 Caroline Bergenfelz
Caroline Bergenfelz Karin Leandersson
Karin Leandersson- 1Department of Translational Medicine, Division of Experimental Infection Medicine, Lund University, Malmö, Sweden
- 2Department of Translational Medicine, Cancer Immunology, Lund University, Skåne University Hospital, Malmö, Sweden
Myeloid-derived suppressor cells (MDSCs) are cells of myeloid lineage with a potent immunosuppressive capacity. They are present in cancer patients as well as in patients with severe inflammatory conditions and infections. MDSCs exist as two main subtypes, the granulocytic (G-MDSCs) and the monocytic (Mo-MDSCs) type, as defined by their surface phenotype and functions. While the functions of MDSCs have been investigated in depth, the origin of human MDSCs is less characterized and even controversial. In this review, we recapitulate theories on how MDSCs are generated in mice, and whether this knowledge is translatable into human MDSC biology, as well as on problems of defining MDSCs by their immature cell surface phenotype in relation to the plasticity of myeloid cells. Finally, the challenge of pharmacological targeting of MDSCs in the future is envisioned.
A Brief History of Myeloid-Derived Suppressor Cells (MDSCs)
Already in 1929, cancer was found to be associated with an aberrant myelopoiesis (1). In the late 1960–80s, experiments revealed leukocytosis, granulocytosis, and extramedullary myelopoiesis in tumor-bearing mice (2–5). This aberrant emergency myelopoiesis was driven by tumor cell-derived colony stimulating factors GM-CSF, G-CSF and M-CSF (5–10), that also promoted cancer cell growth (8). During the same time period, the “left shift” test was established as a clinical test in patients with severe bacterial infections. “Left shift” is defined as an increased ratio of immature myelocytes, metamyelocytes and band neutrophils (i.e., shifted to the left of the differentiation model) in blood smears from patients (11–14). A similar “left shift” is also proposed in patients with sterile inflammation and cancer, although not necessarily associated with as severe leukocytosis (15, 16). The leukocytosis in sepsis patients is a normal feedback regulation to replace consumed neutrophils, and is likely caused by similar factors that cause the aberrant myelopoiesis in cancer, including colony stimulating factors, other growth factors and secondary host responses such as damage associated molecular patterns (DAMPs) (16). The first studies showing that the increased systemic immature myeloid cells in tumor-bearing mice where immunosuppressive (“natural suppressor cells”), came in the late 1970s (3, 17–19) but not until 1996 this was first shown in humans (20). Over the following years, the definition of subpopulations and mechanisms of action were heavily investigated (21) and the consensus terminology myeloid-derived suppressor cells (MDSCs) was established in 2007 (22). Today, MDSCs are divided into two main subtypes; the granulocytic-MDSCs [G-MDSCs or polymorphonuclear (PMN)-MDSCs] and monocytic-MDSCs (Mo-MDSCs). A third subpopulation has also been proposed, the early-stage MDSC (eMDSC) that lacks both CD14 and CD15 expression, which will not be covered in this review (23). All subpopulations above are excellently reviewed from different angels in previous publications (15, 16, 21).
Defining Myeloid-Derived Suppressor Cells (MDSCs) in Mice and Men
The current definition of MDSCs is that that they should be of myeloid origin and enriched in mice/patients with cancer or severe disease, display an immature surface phenotype and with the key defining trait being their potent immunosuppressive capacity (23–26). Using these criteria, they are further divided into Mo-MDSCs and G-MDSCs (26–28). In this review we will use the nomenclature G-MDSC, and not PMN-MDSCs, since this latter population consists of cells with heterogenous morphology and not only polymorphonuclear cells (29, 30). In mice Mo-MDSCs are defined by the surface phenotype CD11b+Ly6G−Ly6Chi and G-MDSCs by CD11b+Ly6G+Ly6Clo (31). In humans Mo-MDSCs are CD14+HLA-DR−/lo and G-MDSCs CD11b+CD15+CD14−CD33+/loCD66b+ cells with a low density [low density granulocytes (LDGs)] (23, 32, 33), and are hence present in the peripheral blood mononuclear (PBMC) fraction of gradient centrifugations. Many markers are still appearing in efforts to further define the human MDSC subsets (34), one being S100A9 (35, 36).
Using these criteria, MDSCs have been studied successfully in mice for many years, and in humans for slightly more than a decade with varying results. In mice, CD11b+Ly6G−Ly6Chi Mo-MDSCs and CD11b+Ly6G+Ly6Clo G-MDSCs with immunosuppressive capacity can be enriched and studied from peripheral blood, secondary lymphoid organs and tumors, with quite consistent results. In humans, using the Mo-MDSC CD14+HLADR−/lo and G-MDSC CD11b+CD15+CD14−CD33+/loCD66b+ cell markers for identification has turned out to be complex. There are multiple reasons for this, some being; (i) Immaturity vs. Plasticity; the problem of defining heterogeneous cell populations using cell surface markers, (ii) Subpopulations vs. Technical issues; the problem of comparing human blood and tissue MDSCs along with the problem of investigating human MDSCs by other means than flow cytometry of PBMCs as source, (iii) In vitro vs. In vivo; as recently suggested, functional studies on human cells are for natural reasons more often performed ex vivo, but all in vitro generated human MDSCs should by all means be defined as “MDSC-like” cells (23). Therefore, questions still remain concerning subsets, origin, and function of human MDSCs. If the debate concerning the true identity of human MDSCs, and subsets thereof, would be of only philosophical character, one could still adhere to the most important notion that they are myeloid cells with an immunosuppressive capacity, and an immature surface phenotype. However, when the question concerns how to be able to target them in cancer patients, the issue of defining human MDSC subsets identity and their origin, is still in need of improvement. Below we will discuss the generation and identity of the different human MDSC subsets and put them in context with their sites of distribution (Figure 1).
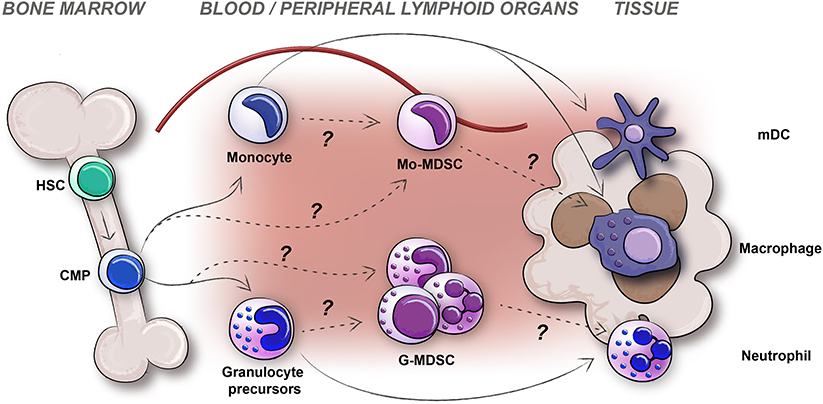
Figure 1. The generation, distribution, and plasticity of human MDSCs subtypes; monocytic-MDSCs (Mo-MDSC) and granulocytic-MDSCs (G-MDSCs) are pictured. Differentiation of myeloid cells in healthy individuals (solid arrows) and potential origin of MDSCs during disease (dashed arrows) are indicated.
