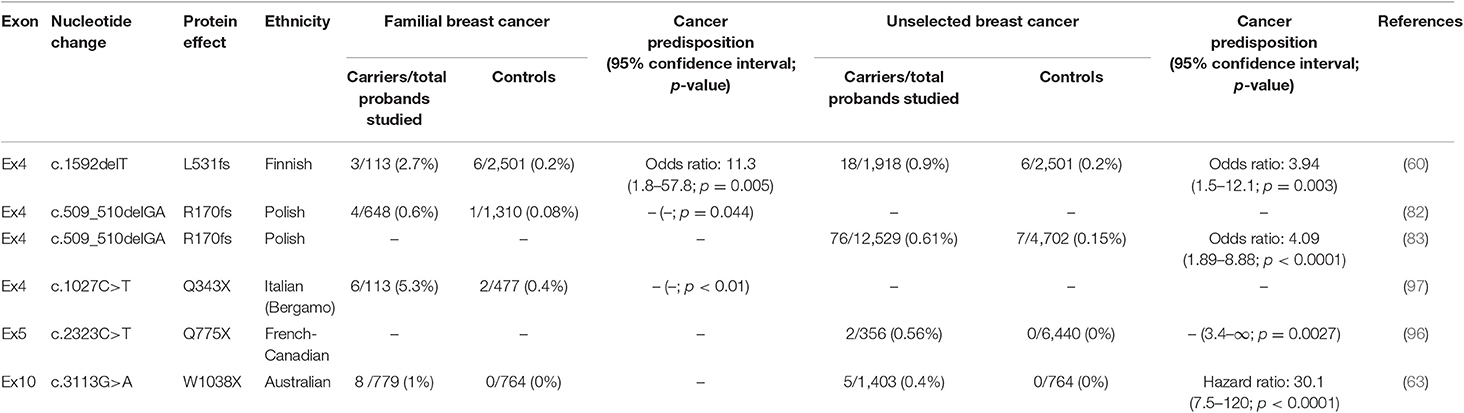
Table 1. Ethnic-specific PALB2 recurrent mutations and related breast cancer predisposition.
Poly (ADP-ribose) polymerases (PARPs) act as DNA damage sensors and regulators and play important roles in the repair of single-stranded DNA breaks through the base-excision repair pathway (102). PARP inhibitor (PARPi) treatment prevents the repair of single-stranded DNA breaks and leads to DSBs in cells. BRCA1/2-deficient cells are unable to repair DSBs via the HR pathway, resulting in cell death (103). Consequently, PARP inhibition is considered a promising strategy for the treatment of BRCA1/2-deficient tumors through synthetic lethality. More recently, the PARPi olaparib and talazoparib have been approved for germline BRCA-mutated (gBRCAm) HER2-negative metastatic breast cancer in clinic (104). Similar to BRCA1/2, PALB2 is an essential component in HR-based DNA repair, and PALB2 loss of function was shown to be synthetic lethal in combination with PARPi (105). Recently, PARPi sensitivity of PALB2 missense variants has been partially elucidated in vitro. Foo et al. identified a PARPi-hypersensitive PALB2 variant (p.L35P) using EUFA1341 cells, an FA-N patient-derived skin fibroblast cell line with biallelic mutations in PALB2 (106). In 2019, Rodrigue et al. (107) utilized siRNA-mediated RNA interference to generate PALB2-depleted HeLa cells, and exogenous siRNA-resistant PALB2 variants were complemented before PARPi sensitivity assay. Cells expressing the PALB2 variants p.T1030I or p.W1140G showed significantly higher olaparib sensitivity than those expressing wild-type PALB2 (107). A concurrent study conducted by Wiltshire et al. revealed four new PARPi-hypersensitive variants in PALB2 (p.L24S, p.I944N, p.A1025R, and p.L1070P) using PALB2-deficient B400 mouse mammary tumor cells (108). Boonen et al. (109) developed a cDNA-based system for the functional analysis of PALB2 variants. By evaluating the ability of PALB2 variants to rescue PARPi sensitivity in PALB2 knockout mouse embryonic stem cells, they identified twelve PALB2 variants (p.Y28C, p.L35P, p.W912G, p.G937R, p.I944N, p.L947S, p.L961P, p.L972Q, p.A1025R, p.T1030I, p.G1043D, and p.L1172P) that showed hypersensitivity to PARPi (109).
