
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 07 February 2020
Sec. Head and Neck Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00087
 Zhexuan Li1†
Zhexuan Li1† Xun Chen2†Ming Wei1Guancheng Liu3Yongquan Tian1Xin Zhang1
Xun Chen2†Ming Wei1Guancheng Liu3Yongquan Tian1Xin Zhang1 Gangcai Zhu4Changhan Chen1Jiangyi Liu5Tiansheng Wang6Gongbiao Lin7Juncheng Wang1
Gangcai Zhu4Changhan Chen1Jiangyi Liu5Tiansheng Wang6Gongbiao Lin7Juncheng Wang1 Gengming Cai7*
Gengming Cai7* Yunxia Lv8*
Yunxia Lv8*Alternative splicing (AS) is an important mechanism that is responsible for the production of protein diversity. An increasing body of evidence has suggested that out-of-control AS is closely related to the genesis and development of cancer. Systematic analysis of genome-wide AS in head and neck squamous cell carcinoma (HNSCC) has not yet been carried out, and consideration of this topic remains at the preliminary stage and requires further investigation. In this study, systemic bioinformatic analysis was carried out on the genome-wide AS events of 555 clinical HNSCC samples from the TCGA database. Firstly, we statistically analyzed the distributions of seven AS event types in HNSCC samples. Then, through univariate survival analysis, we observed the relationship between AS and the prognosis of the disease and found that 437 intersections of AS events were significantly related to overall survival. Among them, 335 cross-genes showed a high degree of consistency in the genes associated with overall survival and recurrence. The overall survival was significantly related to AS events. Besides, the frequency of overall survival-related ES events was evidently reduced, while the AP and the AT events were increased. In addition, AT events accounted for the largest proportion. Further, multiple regression model analysis proved that AS could become a new classification method for HNSCC, and KEGG enrichment analysis proved that most genes and proteins interacting with AS events had different biological functions and were associated with a variety of diseases. Finally, through the selection of characteristic HNSCC genes and the construction of a prognostic model, seven cross-genes related to survival and recurrence were screened out, and these characteristic genes were verified by multivariate survival model analysis so as to classify the prognosis at different splicing times and gene expression levels. These results have laid a solid foundation for our further research and play a decisive role in showing the correlation of AS with the prognosis of HNSCC.
HNSCC is a kind of head and neck malignant tumor that is commonly seen in the clinic, accounting for about 5–10% of systemic malignant tumors, with an average morbidity of about 10-5/100,000 (1). In recent years, the morbidity of such tumors with high malignant grade shows an increasing trend. Clinically advanced cases make up about 50%, despite the rapid development of medical technology, as well as the increasingly improved early diagnosis technique of HNSCC (2–4). Over the past 20 years, great achievements have been made in the surgical method (5), radiotherapy (6), and chemotherapy (7); however, the 5-years survival rate for HNSCC, especially for the advanced patients, has not yet been remarkably improved (8). This ineffectiveness of current therapies underscores the urgent need for new discoveries leading to more effective treatment pathways.
The cancer genome atlas (TCGA) is a collection of genomic information about a variety of tumor types, including GBM, designed to classify and identify genomic changes that cause cancer in order to create a comprehensive cancer genome atlas (9). In the past 10 years, the TCGA project has generated a lot of genomics and proteomics data; these data represent the 33 types of cancer in more than 11,000 tumor molecular structures, mainly concentrated on the assessment of somatic non-synonymous protein change mutation and the mutation of gene expression (10, 11). In this paper, access to and download from the TCGA database and global gene expression profile analysis and database mining were conducted to find the potential association between genes and the overall survival rate of multiple malignancies.