Human Peripheral Blood G-MDSCs
G-MDSCs are a heterogeneous population of cells of the granulocytic lineage. In mice, the surface marker definition is CD11b+Ly6G+Ly6Clo, while in human the definition is CD11b+CD15+CD14−CD33+/loCD66b+ cells with a low density (LDGs) (23, 30). As for all MDSCs, the most critical trait is their immunosuppressive activity. For G-MDSCs, suppression of immune responses is conveyed in an antigen-specific manner, and mediated by secreted factors such as reactive oxygen species (ROS) and G-CSF, and enzymatic mediators like Arginase I (ARG1), although the Arginase function is reported with varying results in humans partly due to inconsistencies in measuring protein levels as compared to enzymatic activity (23, 37, 38). The functional aspects of G-MDSCs, have been excellently reviewed elsewhere and will therefore not be covered in detail here (30, 39). The generation of human G-MDSCs is still debated, mainly since the morphology of human G-MDSCs present a heterogeneous population of cells ranging from immature neutrophils to mature polymorphonuclear (PMN) neutrophils (Figure 2) (29, 32, 38, 40, 41). The “left shift” (11–14), or emergency myelopoiesis exporting immature myeloid granulocytes, may be considered when investigating the morphology and generation of isolated human peripheral blood G-MDSCs (Figure 2). According to previous literature, PMN shaped G-MDSCs (Box 1) can be discriminated from steady-state neutrophils based on a PMN morphology with fewer granules (23). However, in humans, the markers CD11b+CD15+CD14−CD33+/loCD66b+ enrich for neutrophils at all maturation stages; from myelocytes to mature neutrophils (Figure 2, Table 1), including cells with fewer granules thus making this distinction difficult (23, 30, 45). Some markers that have been identified to distinguish immature neutrophils from the PMN shaped G-MDSCs are CD10, CD13, CD16, and CD38 which all represent different stages of neutrophil maturation (Table 1), thus supporting that the PMN shaped G-MDSCs are more mature (46–52). However, as discussed below, there are also studies suggesting that immunosuppressive G-MDSCs with an immature surface phenotype and morphology, could derive from de-differentiated or reprogrammed mature neutrophils into immunosuppressive G-MDSCs (29, 53, 54). The traditional view that immunosuppressive bona fide G-MDSC are immature cells, is being challenged by current literature indicating that mature cells may also be immunosuppressive. The immature neutrophils (the non-PMN G-MDSCs in Figure 2, Table 1, Box 1), make up ~5–15% of all LDGs in the peripheral blood of cancer patients, probably varying with cancer type and stage (55). Whether the immature neutrophils are more immunosuppressive than the PMN shaped G-MDSCs, thus representing the bona fide G-MDSCs, is currently debated (30, 38, 55). There is also a possibility that the immature neutrophils, or subsets thereof, may be mature cells of some other lineage, exemplified by fibrocytes (56). Immature neutrophils are proposed to have a longer half-life and therefore also to survive longer in tissues and tumors, as mentioned below (57). The difference between immature neutrophils and the more mature PMN shaped G-MDSCs regarding function is not clear, but ARG1/iNOS may be mediators preferably used by the immature neutrophil G-MDSCs, as compared to their PMN shaped counterpart (30, 52). Lately, lectin-type oxidized LDL receptor 1 (LOX1) has been suggested as a marker that may identify human G-MDSCs at the functional level (47, 52, 58).
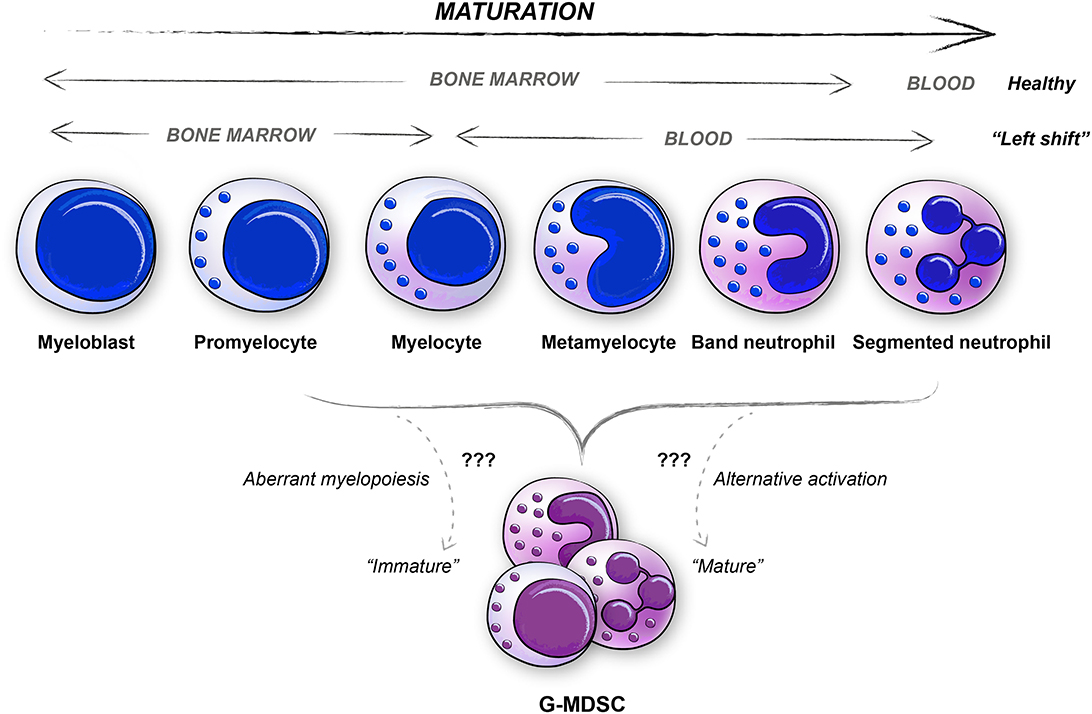
Figure 2. Similarities between neutrophil maturation stages and human G-MDSCs. The various differentiation stages of neutrophils are depicted, both for healthy subjects and for patients with severe diseases as represented by the “Left shift.” Human G-MDSCs subpopulations are represented in most neutrophil maturation stages, ranging from promyelocyte to mature neutrophil (PMN, polymorphonuclear cells).
Box 1. Explanation to nomenclatures used in this review.
Immature neutrophil G-MDSCs: G-MDSCs that are derived from immature neutrophils or G-MDSCs that represent immature neutrophils with non-PMN shaped nuclei and with immunosuppressive activity.
PMN shaped G-MDSCs: G-MDSCs that are derived from mature or activated neutrophils or G-MDSCs that represent mature or activated neutrophils with PMN shaped nuclei and with immunosuppressive activity.

Table 1. Selected surface phenotypes during neutrophil differentiation, with expression levels of indicated markers in specific neutrophil subsets indicated (30, 42–44).