In spite of the lack of clinical evidence for PARPi treatment efficacy in PALB2-deficient breast cancer patients, the response of some other PALB2-deficient solid tumors to PARPi in clinical/preclinical studies have been remarkable. A preclinical study of BMN673 (talazoparib) for the panel of the Pediatric Preclinical Testing Program showed that a maintained complete response was observed in vivo in a Wilms tumor xenograft model, characterized by a truncating mutation in PALB2 (p.Y1108fs) (110). de Bono et al. (111) conducted a phase I trial of the PARPi talazoparib in patients with advanced solid tumors, and reported an objective response rate of 20% in 10 pancreatic cancer patients treated with 1.0 mg/day talazoparib. Of the two patients who showed a partial response, one harbored a mutation in BRCA2, and the other harbored a mutation in PALB2 (111). These results suggest that PARPi exerts a synthetic lethal effect in PALB2-deficient tumors. Several clinical trials of PARPi are currently in progress for breast cancer with mutations in DNA repair genes, including PALB2. A phase II trial is underway for the evaluation of PARPi olaparib monotherapy in the treatment of metastatic breast cancer patients harboring germline/somatic mutations in non-BRCA1/BRCA2 DNA repair genes (NCT03344965). A different phase II clinical trial of the PARPi talazoparib is being performed for non-BRCA1/BRCA2-mutated patients with advanced triple-negative breast cancer and HR deficiency or advanced HER2-negative solid tumors harboring a germline/somatic mutation in a HR pathway gene, such as PALB2 (NCT02401347). The outcomes of these trials are expected to expand the potential applications for PARPi therapy.
Overall, these data indicate that PALB2 status should be assessed and included in genetic counseling and patient treatment regimens for best clinical outcome.
The Challenges of PALB2 Research in Clinical Application
Population-based screening has identified numerous PALB2 variants, and the frequency-penetrance profiles of some ethnic-specific PALB2 PVs have been described. At least 604 distinct variants in PALB2 have been discovered according to an established database (https://databases.lovd.nl/shared/variants/PALB2/unique); however, only ~140 of the variants are thought to be pathogenic, whereas more than 400 are missense variants of unknown significance (VUSs). The lack of verification toward these VUSs challenges genetic counseling (27). Here, we summarize the breast cancer-associated missense variants of PALB2 that have been functionally verified (Figure 2). Pathogenic PALB2 missense variants are mainly located in the N- and C-terminus. In the PALB2 N-terminus, p.L35P (c.104T>C) disrupts BRCA1-PALB2 interaction and abolishes the HR activity of PALB2, resulting in sensitivity to the PARPi (106). Moreover, p.P8L (c.23C>T), p.K18R (c.53A>G), p.L24S (c.71T>C), p.Y28C (c.83A>G), and p.R37H (c.110G>A) compromise HR activity of PALB2 and are suggested to be pathogenic (106–108). In the PALB2 C-terminus, p.W912G, p.G937R, p.L939W, p.I944N (c.2831T>A), p.L947F (c.2841G>T), p.L947S (c.2840T>C), p.L961P, p.L972Q, p.A1025R, p.T1030I (c.3089C>T), p.I1037T, p.G1043D, p.L1070P (c.3209T>C), p.P1088S (c.3262C>T), p.W1140G (c.3418T>C), p.L1143P, and p.L1172P promoted a decrease in the HR activity of PALB2 (20, 107–109, 112). Among these mutations, p.L939W, p.A1025R, p.T1030I, p.P1088S, and p.L1143P disrupt BRCA2-PALB2 interaction; p.W912G, p.G937R, p.I944N, p.L961P, p.L972Q, p.T1030I, p.I1037T, p.G1043D, and p.L1172P are associated with PALB2 protein instability; and p.I944N, p.L947F, p.L947S, p.T1030I, p.L1070P, and p.W1140G result in the mislocalization of PALB2 to the cytoplasm. However, p.L35P remains the only recognized PALB2 pathogenic missense variant validated by systematic in vitro functional assays. Further functional analysis and people-based screening data are needed to properly evaluate the pathogenicity of PALB2 VUSs.