Protein diversity is crucial for the obvious regulatory and functional complexities of eukaryocytes. Pre-mRNA AS is a general mechanism that uses a limited set of genes to produce mRNA isomers (12). AS is a process in which the introns of most human multi-exon genes are deleted and specific exons are alternatively included or excluded (13, 14). Apart from protein diversity, the mRNA isomer translation level can also be down-regulated by introducing AS, leading to degradation of the early termination codon (15). Therefore, AS is an important process; changes in the splicing pattern are closely correlated with protein functions, and it is involved in multiple human physiological functions, such as hematopoiesis (16), brain development (17), and muscular activity (18). Differences in the gene expression levels of splicing regulators have been observed in many cancers, and these proteins generally influence the splicing patterns of many genes that play a role in certain cancer-specific biological pathways, including cell cycle progression, cell proliferation and migration, and RNA processing. The prevalence of AS is currently estimated at the genome-wide level using high-throughput methods, including microarrays, expressed sequence tag analysis, and sequencing (13). The AS process can explain the difference between estimates of the number of genes and the number of proteins.
The protein diversity of cancer genome analysis is the key to the regulation and functional complexity of eukaryotic cells. Individual changes in regulatory binding sites or changes in protein-coding sequences may have strong functional effects (10). However, an increasing number of recent studies have demonstrated that AS is also markedly correlated with cancer genesis and development (19, 20). Out-of-control AS participates in multiple carcinogenic processes, including proliferation (21), apoptosis suppression (22), angiogenesis (23), immune escape (21), and metastasis (24). For example, exon-skipping events in MST1R are controlled by SF2/ASF through AS of the RON proto-oncogene (25). Convergence of Acquired Mutations and Alternative Splicing of CD19 lead to the activity of splicing factor SRSF3 was destroyed, AS also affects the immunotherapy of leukemia (26). Met exon 14 was observed in some lung cancer patients, resulting in a deletion of the protein region that inhibits its kinase catalytic activity (27). More recently, analyses of AS have also shown prognostic value for a variety of cancer types, including non-small cell lung cancer (15), ovarian cancer (28), breast cancer (29), uveal melanoma (30), and glioblastoma (31). However, the alternative splice associated with survival in head and neck squamous cell carcinoma (HNSCC) has not been extensively studied. Nonetheless, few studies on head and neck squamous cell carcinoma (HNSCC) have been reported, and research on the subject remains at the preliminary stage (19). This paper aimed to carry out systemic bioinformatic analysis of the HNSCC clinical samples from the TCGA database and open up a novel path for exploring the pathogenesis, prognosis biomarkers, and therapeutic targets of HNSCC from the point of view of AS, thus more accurately guiding clinical treatment and judging prognosis.
RNA-seq AS event data for HNSCC were obtained from the TCGASpliceSeq database (32), which covered a total of 555 samples, including 43 normal samples. Additionally, the RNA-seq expression profile data of HNSCC and para-carcinoma were obtained from the TCGA database (33), which involved 546 samples, including 44 normal samples. Moreover, all clinical follow-up data (including 527 samples) were downloaded from the TCGA database. The data processing and statistical modeling are crucial and vital, as the information extracted from the raw data will depend on these steps. A better understanding of data processing and statistical algorithms and methods are important to achieve statistically relevant and optimal biological information. In order to obtain data with high accuracy, integrity, and consistency, we pre-processed TCGA data (34). The data were pre-processed as follows: the RNA-Seq expression profile FPKM dataset was downloaded and further converted into TPM data; at the same time, the ID was transformed using the genome file of GENCODE (GRCh38.p2) (35), and the protein-encoding genes were obtained. A total of 498 common samples in TCGASpliceSeq and RNA-Seq were then enrolled in this study, and a total of 19,754 genes with expression values were obtained as the total gene set for this study. The overall analysis process is presented in Figure 1A.