Presently, there is no firm evidence that human PMN shaped G-MDSCs are anything else than activated neutrophils. Mature activated neutrophils may also acquire a low density and thus be isolated in the LDG/PBMC fraction of human peripheral blood (59). Activated neutrophils can be immunosuppressive by inhibiting T cell proliferation via ROS. Neutrophil extracellular traps (NETs) should, however, inevitably induce neutrophil cell death, although with a slight delay (60). Nevertheless, since G-MDSCs theoretically should live longer than activated neutrophils, a unique PMN shaped G-MDSC population has been proposed (23, 61). There are contradicting findings available from gene expression profiles of isolated cancer patient derived G-MDSCs, concerning whether G-MDSCs are activated neutrophils or unique G-MDSC cell populations, a fact that mirrors the complexity of investigating this heterogeneous population of cells in different indications. Indeed, the isolation procedure and choice of neutrophil source, as well as the inter- and intra-patient variation in numbers of immature neutrophils as compared to PMN shaped G-MDSCs, will unequivocally lead to unique profiles for each study (47, 62). Newly introduced methods like multiparameter, multidimensional imaging, single cell RNA Sequencing and mass cytometry by time of flight (CyTOF), will hopefully lead to a better understanding of the heterogeneity of G-MDSCs and their unique subtypes. Indeed, using multidimensional imaging, LOX1+ G-MDSCs were recently found to co-express the neutrophil activation marker MPO (58, 63).
The distinctive function of PMN shaped G-MDSCs should be debated. Even though PMN shaped G-MDSCs are immunosuppressive, they could still be classified as conventional activated neutrophils, or as neutrophils with an alternative activation. Indeed, a high Neutrophil to Lymphocyte Ratio (NLR) in cancer patients, is associated with worse prognosis (64). Deciphering the immunosuppressive mechanisms of action of PMN shaped G-MDSCs will undoubtedly be relevant for understanding their origin and nature (47, 52, 58). Of relevance, LDGs with similar surface phenotype as G-MDSCs are isolated from patients with autoimmune disorders, with the important difference that these cells are pro-inflammatory (41). As for all MDSCs, only cells with a potent immunosuppressive capacity may be defined as MDSCs.
The immature neutrophils, produced as a response to tumor-induced stress and secreted colony stimulating factors GM-CSF, G-CSF, and M-CSF (5–10), could represent unique immature G-MDSC subpopulations. Their mechanisms of action, and also their capacity to differentiate into PMN shaped G-MDSCs, or neutrophils, will be important to delineate. An interesting and important issue for the future, is whether treating cancer patients with G-CSF for neutropenia, could affect the patients negatively in terms of G-MDSC enrichment, or not (65).
Human G-MDSCs in Tumors
In mice, tumor infiltrating G-MDSCs are classically defined by the Ly6G marker. In humans, an equivalent marker has not yet been defined, since many candidate markers (e.g., CD15 or CD66b) are expressed on immature as well as mature neutrophils (Table 1). Indeed, immature and mature neutrophils are found both in the circulation of cancer patients (66, 67), and in human tumors (58, 68, 69), probably at varying density depending on tumor type and stage (58, 66–69). Of relevance, observations concerning migration and accumulation of immature and mature neutrophils in tumors have been made, where immature neutrophils have a reduced migratory capacity, but may still be able to accumulate at metastatic sites (40, 70, 71). Immature neutrophils have also been shown to survive longer in tumors (57). The immature and mature neutrophils in tumors may have different biology, as described above, and will thus affect disease severity differently (30). To define them as G-MDSCs, their immunosuppressive function is of outmost importance. The fact that a diversity of neutrophils is found in tumors have promoted researchers to define them as classical (N1) and alternative (N2) neutrophils (30, 53, 72). This has, however, only been experimentally shown in mice, and will still have to be determined in human tumors (41). Whether tumor infiltrating neutrophils derive from the immature neutrophil G-MDSCs as proposed in mice (66), or if they are reprogrammed or alternatively TGFβ activated N2 neutrophils (53, 73) will be interesting to follow. Until then, tumor infiltrating G-MDSCs may theoretically be grouped as immunosuppressive neutrophils (23). Novel methods as single cell RNA Seq, multiparameter immunofluorescence and mass cytometry by time of flight (CyTOF) will be valuable tools to decipher different subpopulations of tumor associated neutrophils (TANs) and G-MDSCs. Recently, using multidimensional imaging, LOX1+ tumor infiltrating G-MDSCs were reported to co-express the neutrophil activation marker MPO, and associate with immunosuppression and a worse prognosis (58, 63). For the future, TANs and G-MDSC may thus be targeted with similar, or vastly different therapeutic approaches.
Human Peripheral Blood Mo-MDSCs
Mo-MDSCs are cells of the myeloid monocyte lineage, but with an HLA-DR−/lo and co-receptor CD86−/lo cell surface phenotype. They are potently immunosuppressive by soluble mediators like PGE2, IL10, TGFβ, and nitric oxide (NO), and enzymatic mediators like ARG1. As for all MDSCs, it is their immunosuppressive function that is key to defining them as Mo-MDSCs, and their functional mechanisms have been described in depth elsewhere (23, 39, 74–76). During leukocytosis, the emergency myelopoiesis is proposed to export immature myeloid cells into the circulation. This holds true for G-MDSCs, as discussed above, but not necessarily for Mo-MDSCs. The morphology of immature monocytic cells (monoblasts and promonocytes) are quite similar to the mature monocytes (77, 78). Also, the surface phenotype of immature monocytic cells (the CD11b+ promonocytes that could be accounted for being Mo-MDSCs), is very similar to mature monocytes (77, 78), including the HLA-DR+ phenotype, which is in contrast to Mo-MDSCs (Table 2). This makes it difficult to postulate that human Mo-MDSCs are immature cells.
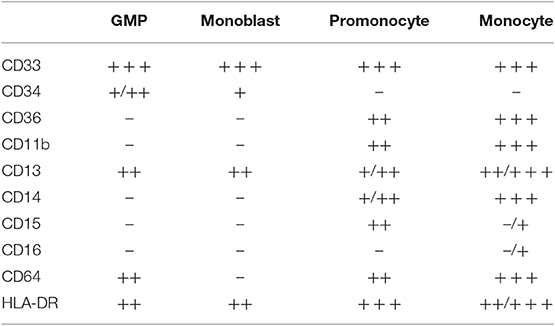
Table 2. Surface phenotypes during monocyte differentiation, with expression levels of indicated markers in specific subpopulations indicated (77, 78).
Mature monocytes come in different versions, with the most typical human monocytes being the classical (CD14++CD16−), non-classical (CD14loCD16+), intermediate (CD14+CD16+) and as recently proposed the immunosuppressive Mo-MDSCs (CD14+HLA-DR−/lo) (79, 80). To date, proof showing that human Mo-MDSCs would be immature, or linked to an increased export of a specific subtype of immunosuppressive monocytes as proposed (79) is, however, still lacking. In contrast, current literature indicate that monocytes are plastic cells that can change surface phenotype and function depending on activation state and the local microenvironment (75, 79). Mo-MDSCs are enriched not only in patients with cancer, but also in patients with severe infections and inflammatory conditions (16). Mo-MDSCs have been proposed to be generated either through affected myelopoiesis, at the stage of activation, or both (61). Independent of which, STAT3 and NFκB inducing signals are required for their generation, and hence also for their key defining immunosuppressive function. STAT3 can be induced through soluble mediators like colony stimulating factors, IL10, IL6, or PGE2; and NFκB through pathogen recognition receptor (PRR) signaling triggered by pathogen associated molecular patterns (PAMPs) or sterile damage associated molecular patterns (DAMPs) (34, 61).
