 Trevor W. Stone
Trevor W. Stone- NDORMS, University of Oxford, Oxford, United Kingdom
The Epithelial-Mesenchymal Transition (EMT) is an important concept in understanding the processes of oncogenesis, especially with respect to the relationship between cell proliferation and metastatic properties such as spontaneous cell motility, chemotaxic migration and tissue invasion. EMT is now recognized as a more complex phenomenon than an all-or-nothing event, in which different components of the EMT may have distinct roles in the physio-pathological regulation of cell function and which may in turn depend on differential interactions with cell constituents and metabolic products. This mini-review summarizes recent work on the induction of cancer properties in parallel with the presence of EMT activities in the presence of serine proteases, with the focus on those tumor suppressors known as “dependence” receptors such as neogenin and Deleted in Colorectal Cancer (DCC). It is concluded that various forms of partial EMT should be given more detailed investigation and consideration as the results could have valuable implications for the development of disease-specific and patient-specific therapies.
Introduction
The concept of Epithelial-Mesenchymal Transition (EMT) was founded on changes in adhesion molecules associated with proliferation, cell dis-association, de-differentiation to a more mesenchymal stem cell character and the establishment of a motile, potentially invasive phenotype (1). EMT was thought necessary to initiate cell departure from a home tissue, a phenomenon seen in embryogenesis and in the metastatic spread of cancers. Recent studies have begun to define “partial” forms of EMT while others propose that EMT is more concerned with the end-stages of migration and invasion, with the reverse process of MET being at least as important as EMT (2, 3). EMT may be critical to the physical stabilization of cells in a metastatic location and in the development of increased resistance to chemotherapeutic drugs. The role of EMT in carcinogenesis may generate new sets of biomarkers for early metastasis detection and novel targets for dedicated drug development.
The complexity of molecular changes associated with EMT was responsible for the proposal of sub-types of EMT, or “partial” EMT, and the possibility that different molecular profiles may relate to the activity and aggressiveness of tumors. Partial EMTs may take many forms, based on different molecular profiles which characterizes different cell types, tissue locations and pathological conditions. The concept predicts that partial EMTs may provide a key for the generation of individual, tissue and disease specificity necessary for developing successful personalized therapy of cancers and other disorders. Defining the factors which induce partial EMTs will be essential if these relationships are to be exploited therapeutically. This mini-review surveys evidence for a role, in these events, of Dependence Receptors and their modulation by serine proteases.
Dependence and Guidance Receptors (DGRs)
The Dependence Receptors include Deleted in Colorectal Carcinoma (DCC) and the structurally related molecule neogenin (49% amino acid identity) (4). The term “dependence” receptors indicates that cell viability depends on ligation of the receptors by extracellular ligands such as the netrin family of extracellular proteins (ligands for DCC and neogenin) (5–8) or the Repulsive Guidance Molecules (RGMs; ligands for neogenin only). When occupied the receptors maintain cell survival, restraining cells from initiating apoptotic death when deprived of their ligands (9, 10) (Figure 1). These same receptors play crucial “guidance” roles, influencing the direction and rate of cell movement and the direction and size of cell outgrowths such as lammellipodia. These actions are seen in embryonic development when the dependence receptors guide progenitor cells to their eventual locations (11, 12), and in the developing and adult nervous system where they influence axonal or dendritic growth as well as the positioning of newly formed neurons (13–16), synaptic contacts and neural plasticity (17, 18). In view of this functional duality, the receptors will be referred to as Dependence and Guidance Receptors (DGRs).
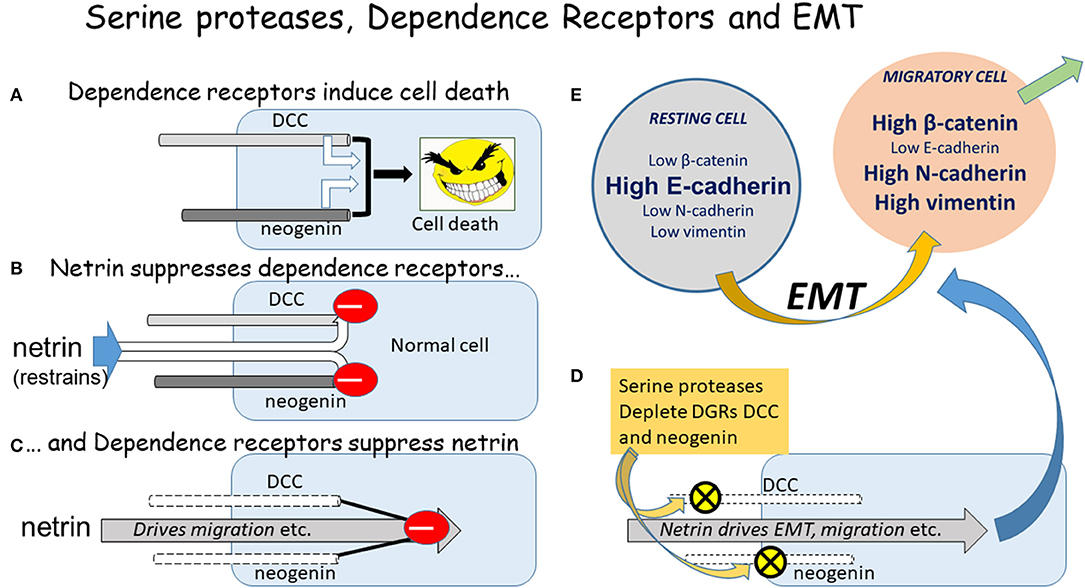
Figure 1. The DGRs, serine proteases, and EMT. A diagrammatic summary of the relationship between the DGRs DCC and neogenin, serine proteases, and the initiation of EMT. (A) If un-restrained by ligand binding, the DGRs promote cell death. (B) The primary ligands of DCC and neogenin are the netrin proteins, which restrain their ability to induce apoptosis. (C) Conversely the DGRs exert a feedback restraint on the netrins which tend to drive cells into an aggressive, pro-migratory mode via the induction of EMT. (D) The serine proteases chymotrypsin and subtilisin deplete DCC and neogenin from cells, allowing netrin to drive EMT and migration. (E) The classical EMT involves changes in the absolute and relative levels of proteins such as those indicated, but the magnitude and direction of change depends on the cell type, inducing stimulus, local microenvironment, and physio-pathological status, yielding a plethora of possible combinations of change which could represent highly selective therapeutic targets.