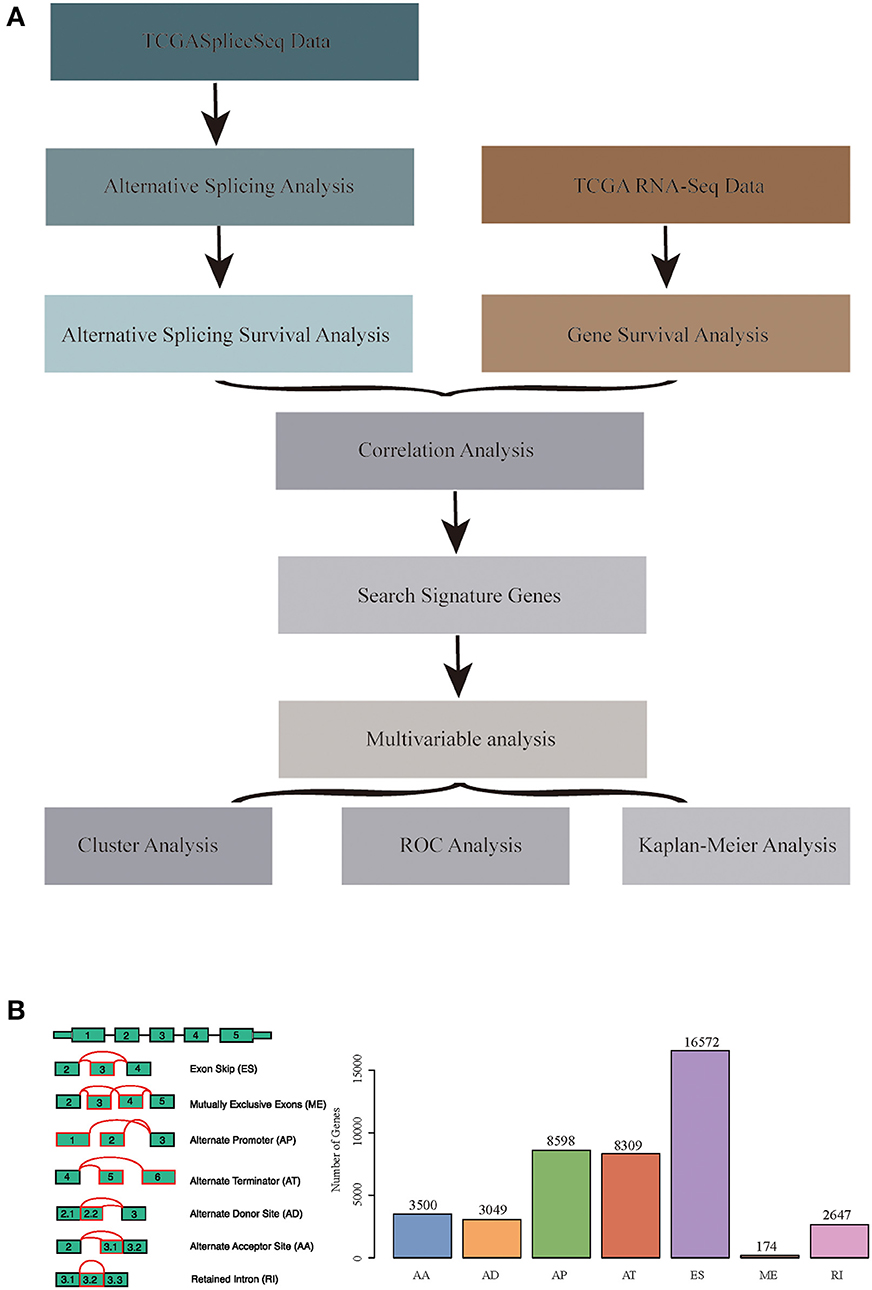
Figure 1. (A) Schematic diagram of the research methodology. (B) Overview of seven types of alternative splicing (AS) events in this study. Illustrations for seven types of AS events: ES, Exon Skip; ME, Mutually Exclusive Exons; RI, Retained Intron; AP, Alternate Promoter; AT, Alternate Terminator; AD, Alternate Donor site; AA, Alternate Acceptor site.
The TCGASpliceSeq database had analyzed the mRNA splicing pattern using SpliceSeq tool (36) based on TCGA RNA-Seq data. There were seven types of AS events, including Exon Skip (ES), Mutually Exclusive Exons (ME), Retained Intron (RI), Alternate Promoter (AP), Alternate Terminator (AT), Alternate Donor site (AD), and Alternate Acceptor site (AA). In addition, the distribution of all coding genes among these seven different types of data was analyzed in the HNSCC samples.