One example of disease-associated activation of monocytes, leading to cells with identical surface phenotype and immunosuppressive function as Mo-MDSCs, are the endotoxin tolerance reprogrammed monocytes present in the peripheral blood of sepsis patients (34, 81–85). Endotoxin tolerance is caused by pathogen and host response signals, with PAMPs as pathogen-induced TLR2/4-NFκB-signals and STAT3-inducing mediators like IL10, TGFβ, or PGE2 as host response signals (81, 86) (Figure 3). The combination of signal transduction lead to activation of alternative transcription factor NFκB complexes, consisting of homodimers of p50/p50 or heterodimers of p52/RelB, instead of the conventional p50/p65 heterodimers (34, 81–85). This leads to, transcriptional activation of IL10 (IL10 ON) and simultaneous inhibition of tumor necrosis factor alpha (TNFα) (TNF OFF) (Figure 3). When this happens, the mature peripheral blood monocytes acquire a CD14+HLA-DR−/loCo-receptor−/lo immunosuppressive and IL10 producing phenotype, suiting the Mo-MDSC criteria (81). Similar mechanisms also occur in a sterile environment, such as in a tumor or systemically in cancer patients, but with DAMPs rather than PAMPs as TLR-ligands (84, 85, 87–90). Downstream mediators of DAMP signaling, like PGE2, IL10, and NGF are already known to induce immunosuppressive myeloid cells (86, 87, 91, 92). One alternative explanation is the potential presence of unique anti-inflammatory endogenous PRR-ligands exclusively inducing transcriptional activation of IL10 ON/TNF OFF (87, 93, 94). To differentiate between pro-inflammatory endogenous alarmins (DAMPs), we proposed to use the term tolerance-associated molecular patterns (TAMPs) for endogenous anti-inflammatory TLR-ligands (87). We have recently found a novel TLR4-ligand (Wnt5a) that is induced upon TLR4-signaling, and that functions as a TAMP activating the IL10 ON/TNF OFF signal in primary human monocytes, resulting in Mo-MDSC-like cells by surface phenotype and function in vitro, a finding that was evolutionarily conserved in Drosophila (95), but not mice (87). In this context, a homeostatic feedback loop downstream of TLR2/4, would be able to regulate the acute pro-inflammatory response, as observed in sepsis. Other endogenous TLR-ligands have previously been observed to promote MDSC generation or to inhibit pro-inflammatory TLR signaling, like HMGB1 and S100B (89, 93, 94, 96). Interestingly, the MDSC marker S100A9 is a DAMP, binding to TLR4 (97).
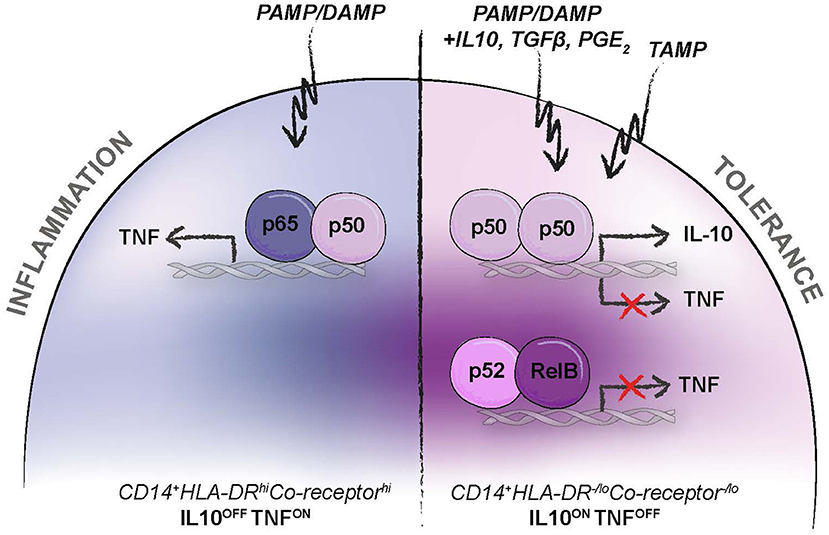
Figure 3. Reprogramming of systemic monocytes into anti-inflammatory monocytes. Human Mo-MDSCs have the same surface phenotype and function as anti-inflammatory monocytes that are reprogrammed by anti-inflammatory mediators (e.g., IL10, PGE2, TGFβ), pro-inflammatory pattern recognition receptor (PRR) ligands (e.g., PAMPs and DAMPs), or by endogenous anti-inflammatory PRR-ligands; tolerance associated molecular pattern (TAMPs), as previously suggested (87). This leads to transcriptional activation of IL10 (IL10 ON), inhibition of TNFα (TNF OFF), and acquisition of a CD14+HLA-DR−/loCo-receptor−/lo Mo-MDSC phenotype.
Whether the plastic differentiation of mature monocytes into immunosuppressive monocytes is reversible or not, is not fully known. In patients with sepsis, the reprogrammed monocytes stay in their immunosuppressive state for up to 10 weeks with secondary infections and death as result, indicating that they may not be able to revert to a pro-inflammatory state again (16, 98). Opposite findings have, however, been presented for macrophages, where anti-inflammatory M2 macrophages where more plastic and differentiated into M1 macrophages upon stimulation with pro-inflammatory mediators (99, 100). It is undoubtfully so that the levels of peripheral blood Mo-MDSCs in cancer patients are associated with disease severity (76). Whether this is linked to an increased export of a certain subtype of immunosuppressive monocytes (79), or to reprogramming of circulating monocytes, is not clear and both may hold true. The latter (reversing reprogramming) may be more difficult to approach in a therapeutic setting.
Human Mo-MDSCs in Tumors
When monocytes enter tissues, they are by definition differentiated into macrophages or monocyte-derived myeloid dendritic cells (mDCs). In an anti-inflammatory tumor microenvironment, this differentiation process is usually skewed into alternatively activated immunosuppressive macrophages of various kind, often exemplified by the simplistic M2 nomenclature, but also into mDCs (75, 101–103). Whether human myeloid cells like Mo-MDSCs also differentiate into macrophages in tumors is not fully known, although Mo-MDSCs have been shown to do so in mice (104). In humans, the hurdle lies with the difficulties to discriminate between the various human myeloid cell subsets with reliable results in tissues, and to be able to define their immunosuppressive function in vivo. In human tumors, various differentiation stages likely exist ranging from monocytes, Mo-MDSCs to tumor associated macrophages (TAMs), where markers are shared with those of Mo-MDSCs (74–76, 105). Also, isolation of myeloid populations from single cell suspensions of human tumors often generate subsets with less clear cell surface phenotypes as compared to myeloid cells from PBMCs. If the definition of Mo-MDSCs involves the CD14+HLA-DR−/lo surface phenotype, it may thus be difficult to identify them in human tumors. If the definition of MDSCs within tumors are amended to describe them as immunosuppressive myeloid cells, tumor-associated MDSCs may be reconsidered and would include immunosuppressive macrophages as well. Still, the potential functional difference between tumor infiltrating Mo-MDSCs and alternatively activated macrophages is not elucidated in humans.