Also of relevance to EMT is the unco-ordinated-5 (unc5) family of DGRs—homologs of the uncoordinated locomotion genes in Caenorhabditis elegans—which also bind netrins as ligands (19). The binding of netrin-1 to DCC normally induces growth toward the focus of netrin production (20). If DCC is absent and netrin binds to unc5, then the result is repulsion: cells are directed away from the site of highest netrin concentration. Interaction between the intracellular domains of DCC and unc5 converts DCC attraction to repulsion (21). It would seem very likely that the migratory drive, direction and eventual target re-location of cells bearing these DGRs will depend on the ratios of attractant and repellent molecular states and may be critical in the establishment of metastases.
Relationships Between DGRs and EMT
Cell dependence on DGRs may have evolved in response to the development of multicellularity and distinct tissues. A cell could develop a genetic mutation spontaneously or induced by an infective organism, toxic compounds or physical trauma, which reduced intercellular adhesion—a necessary early stage of cell dis-association needed for EMT. A cell which becomes detached from the home tissue will then enter the extracellular medium as a free-living, migratory cell. The potential for early stage cancerous cells to establish metastatic colonies is clear. However, after leaving the home tissue the fall in ligand concentration for the cell's DGRs induces the unbound receptors to promote caspase-mediated apoptotic death (anoikis) (9, 22), a result which has been considered a safety mechanism to minimize or prevent the development of metastases.
In some cases, cells could become established at sites where a sufficient concentration of netrin isoforms allows them to create a new micro-colony (metastasis), since many tissues are able to synthesize and secrete these proteins (23–26). This may require the migration of a minimum number of cells which, once established, could produce enough ligand to maintain the metastatic colony. If this hypothesis were correct, it would suggest the need to inhibit tumor development as early as possible to prevent metastatic deposits achieving a size at which their ligand production offers continuing survival.
We have shown that DGR function may be regulated by chymotryptic serine proteases which deplete cells of their DGRs, causing increased cell migration (27), actions that may contribute to dietary effects on cancer incidence (28, 29). Subsequently, the effects of this DGR loss on the induction and progression of EMT were considered to identify any relationship between them, as discussed in the following sections in which the major EMT proteins E-cadherin, N-cadherin, β-catenin and vimentin were investigated (Figure 1) (30).
E-Cadherin
E-cadherin is localized primarily to cell membranes where it is involved in the formation of intercellular adherens junctions. It is considered a marker for resting, physiologically stable epithelial cells and its levels fall as cells de-differentiate before adopting the metabolic and morphological characteristics of migratory cells entering the EMT (31–35). In contrast, N-cadherin expression usually increases with the approaching onset of EMT and migration (36). The depletion of E-cadherin has been associated with carcinogenesis and metastasis formation (34, 37–39) and there is moderate expression in potentially metastatic cells. It has been shown that high levels of E-cadherin occur in metastases but it is absent from many primary tumors (40, 41).
β-Catenin
E-cadherin is usually associated with β-catenin and loss of the former releases β-catenin to enter the cytosol and nucleus where, as part of the wingless (Wnt) pathway, it regulates gene expression to influence proliferation and migration (31, 38, 41–54). It has been considered as the most significant protein characterizing EMT (55–60). However, β-catenin activation has also been linked to an inhibition of cell growth (61) and in some cases has little effect on cell behavior (62) while around 30% of mammary cancer cells had little or no β-catenin, raising questions about the extent and timing of its involvement in tumor development (63). β-catenin may have other functions in the cell independent of proliferation or migration, including the maintenance of epithelial structure and differentiation (51).
Vimentin
Vimentin is a microfilament protein involved in cytoskeletal function. As EMT develops, vimentin levels increase with cytoskeletal reorganization required for migration (64–68). In the absence of vimentin, migration is significantly reduced (69) as the E-cadherin/vimentin ratio may determine the switch from low to high invasion capacity. Transforming Growth Factor-β, a major regulator of EMT, may regulate vimentin expression and the decrease in E-cadherin (70, 71). However, vimentin is not always increased in migratory cells (72) and simple changes in incubation conditions, such as reducing the proportion of serum, can induce vimentin expression (27, 30). Vimentin expression might therefore be linked to the differentiation state of cells rather than their migratory status or capacity (73).
Serine Proteases, DGRs, and Partial EMT
Contradictory results have also been described when cells were exposed to serine proteases such as mammalian chymotrypsin or the bacterial chymotryptic enzyme subtilisin (27, 30). These proteases reduced expression of DCC and neogenin, with changes in the EMT proteins that were not consistent with normal views on their relationship to EMT. In particular, while levels of E-cadherin were reduced with the onset of migration, β-catenin levels were unaffected or substantially reduced, observations also obtained by other groups (45, 74). Vimentin expression was increased, as would be predicted for a compound involved in cytoskeletal adaptations to a change in differentiation state, dissociation and migration.
This pattern of protein changes associated with a loss of DGRs is clearly different from classical descriptions of EMT, suggesting that DGR loss should be added to the list of phenomena able to induce a “partial” EMT. Similar results have been obtained by silencing the neogenin gene in the absence of other primary modifications (75). This selective loss of neogenin also provoked changes in EMT proteins which were not fully consistent with conventional views of EMT leading to the conclusion that (from genetic and morphological data), a “partial EMT” was produced. Importantly, the results not only extend our own work but also confirm that the serine protease effects were probably produced solely by actions involving DGRs.
With the growing recognition of partial EMTs (76–83) some initiating factors are being identified including phosphatidyl-inositol-3-kinase (84), TGF-β (85–87), galectins (88), or altered expression of PTEN (89). Some groups define partial EMT with respect to mismatch between cell protein profile and phenotypic behavior (90) while others are attempting to define RNA fragments which regulate protein expression. These include miR-376a, miR-381, and miR-128 for which evidence indicates a significant relationship to tumor diagnosis and progression (91).