Conclusions and Perspectives
To date, the specific structures, multifaceted functions, and complex regulatory networks of PALB2 have been elaborated by multiple studies. PALB2 is a crucial regulator in maintaining genome integrity, while its dysfunction leads to breast cancer predisposition. The clinical relevance of PALB2 has been partially described, and PALB2 is reported to be a high-risk breast cancer susceptibility gene comparable to BRCA2 (16). With the identification of deleterious PALB2 recurrent mutations and PARPi, individualized risk assessment and precision medicine for PALB2 mutation-associated breast cancer become possible. Nevertheless, caution is warranted before promoting specific treatments, such as preventive surgery, as the existing experimental and clinical data are not sufficient. Moreover, plenty of PALB2 VUSs emerged in large-scale PALB2 screenings; however, their pathogenicity remains undefined, thereby precluding their clinical application. Altogether, to deliver individualized precision medicine, further long-term, population-based PALB2 mutation studies combined with systematic functional verification are required.
Author Contributions
YC and SW designed the subject of the review. SW, JZ, and KZ wrote the manuscript. SW, HC, ML, YL, and YS compiled the figures. JZ and YC reviewed the manuscript. All the authors read and approved the final manuscript.
Funding
This work was supported by the Key Program of the Natural Science Foundation of Zhejiang Province (LZ16H160002), the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents, the National Natural Science Foundation of China (81702866), and the Preclinical and Multi-center Basket Clinical Trial of the Multi-kinase Inhibitor TT-00420 (2019ZX09301158).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ATM, ataxia telangiectasia mutated protein; ATR, ATM and Rad3-related kinase; CDK, cyclin-dependent kinase; ChAM, chromatin-association motif; CI, confidence interval; DSBs, DNA double-strand breaks; FA, Fanconi anemia; FPC, familial pancreatic cancer; H3K36me3, lysine 36-trimethylated histone H3; HR, homologous recombination; MBC, male breast cancer; MMC, mitomycin C; MRN, Mre11–RAD50–Nbs1 complex; NES, nuclear export sequence; OR, odds ratio; PALB2, partner and localizer of BRCA2; PARP, poly (ADP-ribose) polymerase; PARPi, PARP inhibitor; PV, pathogenic variant; ROS, reactive oxygen species; RPA, replication protein A; SETD2, SET domain containing 2; SSA, single-strand annealing; ssDNA, single-stranded DNA; VUSs, variants of unknown significance.
References
1. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. (2006) 22:719–29. doi: 10.1016/j.molcel.2006.05.0222
2. Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. (2009) 7:1110–8. doi: 10.1158/1541-7786.MCR-09-0123
3. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. (2009) 106:7155–60. doi: 10.1073/pnas.0811159106
4. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. (2009) 19:524–9. doi: 10.1016/j.cub.2009.02.018
5. Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. (2007) 39:159–61. doi: 10.1038/ng1942
6. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. (2007) 39:165–7. doi: 10.1038/ng1959
7. Ohmoto A, Yachida S, Morizane C. Genomic features and clinical management of patients with hereditary pancreatic cancer syndromes and familial pancreatic cancer. Int J Mol Sci. (2019) 20:E561. doi: 10.3390/ijms20030561
8. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. (2016) 2:482–90. doi: 10.1001/jamaoncol.2015.5495
9. Braym F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
10. Ollier M, Radosevic-Robin N, Kwiatkowski F, Ponelle F, Viala S, Privat M, et al. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. Am J Cancer Res. (2015) 5:2113–26.