In mice, markers for Gr1 (Ly6C and Ly6G) are used to define MDSCs in tumors, and F4/80 to discriminate them from macrophages. Human Mo-MDSCs are still defined by a CD14+HLA-DR−/lo surface phenotype. The immunosuppressive activity of Mo-MDSCs, which is critical for their definition, is for natural reasons more difficult, yet not less important, to assess in human tumors (74–76, 105). Furthermore, the dull CD14+HLA-DR−/lo surface phenotype is often difficult to detect with accuracy using flow cytometry, resulting in a mixture of myeloid cells from tumor single cell suspensions. Since Mo-MDSCs by definition should have an immature myeloid cell phenotype, the macrophage maturation markers F4/80 in mice and CD68 in humans, should not be expressed (23). However, CD68 is expressed at various levels in macrophages, even when using established clinical pathology diagnostic markers (Figure 4), and it may therefore be difficult to discriminate Mo-MDSCs from TAMs by the use of conventional immunohistochemistry at present. Similar issues become clear when discussing the M2 TAM marker CD163. CD163 is expressed on a range of human anti-inflammatory myeloid cells producing IL10 (106–109) and is also upregulated by IL10, glucocorticoids and M-CSF (110). To complicate things, some tumor infiltrating CD163+ cells express low to negligible levels of the pan-macrophage marker CD68 (107, 111). Similarly, CD163 may be expressed on a subset of circulating anti-inflammatory CD14+HLA-DR−/lo monocytes, Mo-MDSCs (112). It may therefore be of relevance to discuss whether CD163 can be expressed on tumor infiltrating Mo-MDSCs, or if all IL10 producing anti-inflammatory myeloid cells in tumors are M2 TAMs because of the CD163 expression. Recently, it was shown that primary human monocytes co-transplanted with xenografts upregulated both CD163 and nuclear S100A9 (113). Nuclear S100A9 has lately appeared as a human Mo-MDSC marker (36, 114). However, also the expression of S100A9, CD68, and CD163 in human myeloid cells seems to vary (115). Novel immunofluorescent multiparameter assays are being developed and a combination of markers like CD14, CD68, CD163, CD206, and S100A9 (108) is probably the best strategy to define Mo-MDSCs in human tumors, but their indispensable immunosuppressive function would need to be determined by other means. Single cell RNA Seq is a promising tool to provide us with more information on the functions of Mo-MDSCs in human tumors and tissues.
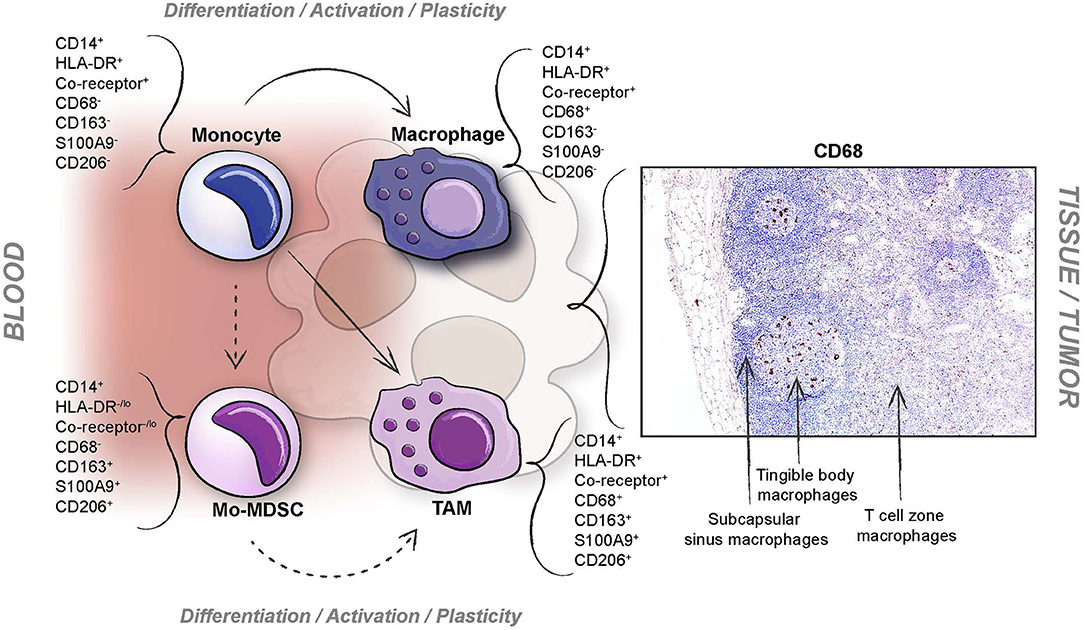
Figure 4. Difficulties when defining human Mo-MDSCs in tumors. Different issues ranging from plasticity of myeloid cells, technical problems concerning investigating multiparameter cell surface phenotypes in myeloid cells isolated from tissues, and definition of subpopulation markers; exemplified by the pan-macrophage marker CD68 which is variably expressed in different macrophage subpopulations (indicated here by a previously unpublished immunohistochemistry image representing staining of human resident lymph node macrophages with varying CD68 levels; using anti-CD68 clone KP1, diluted 1:1000, Dako, Glostrup, Denmark).
Conclusions
An imbalance in myelopoiesis reflects the biology of MDSCs well (Figure 5). All severe inflammatory conditions, including infection and cancer, affect MDSC generation. The challenge in terms of cancer is the strong interdependence between MDSCs and cancer cells. Cancer cells secrete factors that induce aberrant myelopoiesis (G-MDSC), as well as affect the myeloid cells already in circulation (Mo-MDSCs). The affected myeloid cells evolve in congruence with the tumor and metastasizing cells, and a constant feed-back loop is generated. In many cancer patients, myeloid cells are also unintentionally targeted during chemotherapy. To overcome this, patients are given G-CSF to boost myelopoiesis, leading to more MDSCs (65). An important challenge for our future knowledge on MDSCs is the translation from mouse to human MDSCs, where the essential immunosuppressive mediators like ARG1 and IL10 show an extreme polymorphism in humans but not mice (37, 116). Currently, drugs targeting all myeloid cells are being developed (75). It is important to stress that pro-inflammatory myeloid cells like lymph node resident macrophages and monocyte derived tumoricidal macrophages (M1 macrophages) and dendritic cells, are also needed for successful anti-tumor immune responses. Therefore, specific targeting of MDSC generating signals (e.g., STAT3), or the immunosuppressive MDSC specific functions (e.g., ARG1), should also be considered. With implementation of novel imaging and single cell analyses techniques, the origin of human MDSCs will undoubtfully be investigated in the near future. This will hopefully lead to answers on how to target human MDSCs as a therapeutic intervention in cancer patients. Such an intervention of the innate immunosuppressive arm, combined with the established check-point inhibition therapies targeting the adaptive immune response, could potentially offer a very potent therapeutic approach against cancer.