DGRs, EMT, and Differentiation
DCC expression declines as tumors advance and cells become more de-differentiated (92, 93). This would be consistent with the suggestion that EMT is a reflection of the differentiation status of cells and might explain the changes in vimentin expression induced by reduced serum-containing media (30, 73). It would account for the loss, rather than increase in β-catenin expression since β-catenin levels fall in cells differentiated by retinoic acid (94). It is also consistent with the need for de-differentiation for cell migration (95).
DCC transfection into cells inhibits proliferation and migration in some cell populations (96–99) but it remains uncertain whether the expression of DGRs such as DCC and neogenin (100) are primary inducers of cancerous change or are secondary contributors to oncogenesis (45, 101–105). The possible association between DCC and the progress of EMT rather than its initiation would be consistent with several studies (103, 106). Perhaps viewing cell differentiation as a key to this uncertainty, rather than the EMT or any individual component of it, may help to resolve the issue (93, 107, 108). DCC is relatively abundant in fully differentiated cells but declines in the early stages of carcinogenesis (109), falling further with advancing tumor stage and reduced patient survival (13, 99, 103, 105, 106, 108, 110–112). Conversely, but very importantly, differentiation induced by a range of factors may only be possible in the presence of DCC (111, 113).
EMT or MET?
A modified view on the link between EMT and differentiation is that EMT may be associated with the termination of cell migration rather than with its initiation. Post-migratory cells undergo changes of EMT proteins which are the reverse of classical EMT changes and which may be crucial for cell re-settlement in a metastatic deposit (114). Interestingly, the loss of protein frazzled (a homolog of DCC) inhibits MET suggesting that DCC may normally drive the non-migratory state of cells by promoting MET, while its progressive decline with tumor growth eventually leads to an apparent EMT and metastatic migration. The netrin ligation of DCC induces a loss of cadherin and the initiation of EMT, raising the possibility that in the absence of netrin, a re-expression of DCC might enhance E-cadherin expression and MET, consolidating metastasis formation (52). Of course, a migrating cell will be identified as a “foreign” cell and will normally be removed by the immune system unless it can re-express DCC. Cells incapable of expressing DCC would remain susceptible to removal unless they migrate to areas sufficiently safely hidden from immune system cells, or where the local concentration of ligand is sufficient to overcome immune attack. These ideas would be consistent with suggestions that DCC increases in later stages of colony formation, when it promotes cell differentiation (with increased E-cadherin). The explanation also accounts for DCC-expressing cells being sensitive to anti-cancer drugs, whereas truly metastatic cells are relatively resistant (2, 3, 115, 116).
Conclusions
These arguments represent a highly simplified view of molecular events underlying changes in cell properties and behavior. In addition to DGRs, other important factors in EMT include growth factors (especially Transforming Growth Factor-β), oxidative stress, micro-RNAs and immune system cells (macrophages, neutrophils) (117–119). Since neutrophils generate large quantities of chymotryptic serine proteases, they are likely sources of the proteases discussed in this mini-review.
The term “partial EMT” conveys the concept that cells are exhibiting changes which are partially characteristic of epithelial cells and partially typical of mesenchymal cells. On the existing evidence, however, there is likely to be a spectrum of such states, probably dependent on the cell type, preceding mutagenic events and the final EMT-initiating stimulus (119). It may be that these proteins do not fully describe the intermediate states and the temporal sequence of cell properties, or the protein changes may represent a non-canonical EMT-like process or an independent mechanism for migration. These alternative possibilities might be more consistent with the suggestion that DCC is more functionally related to the stabilization and differentiation aspects of EMT or MET than to migration and metastasis. Indeed, vimentin was the only EMT protein whose expression increased with the increased migration generated by serine proteases. Other descriptions of mixed EMT protein changes include the “transition state” in brain metastases exhibiting atypical expression of E-cadherin and vimentin (32). Thus, vimentin may be a more selective component to study in relation to EMT (120).
Several transcription factors once thought to be essential for EMT are dispensible since, for example, Twist and Snail do not suppress cell invasiveness (2). In addition, EMT proteins may have actions which are unrelated to EMT (121). Overall, however, EMT may contribute to aspects of cell function with changes in protein expression which respond differently to changes in cell status. This could yield cells with a variety of partial “EMT signatures” unique to that cell population, disease and pathology. It is difficult to assess whether EMT is more usefully viewed in terms of the “partial” concept of organizational discontinuity rather than as a continuum of activity (78, 122), since even a continuum of molecular changes may generate discontinuous functional events with different thresholds for switching cell behavior with changes in protein signature. This will be difficult to clarify if those thresholds involve critical ratios of several compounds which individually appear to vary continuously. Nevertheless, developing these could facilitate the generation of new anti-cancer therapies with a high degree of specificity and safety for individual patients, cancer type and stage, depending on the cell signature involved. This may be especially important since partial EMTs may be associated with more aggressive and chemoresistant tumors (123–126) although the contrary view has been expressed, emphasizing the need for further work (127).
