 Yolla Haibe1†
Yolla Haibe1† Malek Kreidieh1†
Malek Kreidieh1† Hiba El Hajj1,2
Hiba El Hajj1,2 Ibrahim Khalifeh3
Ibrahim Khalifeh3 Deborah Mukherji1
Deborah Mukherji1 Sally Temraz1
Sally Temraz1 Ali Shamseddine1*
Ali Shamseddine1*- 1Division of Hematology/Oncology, Department of Internal Medicine, American University of Beirut-Medical Center, Beirut, Lebanon
- 2Department of Experimental Pathology, Immunology and Microbiology, American University of Beirut-Medical Center, Beirut, Lebanon
- 3Department of Pathology and Laboratory Medicine, American University of Beirut Medical Center, Beirut, Lebanon
Tumor growth and metastasis rely on tumor vascular network for the adequate supply of oxygen and nutrients. Tumor angiogenesis relies on a highly complex program of growth factor signaling, endothelial cell (EC) proliferation, extracellular matrix (ECM) remodeling, and stromal cell interactions. Numerous pro-angiogenic drivers have been identified, the most important of which is the vascular endothelial growth factor (VEGF). The importance of pro-angiogenic inducers in tumor growth, invasion and extravasation make them an excellent therapeutic target in several types of cancers. Hence, the number of anti-angiogenic agents developed for cancer treatment has risen over the past decade, with at least eighty drugs being investigated in preclinical studies and phase I-III clinical trials. To date, the most common approaches to the inhibition of the VEGF axis include the blockade of VEGF receptors (VEGFRs) or ligands by neutralizing antibodies, as well as the inhibition of receptor tyrosine kinase (RTK) enzymes. Despite promising preclinical results, anti-angiogenic monotherapies led only to mild clinical benefits. The minimal benefits could be secondary to primary or acquired resistance, through the activation of alternative mechanisms that sustain tumor vascularization and growth. Mechanisms of resistance are categorized into VEGF-dependent alterations, non-VEGF pathways and stromal cell interactions. Thus, complementary approaches such as the combination of these inhibitors with agents targeting alternative mechanisms of blood vessel formation are urgently needed. This review provides an updated overview on the pathophysiology of angiogenesis during tumor growth. It also sheds light on the different pro-angiogenic and anti-angiogenic agents that have been developed to date. Finally, it highlights the preclinical evidence for mechanisms of angiogenic resistance and suggests novel therapeutic approaches that might be exploited with the ultimate aim of overcoming resistance and improving clinical outcomes for patients with cancer.
Introduction
Angiogenesis
Angiogenesis is the process of formation of new blood vessels from pre-existing vessels. It is a highly regulated process that involves migration, growth, and differentiation of endothelial cells (ECs). This regulated mechanism is crucial in embryonic development, wound healing, and reproduction (1). Nonetheless, alterations in any of its regulatory pathways may lead to metabolic diseases, cardiovascular disorders, diabetic retinopathy, psoriasis, systemic lupus erythematosus, and importantly tumor growth and metastasis (2–5).
In the avascular phase, tumor growth is usually restricted in size due to a balance between pro-angiogenic and anti-angiogenic factors that control vascular homeostasis (6). Beyond a few millimeters in size, solid tumors build, and increase their own blood supply to provide adequate oxygen and nutrients (Figure 1). This process, referred to as the angiogenic switch, from an avascular state to an angiogenic phase, is crucial for tumors to grow and continue unrestricted proliferation (7). Hence, unlike normal physiological processes favoring negative regulation of angiogenesis, tumors favor its upregulation.
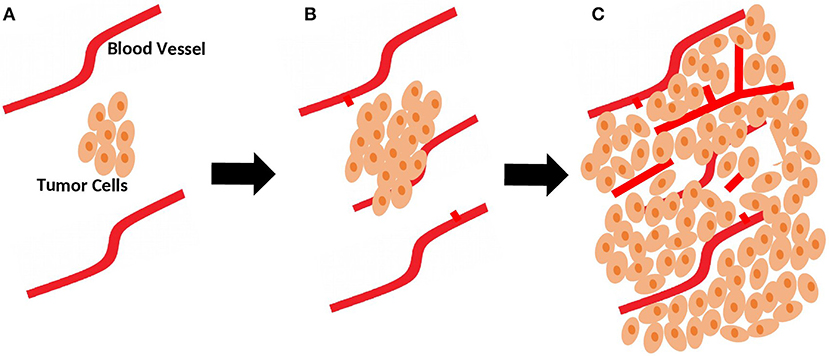
Figure 1. Role of sprouting angiogenesis in tumor growth. (A) During early stages of development, tumor is still small in size and relies on local existing blood vessels for oxygen and nutrients supply. (B) As the tumor grows, sprouting of new vessels from local existing blood vessels occurs to fulfill the need for more oxygen and nutrients supplies. (C) Sprouting angiogenesis results in a more complex network of vasculature to provide adequate blood supply for the growing tumor.
Multiple non-mutually exclusive mechanisms have been described as major players in tumor neovascularization. These include sprouting angiogenesis, non-sprouting angiogenesis, vasculogenesis, vasculogenic mimicry, and intussusception. Sprouting angiogenesis, however, remains the most well-studied mechanism used by tumor cells to produce their vasculature (8). Due to the importance of this latter process in tumor cell growth, invasion, and extravasation, different angiogenesis inhibitors (AIs) have been developed.
In this review, we will discuss the different driver molecules promoting angiogenesis in cancer. These include the angiogenic or angiostatic chemokines, the contribution of the endothelial progenitor cells (EPCs), the tumor vasculogenic mimicry, the markers for tumor-derived ECs, and pericytes. We will also provide an overview on the clinically tested anti-angiogenic drugs slowing down angiogenesis and leading to tumor starvation. Finally, the resistance mechanisms arising in cancer cells against these drugs and the potential therapeutic solutions will be discussed.
Angiogenesis: Pathophysiology During Tumor Growth
Unlike normal angiogenesis and neovascularization, tumor angiogenesis is an uncontrolled and disorganized process. It results in vessels with thin walls, incomplete basement membranes, and atypical pericytes (8). Since the needs of rapid tumor cell proliferation surpass the capacity of host vasculature, hypoxia and low supplies of nutrients characterize early stages of tumor development. Hypoxia triggers the expression of pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) (9–11).
Matrix metalloproteinases (MMPs) secreted by tumor cells degrade the basement membrane as a first essential step to initiate angiogenesis (12). This alters cell-cell interactions and facilitates the migration of ECs through the created gap into the tumor mass, which in turn results in the proliferation and formation of new blood vessels, followed by vessel pruning and pericyte stabilization (Figure 2).
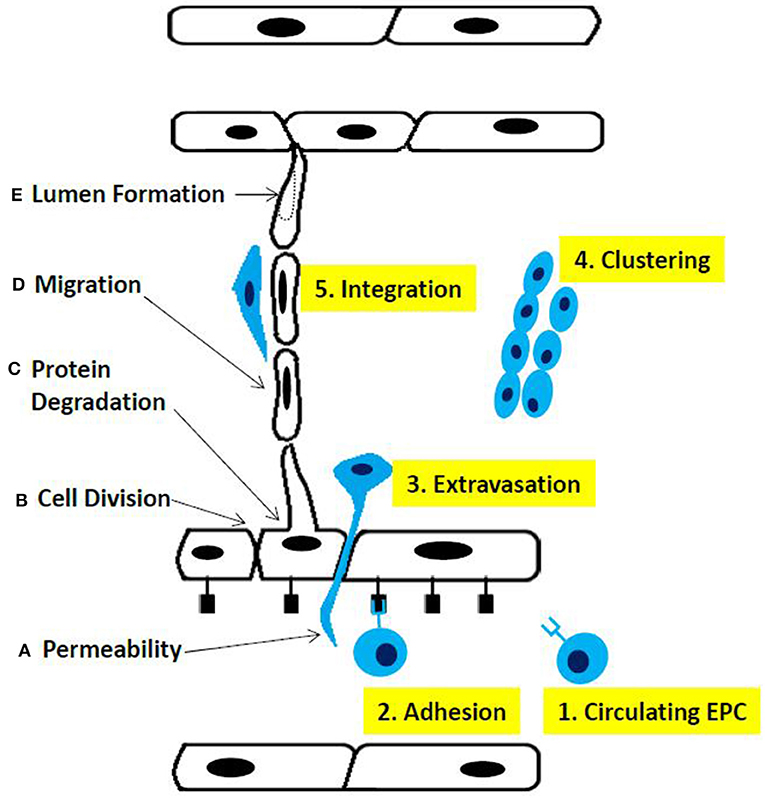
Figure 2. Phases of sprouting angiogenesis. (A) increased permeability across the endothelial cell layer, (B) cell division, (C) proteolysis of basement membrane components, (D) migration ofthe endothelial cells, and (E) lumen fonnation. Altematively, (1) circulating endothelial progenitor cells contribute to the sprouting mechanism, (2) adhere to endothelial cells, (3) extravagate through the endothelial cell layer, (4) cluster together, and (5) integrate into the sprout fonned by endothelial cells.
Angiogenesis: Regulation
Angiogenesis is a tightly balanced mechanism regulated by both pro-angiogenic and anti-angiogenic factors (13). In malignant tumors, this balance is shifted toward a pro-angiogenic milieu to maintain sustainable angiogenic processes (14). Involved soluble growth factors include VEGF, PDGF, fibroblast growth factor (FGF)-2, angiopoietins (Angs), transforming growth factors (TGFs)- beta and alpha, and epidermal growth factors (EGF). Insoluble membrane-bound factors include integrins, ephrins, cadherins, MMPs, and hypoxia inducible factor-1 (HIF-1).
From these, VEGF was broadly studied and shown to significantly contribute to the induction and progression of angiogenesis (15). We will start by listing the different members of the VEGF family. In the following sections, a general overview on the role of the other angiogenic factors in normal and tumor angiogenesis will be described. In addition, direct and indirect angiogenesis inhibitory mechanisms will be discussed.
Vascular Endothelial Growth Factor Family
The VEGF family comprises seven members, VEGFs A to F and placenta growth factor (PGF) (16). These members are ligands that interact with multiple receptors present on the vascular endothelium (17) (Figure 3).
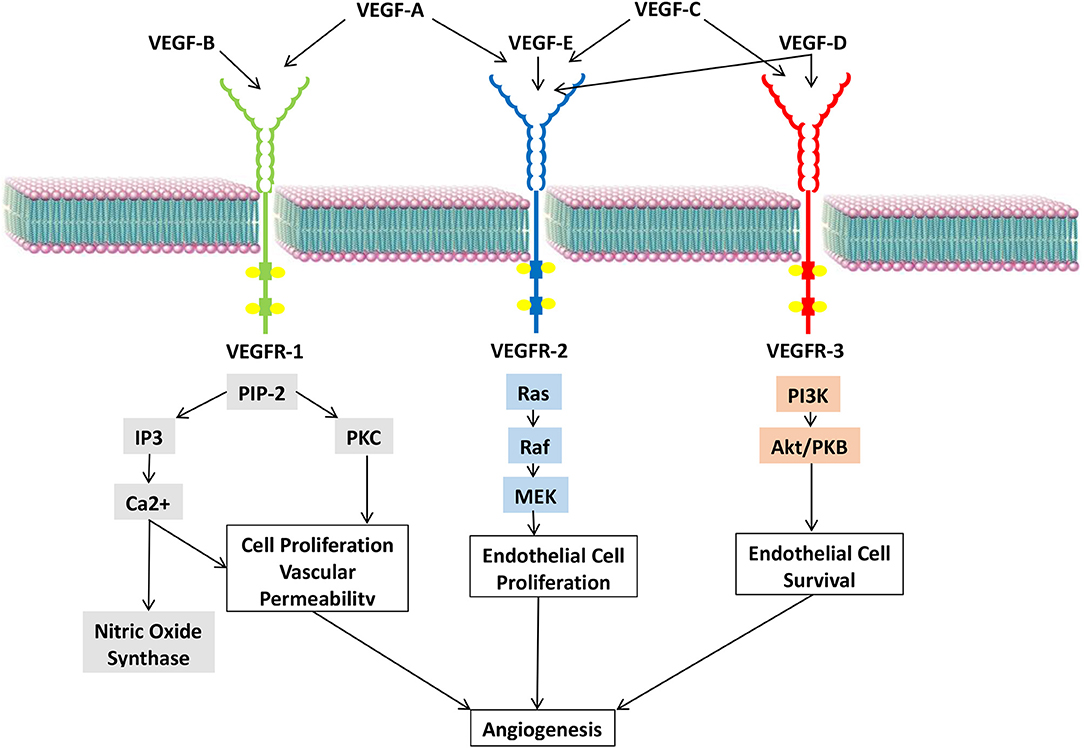
Figure 3. VEGF ligands, their receptors, and respective signaling pathways.
Vascular Endothelial Growth Factor A
VEGF-A is the most potent angiogenic factor that is encoded by a gene located on the short arm of chromosome six (18). Its interaction with the transmembrane tyrosine kinase receptors, VEGF receptors (VEGFRs)-1 and 2, and their co-receptors, NRPs-1 and 2, present on vascular ECs results in the dimerization and phosphorylation of intracellular receptors (19). This further activates downstream signaling cascades involving phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), mitogen-activated protein kinase (MAPK), and extracellular regulated kinase (ERK) (20, 21).
VEGF-A expression is stimulated by hypoxia, growth factors, and cytokines such as IL-1, EGFs, PDGFs, and tumor necrosis factor (TNF)-α (16). It was noted in most solid tumors and some hematologic malignancies (20). VEGF-A is considered the backbone of angiogenesis during physiologic as well as pathologic processes. The deletion of one or both VEGF-A alleles in mouse pre-clinical models resulted in either vascular abnormalities or complete absence of vasculature leading to death (22). Interestingly, a striking positive correlation between the level of VEGF-A expression, tumor progression, and cancer patients' survival was observed (23, 24).
Vascular Endothelial Growth Factor B
VEGF-B is encoded by a gene located on chromosome eleven. It differs from VEGF-A by its promotor region (25, 26). It was found to be upregulated in many types of tumors including prostate, kidney, and colorectal cancers (CRCs) (27, 28). Since the VEGF-B promoter lacks the HIF-1 and AP-1 sites found in the VEGF-A promotor, stimuli such as hypoxia or cold do not induce VEGF-B expression (29, 30).
A study was conducted to explore the role of VEGF-B in cancer development. Results revealed that VEGF-B-deficient transgenic mice with pancreatic endocrine adenocarcinoma had larger tumors compared to transgenic expression of VEGF-B but no difference in tumor vasculature (31). In addition, knockout studies have highlighted the role of VEGF-B in inflammatory angiogenesis and regeneration of coronary collaterals through arteriogenesis (32, 33).
Vascular Endothelial Growth Factors C, D, and E
The VEGF-C encoding gene is located on chromosome four (34–36). Experiments performed on transgenic mice demonstrated the ability of VEGF-C to induce selective lymphangiogenesis without accompanying angiogenesis (37). Several studies showed a positive correlation between VEGF-C expression, lymphatic invasion, metastasis, and survival in cancer patients. For instance, while the 2-year survival rate of patients with uterine cervical cancers with high VEGF-C level in metastatic lymph nodes was 38%, that of patients with normal levels was 81% (38, 39).