Differences in the AS of genes may result in gene diversity, while changes in the gene expression level would affect patient survival. Survival analysis is a method for analyzing and deducing the survival time of an organism or a person based on the data obtained from experiment or investigation so as to examine the relationships of survival time and outcome with numerous influence factors as well as their degrees. It is also referred to as survival rate analysis.
In this study, the different gene-splicing events obtained from disease samples were subjected to univariate survival analysis using the “survival” package in R (37). Genes satisfying the significant level of p < 0.05 were selected as the prognosis differential AS events.
AS can affect the protein diversity translated by genes. In this study, the prognosis-related AS events were selected, and the gene distribution among these genes was determined so as to analyze the gene distribution among the various types of prognosis-related AS events.
To observe whether AS events could serve as prognosis factors, the 20 most significant genes among the AS types were selected for multivariate regression model analysis so as to observe their classification of prognosis.
To observe the gene associations among the various types of AS events that were markedly correlated with prognosis, these genes were mapped to the String database (38), respectively. Then, the interactions of these genes were obtained using score >0.4, and Cytoscope was used for visualization.
To observe the gene functions in various types of AS events that were markedly correlated with prognosis, KEGG enrichment analysis was carried out on the AS genes evidently correlated with prognosis in each type using the “clusterprofile” package in R, so as to observe the enrichment pathways of these genes.
TCGA RNA-Seq expression profile data were used for univariate survival analysis of each gene to observe the relationships between gene expression and prognosis in AS events that were markedly correlated with prognosis; in addition, the influence of the expression profiles of genes involved in AS events on prognosis was also examined.
Genes with a Pearson correlation coefficient between gene expression and AS events of >0.2 or <-0.2 were selected as the prognosis feature genes.
To construct the prognosis-predicting indexes that were suitable for HNSCC patients and to facilitate clinical practice, the prognosis feature genes were selected to construct the multivariate survival model, so as to observe the classification of prognosis by these prognosis feature genes at AS event and expression profile levels.