Author Contributions
TS performed the literature search and wrote the mini-review.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. (1995) 154:8–20. doi: 10.1159/000147748
2. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. (2015) 527:525–30. doi: 10.1038/nature16064
3. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. (2015) 527:7579–89. doi: 10.1038/nature15748
4. Phan KD, Croteau LP, Kam JWK, Kania A, Cloutier JF, Butler SJ. Neogenin may functionally substitute for DCC in chicken. PLOS ONE. (2011) 6:e22072. doi: 10.1371/journal.pone.0022072
5. Mehlen P, Goldschneider D. Dependence receptors: a new paradigm in cell signalling and cancer therapy. Oncogene. (2010) 29:1865–82. doi: 10.1038/onc.2010.13
6. Mehlen P, Llambi F. Role of netrin-1 and netrin-1 dependence receptors in colorectal cancers. Brit J Cancer. (2005) 93:1–6. doi: 10.1038/sj.bjc.6602656
7. Rodrigues S, De Wever O, Bruyneel E, Rooney RJ, Gespach C. Opposing roles of netrin-1 and the dependence receptor DCC in cancer cell invasion, tumor growth and metastasis. Oncogene. (2007) 26:5615–25. doi: 10.1038/sj.onc.1210347
8. Sun KLW, Correia JP, Kennedy TE. Netrins: versatile extracellular cues with diverse functions. Development. (2011) 138:2153–69. doi: 10.1242/dev.044529
9. Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, et al. The dependence receptor DCC defines an alternative mechanism for caspase activation. Proc Nat Acad Sci USA. (2001) 98:3416–21. doi: 10.1073/pnas.051378298
10. Zhang QS, Liang F, Ke Y, Huo YP, Li MC, Li YY, et al. Overexpression of neogenin inhibits cell proliferation and induces apoptosis in human MDA-MB-231 breast carcinoma cells. Oncol Rep. (2015) 34:258–64. doi: 10.3892/or.2015.4004
11. Kang DS, Yang YR, Lee C, Park BW, Park KIL, Seo JK, et al. Netrin-1/DCC-mediated PLC gamma 1 activation is required for axon guidance and brain structure development. EMBO Rep. (2018). 11:e46250. doi: 10.15252/embr.201948117
12. Welniarz Q, Morel MP, Pourchet O, Gallea C, Lamy JC, Cincotta M, et al. Non cell-autonomous role of DCC in the guidance of the corticospinal tract at the midline. Sci Rep. (2017) 7:410. doi: 10.1038/s41598-017-00514-z
13. Duman-Scheel M. Deleted in colorectal cancer (DCC) pathfinding: axon guidance gene finally turned tumor suppressor. Curr Drug Targets. (2012) 13:1445–53. doi: 10.2174/138945012803530215
14. Bernhardt NR, Memic F, Gezelius H, Thiebes AL, Vallstedt A, Kullander K. DCC mediated axon guidance of spinal interneurons is essential for normal locomotor central pattern generator function. Dev Biol. (2012) 366:279–89. doi: 10.1016/j.ydbio.2012.03.017
15. Finci L, Zhang Y, Meijers R, Wang J-H. Signaling mechanism of the netrin-1 receptor DCC in axon guidance. Progr Biophys Mol Biol. (2015) 118:153–60. doi: 10.1016/j.pbiomolbio.2015.04.001
16. Junge HJ, Yung AR, Goodrich LV, Chen Z. Netrin1/DCC signaling promotes neuronal migration in the dorsal spinal cord. Neural Devel. (2016) 11:19. doi: 10.1186/s13064-016-0074-x
17. Horn KE, Glasgow SD, Gobert D, Bull SJ, Luk T, Girgis J, et al. DCC expression by neurons regulates synaptic plasticity in the adult brain. Cell Rep. (2013) 3:173–85. doi: 10.1016/j.celrep.2012.12.005
18. Stone TW, Darlington LG, Forrest CM. Dependence receptor involvement in subtilisin-induced long-term depression and in long-term potentiation. Neuroscience. (2016) 336:49–62. doi: 10.1016/j.neuroscience.2016.08.043
19. Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. (1990) 4:61–85. doi: 10.1016/0896-6273(90)90444-K
20. Fitamant J, Guenebeaud C, Coissieux MM, Guix C, Trilleux I, Scoazec JY, et al. Netrin-1 expression confers a selective advantage for tumor cell survival in metastatic breast cancer. Proc Nat Acad Sci USA. (2008) 105:4850–5. doi: 10.1073/pnas.0709810105
21. Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M, Stein E. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell. (1999) 97:927–41. doi: 10.1016/S0092-8674(00)80804-1
22. Chen J, Du H, Zhang Y, Chen H, Zheng M, Lin P, et al. Netrin-1 prevents rat primary cortical neurons from apoptosis via the DCC/ERK pathway. Front Cell Neurosci. (2017) 11:387. doi: 10.3389/fncel.2017.00387
23. Glasgow SD, Labrecque S, Beamish IV, Aufmkolk S, Gibon J, Han D, et al. Activity-dependent netrin-1 secretion drives synaptic insertion of GluA1-containing AMPA receptors in the hippocampus. Cell Rep. (2018) 25:168–82. doi: 10.1016/j.celrep.2018.09.028
24. Passacquale G, Phinikaridou A, Warboys C, Cooper M, Lavin B, Alfieri A, et al. Aspirin-induced histone acetylation in endothelial cells enhances synthesis of the secreted isoform of netrin-1 thus inhibiting monocyte vascular infiltration. Brit J Pharmacol. (2015) 172:3548–64. doi: 10.1111/bph.13144
25. Mohamed R, Jayakumar C, Ranganathan PV, Ganapathy V, Ramesh G. Kidney proximal tubular epithelial-specific overexpression of netrin-1 suppresses inflammation and albuminuria through suppression of COX-2-mediated PGE2 production in streptozotocin-induced diabetic mice. Amer J Pathol. (2012) 181:1991–2002. doi: 10.1016/j.ajpath.2012.08.014
26. Ratcliffe EM, Fan L, Mohammed TJ, Anderson M, Chalazonitis A, Gershon MD. Enteric neurons synthesize netrins and are essential for the development of the vagal sensory innervation of the fetal gut. Devel Neurobiol. (2011) 71:362–73. doi: 10.1002/dneu.20869
27. Forrest CM, McNair K, Vincenten MC, Darlington LG, Stone TW. Selective depletion of tumour suppressors deleted in colorectal cancer (DCC) and neogenin by environmental and endogenous serine proteases: linking diet and cancer. BMC Cancer. (2016) 16:772. doi: 10.1186/s12885-016-2795-y
28. Stone TW, Darlington LG. Microbial carcinogenic toxins and dietary anti-cancer protectants. Cell Mol Life Sci. (2017) 74:2627–43. doi: 10.1007/s00018-017-2487-z
29. Stone TW, McPherson M, Darlington LG. Obesity and cancer: existing and new hypotheses for a causal connection. EBioMedicine. (2018) 30:14–28. doi: 10.1016/j.ebiom.2018.02.022
30. McNair K, Forrest CM, Vincenten M, Darlington LG, Stone TW. Serine protease modulation of dependence receptors and EMT protein expression. Cancer Biol Therap. (2019) 20:349–67 doi: 10.1080/15384047.2018.1529109
31. Lopes FF, da Costa Miguel MC, Amaral Pereira ALA, Nogueira da Cruz MCF, Freitas RDA, Pinto LP, et al. Changes in immune-expression of E-cadherin and beta-catenin in oral squamous cell carcinoma with and without nodal metastasis. Ann Diagn Pathol. (2009) 13:22–9. doi: 10.1016/j.anndiagpath.2008.07.006
32. Jeevan DS, Cooper JB, Braun A, Murali R, Jhanwar-Unival M. Molecular pathway mediating metastases to the brain via EMT: genes, proteins and functional analysis. Anticancer Res. (2016) 36:523–32.