VEGF-D is closely related to VEGF-C with which it shares 61% homology (40). Similar to VEGF-C, VEGF-D can bind and activate the VEGFRs 2 and 3 (41, 42). Depending on the activated receptor, separate downstream cascades are activated to induce the growth and proliferation of ECs in the vascular and lymphatic systems (43). As such, VEGF-D activity is crucial for hypoxia-induced vascular development (44) in melanoma, lung, breast, pancreatic, and esophageal cancer (43, 45–48).
VEGF-E is a potent angiogenic factor. Its isoform, VEGF-E nz-7, binds with high affinity to VEGFR-2 to stimulate efficient angiogenesis and increase vascular permeability (49).
Placental Growth Factor
PlGF is a member of the VEGF subfamily that binds to VEGFR-1 and its co-receptors, NRP-1 and 2. PlGF/VEGFR-1 signaling activates the downstream PI3K/Akt and p38 MAPK pathways independent of VEGFA signaling (50, 51). This stimulates the growth and migration of ECs, macrophages, and tumor cells (52, 53).
Upregulation of PlGF expression has been observed in tumors resistant to anti-VEGF therapy suggesting that PlGF might serve as a promising therapeutic target in this setting (54–57). In addition, PlGF knockout (pgf−/−) mice were noted to have normal embryonic angiogenesis and impaired pathological angiogenesis following exposure of their tumors to ischemia (58). This suggests that by neutralizing PlGF, pathological angiogenesis can be inhibited without affecting normal blood vessels (59).
Currently Approved Anti-angiogenic Therapies
Since sprouting angiogenesis plays an essential role in tumor growth, invasion, progression, and metastasis, targeting this process may potentially halt the growth and spread of cancer (60). Table 1 lists antiangiogenic agents approved for clinical use and their targets.
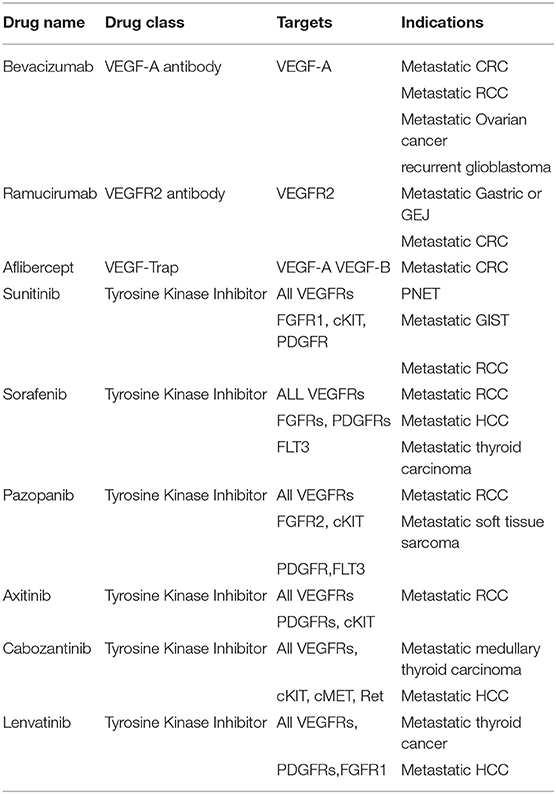
Table 1. List of some FDA-approved anti-angiogenic agents.
Angiogenesis inhibitors (AIs) are classified into direct and indirect agents. Direct endogenous inhibitors target vascular ECs and include endostatin, arrestin, and tumstatin. Unfortunately, phase II or III clinical trials did not result in significant effects on patients (14, 61). In the last decade, a number of molecules have been described, including semaphorins, netrins, slits, and others (62–64). Netrin-1, Netrin-4, and their receptors can have a repulsive or attractive signals in angiogenesis, partially via the regulation of VEGF signaling. There are still some contradictions reported on the positive and negative role of Netrin-1 in regulation of angiogenesis, and studies are still on going to identify its exact role in angiogenesis. Semaphorin-3A and Semaphorin-3E have negative effects on angiogenesis in central nervous system (CNS) and non-CNS tissues.
Indirect AIs target tumor cells or tumor associated stromal cells and include several types (14) (Table 2). They prevent the expression of pro-angiogenic factors or block their activity.
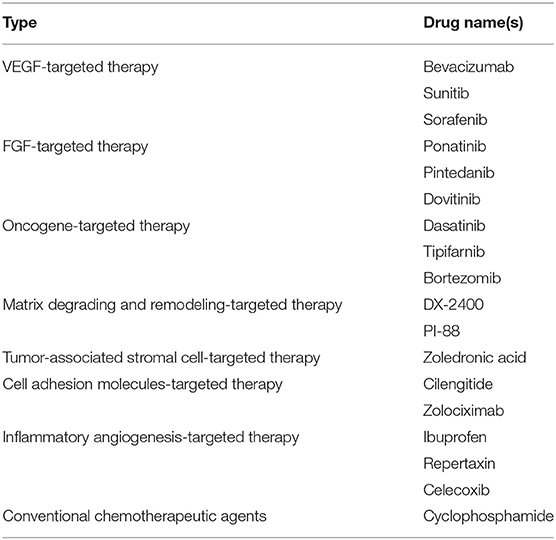
Table 2. List of indirect angiogenesis inhibitors.
Among the AIs, VEGF inhibitors were extensively studied and reached phase III clinical trials. They caused a modest increase in overall survival (OS) (65). Bevacizumab (BVZ), a humanized anti-VEGF monoclonal antibody, was the first drug to be approved by the Food and Drug Administration (FDA) for the treatment of metastatic colon, ovarian, renal, non-squamous cell lung cancer (NSCLC), and glioblastoma mutliforme (GBM) (66, 67). It failed to show clinical significance when used as monotherapy, except in GBM. In contrast, its clinical benefits were evident in association with other chemotherapeutic agents. For instance, since the tumor vasculature induced by VEGF is usually tortuous and dysfunctional, the use of BVZ was thought to normalize the blood vessel texture. It was also hypothesized that the combination of BVZ and chemotherapy increases the delivery of the chemotherapeutic agent to the cancer tissue by increasing its blood flow (68, 69). However, contrary evidence was reported by a decrease in cytotoxic drug delivery to tumors following treatment with AIs (70). Such inconsistency could be due to differences in blood vessel setups among various cancer types (71, 72). BVZ combined with chemotherapy was also studied in the adjuvant setting in colorectal cancer (CRC), but it failed to prove any clinical significance compared to chemotherapy alone in two phase III clinical trials (73–75).
Aflibercept is a soluble VEGF decoy receptor that consists of the extracellular domains of VEGFRs 1 and 2 and the Fc portion of human IgG1. It was FDA approved for the treatment of metastatic CRC in combination with 5-fluorouracil, leucovorin, and irinotecan in 2012 (76). Owing to its structure, Aflibercept can neutralize both, VEGF and PlGF (77). Compared to treatment with BVZ, the use of Aflibercept in patient-derived xenograft models resulted in higher tumor suppressive activity (78). Unfortunately, neutralizing both, PlGF and VEGF, had a minimal effect on tumor suppression in vivo (79). In a phase I clinical trial, relapsing GBM patients treated with BVZ monotherapy were compared to those treated with the combination of an anti-PlGF agent and BVZ. Similar results were obtained with no added benefit in the combination arm (80).
Unlike BVZ and Aflibercept, tyrosine kinase inhibitors, which are small molecules able to interact with the kinase domain on the VEGFRs, showed a remarkable clinical benefit when used as single agents, and with no added value when combined with chemotherapy. This was reported in the treatment of renal cell carcinoma (RCC), hepatocellular carcinoma (HCC), thyroid cancer, gastrointestinal stromal tumor (GIST), and pancreatic neuroendocrine tumor (PNET) (81).
Mechanisms of Resistance to Anti-Angiogenic Therapies and Ways to Overcome them
Although anti-angiogenesis therapies may prolong progression-free survival (PFS), they have limited impact on overall survival (OS) and do not constitute a permanent cure in RCC, CRC, or breast cancer (73, 75, 82, 83). This limited clinical significance might be due to different innate and acquired molecular resistance mechanisms with no clear genetic explanations (65). Hypoxia plays an important role in tumor resistance to chemotherapeutic agents favoring more aggressive metastatic disease and hence worse prognosis. HIF-1 plays a critical role in resistance to anti-angiogenic therapy and is the main survival factor used by cancer cells to adapt to oxygen deprivation (84, 85). In this section, an overview on different mechanisms of resistance to anti-angiogenic therapies in the clinical and preclinical settings will be discussed (Figure 4) and the ways to overcome them will be provided (Table 3). Some of these mechanisms are likely influenced by hypoxia. These include the production of alternative proangiogenic factors, the recruitment of BM-derived cells, the vasculogenic mimicry, as well as the increased tumor cell invasiveness and metastatic behavior.
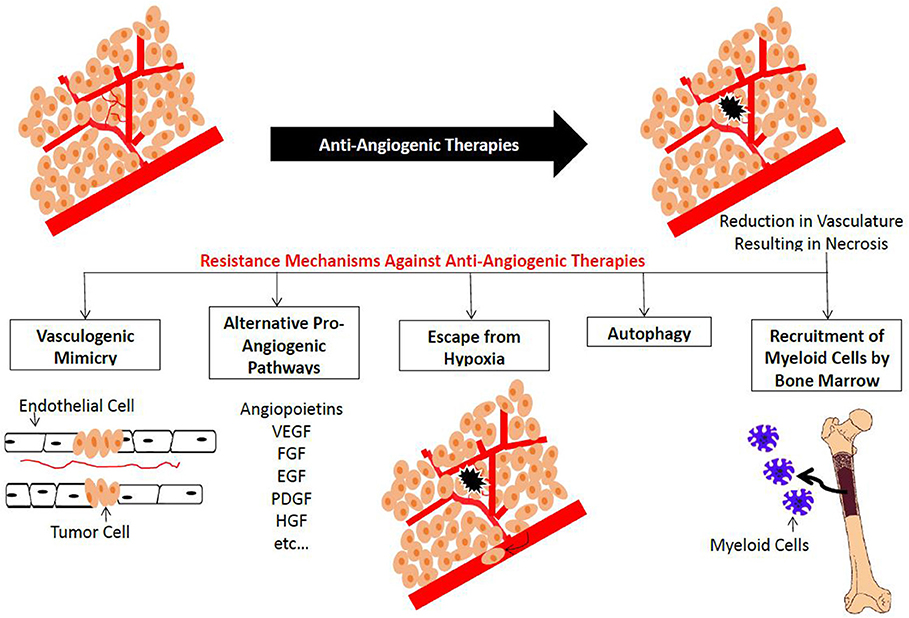
Figure 4. Summary of plausible resistance mechanisms to Anti-angiogenic Agents. Treatment with anti-angiogenic agents results in a reduction in the blood vessel network. This new hypoxic condition results in the activation of vascular mimicry, altemative pro-angiogenic pathways, recruitment of bone man·ow-derived EC precursors and myeloid cells, as well as cell survival mechanisms such as autophagy.

Table 3. List of mechanisms of resistance to anti-angiogenic therapies and ways to target them along with the outcomes associated with each approach.
Hypoxia Caused by Anti-angiogenic Therapies
Treatment with anti-angiogenic agents results in vascular regression and intra-tumoral hypoxia. Several studies have made use of pimonidazole injections, to demonstrate an increase in hypoxic regions in primary tumors following anti-angiogenic treatment (86, 89, 115). Further analysis showed a concomitant increase in HIF-1a expression during treatment.
HIF-1a and hypoxia are known drivers of EMT, a process that promotes tumor metastasis. Upregulation of EMT-related genes, such as Twist and Snail, have been noted following anti-angiogenic treatment. This is in addition to the loss of the epithelial marker, E-cadherin, and the induction of the mesenchymal marker, vimentin (86, 116). Hypoxic environments also induce upregulation of VEGF expression through the upstream transcription factor HIF-1a (117). These factors cause tumors to acquire more angiogenic and invasive capacities, thus promoting metastasis (118).
Effect of Hypoxia on the Hepatocyte Growth Factor/Tyrosine Protein Kinase Met Pathway
The increase in tumor invasiveness and metastasis in response to AI-induced hypoxia from anti-angiogenic therapies can be explained by the over-expression of the tyrosine protein kinase, c-MET. For instance, in vitro studies revealed a direct positive effect of hypoxia on c-MET and phospho-c-Met expression (87). Other studies confirmed that this promotion of c-MET transcription that follows hypoxic conditions occurs via the direct regulation of HIF-1 (119).
The HGF/c-MET pathway is one of the most investigated signaling pathways in tumors resistant to anti-VEGF therapy. Binding of HGF to c-MET activates MAPK/ERK cascades, STAT3 pathway, PI3K/Akt axis, and/or NF-κB inhibitor-α kinase (IKK)-NF-κB complex (119–121). This usually promotes tumor growth and invasiveness.
VEGF exerts a negative feedback on c-MET activation in a GBM mouse model, resulting in the direct suppression of tumor invasion (122). For instance, compared to GBM patients who were not treated with BVZ, those treated with BVZ had more recurrence rates and their tumors had an upregulation in c-MET expression (123). This increased invasiveness of GBM after BVZ treatment was recently linked to inhibitory actions of VEGF and to the increase in c-Met and phospho-c-Met expression upon treatment (122).
MET activation in response to hypoxia can occur in endothelial cells, as well as in tumor cells or other cells of the tumor microenvironment. In fact, in one study (124) this had very diverse functional impacts.
Blocking c-MET to Overcome Resistance to Anti-vascular Endothelial Growth Factor Treatment
To overcome the c-MET protein overexpression that occurs with the neutralization of VEGF by BVZ, the addition of a c-MET inhibitor would be helpful. In the phase III METEOR trial, the administration of the inhibitor of tyrosine kinases including MET, Cabozantinib, after previous vascular endothelial growth factor receptor-targeted therapy in patients with advanced RCC resulted in improved survival (125).
Effect of Hypoxia on β1 Integrin Expression
It is thought that the hypoxic microenvironment generated during anti-angiogenic therapy induces HIF-1α expression, thus stimulating β1 integrin expression. β1 integrin is the member that is mostly implicated in cancer treatment resistance, especially that its expression has been upregulated in clinical specimens of BVZ-resistant GBM tumors (126–128). The expression levels of integrins are correlated with disease progression and poor survival of patients (129, 130). Upon interacting with c-MET, integrins ultimately enhance tumor cell invasiveness (113, 131, 132).
Blocking β1 Integrin to Overcome Resistance to Anti-vascular Endothelial Growth Factor Treatment
Several preclinical studies have demonstrated benefit from β1 integrin blockade in BVZ-resistant and non-resistant GBM tumors in xenograft models (113, 114).
Increased Tumor Invasiveness and Metastasis
Despite their overall inhibition of tumor growth, therapeutic AIs were associated with increased local invasiveness and distant metastasis. These phenomena seem to be major contributors to resistance against anti-angiogenesis therapies. They were first described by Ebos et al. and Paez-Ribes et al. in different preclinical models (115, 133).
Angiogenesis blockade enhances tumor invasiveness. For instance, RCC cells demonstrated an accelerated growth capacity and an invasive profile following treatment with BVZ (134). Similarly, GBM cells in mouse models developed enhanced invasiveness following VEGF inhibition (115).