11. Turnbull C, Sud A, Houlston RS. Cancer genetics, precision prevention and a call to action. Nat Genet. (2018) 50:1212–8. doi: 10.1038/s41588-018-0202-0
12. Cui J, Antoniou AC, Dite GS, Southey MC, Venter DJ, Easton DF, et al. After BRCA1 and BRCA2-what next? Multifactorial segregation analyses of three-generation, population-based Australian families affected by female breast cancer. Am J Hum Genet. (2001) 68:420–31. doi: 10.1086/318187
13. Thompson D, Easton D. The genetic epidemiology of breast cancer genes. J Mammary Gland Biol Neoplasia. (2004) 9:221–36. doi: 10.1023/B:JOMG.0000048770.90334.3b
14. Melchor L, Benitez J. The complex genetic landscape of familial breast cancer. Hum Genet. (2013) 132:845–63. doi: 10.1007/s00439-013-1299-y
15. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. (2017) 3:1190–6. doi: 10.1001/jamaoncol.2017.0424
16. Castera L, Harter V, Muller E, Krieger S, Goardon N, Ricou A, et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet Med. (2018) 20:1677–86. doi: 10.1038/s41436-018-0005-9
17. Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife. (2017) 6:e21350. doi: 10.7554/eLife.21350
18. Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. Embo Rep. (2009) 10:990–6. doi: 10.1038/embor.2009.126
19. Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, et al. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase eta in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. (2014) 6:553–64. doi: 10.1016/j.celrep.2014.01.009
20. Park JY, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, et al. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. (2014) 33:4803–12. doi: 10.1038/onc.2013.421
21. Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, et al. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. Elife. (2017) 6:e20922. doi: 10.7554/eLife.20922
22. Pauty J, Couturier AM, Rodrigue A, Caron MC, Coulombe Y, Dellaire G, et al. Cancer-causing mutations in the tumor suppressor PALB2 reveal a novel cancer mechanism using a hidden nuclear export signal in the WD40 repeat motif. Nucleic Acids Res. (2017) 45:2644–57. doi: 10.1093/nar/gkx011
23. Bleuyard JY, Buisson R, Masson JY, Esashi F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. Embo Rep. (2012) 13:135–41. doi: 10.1038/embor.2011.243
24. Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. (2010) 17:1247–54. doi: 10.1038/nsmb.1915
25. Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol. (2010) 17:1255–9. doi: 10.1038/nsmb.1916
26. Deveryshetty J, Peterlini T, Ryzhikov M, Brahiti N, Dellaire G, Masson JY, et al. Novel RNA and DNA strand exchange activity of the PALB2 DNA binding domain and its critical role for DNA repair in cells. Elife. (2019) 8:e44063. doi: 10.7554/eLife.44063
27. Ducy M, Sesma-Sanz L, Guitton-Sert L, Lashgari A, Gao Y, Brahiti N, et al. The tumor suppressor PALB2: inside out. Trends Biochem Sci. (2019) 44:226–40. doi: 10.1016/j.tibs.2018.10.008
28. Sy SM, Huen MS, Chen J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J Biol Chem. (2009) 284:21127–31. doi: 10.1074/jbc.C109.023937
29. Bertram MJ, Pereira-Smith OM. Conservation of the MORF4 related gene family: identification of a new chromo domain subfamily and novel protein motif. Gene. (2001) 266:111–21. doi: 10.1016/S0378-1119(01)00372-9
30. Bowman BR, Moure CM, Kirtane BM, Welschhans RL, Tominaga K, Pereira-Smith OM, et al. Multipurpose MRG domain involved in cell senescence and proliferation exhibits structural homology to a DNA-interacting domain. Structure. (2006) 14:151–8. doi: 10.1016/j.str.2005.08.019
31. Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, et al. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. (2006) 34:6621–8. doi: 10.1093/nar/gkl989
32. Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. (2005) 280:35261–71. doi: 10.1074/jbc.M504012200
33. Bleuyard JY, Fournier M, Nakato R, Couturier AM, Katou Y, Ralf C, et al. MRG15-mediated tethering of PALB2 to unperturbed chromatin protects active genes from genotoxic stress. Proc Natl Acad Sci USA. (2017) 114:7671–6. doi: 10.1073/pnas.1620208114
34. Hayakawa T, Zhang F, Hayakawa N, Ohtani Y, Shinmyozu K, Nakayama J, et al. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. J Cell Sci. (2010) 123:1124–30. doi: 10.1242/jcs.060178
35. Martrat G, Maxwell CM, Tominaga E, Porta-de-la-Riva M, Bonifaci N, Gomez-Baldo L, et al. Exploring the link between MORF4L1 and risk of breast cancer. Breast Cancer Res. (2011) 13:R40. doi: 10.1186/bcr2862