33. Rosado P, Lequerica-Fernandez P, Fernandez S, Allonca E, Villallain L, de Vicente JC. E-cadherin and beta-catenin expression in well-differentiated and moderately-differentiated oral squamous cell carcinoma: relations with clinical variables. Br J Oral Maxillo Surg. (2013) 51:149–56. doi: 10.1016/j.bjoms.2012.03.018
34. Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha(5)-integrin, which is a therapeutic target. Cancer Res. (2008) 68:2329–39. doi: 10.1158/0008-5472.CAN-07-5167
35. Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. (2008) 68:989–97. doi: 10.1158/0008-5472.CAN-07-2017
36. Duband JL, Monier F, Delannet M, Newgreen D. Epithelium-mesenchyme transition during neural crest development. Acta Anat. (1995) 154:63–78. doi: 10.1159/000147752
37. Faleiro-Rodrigues C, Macedo-Pinto IM, Maia SS, Vieira RH, Lopes CS. Biological relevance of E-cadherin-catenin complex proteins in primary epithelial ovarian tumours. Gynecol Obstet Invest. (2005) 60:75–83. doi: 10.1159/000084614
38. Matos AJF, Lopes C, Carvalheira J, Santos M, Rutteman GR, Gartner F. E-cadherin expression in canine malignant mammary tumours: Relationship to other clinico-pathological variables. J Comp Pathol. (2006) 134:182–9. doi: 10.1016/j.jcpa.2005.10.004
39. Daugaard I, Sanders KJ, Idica A, Vittayarukskul K, Hamdorf M, Krog JD, et al. miR-151a induces partial EMT by regulating E-cadherin in NSCLC cells. Oncogenesis. (2017) 6:e366. doi: 10.1038/oncsis.2017.66
40. Fite K, Gomez-Cambronero J. Down-regulation of MicroRNAs (MiRs) 203, 887, 3619 and 182 prevents vimentin-triggered, phospholipase D (PLD)-mediated cancer cell invasion. J Biol Chem. (2016) 291:719–30. doi: 10.1074/jbc.M115.686006
41. Zhao P, Guo SJ, Tu T, Di LJ, Zha XJ, et al. Grh13 induces human epithelial tumor cell migration and invasion via down-regulation of E-cadherin. Acta Biochim Biophys Acta. (2016) 48:266–74. doi: 10.1093/abbs/gmw001
42. Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1expression and cancer progression. Proc Natl Acad Sci USA. (2000) 97:4262–6. doi: 10.1073/pnas.060025397
43. Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Nat Acad Sci USA. (2011) 108:19204–9. doi: 10.1073/pnas.1108977108
44. Bakker ERM, Hoekstra E, Franken PF, Helvenstein W, van Deurzen CHM, van Veelen W, et al. β-catenin signalling dosage dictates tissue-specific tumor predisposition in APC-driven cancer. Oncogene. (2013) 32:4579–85. doi: 10.1038/onc.2012.449
45. Bamias A, Yu Z, Weinberger PM, Markakis S, Kowalski D, Camp RL, et al. Automated quantitative analysis of DCC tumor suppressor protein in ovarian cancer tissue microarray shows association with beta-catenin levels and outcome in patients with epithelial ovarian cancer. Ann Oncol. (2006) 17:1797–802. doi: 10.1093/annonc/mdl310
46. Dorfman T, Pollak Y, Sohotnik R, Coran AG, Bejar J, et al. Enhanced intestinal epithelial cell proliferation in diabetic rats correlates with beta-catenin accumulation. J Endocrinol. (2015) 226:135–43. doi: 10.1530/JOE-14-0725
47. Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ, Lee KH, et al. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/beta-catenin signaling pathway. Carcinogenesis. (2013) 34:530–8. doi: 10.1093/carcin/bgs371
48. Liu Y, Ye X, Zhang J-B, Ouyang H, Shen Z, Wu Y, et al. PROX1 promotes hepatocellular carcinoma proliferation and sorafenib resistance by enhancing beta-catenin expression and nuclear translocation. Oncogene. (2015) 34:5524–35. doi: 10.1038/onc.2015.7
49. Qiu YM, Yuan RF, Zhang SH, Chen LF, Huang D, Hao HB, et al. Rock2 stabilizes beta-catenin to promote tumor invasion and metastasis in colorectal cancer. Biochim Biophys Res Comm. (2015) 467:629–37. doi: 10.1016/j.bbrc.2015.10.103
50. Fang L, Zhu Q, Neuenschwander M, Specker E, Wulf-Goldenberg A, Weis WI, et al. A small-molecule antagonist of the beta-Catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. (2016) 76:891–901. doi: 10.1158/0008-5472.CAN-15-1519
51. Restucci B, Maiolino P, Martano M, Esposito G, De Filippis D, Borzacchiello G, et al. Expression of beta-catenin, E-cadherin and APC in canine mammary tumors. Anticancer Res. (2007) 27:3083–9.