Treatment with AIs also promotes tumor metastatic potential. Treatment with sunitinib has been shown to result in vascular changes that include decreased adherens junction protein expression, reduced basement membrane and pericyte coverage, and increased leakiness (89, 91, 135, 136). These phenotypic changes were observed in both, tumor vessels and normal organ vessels, so they tend to facilitate local intravasation and extravasation of tumor cells, resulting in metastatic colonization (136).
Factors Promoting or Affecting Tumor Invasiveness and Metastasis
Increased metastasis and enhanced invasiveness in response to anti-angiogenesis therapy are variable and depend on the treatment type, dose, and schedule. Singh et al. observed that sunitinib and anti-VEGF antibody monotherapy had different effects on mouse tumor models. While treatment with sunitinib enhanced the aggressiveness of tumor cells, using an anti-VEGF antibody did not (91). This was supported by Chung et al. who compared the efficacy of different RTK inhibitors and antibody therapies in murine models (135). While pretreatment with imatinib, sunitinib, or sorafenib enhanced lung metastasis following the injection of 66c14 cells, using an anti-VEGFR2 antibody inhibited the formation of lung nodules (135). Altogether, these results prove that the increased metastasis and enhanced invasiveness that result from use of AIs are largely dependent on treatment type.
Dosing and scheduling of administration of AIs can also induce resistance. Indeed, treatment with short-term and high-dosage sunitinib (120 mg/kg per day) before and after intravenous breast tumor cell inoculation into severe combined immune-deficient mice had the most deleterious effects (133). The high-dose of sunitinib increased tumor growth and enhanced metastasis to the liver and lung, resulting in reduced survival. Although similar results were observed using sorafenib, contradictory results were reported with sunitinib in different studies (115, 133). In fact, treatment with high-dose sunitinib before intravenous inoculation of tumor cells increased metastatic potential of lung cancer cells but not of RCC cells. In contrast, treatment with low-dosage sunitinib (30 and 60 mg/kg per day) did not stimulate metastasis (136).
It was documented that hypoxia and EMT also contribute to the increased invasiveness and metastasis of tumors, and c-Met, Twist, and HIF-1a are the key molecular players (11, 116). In contrast, semaphorin 3A (Sema3A), an endogenous anti-angiogenic molecule, is frequently lost in tumors, resulting in increased invasiveness and metastasis (137).
Overcoming Resistance by Targeting Increased Tumor Invasiveness and Metastasis
Different inhibitors of c-Met were tested in preclinical studies and demonstrated promising effects. Crizotinib, a dual c-Met and ALK inhibitor, was effective in reverting sunitinib-induced invasion and metastasis in different models (86–88). Interestingly, this resulted in a reduction in the expression of EMT markers such as Vimentin, Snail, and N-cadherin downstream of c-Met (86, 87). By blocking c-Met and silencing Twist, the master regulator of EMT (138), metastasis was almost fully abrogated in both wild-type and pericyte-depleted tumors (86).
Sunitinib-treated transgenic mice tumors that were subjected to adenoviral Sema3A expression witnessed an impressive increase of 10 weeks in median survival and a reduction in metastasis and hypoxia (89). Normalization of the tumor vasculature was evident, and the expression of EMT markers, including c-Met, were reduced.
Rovida et al. investigated the use of conventional chemotherapeutics to counteract sunitinib-induced metastasis. Gemcitabine and topotecan, but not paclitaxel, cisplatin, and doxorubicin, were effective in reverting sunitinib-induced metastasis and in reducing primary tumor growth (90). Mechanistically, topotecan was shown to inhibit HIF-1a accumulation, thereby preventing hypoxia-driven invasiveness. Gemcitabine was moderately effective in combination with anti-VEGF antibody therapy in an established pancreatic ductal adenocarcinoma model but had no effect in a preventive setting (91).
Redundancy in Angiogenic Signaling Pathways
Initially, the primary focus in angiogenesis blockade was to target VEGF, which is the best known angio-stimulatory protein family responsible for EC activation and functional vessel formation and stabilization. Cancers that are highly dependent on the induction of angiogenesis by VEGF, were the best responders to anti-VEGF agents. These include CRC, RCC, and neuroendocrine tumors (139).
Cancers relying on angiogenic factors other than VEGF are less susceptible to anti-VEGF agents and include malignant melanoma, pancreatic cancer, breast cancer, and prostate cancer (98). The presence of several anti-VEGF resistant cancers suggests alternative angiogenic pathways. These involve Ang-1, EGF, FGF, granulocyte colony-stimulating factor (G-CSF), hepatocyte growth factor (HGF), insulin-like growth factor, PDGF, PGF, stromal cell-derived factor-1 (SDF-1), and TGF (140). Except for P1GF, which binds VEGF receptors, most angiogenic factors signal through specific transmembrane receptors, which are expressed on ECs (141). This variety of growth factors culminates in a plethora of pathways that tumor cells can exploit to induce angiogenesis.
Results from preclinical models and clinical trials suggest that inhibition of a specific growth factor can induce the expression of others (140, 141). In a study by Willett et al. in which rectal cancer patients were treated with BVZ, significantly increased plasma levels of PlGF were noted 12 days following the start of treatment (142). In a phase II study by Kopetz et al. in which metastatic CRC patients were treated with a combination of FOLFIRI and BVZ, the levels of several angiogenic factors including PlGF and HGF were found to increase before disease progression (54). Similarly, the levels of FGF2 and PlGF increased in GBM patients following treatment with cediranib, a pan-VEGF receptor tyrosine kinase inhibitor (71, 143). Similarly, treatment of transgenic mouse models of pancreatic tumors with an anti-VEGFR2 antibody for a prolonged period of time, associated with an increase in the expression of the pro-angiogenic growth factors, Ang-1, Ephrin-A1, Ephrin-A2, and FGF1, FGF2a, resulting in transient tumor growth delay and modest survival benefit (98, 144).
Redundancy in angiogenic signaling and potential in malignant tissues is nowadays more studied. In addition, the therapeutic effect of targeting a single angiogenic growth factor or its receptor became limited due to intrinsic resistance. This resistance arose either from redundancy in activated pathways or alternative growth factor signaling pathways. Thus, targeting multiple growth factors simultaneously or sequentially would be a successful approach to overcome such resistance. In the following subsection, we discuss potential angiogenic factors that might play a role in the escape from anti-VEGF treatment. We also shed light on results of studies evaluating the effects of targeting one or more of these factors on overcoming resistance to anti-VEGF therapies.
Angiopoietin
Role of angiopoietin in the escape from anti-vascular endothelial growth factor treatment
Ang-Tie signaling system is a vascular-specific RTK pathway that regulates vascular permeability and blood vessel development and remodeling through Ang-1 and Ang-2. Ang-1 binds to the Tie2 receptor on the M2 subpopulation of monocytes, HSCs, and ECs of blood and lymphatic vessels. This activates the Ang-Tie pathway and results in the maturation or stabilization of blood vessels (145). In contrast, Ang-2 blocks this pathway resulting in the remodeling or initiation of vascular sprouts following exposure to VEGF (146). Upregulation of Ang-2 expression was described in many types of cancers and presumable contributes to resistance against anti-VEGF therapy (147–151). For example, in CRC patients, elevated serum Ang-2 levels were associated with a poor response to BVZ treatment (152).
Targeting angiopoietin to overcome resistance to anti-vascular endothelial growth factor treatment
Blockade of both, VEGF and Ang2, in preclinical studies suppressed revascularization and tumor progression of cancers resistant to anti-VEGF therapy (92–95). However, results of ongoing clinical trials evaluating the efficacy of the humanized bi-specific monoclonal antibody against VEGF-A and Ang-2, vanucizumab, are still pending (153, 154).
Bombina Variegate Peptide 8 (Bv8)
Role of bombina variegate peptide 8 in the escape from anti-vascular endothelial growth factor treatment
Tumor-infiltrating T helper type 17 (Th17) cells produce interleukin-17 (IL-17), initiating a paracrine network to confer resistance to anti-VEGF therapy (38). IL-17 induces G-CSF secretion by tumor cells through nuclear factor κB (NF-κB) and ERK signaling (155). The increase in G-CSF induces the expression of Bv8, also known as prokineticin-2, in the bone marrow. Bv8 is a pro-angiogenic growth factor that was initially purified from the skin secretion of a yellow-bellied toad. It binds to the G-protein coupled prokineticin receptor (PROKR) and activates the downstream MAPK/ERK pathway (156, 157). As such, Bv8 promotes differentiation of myeloid-derived (suppressor) stem (remove word stem) cells (MDSCs) and induces their mobilization to the peripheral blood and infiltration into the tumor microenvironment. This culminates in the promotion of angiogenesis and results in the escape from anti-VEGF therapy (158–161).
Targeting bombina variegate peptide 8 to overcome resistance to anti- vascular endothelial growth factor treatment
Treatment with the Bv8 antagonist, PKRA7, suppressed tumor formation in vivo by inhibiting angiogenesis in GBM and infiltration of MDSCs in pancreatic cancer (96). Neutralization of Bv8 and upstream G-CSF using monoclonal antibodies also resulted in tumor suppression (162). Results of ongoing clinical trials evaluating combination regimens using Bv8 inhibitors with or without other anti-angiogenic reagents are still pending.
Fibroblast Growth Factor (FGF)
Role of fibroblast growth factor in the escape from anti-vascular endothelial growth factor treatment
The FGF family consists of 22 members. Four of these are intracellular cofactors of voltage-gated sodium channels, while the remaining 18 members are secretory proteins that bind to RTK–FGF receptors (FGFRs) (163). FGFR is expressed on tumor cells and several types of stromal cells, including cancer-associated fibroblasts (CAFs), ECs, and tumor-infiltrating myeloid cells (164).
Binding of FGF to RTK–FGFR activates the downstream pathways such as MAPK/ERK, PI3K/Akt, STAT, and diacylglycerol (DAG)/protein kinase C (PKC) (165–168). One of the roles of this signaling pathway is cancer development and progression through the amelioration of angiogenesis (164, 169). Indeed, upregulation of FGF2 expression correlated with resistance to anti-VEGF agents in several tumors resistant, especially those exposed to hypoxic environments (54, 71, 98, 170).
Targeting fibroblast growth factor to overcome resistance to anti- vascular endothelial growth factor treatment
Simultaneous blockade of VEGF and FGF signaling pathways was very beneficial in many preclinical models of cancer (98, 171–173). Combining the FGFR inhibitor, PD173074, with BVZ in xenografted mouse models with head and neck squamous cell carcinoma (HNSCC) completely abolished tumor growth (97). FGF blockade using the soluble FGF receptor, FGF-trap, was combined with an VEGFR2 inhibitor, and yielded comparable results in late stage pancreatic islet tumors (98). Unfortunately, in the clinical setting, patients with recurrence following anti-VEGF therapy did not benefit from the dual blockade of VEGFR and FGFR by dovitinib or nintedanib (99, 100).
Platelet-Derived Growth Factor
Role of platelet-derived growth factor in the escape from anti-vascular endothelial growth factor treatment
The PDGF family consists of four homodimers and one heterodimer. Binding of the PDGF dimers to tyrosine kinase PDGF receptor (PDGFR) results in the activation of downstream signal transduction pathways, such as PI3K and PLCγ (174). This plays an important role in mesenchymal cell growth and motility during embryonic development and tissue repair (175). When PDGF signaling is over-active in the tumor microenvironment, angiogenesis and tumor growth are promoted (176). Upregulation of PDGF-C expression was observed in vivo in CAFs infiltrating into tumors resistant to anti-VEGF therapy (101).
Targeting platelet-derived growth factor to overcome resistance to anti- vascular endothelial growth factor treatment
Sunitinib has many targets, including VEGFR and PDGFR. Following its FDA approval in 2006 for the treatment of metastatic RCC, it was assumed that combining PDGF and VEGF blockades might offer an additional therapeutic benefit (101). Several studies were initiated to evaluate the safety and efficacy of this combination (177). Unfortunately, combining BVZ with imatinib, which inhibits PDGF-R in addition to other tyrosine kinases such as Abl and Kit, was toxic and not effective treatment against RCC (102–104).
Transforming Growth Factor-β
Role of transforming growth factor-β in the escape from anti-vascular endothelial growth factor treatment
The TGF-β/Activin and bone morphogenetic protein (BMP) are the two main branches of the TGF-β superfamily. When TGF-β binds its type II receptors, it activates type I receptors and results in the phosphorylation of the receptor-regulated Smads (R-Smads) corresponding to each branch. R-Smads then complex with the common partner Smad4 (Co-Smad4) and work as transcription factors (178).
TGF-β signaling regulates cellular growth, differentiation, and apoptosis (179). Although signaling has tumor suppressive effects during the early stage, it switches toward malignant conversion and tumor progression at later stages (180, 181). It activates the production of extracellular matrix (ECM) by fibroblasts and stimulates tube formation by ECs, thus inducing angiogenesis (182–184).
Tumor tissues express higher levels of TGF-β and these levels can be correlated with patient survival (185–187). Upregulation of TGF-β expression was also observed in glioma models resistant to anti-VEGF therapy (188). This suggests a role of TGF-β in the acquired resistance to anti-angiogenic therapy.
Targeting transforming growth factor-β to overcome resistance to anti- vascular endothelial growth factor treatment
Several preclinical studies revealed the anti-angiogenic benefits when inhibiting TGFβ in CRC, HCC, and GBM xenografts (189–191). This offers the rationale to combine TGFβ inhibitors with anti-VEGF agents (192). In that sense, combining galunisertib, a small molecule inhibitor of TGFβRI, with sorafenib and ramucirumab in HCC is currently under evaluation (189, 193). Similarly, the combination of an anti-TGFβ monoclonal antibody, PF-03446962, with regorafenib in CRC is also under investigation (194).
Matrix Metalloproteinases
Role of matrix metalloproteinases in the escape from anti-vascular endothelial growth factor treatment
MMPs play an important role in angiogenesis and in different stages of cancer (195, 196). They are divided into six categories (Table 4) (197). MMP can promote or inhibit angiogenesis. For instance, the secreted MMP-9 plays an important role in the angiogenic switch process and in releasing VEGF from the ECM (1, 198). The membrane type MMP-1 induces degradation and remodeling of matrix during vascular injury and is responsible for invasion and migration of ECs and formation of capillaries (199–201). On the other hand, MMPs such as MMP-3, 7, 12, 13, and 20, inhibit angiogenesis through endostatin and angiostatin production. Endostatin that blocks the activation of pro-MMP-9 and inhibits capillary formation of Deryugina and Quigley (202).
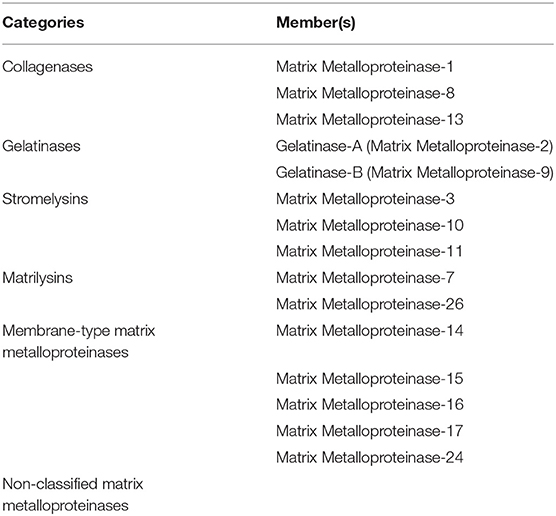
Table 4. Categories of Matrix Metalloproteinase-1 and their corresponding members.