36. Gardini A, Baillat D, Cesaroni M, Shiekhattar R. Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J. (2014) 33:890–905. doi: 10.1002/embj.201385567
37. Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, et al. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. (2014) 206:493–507. doi: 10.1083/jcb.201404111
38. Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. (2012) 32:1506–17. doi: 10.1128/MCB.06271-11
39. Iliakis G, Wang Y, Guan J, Wang H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene. (2003) 22:5834–47. doi: 10.1038/sj.onc.1206682
40. Menzel T, Nahse-Kumpf V, Kousholt AN, Klein DK, Lund-Andersen C, Lees M, et al. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. Embo Rep. (2011) 12:705–12. doi: 10.1038/embor.2011.99
41. Simhadri S, Vincelli G, Huo Y, Misenko S, Foo TK, Ahlskog J, et al. PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response. Oncogene. (2019) 38:1585–96. doi: 10.1038/s41388-018-0535-2
42. Sy SM, Huen MS, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem. (2009) 284:18302–10. doi: 10.1074/jbc.M109.016717
43. Buisson R, Masson JY. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. (2012) 40:10312–23. doi: 10.1093/nar/gks807
44. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. (2004) 24:708–18. doi: 10.1128/MCB.24.2.708-718.2004
45. Song F, Li M, Liu G, Swapna GVT, Daigham NS, Xia B, et al. Antiparallel coiled-coil interactions mediate the homodimerization of the DNA damage-repair protein PALB2. Biochemistry. (2018) 57:6581–91. doi: 10.1021/acs.biochem.8b00789
46. Guo Y, Feng W, Sy SM, Huen MS. ATM-dependent phosphorylation of the Fanconi anemia protein PALB2 promotes the DNA damage response. J Biol Chem. (2015) 290:27545–56. doi: 10.1074/jbc.M115.672626
47. Ahlskog JK, Larsen BD, Achanta K, Sorensen CS. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. Embo Rep. (2016) 17:671–81. doi: 10.15252/embr.201541455
48. Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, et al. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell. (2017) 65:336–46. doi: 10.1016/j.molcel.2016.12.007
49. Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. (2015) 528:422–6. doi: 10.1038/nature16142
50. Zong D, Adam S, Wang Y, Sasanuma H, Callen E, Murga M, et al. BRCA1 Haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Mol Cell. (2019) 73:1267–81.e7. doi: 10.1016/j.molcel.2018.12.010
51. Porro A, Sartori AA. Context matters: RNF168 connects with PALB2 to rewire homologous recombination in BRCA1 haploinsufficiency. Mol Cell. (2019) 73:1089–91. doi: 10.1016/j.molcel.2019.03.005
52. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. (2009) 668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013
53. Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. (2007) 8:735–48. doi: 10.1038/nrg2159
54. Gueiderikh A, Rosselli F, Neto JBC. A never-ending story: the steadily growing family of the FA and FA-like genes. Genet Mol Biol. (2017) 40:398–407. doi: 10.1590/1678-4685-gmb-2016-0213
55. Knies K, Inano S, Ramirez MJ, Ishiai M, Surralles J, Takata M, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. (2017) 127:3013–27. doi: 10.1172/JCI92069
56. Bick G, Zhang F, Meetei AR, Andreassen PR. Coordination of the recruitment of the FANCD2 and PALB2 Fanconi anemia proteins by an ubiquitin signaling network. Chromosoma. (2017) 126:417–30. doi: 10.1007/s00412-016-0602-9
57. Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. (2010) 70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012
58. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. (2017) 317:2402–16. doi: 10.1001/jama.2017.7112
59. Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. (2013) 105:812–22. doi: 10.1093/jnci/djt095
60. Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. (2007) 446:316–9. doi: 10.1038/nature05609
61. Erkko H, Dowty JG, Nikkila J, Syrjakoski K, Mannermaa A, Pylkas K, et al. Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res. (2008) 14:4667–71. doi: 10.1158/1078-0432.CCR-08-0210
62. Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. (2003) 72:1117–30. doi: 10.1086/375033
63. Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. (2010) 12:R109. doi: 10.1186/bcr2796
64. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. New Engl J Med. (2014) 371:497–506. doi: 10.1056/NEJMc1410673