52. Lee SJ, Jung YH, Oh SY, Yong MS, Ryu JM, Han HJ. Netrin-1 Induces MMP-12-dependent E-cadherin degradation via the distinct activation of PKC Alpha and FAK/Fyn In promoting mesenchymal stem cell motility. Stem Cells Dev. (2014) 23:1870–82. doi: 10.1089/scd.2013.0632
53. Fagotto F. The cellular basis of tissue separation. Development. (2014) 141:3303–18. doi: 10.1242/dev.090332
54. Fagotto F. Nuclear beta-catenin under control. Dev Cell. (2015) 33:625–6. doi: 10.1016/j.devcel.2015.06.005
55. Tian X, Zhang J, Tan TK, Lyons JG, Zhao H, Niu B, et al. Association of beta-catenin with P-Smad3 but not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-beta 1. J Cell Sci. (2013) 126:67–76. doi: 10.1242/jcs.103036
56. Hong CS, Jeong O, Piao Z, Guo C, Jung MR, Choi C, et al. HOXB5 induces invasion and migration through direct transcriptional up-regulation of beta-catenin in human gastric carcinoma. Biochem J. (2015) 472:393–403. doi: 10.1042/BJ20150213
57. Leem SH, Li XJ, Park MH, Park BH, Kim SM. Genome-wide transcriptome analysis reveals inactivation of Wnt/beta-catenin by 3,3 '-diindolylmethane inhibiting proliferation of colon cancer cells. Int J Oncol. (2015) 47:918–26. doi: 10.3892/ijo.2015.3089
58. Leal LF, Bueno AC, Gomes DC, Abduch R, de Castro M, Antonini SR. Inhibition of the Tcf/beta-catenin complex increases apoptosis and impairs adrenocortical tumor cell proliferation and adrenal steroidogenesis. Oncotarget. (2015) 6:43016–32. doi: 10.18632/oncotarget.5513
59. Pramanik KC, Fofaria NM, Gupta P, Ranjan A, Kim SH, Srivastava SK. Inhibition of beta-catenin signaling suppresses pancreatic tumor growth by disrupting nuclear beta-catenin/TCF-1 complex: Critical role of STAT-3. Oncotarget. (2015) 6:11561–74. doi: 10.18632/oncotarget.3427
60. Siemens H, Neumann J, Jackstadt R, Mansmann U, Horst D, Kirchner T, et al. Detection of miR-34a promoter methylation in combination with elevated expression of c-met and beta-catenin predicts distant metastasis of colon cancer. Clin Cancer Res. (2013) 19:710–21. doi: 10.1158/1078-0432.CCR-12-1703
61. Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM, et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/beta-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Nat Acad Sci USA. (2014) 111:5349–54. doi: 10.1073/pnas.1317731111
62. Sekine S, Shibata T, Sakamoto M, Hirohashi S. Target disruption of the mutant beta-catenin gene in colon cancer cell line HCT116: preservation of its malignant phenotype Oncogene. (2002) 21:5906–11. doi: 10.1038/sj.onc.1205756
63. Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, et al. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. (2011) 24:209–31. doi: 10.1038/modpathol.2010.205
64. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. (2011) 68:3033–46. doi: 10.1007/s00018-011-0735-1
65. Satelli A, Hu JM, Xia XQ, Li SL. Potential function of exogenous vimentin on the activation of Wnt signalling pathway in cancer cells. J Cancer. (2016) 7:1824–32. doi: 10.7150/jca.15622
66. Hendrix MJC, Seftor EA, Chu YW, Trevor KT, Seftor REB. Role of intermediate filaments in migration, invasion and metastasis. Cancer Metast Rev. (1996) 15:507–25. doi: 10.1007/BF00054016
67. Hendrix MJC, Seftor EA, Seftor REB, Trevor KT. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behaviour. Am J Pathol. (1997) 150:483–95.
68. Yoneyama MS, Hatakeyama S, Habuchi T, Inoue T, Nakamura T, Funyu T, et al. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Europ J Cell Biol. (2014) 93:157–69. doi: 10.1016/j.ejcb.2014.03.002
69. Ozguven BY, Karaçetin D, Kabukçuoglu F, Taskin T, Yener S. Immunohistochemical study of E-cadherin and beta-catenin expression in colorectal carcinomas. Pol J Pathol. (2011) 62:19–24.
70. Li X, Yang J, Wang X, Li X, Liang J, Xing H. Role of TWIST2, E-cadherin and Vimentin in epithelial ovarian carcinogenesis and prognosis and their interaction in cancer progression. Eur J Gynaecol Oncol. (2016) 37:100–8.
71. Trivanovic D, Jaukovic A, Krstic J, Nikolic S, Djordjevic IO, Kukolj T, et al. Inflammatory cytokines prime adipose tissue mesenchymal stem cells to enhance malignancy of MCF-7 breast cancer cells via Transforming Growth Factor-beta 1. IUBMB Life. (2016) 68:190–200. doi: 10.1002/iub.1473
72. Messi E, Florian MC, Caccia C, Zanisi M, Maggi R. Retinoic acid reduces human neuroblastoma cell migration and invasiveness: effects on DCX, LISI, neurofilaments-68 and vimentin expression. BMC Cancer. (2008) 8:30. doi: 10.1186/1471-2407-8-30
73. Polioudaki H, Agelaki S, Chiotaki R, Politaki E, Mavroudis D, Matikas A, et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. (2015) 15:399. doi: 10.1186/s12885-015-1386-7
74. Lin VCL, Jin RX, Tan PH, Aw SE, Woon CT, Bay BH. Progesterone induces cellular differentiation in MDA-MB-231 breast cancer cells transfected with progesterone receptor complementary DNA. Am J Pathol. (2003) 162:1781–7. doi: 10.1016/S0002-9440(10)64313-1
75. Chaturvedi V, Fournier-Level A, Cooper HM, Murray MJ. Loss of Neogenin1 in human colorectal carcinoma cells causes a partial EMT and wound-healing response. Sci Rep. (2019) 9:4110. doi: 10.1038/s41598-019-40886-y
76. Campbell K, Rossi F, Adams J, Pitsidianaki I, Barriga FM, Garcia-Gerique L, et al. Collective cell migration and metastases induced by an epithelial-to-mesenchymal transition in Drosophila intestinal tumors. Nat Commun. (2019) 24:2311. doi: 10.1038/s41467-019-10269-y
77. Aiello NM, Kang Y. Context-dependent EMT programs in cancer metastasis. J Exp Med. (2019) 216:1016–26. doi: 10.1084/jem.20181827