Targeting matrix metalloproteinases to overcome resistance to anti- vascular endothelial growth factor treatment
Targeting MMPs released by bone marrow derived cells (BMDCs) prevents the release of sequestered growth factors in the ECM, and can help overcoming resistance to anti-angiogenic therapy (203). Despite the fact that doing so has proven some clinical efficacy in patients with advanced and refractory solid tumors in a phase I clinical trial (105), most MMP inhibitors failed to offer any clinical benefit (204). Few agents are still being developed and evaluated. Results from an ongoing phase II clinical trial evaluating one MMP inhibitor in patients with Kaposi's sarcoma are still pending (205).
Recruitment of Bone Marrow-Derived Cells
Long-term administration of AIs up-regulates HIF-1α and induces hypoxia in the tumor microenvironment by over-pruning blood vessels (206). Hypoxic conditions due to anti-angiogenic therapy result in the expansion and recruitment of myeloid cells and CAFs into the tumor environment. The presence of these BMDCs in the tumor microenvironment leads to a weakened antitumor response and an immunosuppressive tumor microenvironment (207). This promotes angiogenesis, tumor growth, EMT transition, and metastasis (208, 209). As a result, it has become evident that myeloid cells and CAFs play a major role in the induction of resistance to anti-angiogenic drugs.
Myeloid Cells
Recruitment of myeloid cells
Myeloid derived suppressor cells (MDSCs), also known as Gr1+ CD11b+ myeloid cells, consist of neutrophils, macrophages, and dendritic cells (DCs). An excessive production of MDSCs was described in cancer patients and tumor-bearing mice (210–213). This was linked to the immunosuppressive and tumor promoting capacities (214, 215). In a study by Shojaei et al., resistant tumors to anti-VEGF treatment had increased mobilization and infiltration of MDSCs into their microenvironments as compared with treatment-sensitive tumors (216).
Neutrophils are considered predictive biomarkers for patients treated with BVZ (217–222). Increased recruitment of neutrophils during anti-VEGF therapy promotes tumor progression and treatment resistance (216). This is mediated by the expression of the calcium-binding protein that regulates cell growth, survival, and motility, S100A4. As such, blocking granulocytes and S100A4 may be beneficial in diminishing anti-angiogenic therapy resistance (223).
Monocytes and macrophages are possibly implicated in resistance to anti-angiogenic therapy as well. Recruitment of these cells to the tumor microenvironment is mediated by different cytokines, including VEGF, chemokine C-C motif ligand 2 (CCL2), and macrophage colony stimulating factor (MCSF) (224, 225). Tumor associated macrophages actively participate in vascular sprouting by functioning as bridging cells between two different tip cells (226–228). They also secrete MMPs, promotingangiogenesis (198, 226, 229, 230). In addition, they can release pro-angiogenic growth factors including TGF-b, VEGF, EGF, and the chemokines, CCL2 and CXCL8 (226, 227, 231–233).
In different murine tumor models, anti-VEGF therapy reduced macrophage infiltration (217, 234–236). However, this was not the case with the tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (TIE2)-expressing macrophages that constitute a specific subset of macrophages. These are usually recruited by HIF1a and tumor-secreted chemokines such as ANG2 in the setting of anti-angiogenic therapy (237–240). They tend to associate with tumor vessels and release proangiogenic growth factors including VEGF (237, 241). As such, macrophages contribute to the resistance against anti-angiogenic therapy. Preclinical studies on models of mammary carcinoma and insulinoma evaluated the effect of inhibiting ANG2 on TIE2-expressing macrophage infiltration and angiogenesis. Although this approach did not block the recruitment of these macrophages, it hindered the upregulation of their TIE2 receptor. This reduced the production of pro-angiogenic growth factors and the association of TIE2 macrophages with blood vessels (242–244). As a result, MDSCs represent promising targets for therapy. Since G-CSF expression stimulated by tumor infiltrating T helper type 17 cells results in MDSC recruitment into the tumor microenvironment, inhibition of Th17 cell function might sensitize tumors to anti-VEGF therapies (155, 207).
Targeting myeloid cells to overcome resistance to anti- vascular endothelial growth factor treatment
Since SDF1 is the major BMDC recruiting factor, targeting its signaling pathway could potentially decrease BMDC infiltration and overcome resistance to anti-angiogenic therapy. In a transgenic mouse model of breast cancer, treatment with an SDF1 neutralizing antibody inhibited MDSC infiltration and angiogenesis (106). Since Bv8 leads to the recruitment of MDSCs into the tumor tissue after VEGF blockade, its inhibition can possibly improve the effect of anti-angiogenic therapy. A recent study showed that the combination of gemcitabine and an anti-Bv8 monoclonal antibody treatment in mice with adenocarcinoma inhibited tumor regrowth, angiogenesis, and metastasis (107). In addition, anti-Bv8 antibodies blocked MDSC recruitment and tumor angiogenesis in an RIP1-Tag2 insulinoma model of pancreatic cancer (245).
Blocking the recruitment of monocytes and macrophages can be another therapeutic opportunity to overcome resistance to anti-angiogenic therapy. In a phase I clinical trial, patients with solid tumors were treated with the human anti-CCL2 monoclonal antibody, carlumab, which targets the monocyte chemotactic protein-1 (MCP1). In addition to causing a drop in free CCL2 levels and a reduction in the level of tumor-infiltrating macrophages, this therapy resulted in a temporary antitumor activity (108). Treatment of RIP1-Tag2 pancreatic neuroendocrine tumors with combined ANG2 and VEGFR2 blockers decreased infiltration of TIE2 expressing monocytes and suppressed revascularization and tumor progression (92). Since macrophages express colony stimulating factor-1 receptor, its targeting is currently being evaluated by several phase I clinical trials (NCT01346358; NCT01004861; NCT01596751). This is supported by results from earlier studies showing a reduced macrophage infiltration into tumor tissue and clinical objective responses following treatment of diffuse-type giant cell tumor patients with the anti-colony-stimulating factor-1 receptor antibody, RG7155 (246).
Macrophage Migration Inhibitory Factor (MIF) suppresses the anti-inflammatory activity of macrophages. TAMs, mainly M2-polarized macrophages, stimulate angiogenesis thus promoting tumor cell migration and progression (247). VEGF increases MIF production in a VEGFR-dependent manner. Compared to tissue specimens of BVZ-sensitive GBM patients, BVZ-resistant ones had a decreased MIF expression and an increased TAM infiltration (248). As such, blocking the VEGF pathway using BVZ can deplete MIF expression. This explains the enhanced recruitment of TAM and M2 in BVZ-resistant GBM tumors. Data is lacking when it comes to evaluating the application of this target in the clinical setting.
Endothelial Progenitor Cells
Recruitment of endothelial progenitor cells
Anti-angiogenic therapy causes hypoxia which results in the activation of HIF1a in tumor cells (249). This causes tumor cells to secrete SDF1 and VEGF,main chemotactic factors for EPCs (209, 215, 250, 251). Upon stimulation of the C-X-C chemokine receptor-7 (CXCR7) by SDF1, EPCs secrete pro-angiogenic cytokines and promote angiogenesis (252, 253). For instance, in multiple myeloma, this occurs through regulating the trafficking of angiogenic mononuclear cells into areas of tumor growth (254). EPCs can also promote angiogenesis by differentiating into ECs and subsequently incorporating into newly forming blood vessels.
Recruitment of Local Stromal Cells
Pericytes
Recruitment of pericytes
Pericytes, also known as Rouget cells, are cells that interact with ECs. They regulate endothelial proliferation and differentiation and modulate vessel diameter and permeability, thus stabilizing the newly formed endothelial tubes (255, 256). In a study by Abramsson et al., paracrine co-signaling via PDGF-B and PDGFR-b played a major role in pericyte recruitment to ECs (257).
Several studies revealed enhanced pericyte recruitment to and coverage of the microvasculature in the tumor after treatment with AIs. Reduction in tumor vascularity following anti-VEGF therapy is accompanied by a tightly pericyte covered vessels (258). For instance, after treatment with sunitinib and the chemotherapy drug, temozolomide, a preclinical malignant glioma model revealed an increased number of vessels covered with pericytes (259). In addition, esophageal and ovarian cancer xenografts showed increased pericyte coverage around vessels following treatment with BVZ (260).
Tumor vessels that are heavily covered by pericytes have a reduced sensitivity for anti-angiogenic therapies (261) As such, the increase in pericyte infiltration was suggested to be a mechanism of resistance to anti-VEGF and anti-VEGFR therapies. By suppressing EC proliferation and by providing survival signals that contribute to the maintenance of ECs, pericytes mediate vascular maturation and stability hence allowing tumor cells to proliferate during the course of an anti-angiogenic therapy (262–264). As a result of protecting ECs from anti-angiogenic agents, pericytes were implicated in clinical resistance to VEGFR inhibitors (249).
While there is a broad consensus on the fact that pericyte-covered vessels are less sensitive to AI, several recent studies have highlighted that tumor vessels typically lack pericyte coverage due to their immaturity and rapid growth phase while normal quiescent vessels are well covered (265–267). This could identify a selective therapeutic window to target abnormal tumor blood vessels, rather than suggesting to target pericyte coverage.
In keeping with that, accumulating evidence supports the idea that—in addition to pruning non-covered vessels- cancer therapies should aim at promoting the establishment of a normal vasculature in tumors in order to favor wide distribution of standard chemotherapeutics and innovative drugs into the tumor mass and improve radiotherapy efficacy. This process is known as “vascular normalization” that many adopt as the future of anti-angiogenic therapy. By therapeutically improving, rather than reducing, the stability and function of tumor blood vessels, these may be exploited for delivery of therapeutics including endogenous anti-cancer immune cells. This would also improve perfusion, reduce hypoxia, and thereby reduce metastasis. Tumor vessel normalization for cancer therapy has been achieved by the application of molecules directly targeting endothelial cells, such as semaphorins (268, 269).
Although ANG1 is a growth factor that provides ECs with survival signals, its introduction in CRC tumor cells displays an anti-angiogenic therapy in one study (270). Although this approach was accompanied by a major increase in tumor microvessel pericyte coverage, it resulted in smaller tumors with less vasculature, suggesting a decreased sensitivity for angiogenesis (270). In a more recent study, tumor-bearing mice were treated with antibodies against ANG2A, and a similar observation was noted (261). Combining the chemotherapeutic agent, topotecan, with pazopanib significantly inhibited tumor growth, despite an increase in the number of vessels that were infiltrated by pericytes (271). Similar results were observed in a preclinical malignant glioma model following treatment with the combination of temozolomide and sunitinib (272).
Targeting pericytes to overcome resistance to anti- vascular endothelial growth factor treatment
Targeting blood vessel maturation by inhibiting pericyte coverage of the tumor vasculature was suggested as a promising strategy, to break the resistance to anti-angiogenic therapies and improve their efficacy. ECs secrete PDGF-B that mediates migration and proliferation of pericytes expressing PDGFR-b (273). Since SDF1, and the heparin-binding EGF-like growth factor also play a major role in pericyte behavior (274), blocking the PDGF pathway alone might not be sufficient to prevent pericyte coverage of vasculature.
Although several studies showed that targeting pericytes and ECs leads to impaired tumor growth and improved efficacy to anti-angiogenic agents, data negating the potentiation of treatment outcome with dual blockade exists (275). For instance, in a study by Nisancioglu et al., treatment of lung cancer in pericyte-deficient PDGF-B (ret/ret) mice with the anti–VEGFA antibody, G6-31, did not have any additional anti-tumor benefit (276).
Other pathways like sphingosine-1-phosphate (S1P)/edg-1, TGF-b1/Alk5, or MMPs should be considered while trying to overcome resistance associated with pericyte coverage (277). As a result, anti-pericyte agents should always be combined with other therapies, including chemotherapeutic agents. For instance, in a preclinical study by Pietras et al., transgenic mouse models of cancer were treated with a combination of the two anti-PDGFR agents, imatinib and SU11248, cyclophosphamide, and/or an anti-VEGFR agent (109). Compared to monotherapies, combination therapies significantly improved anti-tumor responses. Of note, the combination of all three approaches resulted in complete responses. Also, treatment of neuroblastoma mouse xenograft models with a combination of metronomic topotecan and pazopanib resulted in a sustained anti-angiogenic effect. but induced resistance mediated by elevated glycolysis (109).
Cancer-Associated Fibroblasts
Recruitment of cancer-associated fibroblasts
CAFs are activated by growth factors released from tumor and inflammatory cells, including TGFb, PDGF, and FGF (169, 278, 279). CAFs also secrete several pro-angiogenic growth factors, including EGF, HGF, and FGF. For instance, VEGF-producing CAFs maintain tumor angiogenesis in VEGF-deficient tumor cells (280).
When CAFs were isolated from a mixture of EL4 tumors resistant to anti-VEGF agents and TIB6 tumors sensitive to anti-VEGF agents, they were able to promote tumor cell proliferation and growth even when VEGF was blocked. When CAFs were isolated from TIB6 tumors sensitive to anti-VEGF agents, no tumor growth was observed (215). This supports the role of CAFs in the acquired resistance to anti-angiogenic therapy. Further analysis revealed an upregulation in the expression of pro-angiogenic genes in CAFs derived from therapy-resistant tumors, and these included PDGF-C and Ang-like protein 2. As a result, it is assumed that a PDGF-C neutralizing antibody could be used in the treatment of tumors refractory to anti-VEGF agents (215).
CAFs can promote tumor growth and angiogenesis through the release of certain growth factors and proteases. For instance, CAFs secrete the chemokine SDF1 which directly stimulates tumor cells and recruits EPCs and other BMDCs into the tumor tissue (250, 251). They also produce proteases, including MMPs that stimulate the release of matrix-bound pro-angiogenic growth factors, thus promoting angiogenesis and resistance to anti-angiogenic agents (281–283).
Targeting cancer-associated fibroblasts to overcome resistance to anti- vascular endothelial growth factor treatment
Targeting CAFs might play a role in overcoming resistance to anti-angiogenic therapy. Treatment of nude mice human HCC xenografts with the anti-FGF2 monoclonal antibody, GAL-F2, inhibited tumor growth and angiogenesis by blocking the effect of the proangiogenic FGF in CAFs. Also, the addition of an anti-VEGF antibody or the tyrosine kinase inhibitor, sorafenib, led to an additive treatment effect (110). Similarly, treatment of patients with recurrent and persistent endometrial cancer with the dual VEGFR/FGFR inhibitor, brivanib, extended their progression-free survival (PFS) by blocking the effect of the proangiogenic VEGF in CAFs. (111). Neutralization of PDGF-C suppressed CAF-mediated tumor progression.
Adoption of Different Neovascularization Modalities
Besides acquiring resistance to angiogenesis inhibition through growth factor redundancy and recruitment of different cells, tumor cells may also escape the effect of AIs by adopting different neovascularization modalities (284–286). These include vascular co-option and vasculogenic mimicry.
Vessel Co-option
Role of vessel co-option in the escape from anti-vascular endothelial growth factor treatment
Vessel co-option refers to the process by which cancer cells incorporate into and grow along pre-existing vessels rather than inducing new vasculature (287). This strategy provides oxygen and nutrients for efficient tumor outgrowth. It was first described in brain tumors arising from well-vascularized brain parenchyma (288). For instance, vessel co-option was also observed in gliomas and other cancer types including lung cancers (289). It was shown to sustain the growth of cerebral metastases from melanomas, liver metastases from breast cancers and NSCLCs, and lung metastases from different primaries (290, 291). Interestingly, vessel co-option is independent of the classic angiogenic switch and doesn't require any angiogenic growth factors. As such, vessel co-opting tumors are usually not sensitive to anti-angiogenic agents. For example, patients with CRC and liver metastases demonstrated a poor response to BVZ therapy due to vessel co-option.