All the AS events in 498 cancer samples were calculated, as shown in Table S1, and the seven AS patterns are presented in Figure 1B, which cover 42849 AS events, including 10,123 genes. The distribution of the seven types of AS events is shown in Figure 2C; as can be seen, one gene might be associated with several types of mRNA AS events. Typically, ES was the main type, accounting for almost 1/3 of all AS events.
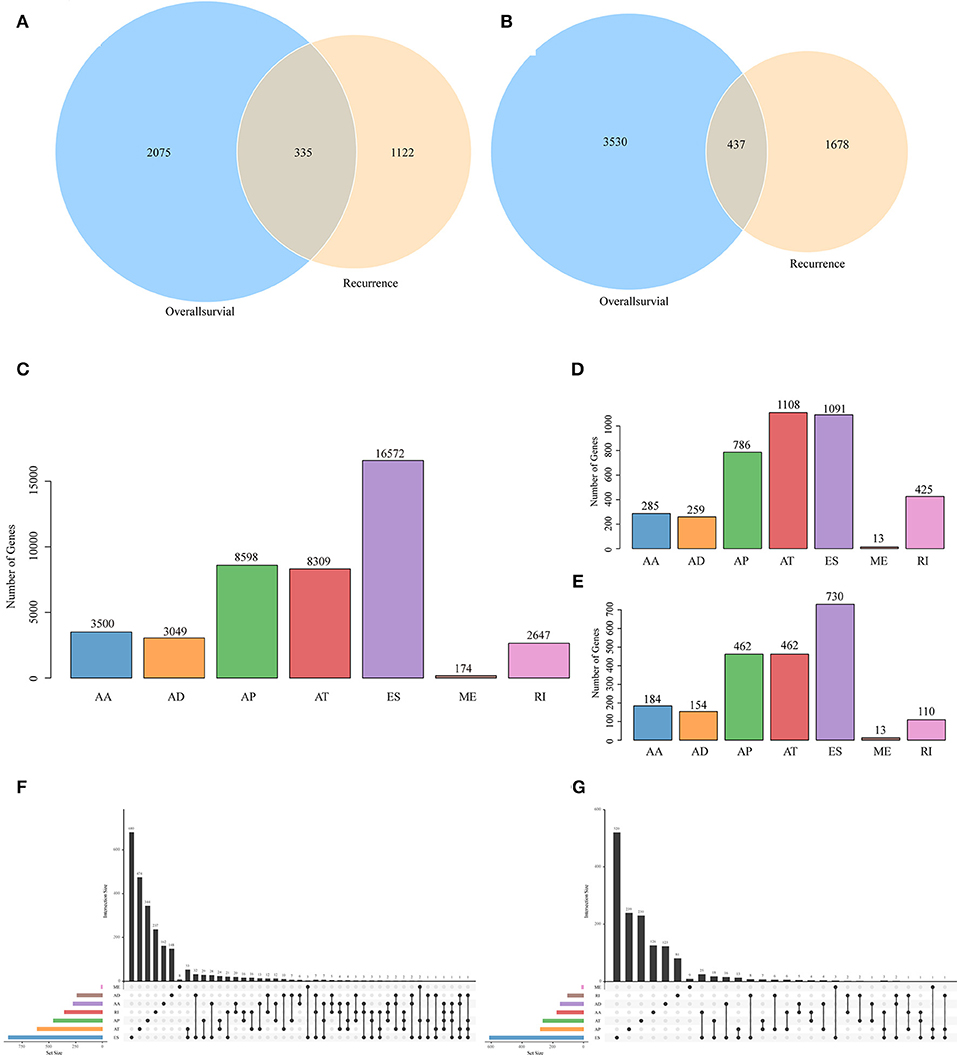
Figure 2. Venn diagram of AS events (A) and involved genes (B) that were significantly correlated with OS and recurrence. (C) Distribution of AS events of seven types. (D) Distribution of OS-associated AS events of seven types. (E) Distribution of recurrence-associated AS events of seven types. The UpSet intersection diagram shows seven types of OS (F)—and recurrence (G)—associated AS events in HNSCC. One gene may have up to four types of alternative splicing associated with patient survival or recurrence.
To observe the relationships between AS and disease prognosis, all clinical follow-up data for the diseases were integrated to form Table S2, and univariate survival analysis was performed on 42849 AS events to examine the relationships between these AS events and the prognosis for HNSCC patients. When selecting p < 0.05, a total of 3697 AS events that were remarkably correlated with survival, involving 2,410 genes, were obtained; in addition, 2115 AS events that were markedly correlated with disease recurrence, covering 1,457 genes, were acquired, as displayed in Table S3. Besides, there were 335 intersections between AS events that were significantly correlated with overall survival and those that were markedly related to recurrence, as presented in Figure 2A. Among them, there were 473 intersected genes among all the involved genes, as shown in Figure 2B, suggesting great consistency between genes involved in overall survival and recurrence-related genes. The numbers of AS events that were markedly correlated with overall survival are plotted in Figure 2D, from which it could be seen that the frequency of overall survival-related ES events was evidently reduced, while the AP and AT events were increased. Besides, among the recurrence related AS events, AT events took up the greatest proportion (Figure 2E), suggesting that most ES events were not correlated with prognosis, while about 10% of AT events were markedly correlated with survival. Furthermore, type analysis was performed on the prognosis-related-gene-selective AS events. Typically, the prognosis-related AS events were selected, and the gene distribution among those genes was calculated (Figures 2F,G). It can be seen from the figure that one gene might be associated with multiple AS events of various types, and these different AS events might be associated with prognosis.