78. Aiello NM, Maddipati R, Norgard RJ, Balli D, Li JY, Yuan S, et al. EMT subtype influences epithelial plasticity and mode of cell migration. Devel Cell. (2018) 45:681–90. doi: 10.1016/j.devcel.2018.05.027
79. Agnoletto C, Corra F, Minotti L, Baldassari F, Crudele F, Cook WJJ, et al. Heterogeneity in circulating tumor cells: the relevance of the stem-cell subset. Cancers. (2019) 11:483. doi: 10.3390/cancers11040483
80. Pallegar NK, Garland CJ, Mahendralingam M, Viloria-Petit AM, Christian SL. A novel 3-dimensional co-culture method reveals a partial mesenchymal to epithelial transition in breast cancer Cells Induced by adipocytes. J Mamm Gland Biol Neoplasia. (2019) 24:85–97. doi: 10.1007/s10911-018-9420-4
81. Puram SV, Parikh AS, Tirosh I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol. (2018) 5:e1448244. doi: 10.1080/23723556.2018.1448244
82. Xu YX, Lee DK, Feng Z, Xu Y, Bu W, Li Y, et al. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc Nat Acad Sci USA. (2017) 114:11494–9. doi: 10.1073/pnas.1618091114
83. Grigore AD, Jolly MK, Jia D, Farach-Carson MC, Levine H. Tumor budding: the name is EMT. Partial EMT J Clin Med. (2016) 5:51–60 doi: 10.3390/jcm5050051
84. Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. (2019) 10:3554. doi: 10.1038/s41467-019-11403-6
85. Almale L, Garcia-Alvaro M, Martinez-Palacian A, Garcia-Bravo M, Lazcanoiturburu N, Addante A, et al. c-Met signaling is essential for mouse adult liver progenitor cells expansion after transforming growth factor-beta-induced epithelial-mesenchymal transition and regulates cell phenotypic switch. Stem Cells. (2019) 37:1108–18. doi: 10.1002/stem.3038
86. Katsuno Y, Meyer DS, Zhang ZY, Shokat KM, Akhurst RJ, Miyazono K, et al. Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. (2019) 12:eaau8544. doi: 10.1126/scisignal.aau8544
87. Zhou T, Luo MC, Cai W, Zhou SY, Feng DY, Xu CD, et al. Runt-related transcription factor 1 (RUNX1) promotes TGF-beta-induced renal tubular epithelial-to-mesenchymal transition (EMT) and renal fibrosis through the PI3K subunit p110 delta. EBioMedicine. (2018) 31:217–25. doi: 10.1016/j.ebiom.2018.04.023
88. Oyanadel C, Holmes C, Pardo E, Retamal C, Shaughnessy R, Smith P, et al. Galectin-8 induces partial epithelial-mesenchymal transition with invasive tumorigenic capabilities involving a FAK/EGFR/proteasome pathway in Madin-Darby canine kidney cells. Mol Biol Cell. (2018) 29:557–74. doi: 10.1091/mbc.E16-05-0301
89. Mei WJ, Lin XZ, Kapoor A, Gu Y, Zhao KC, Tang DM. The contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers. (2019) 11:434. doi: 10.3390/cancers11040434
90. Siar CH, Ng KH. Epithelial-to-mesenchymal transition in ameloblastoma: focus on morphologically evident mesenchymal phenotypic transition. Pathology. (2019) 51:494–501. doi: 10.1016/j.pathol.2019.04.004
91. Bure IV, Nemtsova MV, Zaletaev DV. Roles of E-cadherin and noncoding RNAs in the epithelial-mesenchymal transition and progression in gastric cancer. Int J Mol Sci. (2019) 20:2870. doi: 10.3390/ijms20122870
92. Vielmetter J, Kayyem JF, Roman JM, Dreyer WJ. Neogenin, an avian cell-surface protein expressed during terminal neuronal differentiation, closely-related to the human tumor-suppressor molecule deleted in colorectal-cancer. J Cell Biol. (1994) 127:2009–20. doi: 10.1083/jcb.127.6.2009
93. Cho KR, Fearon ER. DCC - linking tumor-suppressor genes and altered cell-surface interactions in cancer. Eur J Cancer. (1995) 31A:1055–60. doi: 10.1016/0959-8049(95)00128-6
94. Elizalde C, Campa VM, Caro M, Schlangen K, Maria A, Aransay AM, et al. Distinct Roles for Wnt-4 and Wnt-11 during retinoic acid-inducedneuronal differentiation. Stem Cells. (2011) 29:141–53. doi: 10.1002/stem.562
95. Wang HM, Unternaehrer JJ. Epithelial-mesenchymal transition and cancer stem cells: at the crossroads of differentiation and dedifferentiation. Dev Dyn. (2019) 248:10–20. doi: 10.1002/dvdy.24678
96. Velcich A, Corner G, Palumbo L, Augenlicht L. Altered phenotype of HT29 colonic adenocarcinoma cells following expression of the DCC gene. Oncogene. (1999) 18:2599–606. doi: 10.1038/sj.onc.1202610
97. Klingelhutz AJ, Hedrick L, Cho KR, McDougall JK. The DCC gene suppresses the malignant phenotype of transformed human epithelial-cells. Oncogene. (1995) 10:1581–6.
98. Kato H, Zhou Y, Asanoma K, Kondo H, Yoshikawa Y, Watanabe K, et al. Suppressed tumorgenicity of human endometrial cancer cells by the restored expression of the DCC gene. Br J Cancer. (2000) 2000:459–66. doi: 10.1054/bjoc.1999.0943
99. Krimpenfort P, Song J-Y, Proost N, Zevenhoven J, Jonkers J, Berns A. Deleted in colorectal carcinoma suppresses metastasis in p53-deficient mammary tumours. Nature. (2012) 482:538–40. doi: 10.1038/nature10790
100. Cole SJ, Bradford D, Cooper HM. Neogenin: a multi-functional receptor regulating diverse developmental processes. Int J Biochem Cell Biol. (2007) 39:1569–75. doi: 10.1016/j.biocel.2006.11.009
101. Lee JE, Kim HJ, Bae JY, Kim SW, Park JS, et al. Neogenin expression may be inversely correlated to the tumorigenicity of human breast cancer. BMC Cancer. (2005) 5:154. doi: 10.1186/1471-2407-5-154
102. Yamashita T, Yaginuma Y, Yamashita K, Fujita M, Ishikawa M. DCC gene alterations in histological types and clinical stages of epithelial ovarian cancer. Int J Oncol. (1996) 9:291–6. doi: 10.3892/ijo.9.2.291
103. Reyes-Mugica M, Lin P, Yokota J, Reale MA. Status of deleted in colorectal cancer gene expression correlates with neuroblastoma metastasis. Lab Invest. (1998) 78:669–75.