An interesting question is whether this process represents an intrinsic resistance mechanism to anti-angiogenic therapies or whether it occurs in response to treatment. According to results from several studies, an increase in vessel co-option tends to follow, rather than precede, the inhibition of angiogenesis (292). For instance, the use of an anti-VEGF antibody in GBM patients resulted in an increase in vessel co-option (293, 294). Similarly, the growth cerebral melanoma metastasis was sustained by vessel co-option following treatment with the anti-angiogenic agent, ZD6474 (290). Nevertheless, more data is needed to check whether this applies to different tumor types and to evaluate its impact in the clinical setting.
Vasculogenic Mimicry
Role of vasculogenic mimicry in the escape from anti-vascular endothelial growth factor treatment
Vasculogenic mimicry refers to the process in which vascular-like structures are formed by tumor cells, after they trans-differentiate and gain features of ECs such as the expression of the endothelial markers, VE-cadherin, TIE1, and ephrin A2 (295, 296). Since no new blood vessels are formed, this phenomenon is different from vasculogenesis and angiogenesis. Nevertheless, the fact that blood can still be transported through the vascular-like networks and tumors can still be well-oxygenated, vasculogenic mimicry strongly associated with poor patient survival. This process was described in different tumor types, including gliomas, malignant melanomas, sarcomas, and breast cancers (284, 297–300).
Since tumor cells trans-differentiate into endothelial-like cells as part of vasculogenic mimicry, it might be assumed that the process can be inhibited by anti-angiogenic agents. However, tumor cells that make use of this phenomenon were not found to develop sensitivity to anti-angiogenic therapies in early studies (301). Instead, they were shown to upregulate this process following treatment with BVZ or induction of hypoxia by several preclinical studies (302, 302, 303). As such, vasculogenic mimicry might serve as an escape mechanism from anti-angiogenic therapies. The idea of combining AIs with chemotherapeutic agents has been suggested but more data is needed to evaluate its impact in the clinical setting.
Targeting vasculogenic mimicry to overcome resistance to anti- vascular endothelial growth factor treatment
Following the emergence of vasculogenic mimicry as an alternative vascular-like network in tumors, researchers have realized the importance of combining angiogenesis inhibition with an anti-tumor cell strategy. This is particularly challenging because the transition of tumor cells into a more stem cell–like phenotype is linked to reduced responsiveness to chemotherapy and radiotherapy.
In an attempt to better understand the regulators of vasculogenic mimicry, several studies tried to recognize the molecular players of this process. Direct targeting of these molecules, including VEGF, is thought to serve as a promising therapeutic approach (302, 303). Other regulators of mimicry were also involved in the plasticity and stem cell-like phenotype of tumor cells. An example is the overexpression of the marker of brain development, NODAL (304–307). In addition, the overexpression of CD44 on vasculogenic tumor cells led to the initiation of the ongoing clinical study (NCT01358903). This trial evaluates the effect of an anti-CD44 agent on the process of vasculogenic mimicry during the treatment of solid tumors.
Conclusion and Future Outlook
The concept of targeting tumor angiogenesis is an important advancement in cancer therapy and has resulted in the development of therapeutic agents such as BVZ, sunitinib, and sorafenib. Benefits of using anti-angiogenesis therapies seem to be limited due to several reasons.
Despite the resulting stabilization of disease and increased PFS, treatment with anti-angiogenic agents may give rise to more resistant tumors with higher patient relapse rates. This lack of clinical benefit could be associated with preexisting resistance or with rapid adaptation to anti-angiogeneic agents. It is clear that multiple mechanisms of resistance against AIs exist, including upregulation of alternative angiogenic factors by tumor cells, involvement of stromal cells, and co-option/mimicry. The fact that the process of angiogenesis is complicated and involves a network of mechanisms suggests that the tumor microenvironment could mediate resistance to AIs (308). In addition, the vascular regression that is caused by AIs could elevate intra-tumoral hypoxia, which in turn, ameliorates resistance to radiotherapy, chemotherapy, and AIs. Also, the regression in tumor vasculature and the reduction in blood flow that result from AIs would impede the delivery of chemotherapeutic agents into tumors. All these complications of AI use would allow for tumor metastasis and would hence serve as practical limitations to drug development (309).
With the progress in several scientific and medical fields and with the growing surge in knowledge about angiogenesis and its resistance mechanisms, new pharmacological strategies ought to be developed in the near future. For instance, new ways of targeting tumor vessels should be designed. This could be made possible by developing novel therapeutics that can either optimize the function of tumor vessels to allow adequate tumor response to cancer therapy or directly target tumor vessels (310).
In addition, in the light of the wide gap between our improving knowledge in the mechanobiology of MSCs and our satisfactory understanding of their clinical implications, novel approaches should be suggested to fill the gap. This could be made possible by engineering MSCs to selectively deliver anti-angiogenic molecules (309).
In addition, the use of combination strategies as a means to target multiple pathways involved in angiogenesis has been suggested to be a promising approach in overcoming resistance to AIs. To date, these either include a combination of multiple anti-angiogenic agents or a combination of anti-angiogenic drugs and other treatment regimens.
This process of selecting the most effective combination regimen is challenging because it requires extensive profiling of angiogenesis signaling pathways and involves a careful patient selection. Not only do combination regimens require regular dose adjustments to enhance efficacy and reduce toxicity, but also they require intermittent monitoring of treatment efficacy through biomarkers. Although combinations of different anti-angiogenic agents might increase treatment benefit, the presence of many alternative pathways can still result in acquired resistance. They can also induce excessive hypoxia that leads to additional resistance. Hence, the initiation of clinical trials to evaluate the efficacy and safety of such new combination strategies seems to be of utmost importance. In addition, the development of genetically engineered animal models whose tumor microenvironment can mimic that of humans could be of so much help in the development of reliable treatment approaches. This, in addition to clinical trials, would enable scientists and clinicians to make use of precision medicine for coming up with effective combinations of AIs and other therapies that would hopefully prevent the early acquisition of resistance or even impede its occurrence (141).
It is likely that the future therapy will make use of genomic, transcriptomic, and proteomic techniques as part of diagnostic profiling. Different therapeutic combinations can then be personalized and matched to current stages of tumor progression. Since tumors have rapid genetic drifts and might rapidly develop resistance to treatment, diagnostic profiling would have to be repeated during the course of treatment (141).
Nanotechnology enables researchers to develop novel nano-therapeutics, but this requires more knowledge about metabolic behaviors of tumor cells and possible physiological barriers or material properties that would improve or impede the efficiency of nano-therapeutics, respectively (311). It can therefore be foreseen that the future of AI-based therapies is heavily dependent on the efforts of basic scientists who can provide a clearer image regarding the response of cancer cells to the agents and on the ability of clinicians to make use of this knowledge to benefit patients (312).
These issues highlight the major challenges for future research. We look forward to the results of ongoing and future clinical trials discussed in this review paper in hopes that outcomes can be improved for all patients with cancers that are resistant to angiogenesis.
Author Contributions
All authors made substantial contributions to study conception and design. YH, MK, HE, IK, DM, ST, and AS have been involved in drafting the manuscript and revising it critically for important intellectual content. All authors have provided final approval of the version to be published.
Conflict of Interest
DM reports honoraria, travel support and institutional research funding from Roche, Pfizer, Novartis, and Amgen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Otrock ZK, Mahfouz RA, Makarem JA, Shamseddine AI. Understanding the biology of angiogenesis: review of the most important molecular mechanisms. Blood Cells Mol Dis. (2007) 39:212–20. doi: 10.1016/j.bcmd.2007.04.001
2. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. (1995) 1:27–31. doi: 10.1038/nm0195-27
3. Adamis AP, Miller JW, Bernal MT, D'Amico DJ, Folkman J, Yeo TK, et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol. (1994) 118:445–50. doi: 10.1016/S0002-9394(14)75794-0
4. Couffinhal T, Kearney M, Witzenbichler B, Chen D, Murohara T, Losordo DW, et al. Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) in normal and atherosclerotic human arteries. Am J Pathol. (1997) 150:1673–85.
5. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
6. Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. (2003) 13:159–67. doi: 10.1016/S1044-579X(02)00133-5
7. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. (2003) 3:401–10. doi: 10.1038/nrc1093
8. Zuazo-Gaztelu I, Casanovas O. Unraveling the role of angiogenesis in cancer ecosystems. Front Oncol. (2018) 8:248. doi: 10.3389/fonc.2018.00248
9. Yang Y, Sun M, Wang L, Jiao B. HIFs, angiogenesis, and cancer. J Cell Biochem. (2013) 114:967–74. doi: 10.1002/jcb.24438
10. Lekas A, Lazaris AC, Deliveliotis C, Chrisofos M, Zoubouli C, Lapas D, et al. Expression of HIF-1α and markers of angiogenesis are not significantly different in triple negative breast cancer compared to other breast cancer molecular subtypes: implications for future therapy. PLoS ONE. (2015) 10:e0129356. doi: 10.1371/journal.pone.0129356
11. Ghattass K, Assah R, El-Sabban M, Gali-Muhtasib H. Targeting hypoxia for sensitization of tumors to radio- and chemotherapy. Curr Cancer Drug Targets. (2013) 13:670–85. doi: 10.2174/15680096113139990004
12. Vandekeere S, Dewerchin M, Carmeliet P. Angiogenesis revisited: an overlooked role of endothelial cell metabolism in vessel sprouting. Microcirculation. (2015) 22:509–17. doi: 10.1111/micc.12229
13. Kazerounian S, Lawler J. Integration of pro- and anti-angiogenic signals by endothelial cells. J Cell Commun Signal. (2018) 12:171–9. doi: 10.1007/s12079-017-0433-3
14. El-Kenawi AE, El-Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharmacol. (2013) 170:712–29. doi: 10.1111/bph.12344
15. Felmeden DC, Blann AD, Lip YH. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J. (2003) 24:586–603. doi: 10.1016/S0195-668X(02)00635-8
16. Otrock ZK, Makarem JA, Shamseddine AI. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol Dis. (2007) 38:258–68. doi: 10.1016/j.bcmd.2006.12.003
17. Olofsson B, Jeltsch M, Eriksson U, Alitalo K. Current biology of VEGF-B VEGF-C. Curr Opin Biotechnol. (1999) 10:528–35. doi: 10.1016/S0958-1669(99)00024-5
18. Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, et al. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med. (1992) 176:1375–9. doi: 10.1084/jem.176.5.1375
19. Ozawa K. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. (2002) 20:4368–80. doi: 10.1200/JCO.2002.10.088
20. Ferrara N, Gerber PH, LeCouter J. The biology of VEGF and its receptors. Nat Med. (2003) 9:669. doi: 10.1038/nm0603-669
21. Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. (2003) 28:488–94. doi: 10.1016/S0968-0004(03)00193-2
22. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. (1996) 380:435. doi: 10.1038/380435a0
23. Larcher F, Murillas R, Bolontrade M, Conti CJ, Jorcano JL. VEGF/VPF overexpression in skin of transgenic mice induces angiogenesis, vascular hyperpermeability and accelerated tumor development. Oncogene. (1998) 17:303. doi: 10.1038/sj.onc.1201928
24. Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. (2002) 29:10–4. doi: 10.1053/sonc.2002.37264
25. Grimmond S, Lagercrantz J, Drinkwater C, Silins G, Townson S, Pollock P, et al. Cloning and characterization of a novel human gene related to vascular endothelial growth factor. Genome Res. (1996) 6:124–31. doi: 10.1101/gr.6.2.124
26. Paavonen K, Horelli-Kuitunen N, Chilov D, Kukk E, Pennanen S, Kallioniemi OP, et al. Novel human vascular endothelial growth factor genes VEGF-B and VEGF-C localize to chromosomes 11q13 and 4q34, respectively. Circulation. (1996) 93:1079–82. doi: 10.1161/01.CIR.93.6.1079
27. Gunningham SP, Currie MJ, Han C, Turner K, Scott PA, Robinson BA, et al. Vascular endothelial growth factor-B and vascular endothelial growth factor-C expression in renal cell carcinomas: regulation by the von Hippel-Lindau gene and hypoxia. Cancer Res. (2001) 61:3206–11.
28. Hanrahan V, Currie MJ, Gunningham SP, Morrin HR, Scott PA, Robinson BA, et al. The angiogenic switch for vascular endothelial growth factor (VEGF)-A, VEGF-B, VEGF-C, and VEGF-D in the adenoma-carcinoma sequence during colorectal cancer progression. J Pathol. (2003) 200:183–94. doi: 10.1002/path.1339
29. Ristimäki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. (1998) 273:8413–8. doi: 10.1074/jbc.273.14.8413
30. Asano A, Kimura K, Saito M. Cold-induced mRNA expression of angiogenic factors in rat brown adipose tissue. J Vet Med Sci. (1999) 61:403–9. doi: 10.1292/jvms.61.403
31. Albrecht I, Kopfstein L, Strittmatter K, Schomber T, Falkevall A, Hagberg CE, et al. Suppressive effects of vascular endothelial growth factor-B on tumor growth in a mouse model of pancreatic neuroendocrine tumorigenesis. PLoS ONE. (2010) 5:e14109. doi: 10.1371/journal.pone.0014109
32. Bellomo D, Headrick JP, Silins GU, Paterson CA, Thomas PS, Gartside M, et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circul Res. (2000) 86:e29–35. doi: 10.1161/01.RES.86.2.e29
33. Mould AW, Tonks ID, Cahill MM, Pettit AR, Thomas R, Hayward NK, et al. Vegfb gene knockout mice display reduced pathology and synovial angiogenesis in both antigen-induced and collagen-induced models of arthritis. Arthritis Rheum. (2003) 48:2660–9. doi: 10.1002/art.11232
34. Joukov V, Kaipainen A, Jeltsch M, Pajusola K, Olofsson B, Kumar V, et al. Vascular endothelial growth factors VEGF-B VEGF-C. J Cell Physiol. (1997) 173:211–5. doi: 10.1002/(SICI)1097-4652(199711)173:2<211::AID-JCP23>3.0.CO;2-H
35. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. (2004) 5:74. doi: 10.1038/ni1013
36. Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, et al. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development. (1996) 122:3829–37.
37. Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. (1997) 276:1423–5. doi: 10.1126/science.276.5317.1423
38. Fujimoto J, Toyoki H, Sato E, Sakaguchi H, Tamaya T. Clinical implication of expression of vascular endothelial growth factor-C in metastatic lymph nodes of uterine cervical cancers. Br J Cancer. (2004) 91:466–9. doi: 10.1038/sj.bjc.6601963
39. He Y, Karpanen T, Alitalo K. Role of lymphangiogenic factors in tumor metastasis. Biochim Biophys Acta Rev Cancer. (2004) 1654:3–12. doi: 10.1016/j.bbcan.2003.07.003
40. Farnebo F, Piehl F, Lagercrantz J. Restricted expression pattern ofvegf-din the adult and fetal mouse: high expression in the embryonic lung. Biochem Biophys Res Commun. (1999) 257:891–4. doi: 10.1006/bbrc.1999.0562
41. Achen MG, Jeltsch M, Kukk E, Mäkinen T, Vitali A, Wilks AF, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. (1998) 95:548–53. doi: 10.1073/pnas.95.2.548
42. Lee J, Gray A, Yuan J, Luoh SM, Avraham H, Wood WI. Vascular endothelial growth factor-related protein: a ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Natl Acad Sci USA. (1996) 93:1988–92. doi: 10.1073/pnas.93.5.1988
43. Akahane M, Akahane T, Matheny SL, Shah A, Okajima E, Thorgeirsson UP. Vascular endothelial growth factor-D is a survival factor for human breast carcinoma cells. Int J Cancer. (2006) 118:841–9. doi: 10.1002/ijc.21420
44. Nilsson I, Rolny C, Wu Y, Pytowski B, Hicklin D, Alitalo K, et al. Vascular endothelial growth factor receptor-3 in hypoxia-induced vascular development. FASEB J. (2004) 18:1507–15. doi: 10.1096/fj.03-1276com
45. Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. (2001) 7:186–91. doi: 10.1038/84635
46. Achen MG, Williams RA, Minekus MP, Thornton GE, Stenvers K, Rogers PA, et al. Localization of vascular endothelial growth factor-D in malignant melanoma suggests a role in tumour angiogenesis. J Pathol. (2001) 193:147–54. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH757>3.0.CO;2-G
47. Von Marschall Z, Scholz A, Stacker SA, Achen MG, Jackson DG, Alves F, et al. Vascular endothelial growth factor-D induces lymphangiogenesis and lymphatic metastasis in models of ductal pancreatic cancer. Int J Oncol. (2005) 27:669–79. doi: 10.3892/ijo.27.3.669
48. Kleespies A, Bruns CJ, Jauch WK. Clinical significance of VEGF-A,-C and-D expression in esophageal malignancies. Oncol Res Treatment. (2005) 28:281–8. doi: 10.1159/000085198
49. Savory LJ, Stacker SA, Fleming SB, Niven BE, Mercer AA. Viral vascular endothelial growth factor plays a critical role in orf virus infection. J Virol. (2000) 74:10699–706. doi: 10.1128/JVI.74.22.10699-10706.2000
50. Maglione D, Guerriero V, Viglietto G, Delli-Bovi P, Persico MG. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc Natl Acad Sci USA. (1991) 88:9267–71. doi: 10.1073/pnas.88.20.9267
51. Park JE, Chen HH, Winer J, Houck KA, Ferrara N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro, in vivo, high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. (1994) 269:25646–54.
52. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. (2005) 438:820–7. doi: 10.1038/nature04186
53. Zhou Y, Tu C, Zhao Y, Liu H, Zhang S. Placental growth factor enhances angiogenesis in human intestinal microvascular endothelial cells via PI3K/Akt pathway: Potential implications of inflammation bowel disease. Biochem Biophys Res Commun. (2016) 470:967–74. doi: 10.1016/j.bbrc.2016.01.073
54. Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol. (2010) 28:453–9. doi: 10.1200/JCO.2009.24.8252
55. Bagley RG, Ren Y, Weber W, Yao M, Kurtzberg L, Pinckney J, et al. Placental growth factor upregulation is a host response to antiangiogenic therapy. Clin Cancer Res. (2011) 17:976–88. doi: 10.1158/1078-0432.CCR-10-2687
56. Willett CG, Duda DG, di Tomaso E, Boucher Y, Ancukiewicz M, Sahani DV, et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: a multidisciplinary phase II study. J Clin Oncol. (2009) 27:3020–6. doi: 10.1200/JCO.2008.21.1771
57. Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM, et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol. (2008) 26:3743–8. doi: 10.1200/JCO.2007.15.5416
58. Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. (2001) 7:575–83. doi: 10.1038/87904
59. Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. (2007) 131:463–75. doi: 10.1016/j.cell.2007.08.038
60. Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. (1971) 133:275–88. doi: 10.1084/jem.133.2.275
61. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. (2008) 8:579–91. doi: 10.1038/nrc2403
62. Sakurai A, Doçi CL, Gutkind JS. Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer. Cell Res. (2012) 22:23–32. doi: 10.1038/cr.2011.198
63. Eichmann A, Makinen T, Alitalo K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. (2005) 19:1013–21. doi: 10.1101/gad.1305405
64. Wälchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, et al. Wiring the vascular network with neural cues: a CNS perspective. Neuron. (2015) 87:271–96. doi: 10.1016/j.neuron.2015.06.038
65. Dahan N, Magidey K, Shaked Y. editors. Resistance to inhibitors of angiogenesis. In: Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Cham: Springer (2018). p. 211–36. doi: 10.1007/978-3-319-67932-7_9
66. Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. (2010) 15:104–11. doi: 10.1634/theoncologist.2009-0250
67. Rosen LS, Jacobs IA, Burkes RL. Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target Oncol. (2017) 12:599–610. doi: 10.1007/s11523-017-0518-1
68. Boere IA, Hamberg P, Sleijfer S. It takes two to tango: combinations of conventional cytotoxics with compounds targeting the vascular endothelial growth factor-vascular endothelial growth factor receptor pathway in patients with solid malignancies. Cancer Sci. (2010) 101:7–15. doi: 10.1111/j.1349-7006.2009.01369.x
69. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
70. Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. (2012) 21:82–91. doi: 10.1016/j.ccr.2011.11.023
71. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. (2007) 11:83–95. doi: 10.1016/j.ccr.2006.11.021
72. Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. (2006) 290:H560–76. doi: 10.1152/ajpheart.00133.2005
73. de Gramont A, Van Cutsem E, Schmoll HJ, Tabernero J, Clarke S, Moore MJ, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT):a phase 3 randomised controlled trial. Lancet Oncol. (2012) 13:1225–33. doi: 10.1016/S1470-2045(12)70509-0
74. Allegra CJ, Yothers G, O'Connell MJ, Sharif S, Petrelli NJ, Lopa SH, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the national surgical adjuvant breast and bowel project C-08 trial. J Clin Oncol. (2013) 31:359–64. doi: 10.1200/JCO.2012.44.4711
75. Allegra CJ, Yothers G, O'Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol. (2011) 29:11–6. doi: 10.1200/JCO.2010.30.0855
76. FDA Approval for Ziv-Aflibercept. Available online at: https://www.cancer.gov/about-cancer/treatment/drugs/fda-ziv-aflibercept (accessed August 3, 2012).
77. Ciombor KK, Berlin J, Chan E. Aflibercept. Clin Cancer Res. (2013) 19:1920–5. doi: 10.1158/1078-0432.CCR-12-2911
78. Chiron M, Bagley RG, Pollard J, Mankoo PK, Henry C, Vincent L, et al. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol Cancer Ther. (2014) 13:1636–44. doi: 10.1158/1535-7163.MCT-13-0753
79. Bais C, Wu X, Yao J, Yang S, Crawford Y, McCutcheon K, et al. PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell. (2010) 141:166–77. doi: 10.1016/j.cell.2010.01.033
80. Lassen U, Chinot OL, McBain C, Mau-Sørensen M, Larsen VA, Barrie M, et al. Phase 1 dose-escalation study of the antiplacental growth factor monoclonal antibody RO5323441 combined with bevacizumab in patients with recurrent glioblastoma. Neuro Oncol. (2015) 17:1007–15. doi: 10.1093/neuonc/nov019
81. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. (2011) 8:210–21. doi: 10.1038/nrclinonc.2011.21
82. Escudier B, Bellmunt J, Négrier S, Bajetta E, Melichar B, Bracarda S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN):final analysis of overall survival. J Clin Oncol. (2010) 28:2144–50. doi: 10.1200/JCO.2009.26.7849
83. Montero AJ, Escobar M, Lopes G, Glück S, Vogel C. Bevacizumab in the treatment of metastatic breast cancer: friend or foe? Curr Oncol Rep. (2012) 14:1–11. doi: 10.1007/s11912-011-0202-z
84. Ma S, Pradeep S, Hu W, Zhang D, Coleman R, Sood A. The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Res. (2018) 7:326. doi: 10.12688/f1000research.11771.1
85. Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat. (2009) 12:74–80. doi: 10.1016/j.drup.2009.03.002
86. Cooke VG, LeBleu VS, Keskin D, Khan Z, O'Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. (2012) 21:66–81. doi: 10.1016/j.ccr.2011.11.024
87. Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. (2012) 2:270–87. doi: 10.1158/2159-8290.CD-11-0240
88. Shojaei F, Simmons BH, Lee JH, Lappin PB, Christensen JG. HGF/c-Met pathway is one of the mediators of sunitinib-induced tumor cell type-dependent metastasis. Cancer Lett. (2012) 320:48–55. doi: 10.1016/j.canlet.2012.01.026
89. Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, et al. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest. (2012) 122:1832–48. doi: 10.1172/JCI58976
90. Rovida A, Castiglioni V, Decio A, Scarlato V, Scanziani E, Giavazzi R, et al. (2013) Chemotherapy counteracts metastatic dissemination induced by antiangiogenic treatment in mice. Mol Cancer Ther. 12:2237–47. doi: 10.1158/1535-7163.MCT-13-0244
91. Singh M, Couto SS, Forrest WF, Lima A, Cheng JH, Molina R, et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J Pathol. (2012) 227:417–30. doi: 10.1002/path.4053
92. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. (2014) 8:696–706. doi: 10.1016/j.celrep.2014.06.059
93. Brown JL, Cao ZA, Pinzon-Ortiz M, Kendrew J, Reimer C, Wen S, et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther. (2010) 9:145–56. doi: 10.1158/1535-7163.MCT-09-0554
94. Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res. (2013) 19:6730–40. doi: 10.1158/1078-0432.CCR-13-0081
95. Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med. (2016) 8:39–57. doi: 10.15252/emmm.201505505
96. Curtis VF, Wang H, Yang P, McLendon RE, Li X, Zhou QY, et al. A PK2/Bv8/PROK2 antagonist suppresses tumorigenic processes by inhibiting angiogenesis in glioma and blocking myeloid cell infiltration in pancreatic cancer. PLoS ONE. (2013) 8:e54916. doi: 10.1371/journal.pone.0054916
97. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol Cancer Res. (2013) 11:1585–96. doi: 10.1158/1541-7786.MCR-13-0358
98. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. (2005) 8:299–309. doi: 10.1016/j.ccr.2005.09.005
99. Semrad TJ, Kim EJ, Tanaka MS, Sands J, Roberts C, Burich RA, et al. Phase II study of dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat Res Commun. (2017) 10:21–6. doi: 10.1016/j.ctarc.2016.12.002
100. Norden AD, Schiff D, Ahluwalia MS, Lesser GJ, Nayak L, Lee EQ, et al. Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J Neurooncol. (2015) 121:297–302. doi: 10.1007/s11060-014-1631-y
101. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell. (2009) 15:21–34. doi: 10.1016/j.ccr.2008.12.004
102. Rock EP, Goodman V, Jiang JX, Mahjoob K, Verbois SL, Morse D, et al. Food and drug administration drug approval summary: sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist. (2007) 12:107–13. doi: 10.1634/theoncologist.12-1-107
103. FDA Approves Sunitinib Malate for Adjuvant Treatment of Renal Cell Carcinoma. (2017). Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm585686.htm.