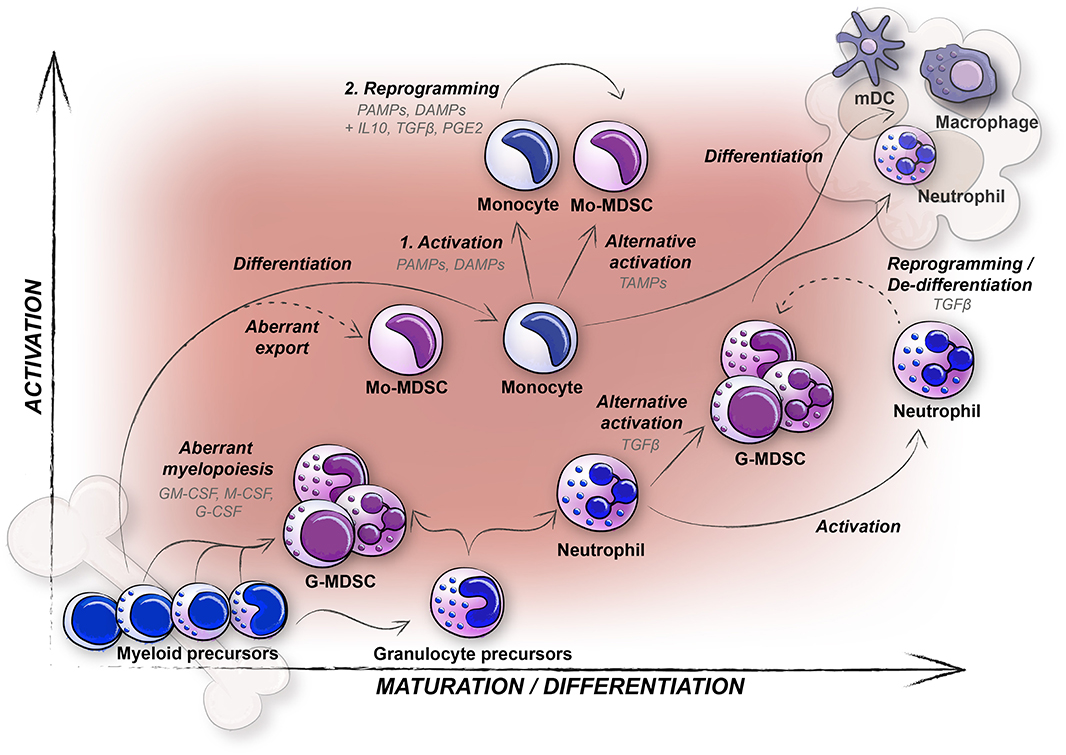
Figure 5. Concluding graphical summary featuring different hypotheses concerning the generation of human MDSCs (Mo-MDSCs and G-MDSCs), in relation to maturation and activation. Mo-MDSCs are predominantly generated from peripheral blood monocytes (solid arrows), and conceivably also through export of immature immunosuppressive monocytes although not yet proven in humans (dashed arrow). In contrast, G-MDSCs likely originate from aberrant myelopoiesis (immature neutrophil G-MDSCs) and alternative activation of mature neutrophils (mature neutrophil (PMN shaped) G-MDSCs, solid arrows). Whether also reprogramming or de-differentiation of mature neutrophils occur among G-MDSCs (dashed arrow) is yet to be determined in humans.
Author Contributions
KL was responsible for writing the first draft. CB was responsible for the first figure drafts. KL and CB then together wrote, revised, and finalized the manuscript.
Funding
This work was generously supported by grants from the Swedish Research Council, The Swedish Cancer Society, the Swedish Society for Medical Research (SSMF), MAS Cancer Foundation and Gyllenstiernska Krapperups Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Lappat EJ, Cawein M. A study of the leukemoid response to transplantable a-280 tumor in mice. Cancer Res. (1964) 24:302–11.
3. Lee MY, Rosse C. Depletion of lymphocyte subpopulations in primary and secondary lymphoid organs of mice by a transplanted granulocytosis-inducing mammary carcinoma. Cancer Res. (1982) 42:1255–60.
4. Oseroff A, Okada S, Strober S. Natural suppressor (NS) cells found in the spleen of neonatal mice and adult mice given total lymphoid irradiation (TLI) express the null surface phenotype. J Immunol. (1984) 132:101–10.
5. Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. (1987) 47:100–5.
6. Robinson WA. Granulocytosis in neoplasia. Ann N Y Acad Sci. (1974) 230:212–8. doi: 10.1111/j.1749-6632.1974.tb14451.x
7. Suda T, Miura Y, Mizoguchi H, Kubota K, Takaku F. A case of lung cancer associated with granulocytosis and production of colony-stimulating activity by the tumour. Br J Cancer. (1980) 41:980–4. doi: 10.1038/bjc.1980.177
8. Berdel WE, Danhauser-Riedl S, Steinhauser G, Winton EF. Various human hematopoietic growth factors (interleukin-3, GM-CSF, G-CSF) stimulate clonal growth of nonhematopoietic tumor cells. Blood. (1989) 73:80–3. doi: 10.1182/blood.V73.1.80.80
9. Matsuguchi T, Okamura S, Kawasaki C, Shimoda K, Omori F, Hayashi S, et al. Constitutive production of granulocyte colony-stimulating factor and interleukin-6 by a human lung cancer cell line, KSNY: gene amplification and increased mRNA stability. Eur J Haematol. (1991) 47:128–33. doi: 10.1111/j.1600-0609.1991.tb00135.x
10. Sawyers CL, Golde DW, Quan S, Nimer SD. Production of granulocyte-macrophage colony-stimulating factor in two patients with lung cancer, leukocytosis, and eosinophilia. Cancer. (1992) 69:1342–6. doi: 10.1002/1097-0142(19920315)69:6<1342::AID-CNCR2820690607>3.0.CO;2-U
11. Robertson OH, Fox JP. The relationship of infecting dosage, leucocytic response, bacteremia, and extent of pulmonary involvement to the outcome of experimental lobar pneumonia in the dog. J Exp Med. (1939) 69:229–46. doi: 10.1084/jem.69.2.229
12. Marsh JC, Boggs DR, Cartwright GE, Wintrobe MM. Neutrophil kinetics in acute infection. J Clin Invest. (1967) 46:1943–53. doi: 10.1172/JCI105684
13. Akenzua GI, Hui YT, Milner R, Zipursky A. Neutrophil and band counts in the diagnosis of neonatal infections. Pediatrics. (1974) 54:38–42.
14. Honda T, Uehara T, Matsumoto G, Arai S, Sugano M. Neutrophil left shift and white blood cell count as markers of bacterial infection. Clin Chim Acta. (2016) 457:46–53. doi: 10.1016/j.cca.2016.03.017
15. Wilcox RA. Cancer-associated myeloproliferation: old association, new therapeutic target. Mayo Clin Proc. (2010) 85:656–63. doi: 10.4065/mcp.2010.0077
16. Schrijver IT, Theroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol. (2019) 10:327. doi: 10.3389/fimmu.2019.00327
17. Bennett JA, Rao VS, Mitchell MS. Systemic bacillus calmette-guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci USA. (1978) 75:5142–4. doi: 10.1073/pnas.75.10.5142
18. Duwe AK, Singhal SK. The immunoregulatory role of bone marrow. I Suppression of the induction of antibody responses to T-dependent and T-independent antigens by cells in the bone marrow. Cell Immunol. (1979) 43:362–71. doi: 10.1016/0008-8749(79)90180-1
19. Slavin S, Strober S. Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR). J Immunol. (1979) 123:942–6.
20. Talmadge JE, Reed EC, Kessinger A, Kuszynski CA, Perry GA, Gordy CL, et al. Immunologic attributes of cytokine mobilized peripheral blood stem cells and recovery following transplantation. Bone Marrow Transplant. (1996) 17:101–9.
21. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. (2013) 13:739–52. doi: 10.1038/nrc3581
22. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. (2007) 67:425. doi: 10.1158/0008-5472.CAN-06-3037
23. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
24. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. (2009) 182:4499–506. doi: 10.4049/jimmunol.0802740
25. Poschke I, Kiessling R. On the armament and appearances of human myeloid-derived suppressor cells. Clin Immunol. (2012) 144:250–68. doi: 10.1016/j.clim.2012.06.003
26. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
27. Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
28. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
29. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci. (2013) 70:3813–27. doi: 10.1007/s00018-013-1286-4
30. Mackey JBG, Coffelt SB, Carlin LM. Neutrophil Maturity in Cancer. Front Immunol. (2019) 10:1912. doi: 10.3389/fimmu.2019.01912
31. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. (2010) 22:238–44. doi: 10.1016/j.coi.2010.01.021
32. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. (2012) 61:1155–67. doi: 10.1007/s00262-012-1294-5
33. Filipazzi P, Huber V, Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother. (2012) 61:255–63. doi: 10.1007/s00262-011-1161-9
34. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget. (2017) 8:3649–65. doi: 10.18632/oncotarget.12278
35. Feng PH, Lee KY, Chang YL, Chan YF, Kuo LW, Lin TY, et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med. (2012) 186:1025–36. doi: 10.1164/rccm.201204-0636OC
36. Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, et al. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology. (2012) 136:176–83. doi: 10.1111/j.1365-2567.2012.03566.x
37. Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol. (2014) 5:479. doi: 10.3389/fimmu.2014.00479
38. Lang S, Bruderek K, Kaspar C, Hoing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res. (2018) 24:4834–44. doi: 10.1158/1078-0432.CCR-17-3726
39. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
40. Brandau S, Trellakis S, Bruderek K, Schmaltz D, Steller G, Elian M, et al. Myeloid-derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol. (2011) 89:311–7. doi: 10.1189/jlb.0310162
41. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
42. Elghetany MT. Surface antigen changes during normal neutrophilic development: a critical review. Blood Cells Mol Dis. (2002) 28:260–74. doi: 10.1006/bcmd.2002.0513
43. Liu L, Shi GP. CD31: beyond a marker for endothelial cells. Cardiovasc Res. (2012) 94:3–5. doi: 10.1093/cvr/cvs108
44. Yang P, Li Y, Xie Y, Liu Y. Different faces for different places: heterogeneity of neutrophil phenotype and function. J Immunol Res. (2019) 2019:8016254. doi: 10.1155/2019/8016254
45. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. (2011) 118:2254–65. doi: 10.1182/blood-2010-12-325753
46. Karakasheva TA, Waldron TJ, Eruslanov E, Kim SB, Lee JS, O'brien S, et al. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. (2015) 75:4074–85. doi: 10.1158/0008-5472.CAN-14-3639
47. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
48. Blazkova J, Gupta S, Liu Y, Gaudilliere B, Ganio EA, Bolen CR, et al. Multicenter systems analysis of human blood reveals immature neutrophils in males and during pregnancy. J Immunol. (2017) 198:2479–88. doi: 10.4049/jimmunol.1601855
49. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and immature CD10(-) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood. (2017) 129:1343–56. doi: 10.1182/blood-2016-04-713206
50. Zhang J, Xu X, Shi M, Chen Y, Yu D, Zhao C, et al. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. (2017) 6:e1258504. doi: 10.1080/2162402X.2016.1258504
51. Karakasheva TA, Dominguez GA, Hashimoto A, Lin EW, Chiu C, Sasser K, et al. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight. (2018) 3:e97022. doi: 10.1172/jci.insight.97022
52. Chai E, Zhang L, Li C. LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression. Cancer Manag Res. (2019) 11:7307–15. doi: 10.2147/CMAR.S210545
53. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
54. Singel KL, Emmons TR, Khan ANH, Mayor PC, Shen S, Wong JT, et al. Mature neutrophils suppress T cell immunity in ovarian cancer microenvironment. JCI Insight. (2019) 4:e122311. doi: 10.1172/jci.insight.122311
55. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
56. Zhang H, Maric I, Diprima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. (2013) 122:1105–13. doi: 10.1182/blood-2012-08-449413
57. Drifte G, Dunn-Siegrist I, Tissieres P, Pugin J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med. (2013) 41:820–32. doi: 10.1097/CCM.0b013e318274647d
58. Si Y, Merz SF, Jansen P, Wang B, Bruderek K, Altenhoff P, et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci Immunol. (2019) 4:eaaw9159. doi: 10.1126/sciimmunol.aaw9159
59. Pember SO, Barnes KC, Brandt SJ, Kinkade JM Jr. Density heterogeneity of neutrophilic polymorphonuclear leukocytes: gradient fractionation and relationship to chemotactic stimulation. Blood. (1983) 61:1105–15. doi: 10.1182/blood.V61.6.1105.1105
60. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
61. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19–25. doi: 10.1016/j.it.2010.10.002
62. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE. (2012) 7:e31524. doi: 10.1371/journal.pone.0031524
63. Rymaszewski AL, Tate E, Yimbesalu JP, Gelman AE, Jarzembowski JA, Zhang H, et al. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers. (2014) 6:1111–27. doi: 10.3390/cancers6021111
64. Faria SS, Fernandes PC Jr, Silva MJ, Lima VC, Fontes W, Freitas-Junior R, et al. The neutrophil-to-lymphocyte ratio: a narrative review. Ecancermedicalscience. (2016) 10:702. doi: 10.3332/ecancer.2016.702
65. Luyckx A, Schouppe E, Rutgeerts O, Lenaerts C, Fevery S, Devos T, et al. G-CSF stem cell mobilization in human donors induces polymorphonuclear and mononuclear myeloid-derived suppressor cells. Clin Immunol. (2012) 143:83–7. doi: 10.1016/j.clim.2012.01.011
66. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
67. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
68. Droeser RA, Hirt C, Eppenberger-Castori S, Zlobec I, Viehl CT, Frey DM, et al. High myeloperoxidase positive cell infiltration in colorectal cancer is an independent favorable prognostic factor. PLoS ONE. (2013) 8:e64814. doi: 10.1371/journal.pone.0064814
69. Singhal S, Bhojnagarwala PS, O'brien S, Moon EK, Garfall AL, Rao AS, et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell. (2016) 30:120–35. doi: 10.1016/j.ccell.2016.06.001
70. Van Eeden SF, Kitagawa Y, Klut ME, Lawrence E, Hogg JC. Polymorphonuclear leukocytes released from the bone marrow preferentially sequester in lung microvessels. Microcirculation. (1997) 4:369–80. doi: 10.3109/10739689709146801
71. Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. (2018) 19:1236–47. doi: 10.1038/s41590-018-0229-5
72. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is required for “emergency” granulopoiesis. Nat Immunol. (2006) 7:732–9. doi: 10.1038/ni1354
73. Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology. (2016) 5:e1232221. doi: 10.1080/2162402X.2016.1232221
74. Bercovici N, Guerin MV, Trautmann A, Donnadieu E. The remarkable plasticity of macrophages: a chance to fight cancer. Front Immunol. (2019) 10:1563. doi: 10.3389/fimmu.2019.01563
75. Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. (2019) 10:1611. doi: 10.3389/fimmu.2019.01611
76. Mengos AE, Gastineau DA, Gustafson MP. The CD14(+)HLA-DR(lo/neg) monocyte: an immunosuppressive phenotype that restrains responses to cancer immunotherapy. Front Immunol. (2019) 10:1147. doi: 10.3389/fimmu.2019.01147
77. Wood B. Multicolor immunophenotyping: human immune system hematopoiesis. Methods Cell Biol. (2004) 75:559–76. doi: 10.1016/S0091-679X(04)75023-2
78. Goasguen JE, Bennett JM, Bain BJ, Vallespi T, Brunning R, Mufti GJ, et al. Morphological evaluation of monocytes and their precursors. Haematologica. (2009) 94:994–7. doi: 10.3324/haematol.2008.005421
79. Cane S, Ugel S, Trovato R, Marigo I, De Sanctis F, Sartoris S, et al. The endless saga of monocyte diversity. Front Immunol. (2019) 10:1786. doi: 10.3389/fimmu.2019.01786
80. Kapellos TS, Bonaguro L, Gemund I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
81. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. (2009) 30:475–87. doi: 10.1016/j.it.2009.07.009
82. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci USA. (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
83. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. (2011) 186:7243–54. doi: 10.4049/jimmunol.1001952
84. Bergenfelz C, Medrek C, Ekstrom E, Jirstrom K, Janols H, Wullt M, et al. Wnt5a induces a tolerogenic phenotype of macrophages in sepsis and breast cancer patients. J Immunol. (2012) 188:5448–58. doi: 10.4049/jimmunol.1103378
85. Bergenfelz C, Larsson AM, Von Stedingk K, Gruvberger-Saal S, Aaltonen K, Jansson S, et al. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS ONE. (2015) 10:e0127028. doi: 10.1371/journal.pone.0127028
86. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. (2009) 15:42–9. doi: 10.1038/nm.1905
87. Mehmeti M, Bergenfelz C, Kallberg E, Millrud CR, Bjork P, Ivars F, et al. Wnt5a is a TLR2/4-ligand that induces tolerance in human myeloid cells. Commun Biol. (2019) 2:176. doi: 10.1038/s42003-019-0432-4
88. Ray A, Chakraborty K, Ray P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front Cell Infect Microbiol. (2013) 3:52. doi: 10.3389/fcimb.2013.00052
89. Li J, Sun J, Rong R, Li L, Shang W, Song D, et al. HMGB1 promotes myeloid-derived suppressor cells and renal cell carcinoma immune escape. Oncotarget. (2017) 8:63290–8. doi: 10.18632/oncotarget.18796
90. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
91. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. (2011) 118:5498–505. doi: 10.1182/blood-2011-07-365825
92. Prencipe G, Minnone G, Strippoli R, De Pasquale L, Petrini S, Caiello I, et al. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. J Immunol. (2014) 192:3345–54. doi: 10.4049/jimmunol.1300825
93. Erridge C, Kennedy S, Spickett CM, Webb DJ. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J Biol Chem. (2008) 283:24748–59. doi: 10.1074/jbc.M800352200
94. Liu T, Xiang A, Peng T, Doran AC, Tracey KJ, Barnes BJ, et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc Natl Acad Sci USA. (2019) 116:23254–63. doi: 10.1073/pnas.1907490116
95. Gordon MD, Dionne MS, Schneider DS, Nusse R. WntD is a feedback inhibitor of Dorsal/NF-kappaB in Drosophila development and immunity. Nature. (2005) 437:746–9. doi: 10.1038/nature04073
96. Sorci G, Giovannini G, Riuzzi F, Bonifazi P, Zelante T, Zagarella S, et al. The danger signal S100B integrates pathogen- and danger-sensing pathways to restrain inflammation. PLoS Pathog. (2011) 7:e1001315. doi: 10.1371/journal.ppat.1001315
97. Kallberg E, Vogl T, Liberg D, Olsson A, Bjork P, Wikstrom P, et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS ONE. (2012) 7:e34207. doi: 10.1371/journal.pone.0034207
98. Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. (2008) 29:617–25. doi: 10.1016/j.ccm.2008.06.010
99. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. (2014) 157:832–44. doi: 10.1016/j.cell.2014.04.016
100. O'neill LA. A metabolic roadblock in inflammatory macrophages. Cell Rep. (2016) 17:625–6. doi: 10.1016/j.celrep.2016.09.085
101. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
102. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. (2016) 7:13720. doi: 10.1038/ncomms13720
103. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. (2017) 47:597. doi: 10.1016/j.immuni.2017.08.018
104. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
105. Kadowaki T, Shimada M, Inagawa H, Kohchi C, Hirashima M, Soma G. Reconsideration of macrophage and dendritic cell classification. Anticancer Res. (2012) 32:2257–61.
106. Sulahian TH, Hogger P, Wahner AE, Wardwell K, Goulding NJ, Sorg C, et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. (2000) 12:1312–21. doi: 10.1006/cyto.2000.0720
107. Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. (2012) 12:306. doi: 10.1186/1471-2407-12-306
108. Saylor J, Ma Z, Goodridge HS, Huang F, Cress AE, Pandol SJ, et al. Spatial mapping of myeloid cells and macrophages by multiplexed tissue staining. Front Immunol. (2018) 9:2925. doi: 10.3389/fimmu.2018.02925
109. Comi M, Avancini D, Santoni De Sio F, Villa M, Uyeda MJ, Floris M, et al. Coexpression of CD163 and CD141 identifies human circulating IL-10-producing dendritic cells (DC-10). Cell Mol Immunol. (2019) 17:95–107. doi: 10.1038/s41423-019-0218-0
110. Yang H, Wang H, Levine YA, Gunasekaran MK, Wang Y, Addorisio M, et al. Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight. (2016) 1:85375. doi: 10.1172/jci.insight.85375
111. Ni C, Yang L, Xu Q, Yuan H, Wang W, Xia W, et al. CD68- and CD163-positive tumor infiltrating macrophages in non-metastatic breast cancer: a retrospective study and meta-analysis. J Cancer. (2019) 10:4463–72. doi: 10.7150/jca.33914
112. Triantafyllou E, Woollard KJ, Mcphail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front Immunol. (2018) 9:2948. doi: 10.3389/fimmu.2018.02948
113. Allaoui R, Bergenfelz C, Mohlin S, Hagerling C, Salari K, Werb Z, et al. Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat Commun. (2016) 7:13050. doi: 10.1038/ncomms13050
114. Dai J, Kumbhare A, Youssef D, Mccall CE, El Gazzar M. Intracellular S100A9 promotes myeloid-derived suppressor cells during late sepsis. Front Immunol. (2017) 8:1565. doi: 10.3389/fimmu.2017.01565
115. Yang J, Anholts J, Kolbe U, Stegehuis-Kamp JA, Claas FHJ, Eikmans M. Calcium-binding proteins S100A8 and S100A9: investigation of their immune regulatory effect in myeloid cells. Int J Mol Sci. (2018) 19:E1833. doi: 10.3390/ijms19071833
Keywords: myeloid-derived suppressor cell, cancer, infection, development, differentiation, maturation, activation, tolerance
Citation: Bergenfelz C and Leandersson K (2020) The Generation and Identity of Human Myeloid-Derived Suppressor Cells. Front. Oncol. 10:109. doi: 10.3389/fonc.2020.00109
Received: 25 November 2019; Accepted: 21 January 2020;
Published: 07 February 2020.
Edited by:
George S. Karagiannis, Albert Einstein College of Medicine, United StatesReviewed by:
Sven Brandau, University of Duisburg-Essen, GermanyPanayotis Verginis, Biomedical Research Foundation of the Academy of Athens, Greece
Copyright © 2020 Bergenfelz and Leandersson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karin Leandersson, a2FyaW4ubGVhbmRlcnNzb24mI3gwMDA0MDttZWQubHUuc2U=