104. Saegusa M, Hashimura M, Hara A, Okayasu I. Loss of expression of the gene deleted in colon carcinoma (DCC) is closely related to histologic differentiation and lymph node metastasis in endometrial carcinoma. Cancer. (1999) 85:53–64. doi: 10.1002/(SICI)1097-0142(19990115)85:2<453::AID-CNCR25>3.0.CO
105. Saegusa M, Machida D, Okayasu I. Loss of DCC gene expression during ovarian tumorigenesis: relation to tumour differentiation and progression. Br J Cancer. (2000) 82:571–8. doi: 10.1054/bjoc.1999.0966
106. Fujita M, Enomoto T, Murata Y. Genetic alterations in ovarian carcinoma: with specific reference to histological subtypes. Mol Cell Endocrinol. (2003) 202:97–9. doi: 10.1016/S0303-7207(03)00069-8
107. Jarjour AA, Durko M, Luk TL, Marcal N, Shekarabi M, Kennedy TE. Autocrine netrin function inhibits glioma cell motility and promotes focal adhesion formation. PLoS ONE. (2011) 6:e25408. doi: 10.1371/journal.pone.0025408
108. Saegusa M, Okayasu I. DCC expression is related to mucinous differentiation but not changes in expression of p21(WAF1/Cip1) and p27(Kip1), apoptosis, cell proliferation and human papillomavirus infection in uterine cervical adenocarcinomas. Br J Cancer. (1999) 80:51–8. doi: 10.1038/sj.bjc.6690320
109. Hedrick L, Cho KR, Fearon ER, Wu TC, Kinzler KW, Vogelstein B. The DCC gene-product in cellular-differentiation and colorectal tumorigenesis. Genes Dev. (1994) 8:1174–83. doi: 10.1101/gad.8.10.1174
110. Castets M, Broutier L, Molin Y, Brevet M, Chazot G, Gadot N, et al. DCC constrains tumour progression via its dependence receptor activity. Nature. (2012) 482:534–7. doi: 10.1038/nature10708
111. Narayanan R, Lawlor KG, Schaapveld RQJ, Cho KR, Vogelstein B, Tran PBV, et al. Antisense RNA to the putative tumor-suppressor gene DCC transforms rat-1 fibroblasts. Oncogene. (1992) 7:553–61.
112. Martin M, Simon-Assmann P, Kedinger M, Martin M, Mangeat P, Real FX, et al. DCC regulates cell adhesion in human colon cancer derived HT-29 cells and associates with ezrin. Eur J Cell Biol. (2006) 85:769–83. doi: 10.1016/j.ejcb.2006.02.013
113. Hedrick L, Cho KR, Boyd J, Risinger J, Vogelstein B. DCC - A tumor-suppressor gene expressed on the cell-surface. Cold Spring Harb Symp Quant Biol. (1992) 57:345–51. doi: 10.1101/SQB.1992.057.01.039
114. Pert M, Gan M, Saint R, Murray MJ. Netrins and Frazzled/DCC promote the migration and mesenchymal to epithelial transition of Drosophila midgut cells. Biol Open. (2015) 4:233–43. doi: 10.1242/bio.201410827
115. Gal R, Sadikov E, Sulkes J, Klein B, Koren R. Deleted in colorectal cancer protein expression as a possible predictor of response to adjuvant chemotherapy in colorectal cancer patients. Dis Colon Rectum. (2004) 47:1216–24. doi: 10.1007/s10350-004-0552-9
116. Shibata D, Reale MA, Lavin P, Silverman M, Fearon ER, Steele G, et al. The DCC protein and prognosis in colorectal cancer. New Engl J Med. (1996) 335:1727–32. doi: 10.1056/NEJM199612053352303
117. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. (2010) 9:4461–8. doi: 10.4161/cc.9.22.14048
118. Ma N, Xiang Y, Zhang Y, Zhao X, Zhou L, Gao X. The balance mediated by miRNAs and the heme oxygenase 1 feedback loop contributes to biological effects. J Cell Biochem. (2013) 114:2637–42. doi: 10.1002/jcb.24631
119. Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nature. (2019) 19:716–32. doi: 10.1038/s41568-019-0213-x
120. Kai K, Iwamoto T, Zhang DW, Shen L, Takahashi Y, Rao A, et al. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci Rep. (2018) 8:9427. doi: 10.1038/s41598-018-27409-x
121. Sekine S, Gutierrez PJA, Lan BYA, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. (2007) 45:361–8. doi: 10.1002/hep.21523
122. Campbell K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr Opin Cell Biol. (2018) 55:30–5. doi: 10.1016/j.ceb.2018.06.008
123. Jia DY, George JT, Tripathi SC, Kundnani DL, Lu MY, Hanash SM, et al. Testing the gene expression classification of the EMT spectrum. Physical Biol. (2019) 16:025002. doi: 10.1088/1478-3975/aaf8d4
124. Papadaki MA, Stoupis G, Theodoropoulos PA, Mavroudis D, Georgoulias V, Agelaki S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol Cancer Ther. (2019) 18:437–47. doi: 10.1158/1535-7163.MCT-18-0584
125. Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. (2018) 164:257–64. doi: 10.1093/jb/mvy047
126. Wallesch M, Pachow D, Blucher C, Firsching R, Warnke JP, Braunsdorf WEK, et al. Altered expression of E-Cadherin-related transcription factors indicates partial epithelial-mesenchymal transition in aggressive meningiomas. J Neurol Sci. (2017) 380:112–21. doi: 10.1016/j.jns.2017.07.009
Keywords: DCC, neogenin, chymotrypsin, subtilisin, serine proteases
Citation: Stone TW (2020) Dependence and Guidance Receptors—DCC and Neogenin—In Partial EMT and the Actions of Serine Proteases. Front. Oncol. 10:94. doi: 10.3389/fonc.2020.00094
Received: 19 October 2019; Accepted: 17 January 2020;
Published: 04 February 2020.
Edited by:
Katarína Smolková, Institute of Physiology (ASCR), CzechiaReviewed by:
Jaroslav Truksa, Institute of Biotechnology (ASCR), CzechiaAlberto Leguina Ruzzi, Institute of Physiology (ASCR), Czechia
Ciro Leonardo Pierri, University of Bari Aldo Moro, Italy
Copyright © 2020 Stone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trevor W. Stone, dHJldm9yLnN0b25lQGtlbm5lZHkub3guYWMudWs=