104. Hainsworth JD, Spigel DR, Sosman JA, Burris HA, Farley C, Cucullu H, et al. Treatment of advanced renal cell carcinoma with the combination bevacizumab/erlotinib/imatinib: a phase I/II trial. Clin Genitourin Cancer. (2007) 5:427–32. doi: 10.3816/CGC.2007.n.030
105. Chiappori AA, Eckhardt SG, Bukowski R, Sullivan DM, Ikeda M, Yano Y, et al. A phase I pharmacokinetic and pharmacodynamic study of s-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin Cancer Res. (2007) 13:2091–9. doi: 10.1158/1078-0432.CCR-06-1586
106. Liu BY, Soloviev I, Chang P, Lee J, Huang X, Zhong C, et al. Stromal cell-derived factor-1/CXCL12 contributes to MMTV-Wnt1 tumor growth involving Gr1+CD11b+ cells. PLoS ONE. (2010) 5:e8611. doi: 10.1371/journal.pone.0008611
107. Hasnis E, Alishekevitz D, Gingis-Veltski S, Bril R, Fremder E, Voloshin T, et al. Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia. (2014) 16:501–10. doi: 10.1016/j.neo.2014.05.011
108. Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. (2013) 71:1041–50. doi: 10.1007/s00280-013-2099-8
109. Pietras K, Hanahan D. A multitargeted, metronomic, and maximum-tolerated dose chemo-switch regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. (2005) 23:939–52. doi: 10.1200/JCO.2005.07.093
110. Wang L, Park H, Chhim S, Ding Y, Jiang W, Queen C, et al. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol Cancer Ther. (2012) 11:864–72. doi: 10.1158/1535-7163.MCT-11-0813
111. Powell MA, Sill MW, Goodfellow PJ, Benbrook DM, Lankes HA, Leslie KK, et al. A phase II trial of brivanib in recurrent or persistent endometrial cancer: an NRG Oncology/Gynecologic Oncology Group Study. Gynecol Oncol. (2014) 135:38–43. doi: 10.1016/j.ygyno.2014.07.083
112. Wakelee H, Zvirbule Z, De Braud F, Kingsley CD, Mekhail T, Lowe T, et al. Efficacy and safety of onartuzumab in combination with first-line bevacizumab- or pemetrexed-based chemotherapy regimens in advanced non-squamous non-small-cell lung cancer. Clin Lung Cancer. (2017) 18:50–9. doi: 10.1016/j.cllc.2016.09.013
113. Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. (2013) 73:3145–54. doi: 10.1158/0008-5472.CAN-13-0011
114. Jahangiri A, Aghi MK, Carbonell WS. Beta1 Integrin: critical path to antiangiogenic therapy resistance and beyond. Cancer Res. (2014) 74:3–7. doi: 10.1158/0008-5472.CAN-13-1742
115. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. (2009) 15:220–31. doi: 10.1016/j.ccr.2009.01.027
116. Jung HY, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. (2014) 21:962–8. doi: 10.1158/1078-0432.CCR-13-3173
117. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. (1996) 16:4604–13. doi: 10.1128/MCB.16.9.4604
118. Hida K, Ohga N, Akiyama K, Maishi N, Hida Y. Heterogeneity of tumor endothelial cells. Cancer Sci. (2013) 104:1391–5. doi: 10.1111/cas.12251
119. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. (2003) 3:347–61. doi: 10.1016/S1535-6108(03)00085-0
120. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. (2003) 4:915–25. doi: 10.1038/nrm1261
121. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. (2010) 11:834–48. doi: 10.1038/nrm3012
122. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. (2012) 22:21–35. doi: 10.1016/j.ccr.2012.05.037
123. Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. (2013) 19:1773–83. doi: 10.1158/1078-0432.CCR-12-1281
124. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. (2010) 70:10090–100. doi: 10.1158/0008-5472.CAN-10-0489
125. Escudier B, Powles T, Motzer RJ, Olencki T, Arén Frontera O, Oudard S, et al. Cabozantinib, a new standard of care for patients with advanced renal cell carcinoma and bone metastases? Subgroup analysis of the METEOR trial. J Clin Oncol. (2018) 36:765–72. doi: 10.1200/JCO.2017.74.7352
126. Cordes N, Park CC. beta1 integrin as a molecular therapeutic target. Int J Radiat Biol. (2007) 83:753–60. doi: 10.1080/09553000701639694
127. Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. (1999) 5:662–8. doi: 10.1038/9511
128. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. (2009) 9:665–74. doi: 10.1038/nrc2714
129. Gotzmann J, Mikula M, Eger A, Schulte-Hermann R, Foisner R, Beug H, et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. (2004) 566:9–20. doi: 10.1016/S1383-5742(03)00033-4
130. Foubert P, Varner JA. Integrins in tumor angiogenesis and lymphangiogenesis. Methods Mol Biol. (2012) 757:471–86. doi: 10.1007/978-1-61779-166-6_27
131. Sidorov M, Jahangiri A, Han S-W, De Lay M, Wagner J, Castro B, et al. 340 c-Met/β1 Integrin: a receptor complex driving invasive glioblastoma resistance to antiangiogenic therapy. Neurosurgery. (2016) 63:199–200. doi: 10.1227/01.neu.0000489829.38251.85
132. Jahangiri A, Nguyen A, Chandra A, Sidorov MK, Yagnik G, Rick J, et al. Cross-activating c-Met/beta1 integrin complex drives metastasis and invasive resistance in cancer. Proc Natl Acad Sci USA. (2017) 114:E8685–94. doi: 10.1073/pnas.1701821114
133. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. (2009) 15:232–9. doi: 10.1016/j.ccr.2009.01.021
134. Grepin R, Guyot M, Jacquin M, Durivault J, Chamorey E, Sudaka A, et al. Acceleration of clear cell renal cell carcinoma growth in mice following bevacizumab/Avastin treatment: the role of CXCL cytokines. Oncogene. (2012) 31:1683–94. doi: 10.1038/onc.2011.360
135. Chung AS, Kowanetz M, Wu X, Zhuang G, Ngu H, Finkle D, et al. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J Pathol. (2012) 227:404–16. doi: 10.1002/path.4052
136. Welti JC, Powles T, Foo S, Gourlaouen M, Preece N, Foster J, et al. Contrasting effects of sunitinib within in vivo models of metastasis. Angiogenesis. (2012) 15:623–41. doi: 10.1007/s10456-012-9291-z
137. Alto LT, Terman JR. Semaphorins and their signaling mechanisms. Methods Mol Biol. (2017) 1493:1–25. doi: 10.1007/978-1-4939-6448-2_1
138. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. (2004) 117:927–39. doi: 10.1016/j.cell.2004.06.006
139. Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. (2012) 72:1909–14. doi: 10.1158/0008-5472.CAN-11-3406
140. Falcon BL, Chintharlapalli S, Uhlik MT, Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol Ther. (2016) 164:204–25. doi: 10.1016/j.pharmthera.2016.06.001
141. van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ, Thijssen VL, Griffioen AW. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol Rev. (2015) 67:441–61. doi: 10.1124/pr.114.010215
142. Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. (2005) 23:8136–9. doi: 10.1200/JCO.2005.02.5635
143. Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. (2010) 28:2817–23. doi: 10.1200/JCO.2009.26.3988
144. Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. (2010) 70:299–308. doi: 10.1158/0008-5472.CAN-09-1845
145. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. (1997) 277:55–60. doi: 10.1126/science.277.5322.55
146. Hanahan D. Signaling vascular morphogenesis and maintenance. Science. (1997) 277:48–50. doi: 10.1126/science.277.5322.48
147. Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. (2013) 319:1271–80. doi: 10.1016/j.yexcr.2013.03.011
148. Helfrich I, Edler L, Sucker A, Thomas M, Christian S, Schadendorf D, et al. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin Cancer Res. (2009) 15:1384–92. doi: 10.1158/1078-0432.CCR-08-1615
149. Sfiligoi C, de Luca A, Cascone I, Sorbello V, Fuso L, Ponzone R, et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int J Cancer. (2003) 103:466–74. doi: 10.1002/ijc.10851
150. Moon WS, Rhyu KH, Kang MJ, Lee DG, Yu HC, Yeum JH, et al. Overexpression of VEGF and angiopoietin 2:a key to high vascularity of hepatocellular carcinoma? Mod Pathol. (2003) 16:552–7. doi: 10.1097/01.MP.0000071841.17900.69
151. Ogawa M, Yamamoto H, Nagano H, Miyake Y, Sugita Y, Hata T, et al. Hepatic expression of ANG2 RNA in metastatic colorectal cancer. Hepatology. (2004) 39:528–39. doi: 10.1002/hep.20048
152. Goede V, Coutelle O, Neuneier J, Reinacher-Schick A, Schnell R, Koslowsky TC, et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br J Cancer. (2010) 103:1407–14. doi: 10.1038/sj.bjc.6605925
153. A Study of Vanucizumab (RO5520985) Alone or in Combination with Atezolizumab in Participants with Locally Advanced or Metastatic Solid Tumors. (2018). Available online: https://clinicaltrials.gov/ct2/show/NCT01688206
154. Study Comparing the Efficacy and Safety of Vanucizumab and FOLFOX with Bevacizmab and FOLFOX in Participants with Untreated Metastatic Colorectal Cancer (McCAVE). (2018). Available online: https://clinicaltrials.gov/ct2/show/NCT02141295
155. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. (2013) 19:1114–23. doi: 10.1038/nm.3291
156. Evans J, Catalano RD, Morgan K, Critchley HO, Millar RP, Jabbour HN. Prokineticin 1 signaling and gene regulation in early human pregnancy. Endocrinology. (2008) 149:2877–87. doi: 10.1210/en.2007-1633
157. Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, et al. The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev. (2011) 32:225–46. doi: 10.1210/er.2010-0007
158. LeCouter J, Lin R, Tejada M, Frantz G, Peale F, Hillan KJ, et al. The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc Natl Acad Sci USA. (2003) 100:2685–90. doi: 10.1073/pnas.0337667100
159. Qu X, Zhuang G, Yu L, Meng G, Ferrara N. Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: key role of Stat3 signaling. J Biol Chem. (2012) 287:19574–84. doi: 10.1074/jbc.M111.326801
160. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA. (2010) 107:21248–55. doi: 10.1073/pnas.1015855107
161. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci USA. (2009) 106:6742–7. doi: 10.1073/pnas.0902280106
162. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. (2007) 450:825–31. doi: 10.1038/nature06348
163. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. (2015) 4:215–66. doi: 10.1002/wdev.176
164. Poon RT, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol. (2001) 19:1207–25. doi: 10.1200/JCO.2001.19.4.1207
165. LaVallee TM, Prudovsky IA, McMahon GA, Hu X, Maciag T. Activation of the MAP kinase pathway by FGF-1 correlates with cell proliferation induction while activation of the Src pathway correlates with migration. J Cell Biol. (1998) 141:1647–58. doi: 10.1083/jcb.141.7.1647
166. Chen Y, Li X, Eswarakumar VP, Seger R, Lonai P. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene. (2000) 19:3750–6. doi: 10.1038/sj.onc.1203726
167. Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. Transformation and stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. (2000) 19:3309–20. doi: 10.1038/sj.onc.1203650
168. Chae YK, Ranganath K, Hammerman PS, Vaklavas C, Mohindra N, Kalyan A, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. (2017) 8:16052–74. doi: 10.18632/oncotarget.14109
169. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. (2010) 10:116–29. doi: 10.1038/nrc2780
170. Mitsuhashi A, Goto H, Saijo A, Trung VT, Aono Y, Ogino H, et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat Commun. (2015) 6:8792. doi: 10.1038/ncomms9792
171. Lee CK, Lee ME, Lee WS, Kim JM, Park KH, Kim TS, et al. Dovitinib (TKI258), a multi-target angiokinase inhibitor, is effective regardless of KRAS or BRAF mutation status in colorectal cancer. Am J Cancer Res. (2015) 5:72–86.
172. André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258):preclinical and clinical data in breast cancer. Clin Cancer Res. (2013) 19:3693–702. doi: 10.1158/1078-0432.CCR-13-0190
173. Burbridge MF, Bossard CJ, Saunier C, Fejes I, Bruno A, Léonce S, et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol Cancer Ther. (2013) 12:1749–62. doi: 10.1158/1535-7163.MCT-13-0075
174. Montmayeur JP, Valius M, Vandenheede J, Kazlauskas A. The platelet-derived growth factor beta receptor triggers multiple cytoplasmic signaling cascades that arrive at the nucleus as distinguishable inputs. J Biol Chem. (1997) 272:32670–8. doi: 10.1074/jbc.272.51.32670
175. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. (2013) 11:97. doi: 10.1186/1478-811X-11-97
176. Francia G, Emmenegger U, Kerbel RS. Tumor-associated fibroblasts as trojan horse mediators of resistance to anti-VEGF therapy. Cancer Cell. (2009) 15:3–5. doi: 10.1016/j.ccr.2008.12.011
177. Roskoski R. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. (2007) 356:323–8. doi: 10.1016/j.bbrc.2007.02.156
178. Massague J. TGF-beta signal transduction. Annu Rev Biochem. (1998) 67:753–91. doi: 10.1146/annurev.biochem.67.1.753
179. Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. (2000) 103:295–309. doi: 10.1016/S0092-8674(00)00121-5
180. Barcellos-Hoff MH, Akhurst RJ. Transforming growth factor-beta in breast cancer: too much, too late. Breast Cancer Res. (2009) 11:202. doi: 10.1186/bcr2224
181. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. (2001) 29:117–29. doi: 10.1038/ng1001-117
182. Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. (1986) 83:4167–71. doi: 10.1073/pnas.83.12.4167
183. Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol. (1988) 106:1375–84. doi: 10.1083/jcb.106.4.1375
184. Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. (2009) 219:449–58. doi: 10.1002/jcp.21706
185. Derynck R, Goeddel DV, Ullrich A, Gutterman JU, Williams RD, Bringman TS, et al. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. (1987) 47:707–12.
186. Chen XL, Chen ZQ, Zhu SL, Liu TW, Wen Y, Su YS, et al. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: a meta-analysis. BMC Cancer. (2017) 17:240. doi: 10.1186/s12885-017-3215-7
187. Dave H, Shah M, Trivedi S, Shukla S. Prognostic utility of circulating transforming growth factor beta 1 in breast cancer patients. Int J Biol Markers. (2012) 27:53–9. doi: 10.5301/JBM.2011.8736
188. Park SY, Piao Y, Jeong KJ, Dong J, de Groot JF. Periostin (POSTN) regulates tumor resistance to antiangiogenic therapy in glioma models. Mol Cancer Ther. (2016) 15:2187–97. doi: 10.1158/1535-7163.MCT-15-0427
189. Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting transforming growth factor (TGF)-betaRI inhibits activation of beta1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology. (2009) 49:839–50. doi: 10.1002/hep.22731
190. Zhang D, Hedlund EM, Lim S, Chen F, Zhang Y, Sun B, et al. Antiangiogenic agents significantly improve survival in tumor-bearing mice by increasing tolerance to chemotherapy-induced toxicity. Proc Natl Acad Sci USA. (2011) 108:4117–22. doi: 10.1073/pnas.1016220108
191. Comunanza V, Bussolino F. Therapy for cancer: strategy of combining anti-angiogenic and target therapies. Front Cell Dev Biol. (2017) 5:101. doi: 10.3389/fcell.2017.00101
192. Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. (2015) 147:22–31. doi: 10.1016/j.pharmthera.2014.11.001
193. Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunotherap Cancer. (2018) 6:47. doi: 10.1186/s40425-018-0356-4
194. Clarke JM, Blobe GC, Strickler JH, Uronis HE, Zafar SY, Morse M, et al. Phase Ib study of regorafenib (rego) and PF-03446962 (PF) in patients with refractory metastatic colorectal cancer (mCRC) (REGAL). J Clin Oncol. (2016) 34 (15_suppl):e15013. doi: 10.1200/JCO.2016.34.15_suppl.e15013
195. Mannello F, Tonti G, Papa S. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr Cancer Drug Targets. (2005) 5:285–98. doi: 10.2174/1568009054064615
196. Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. (1997) 89:1260–70. doi: 10.1093/jnci/89.17.1260
197. Jabłonska-Trypuć A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J Enzyme Inhib Med Chem. (2016) 31:177–83. doi: 10.3109/14756366.2016.1161620
198. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. (2000) 2:737–44. doi: 10.1038/35036374
199. Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. (1998) 95:365–77. doi: 10.1016/S0092-8674(00)81768-7
200. Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. (2004) 167:757–67. doi: 10.1083/jcb.200405001
201. Oblander SA, Zhou Z, Gálvez BG, Starcher B, Shannon JM, Durbeej M, et al. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev Biol. (2005) 277:255–69. doi: 10.1016/j.ydbio.2004.09.033
202. Deryugina EI, Quigley JP. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim Biophys Acta. (2010) 1803:103–20. doi: 10.1016/j.bbamcr.2009.09.017
203. Fingleton B. Matrix metalloproteinases: roles in cancer and metastasis. Front Biosci. (2006) 11:479–91. doi: 10.2741/1811
204. Bauvois B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim Biophys Acta. (2012) 1825:29–36. doi: 10.1016/j.bbcan.2011.10.001
205. Cianfrocca M, Lee S, Von Roenn J, Rudek MA, Dezube BJ, Krown SE, et al. Pilot study evaluating the interaction between paclitaxel and protease inhibitors in patients with human immunodeficiency virus-associated kaposi's sarcoma: an Eastern Cooperative Oncology Group (ECOG) and AIDS Malignancy Consortium (AMC) trial. Cancer Chemother Pharmacol. (2011) 68:827–33. doi: 10.1007/s00280-010-1509-4
206. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. (2011) 91:1071–121. doi: 10.1152/physrev.00038.2010
207. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. (2008) 267:204–15. doi: 10.1016/j.canlet.2008.03.028
208. Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. (2007) 28:299–307. doi: 10.1016/j.it.2007.05.007
209. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. (2004) 10:858–64. doi: 10.1038/nm1075
210. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
211. Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. (2006) 16:53–65. doi: 10.1016/j.semcancer.2005.07.005
212. Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. (2008) 222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x
213. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. (2009) 58:49–59. doi: 10.1007/s00262-008-0523-4
214. Shojaei F, Ferrara N. Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res. (2008) 68:5501–4. doi: 10.1158/0008-5472.CAN-08-0925
215. Crawford Y, Ferrara N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol Sci. (2009) 30:624–30. doi: 10.1016/j.tips.2009.09.004
216. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. (2007) 25:911–20. doi: 10.1038/nbt1323
217. Roland CL, Dineen SP, Lynn KD, Sullivan LA, Dellinger MT, Sadegh L, et al. Inhibition of vascular endothelial growth factor reduces angiogenesis and modulates immune cell infiltration of orthotopic breast cancer xenografts. Mol Cancer Ther. (2009) 8:1761–71. doi: 10.1158/1535-7163.MCT-09-0280
218. Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, et al. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. (2012) 14:1379–92. doi: 10.1093/neuonc/nos158
219. Cetin B, Kaplan MA, Berk V, Ozturk SC, Benekli M, Isikdogan A, et al. Prognostic factors for overall survival in patients with metastatic colorectal carcinoma treated with vascular endothelial growth factor-targeting agents. Asian Pac J Cancer Prev. (2012) 13:1059–63. doi: 10.7314/APJCP.2012.13.3.1059
220. Botta C, Barbieri V, Ciliberto D, Rossi A, Rocco D, Addeo R, et al. Systemic inflammatory status at baseline predicts bevacizumab benefit in advanced non-small cell lung cancer patients. Cancer Biol Ther. (2013) 14:469–75. doi: 10.4161/cbt.24425
221. Dirican A, Varol U, Kucukzeybek Y, Alacacioglu A, Erten C, Somali I, et al. Treatment of metastatic colorectal cancer with or without bevacizumab: can the neutrophil/lymphocyte ratio predict the efficiency of bevacizumab? Asian Pac J Cancer Prev. (2014) 15:4781–6. doi: 10.7314/APJCP.2014.15.12.4781
222. Passardi A, Scarpi E, Cavanna L, Dall'Agata M, Tassinari D, Leo S, et al. Inflammatory indexes as predictors of prognosis and bevacizumab efficacy in patients with metastatic colorectal cancer. Oncotarget. (2016) 7:33210–9. doi: 10.18632/oncotarget.8901
223. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. (2014) 20:187–98. doi: 10.1158/1078-0432.CCR-13-1279
224. Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. (2013) 2013:187204. doi: 10.1155/2013/187204
225. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. (2009) 86:1065–73. doi: 10.1189/jlb.0609385
226. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
227. Schmidt T, Carmeliet P. Blood-vessel formation: bridges that guide and unite. Nature. (2010) 465:697–9. doi: 10.1038/465697a
228. Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. (2010) 116:829–40. doi: 10.1182/blood-2009-12-257832
229. Coussens LM, Werb Z. Inflammation and cancer. Nature. (2002) 420:860–7. doi: 10.1038/nature01322
230. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
231. Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. (2008) 8:618–31. doi: 10.1038/nrc2444
232. Hotchkiss KA, Ashton AW, Klein RS, Lenzi ML, Zhu GH, Schwartz EL. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res. (2003) 63:527–33.
233. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
234. Salnikov AV, Heldin NE, Stuhr LB, Wiig H, Gerber H, Reed RK, et al. Inhibition of carcinoma cell-derived VEGF reduces inflammatory characteristics in xenograft carcinoma. Int J Cancer. (2006) 119:2795–802. doi: 10.1002/ijc.22217
235. Lynn KD, Roland CL, Brekken RA. VEGF and pleiotrophin modulate the immune profile of breast cancer. Cancers. (2010) 2:970–88. doi: 10.3390/cancers2020970
236. Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, et al. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. (2008) 68:4340–6. doi: 10.1158/0008-5472.CAN-07-6705
237. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. (2005) 8:211–26. doi: 10.1016/j.ccr.2005.08.002
238. Sica A, Porta C, Morlacchi S, Banfi S, Strauss L, Rimoldi M, et al. Origin and functions of tumor-associated myeloid cells (TAMCs). Cancer Microenviron. (2012) 5:133–49. doi: 10.1007/s12307-011-0091-6
239. Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. (2007) 178:7405–11. doi: 10.4049/jimmunol.178.11.7405
240. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. (2007) 109:5276–85. doi: 10.1182/blood-2006-10-053504
241. De Palma M, Naldini L. Angiopoietin-2 TIEs up macrophages in tumor angiogenesis. Clin Cancer Res. (2011) 17:5226–32. doi: 10.1158/1078-0432.CCR-10-0171
242. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. (2011) 19:512–26. doi: 10.1016/j.ccr.2011.02.005
243. Clarke JM, Hurwitz HI. Understanding and targeting resistance to anti-angiogenic therapies. J Gastrointest Oncol. (2013) 4:253–63. doi: 10.3978/j.issn.2078-6891.2013.036
244. Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. (2010) 70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012
245. Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci USA. (2008) 105:2640–5. doi: 10.1073/pnas.0712185105
246. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
247. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
248. Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene. (2017) 36:3749–59. doi: 10.1038/onc.2017.1
249. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. (2008) 8:592–603. doi: 10.1038/nrc2442
250. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
251. Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. (2006) 124:175–89. doi: 10.1016/j.cell.2006.08.015
252. Dai X, Tan Y, Cai S, Xiong X, Wang L, Ye Q, et al. The role of CXCR7 on the adhesion, proliferation and angiogenesis of endothelial progenitor cells. J Cell Mol Med. (2011) 15:1299–309. doi: 10.1111/j.1582-4934.2011.01301.x
253. Yan X, Cai S, Xiong X, Sun W, Dai X, Chen S, et al. Chemokine receptor CXCR7 mediates human endothelial progenitor cells survival, angiogenesis, but not proliferation. J Cell Biochem. (2012) 113:1437–46. doi: 10.1002/jcb.24015
254. Azab AK, Sahin I, Moschetta M, Mishima Y, Burwick N, Zimmermann J, et al. CXCR7-dependent angiogenic mononuclear cell trafficking regulates tumor progression in multiple myeloma. Blood. (2014) 124:1905–14. doi: 10.1182/blood-2014-02-558742
255. Teicher BA, Ellis LM. Antiangiogenic Agents in Cancer Therapy, Totowa, NJ: Humana Press (2008) doi: 10.1007/978-1-59745-184-0
256. Shepro D, Morel NM. Pericyte physiology. FASEB J. (1993) 7:1031–8. doi: 10.1096/fasebj.7.11.8370472
257. Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. (2003) 112:1142–51. doi: 10.1172/JCI200318549
258. Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. (2007) 96:1788–95. doi: 10.1038/sj.bjc.6603813
259. Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. (2009) 5:610–20. doi: 10.1038/nrneurol.2009.159
260. Pinto MP, Sotomayor P, Carrasco-Avino G, Corvalan AH, Owen GI. Escaping antiangiogenic therapy: strategies employed by cancer cells. Int J Mol Sci. (2016) 17:E1489. doi: 10.3390/ijms17091489
261. Thomas M, Kienast Y, Scheuer W, Bähner M, Kaluza K, Gassner C, et al. A novel angiopoietin-2 selective fully human antibody with potent anti-tumoral and anti-angiogenic efficacy and superior side effect profile compared to pan-angiopoietin-1/-2 inhibitors. PLoS ONE. (2013) 8:e54923. doi: 10.1371/journal.pone.0054923
262. Orlidge A, D'Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol. (1987) 105:1455–62. doi: 10.1083/jcb.105.3.1455
263. Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. (2004) 6:553–63. doi: 10.1016/S1535-6108(04)00305-8
264. McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J Clin Invest. (2007) 117:2114–22. doi: 10.1172/JCI31334
265. Martin JD, Seano G, Jain RK. Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol. (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
266. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol. (2018) 15:310–24. doi: 10.1038/nrclinonc.2018.9
267. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. (2018) 15:325–40. doi: 10.1038/nrclinonc.2018.29
268. Gioelli N, Maione F, Camillo C, Ghitti M, Valdembri D, Morello N, et al. A rationally designed NRP1-independent superagonist SEMA3A mutant is an effective anticancer agent. Sci Transl Med. (2018) 10:eaah4807. doi: 10.1126/scitranslmed.aah4807
269. Serini G, Bussolino F, Maione F, Giraudo E. Class 3 semaphorins: physiological vascular normalizing agents for anti-cancer therapy. J Intern Med. (2013) 273:138–55. doi: 10.1111/joim.12017
270. Stoeltzing O, Ahmad SA, Liu W, McCarty MF, Wey JS, Parikh AA, et al. Angiopoietin-1 inhibits vascular permeability, angiogenesis, and growth of hepatic colon cancer tumors. Cancer Res. (2003) 63:3370–7.
271. Kumar S, Mokhtari RB, Oliveira ID, Islam S, Toledo SR, Yeger H, et al. Tumor dynamics in response to antiangiogenic therapy with oral metronomic topotecan and pazopanib in neuroblastoma xenografts. Transl Oncol. (2013) 6:493–503. doi: 10.1593/tlo.13286
272. Czabanka M, Bruenner J, Parmaksiz G, Broggini T, Topalovic M, Bayerl SH, et al. Combined temozolomide and sunitinib treatment leads to better tumour control but increased vascular resistance in O6-methylguanine methyltransferase-methylated gliomas. Eur J Cancer. (2013) 49:2243–52. doi: 10.1016/j.ejca.2013.02.019
273. Reinmuth N, Liu W, Jung YD, Ahmad SA, Shaheen RM, Fan F, et al. Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. FASEB J. (2001) 15:1239–41. doi: 10.1096/fj.00-0693fje
274. Stratman AN, Davis MJ, Davis GE. VEGF and FGF prime vascular tube morphogenesis and sprouting directed by hematopoietic stem cell cytokines. Blood. (2011) 117:3709–19. doi: 10.1182/blood-2010-11-316752
275. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. (2003) 111:1287–95. doi: 10.1172/JCI200317929
276. Nisancioglu MH, Betsholtz C, Genové G. The absence of pericytes does not increase the sensitivity of tumor vasculature to vascular endothelial growth factor-A blockade. Cancer Res. (2010) 70:5109–15. doi: 10.1158/0008-5472.CAN-09-4245
277. Chantrain CF, Henriet P, Jodele S, Emonard H, Feron O, Courtoy PJ, et al. Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur J Cancer. (2006) 42:310–8. doi: 10.1016/j.ejca.2005.11.010
278. Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. (2001) 61:550–5.
279. Mueller MM, Fusenig NE. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. (2004) 4:839–49. doi: 10.1038/nrc1477
280. Dong J, Grunstein J, Tejada M, Peale F, Frantz G, Liang WC, et al. VEGF-null cells require PDGFR α signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. (2004) 23:2800–10. doi: 10.1038/sj.emboj.7600289
281. Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Ann Rev Cell Biol. (1993) 9:541–73. doi: 10.1146/annurev.cb.09.110193.002545
282. Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW., et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. (1999) 98:137–46. doi: 10.1016/S0092-8674(00)81009-0
283. Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. (2005) 120:303–13. doi: 10.1016/j.cell.2004.12.018
284. Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. (2007) 26:489–502. doi: 10.1007/s10555-007-9094-7
285. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T., et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. (1997) 275:964–66. doi: 10.1126/science.275.5302.964
286. Djonov V, Schmid M, Tschanz SA, Burri PH. Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circul Res. (2000) 86:286–92. doi: 10.1161/01.RES.86.3.286
287. Coelho AL, Gomes MP, Catarino RJ, Rolfo C, Lopes AM, Medeiros RM, et al. Angiogenesis in NSCLC: is vessel co-option the trunk that sustains the branches? Oncotarget. (2017) 8:39795–804. doi: 10.18632/oncotarget.7794
288. Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, et al. Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med. (2013) 2:427–36. doi: 10.1002/cam4.105
289. Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E., et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. (2008) 13:206–20. doi: 10.1016/j.ccr.2008.01.034
290. Leenders WP, Küsters B, Verrijp K, Maass C, Wesseling P, Heerschap A., et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. (2004) 10:6222–30. doi: 10.1158/1078-0432.CCR-04-0823
291. Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, et al. Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J Natl Cancer Inst. (2016) 108:djw030. doi: 10.1093/jnci/djw030
292. Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E., et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med. (2016) 22:1294–1302. doi: 10.1038/nm.4197
293. Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF., et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. (2000) 2:306–14. doi: 10.1038/sj.neo.7900102
294. Qian CN, Tan MH, Yang JP, Cao Y. Revisiting tumor angiogenesis: vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin J Cancer. (2016) 35:10. doi: 10.1186/s40880-015-0070-2
295. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. (1999) 155:739–52. doi: 10.1016/S0002-9440(10)65173-5
296. Ge H, Luo H. Overview of advances in vasculogenic mimicry - a potential target for tumor therapy. Cancer Manag Res. (2018) 10:2429–37. doi: 10.2147/CMAR.S164675
297. Hillen F, Baeten CI, van de Winkel A, Creytens D, van der Schaft DW, Winnepenninckx V., et al. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol Immunother. (2008) 57:97–106. doi: 10.1007/s00262-007-0353-9
298. van der Schaft DW, Hillen F, Pauwels P, Kirschmann DA, Castermans K, Egbrink MG., et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. (2005) 65:11520–8. doi: 10.1158/0008-5472.CAN-05-2468
299. Sood AK, Seftor EA, Fletcher MS, Gardner LM, Heidger PM, Buller RE., et al. Molecular determinants of ovarian cancer plasticity. Am J Pathol. (2001) 158:1279–88. doi: 10.1016/S0002-9440(10)64079-5
300. Kuczynski EA. Mechanisms of reversible sorafenib resistance in hepatocellular carcinoma (Doktorarbeit). Doktorarbeit University of Toronto, Toronto, ON, Canada (2016).
301. van der Schaft DW, Seftor RE, Seftor EA, Hess AR, Gruman LM, Kirschmann DA., et al. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J Natl Cancer Inst. (2004) 96:1473–7. doi: 10.1093/jnci/djh267
302. Xu Y, Li Q, Li XY, Yang QY, Xu WW, Liu GL. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J Exp Clin Cancer Res. (2012) 31:16. doi: 10.1186/1756-9966-31-16
303. Sun B, Zhang D, Zhang S, Zhang W, Guo H, Zhao X. Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett. (2007) 249:188–97. doi: 10.1016/j.canlet.2006.08.016
304. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. (2003) 3:411–21. doi: 10.1038/nrc1092
305. Topczewska JM, Postovit LM, Margaryan NV, Sam A, Hess AR, Wheaton WW., et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med. (2006) 12:925–32. doi: 10.1038/nm1448
306. Paulis YW, Soetekouw PM, Verheul HM, Tjan-Heijnen VC, Griffioen AW. Signalling pathways in vasculogenic mimicry. Biochim Biophys Acta. (2010) 1806:18–28. doi: 10.1016/j.bbcan.2010.01.001
307. Chen HF, Huang CH, Liu CJ, Hung JJ, Hsu CC, Teng SC., et al. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun. (2014) 5:4697. doi: 10.1038/ncomms5697
308. Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB., et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor–resistant human lung adenocarcinoma. J Clin Investig. (2011) 121:1313–28. doi: 10.1172/JCI42405
309. Javan MR, Khosrojerdi A, Moazzeni SM. New insights into implementation of mesenchymal stem cells in cancer therapy: prospects for anti-angiogenesis treatment. Front Oncol. (2019) 9:840. doi: 10.3389/fonc.2019.00840
310. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. (2019) doi: 10.1007/s00018-019-03351-7. [Epub ahead of print].
311. Abdalla AME, Xiao L, Ullah MW, Yu M, Ouyang C, Yang G. Current challenges of cancer anti-angiogenic therapy and the promise of nanotherapeutics. Theranostics. (2018) 8:533–48. doi: 10.7150/thno.21674
Keywords: VEGF, VEGF-R, bevacizumab, colorectal cancer, angiogenesis, resistance mechanisms
Citation: Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S and Shamseddine A (2020) Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 10:221. doi: 10.3389/fonc.2020.00221
Received: 20 June 2019; Accepted: 10 February 2020;
Published: 27 February 2020.
Edited by:
Erika Ruiz-Garcia, National Institute of Cancerology (INCan), MexicoReviewed by:
Luca Tamagnone, Institute for Cancer Research and Treatment (IRCC), ItalyLasse Dahl Ejby Jensen, Linköping University, Sweden
Copyright © 2020 Haibe, Kreidieh, El Hajj, Khalifeh, Mukherji, Temraz and Shamseddine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali Shamseddine, YXMwNCYjeDAwMDQwO2F1Yi5lZHUubGI=
†These authors have contributed equally to this work and share first authorship