65. Southey MC, Goldgar DE, Winqvist R, Pylkas K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. (2016) 53:800–11. doi: 10.1136/jmedgenet-2016-103839
66. Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun. (2018) 9:4083. doi: 10.1038/s41467-018-06581-8
67. Decker B, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J Med Genet. (2017) 54:732–41. doi: 10.1158/1538-7445.AM2016-4309
68. Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol. (2019) Jco1901907. doi: 10.1200/JCO.19.01907
69. Ly D, Forman D, Ferlay J, Brinton LA, Cook MB. An international comparison of male and female breast cancer incidence rates. Int J Cancer. (2013) 132:1918–26. doi: 10.1002/ijc.27841
70. Korde LA, Zujewski JA, Kamin L, Giordano S, Domchek S, Anderson WF, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol. (2010) 28:2114–22. doi: 10.1200/JCO.2009.25.5729
71. Rizzolo P, Silvestri V, Tommasi S, Pinto R, Danza K, Falchetti M, et al. Male breast cancer: genetics, epigenetics, and ethical aspects. Ann Oncol. (2013) 24 (Suppl. 8):viii75–82. doi: 10.1093/annonc/mdt316
72. Adank MA, van Mil SE, Gille JJ, Waisfisz Q, Meijers-Heijboer H. PALB2 analysis in BRCA2-like families. Breast Cancer Res Treat. (2011) 127:357–62. doi: 10.1007/s10549-010-1001-1
73. Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. (2011) 71:2222–9. doi: 10.1158/0008-5472.CAN-10-3958
74. Ding YC, Steele L, Kuan CJ, Greilac S, Neuhausen SL. Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res Treat. (2011) 126:771–8. doi: 10.1007/s10549-010-1195-2
75. Vietri MT, Caliendo G, Casamassimi A, Cioffi M, De Paola ML, Napoli C, et al. A novel PALB2 truncating mutation in an Italian family with male breast cancer. Oncol Rep. (2015) 33:1243–7. doi: 10.3892/or.2014.3685
76. Silvestri V, Zelli V, Valentini V, Rizzolo P, Navazio AS, Coppa A, et al. Whole-exome sequencing and targeted gene sequencing provide insights into the role of PALB2 as a male breast cancer susceptibility gene. Cancer. (2017) 123:210–8. doi: 10.1002/cncr.30337
77. Pritzlaff M, Summerour P, McFarland R, Li S, Reineke P, Dolinsky JS, et al. Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat. (2017) 161:575–86. doi: 10.1007/s10549-016-4085-4
78. Rizzolo P, Zelli V, Silvestri V, Valentini V, Zanna I, Bianchi S, et al. Insight into genetic susceptibility to male breast cancer by multigene panel testing: results from a multicenter study in Italy. Int J Cancer. (2019) 145:390–400. doi: 10.1158/1538-7445.AM2018-1236
79. Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. (2012) 23 (Suppl. 6):vi7–12. doi: 10.1093/annonc/mds187
80. Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. (2009) 15:3214–22. doi: 10.1158/1078-0432.CCR-08-3128
81. Garcia MJ, Fernandez V, Osorio A, Barroso A, Llort G, Lazaro C, et al. Analysis of FANCB and FANCN/PALB2 Fanconi anemia genes in BRCA1/2-negative Spanish breast cancer families. Breast Cancer Res Treat. (2009) 113:545–51. doi: 10.1007/s10549-008-9945-0
82. Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, et al. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. (2010) 11:20. doi: 10.1186/1471-2350-11-20
83. Cybulski C, Kluzniak W, Huzarski T, Wokolorczyk D, Kashyap A, Jakubowska A, et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol. (2015) 16:638–44. doi: 10.1016/S1470-2045(15)70142-7
84. Shimelis H, LaDuca H, Hu C, Hart SN, Na J, Thomas A, et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J Natl Cancer Inst. (2018) 110:855–62. doi: 10.1093/jnci/djy106
85. Deng M, Chen HH, Zhu X, Luo M, Zhang K, Xu CJ, et al. Prevalence and clinical outcomes of germline mutations in BRCA1/2 and PALB2 genes in 2769 unselected breast cancer patients in China. Int J Cancer. (2019) 145:1517–28. doi: 10.1002/ijc.32184
86. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. (2009) 324:217. doi: 10.1126/science.1171202
87. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. (2010) 78:490–4. doi: 10.1111/j.1399-0004.2010.01425.x
88. Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget. (2016) 7:74227–35. doi: 10.18632/oncotarget.12490
89. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. (2011) 108:18032–7. doi: 10.1073/pnas.1115052108
90. Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. (2014) 5:3156. doi: 10.1038/ncomms4156
91. Sahasrabudhe R, Lott P, Bohorquez M, Toal T, Estrada AP, Suarez JJ, et al. Germline mutations in PALB2, BRCA1, and RAD51C, which regulate DNA recombination repair, in patients with gastric cancer. Gastroenterology. (2017) 152:983–6.e6. doi: 10.1053/j.gastro.2016.12.010
92. Fewings E, Larionov A, Redman J, Goldgraben MA, Scarth J, Richardson S, et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol Hepatol. (2018) 3:489–98. doi: 10.1016/S2468-1253(18)30079-7
93. Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. (2018) 173:355–70.e14. doi: 10.1158/1538-7445.AM2018-5359
94. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. New Engl J Med. (2016) 375:443–53. doi: 10.1056/NEJMc1611137
95. Thompson ER, Rowley SM, Li N, McInerny S, Devereux L, Wong-Brown MW, et al. Panel testing for familial breast cancer: calibrating the tension between research and clinical care. J Clin Oncol. (2016) 34:1455–9. doi: 10.1200/JCO.2015.63.7454
96. Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. (2007) 9:R83. doi: 10.1186/bcr1828
97. Catucci I, Peterlongo P, Ciceri S, Colombo M, Pasquini G, Barile M, et al. PALB2 sequencing in Italian familial breast cancer cases reveals a high-risk mutation recurrent in the province of Bergamo. Genet Med. (2014) 16:688–94. doi: 10.1038/gim.2014.13
98. Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. (2016) 18:823–32. doi: 10.1038/gim.2016.21
99. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. (2017) 123:1721–30. doi: 10.1002/cncr.30498
100. Kraus C, Hoyer J, Vasileiou G, Wunderle M, Lux MP, Fasching PA, et al. Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int J Cancer. (2017) 140:95–102. doi: 10.1002/ijc.30428
101. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. (2019). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed February 25, 2020).
102. Wei H, Yu X. Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. (2016) 14:131–9. doi: 10.1016/j.gpb.2016.05.001
103. Sachdev E, Tabatabai R, Roy V, Rimel BJ, Mita MM. PARP Inhibition in cancer: an update on clinical development. Target Oncol. (2019) 14:657–79. doi: 10.1007/s11523-019-00680-2
104. Keung MYT, Wu Y, Vadgama JV. PARP inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J Clin Med. (2019) 8:E435. doi: 10.3390/jcm8040435
105. Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. (2013) 19:5003–15. doi: 10.1158/1078-0432.CCR-13-1391
106. Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. (2017) 36:4161–70. doi: 10.1038/onc.2017.46
107. Rodrigue A, Margaillan G, Torres Gomes T, Coulombe Y, Montalban G, da Costa ESCS, et al. A global functional analysis of missense mutations reveals two major hotspots in the PALB2 tumor suppressor. Nucleic Acids Res. (2019) 47:10662–77. doi: 10.1093/nar/gkz780
108. Wiltshire T, Ducy M, Foo TK, Hu C, Lee KY, Belur Nagaraj A, et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genet Med. (2019). doi: 10.1038/s41436-019-0682-z (accessed October 21, 2019).
109. Boonen R, Rodrigue A, Stoepker C, Wiegant WW, Vroling B, Sharma M, et al. Functional analysis of genetic variants in the high-risk breast cancer susceptibility gene PALB2. Nat Commun. (2019) 10:5296. doi: 10.1038/s41467-019-13194-2
110. Smith MA, Hampton OA, Reynolds CP, Kang MH, Maris JM, Gorlick R, et al. Initial testing (stage 1) of the PARP inhibitor BMN 673 by the pediatric preclinical testing program: PALB2 mutation predicts exceptional in vivo response to BMN 673. Pediatr Blood Cancer. (2015) 62:91–8. doi: 10.1002/pbc.25201
111. de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor Talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. (2017) 7:620–9. doi: 10.1158/2159-8290.CD-16-1250
Keywords: PALB2, homologous recombination, breast cancer, precision medicine, pathogenic variants
Citation: Wu S, Zhou J, Zhang K, Chen H, Luo M, Lu Y, Sun Y and Chen Y (2020) Molecular Mechanisms of PALB2 Function and Its Role in Breast Cancer Management. Front. Oncol. 10:301. doi: 10.3389/fonc.2020.00301
Received: 01 December 2019; Accepted: 20 February 2020;
Published: 28 February 2020.
Edited by:
Gulisa Turashvili, University of Toronto, CanadaReviewed by:
Jean-Yves Masson, Laval University, CanadaBing Xia, Rutgers Cancer Institute of New Jersey, United States
Claudia Wiese, Colorado State University, United States
Copyright © 2020 Wu, Zhou, Zhang, Chen, Luo, Lu, Sun and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiding Chen, eWRjaGVuJiN4MDAwNDA7emp1LmVkdS5jbg==
†These authors have contributed equally to this work