To observe whether the selective AS events could be used as prognosis factors, the 20 most significant genes of each AS type were selected from all prognosis-related AS events for multivariate regression model analysis. Similarly, the significant correlation between AS and genes was observed and the 20 most significant genes were also selected for multivariate regression model analysis so as to observe their classification of prognosis. As could be seen from Figure 3, the seven types of AS events had large areas under the curve (AUC) for prognosis classification, among which, the AA type AS displayed the best overall survival, while the AA and ES types had the best performance among AS significantly correlated with recurrence. Moreover, the top 20 most significant genes were selected from each type of AS event, and the forest maps of these genes are presented in Figures S1, S2. It can be seen that there were 16 genes in the AA splicing type with a hazard ratio (HR) of <1, and four with HR of >1; consistently, there were 16 genes in recurrence-related splicing with a HR of <1 and four with HR of >1. These findings revealed that AS might serve as a new classification method for HNSCC.
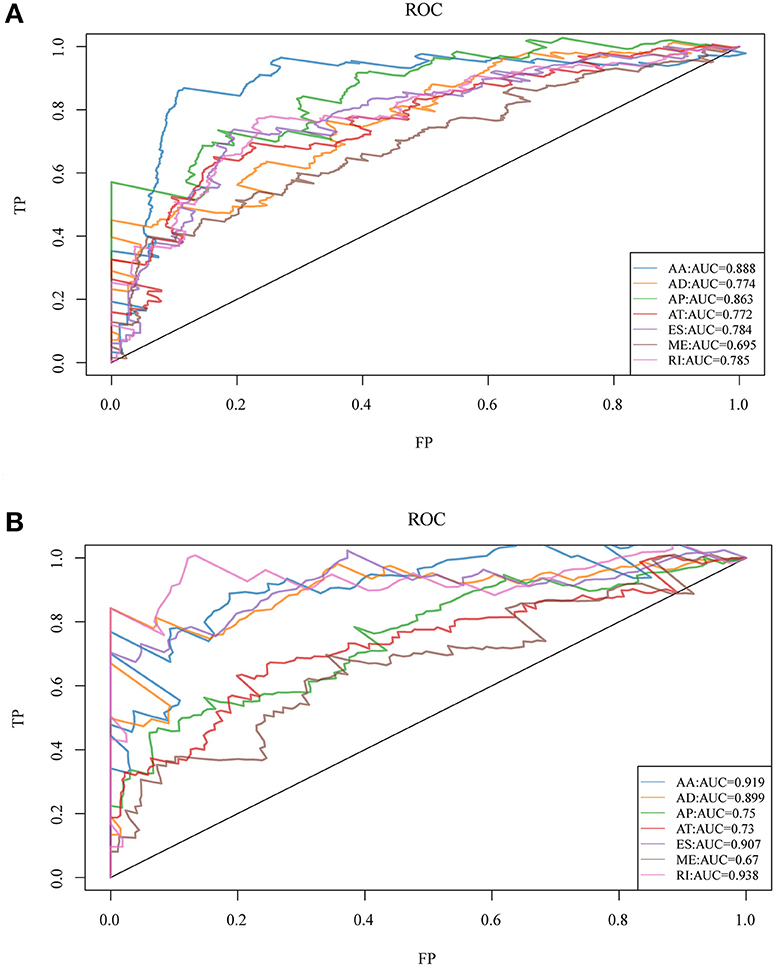
Figure 3. ROC curves for OS (A) and recurrence (B)-associated AS events of each type for HNSCC patients.
To observe the gene associations among different types of AS events that were apparently correlated with prognosis, these genes were mapped to the String database using a score of >0.4. The gene interactions were obtained to construct the gene interaction network among the various types of AS events that were markedly correlated with prognosis, and Cytoscope was used for visualization. As can be observed from Figures 4A,B, AP and AT displayed most interactions, and most genes in the prognosis-related AS events were associated with protein interactions, revealing that most of these genes were involved in different biological functions. Furthermore, to observe gene function in various types of AS events that were significantly correlated with prognosis, KEGG enrichment analysis was performed on the AS genes in each type that were significantly correlated with prognosis. The results are shown in Figures 4C,D, from which it can be seen that these genes were enriched in multiple disease-related pathways, suggesting that these genes were involved in numerous biological functions.
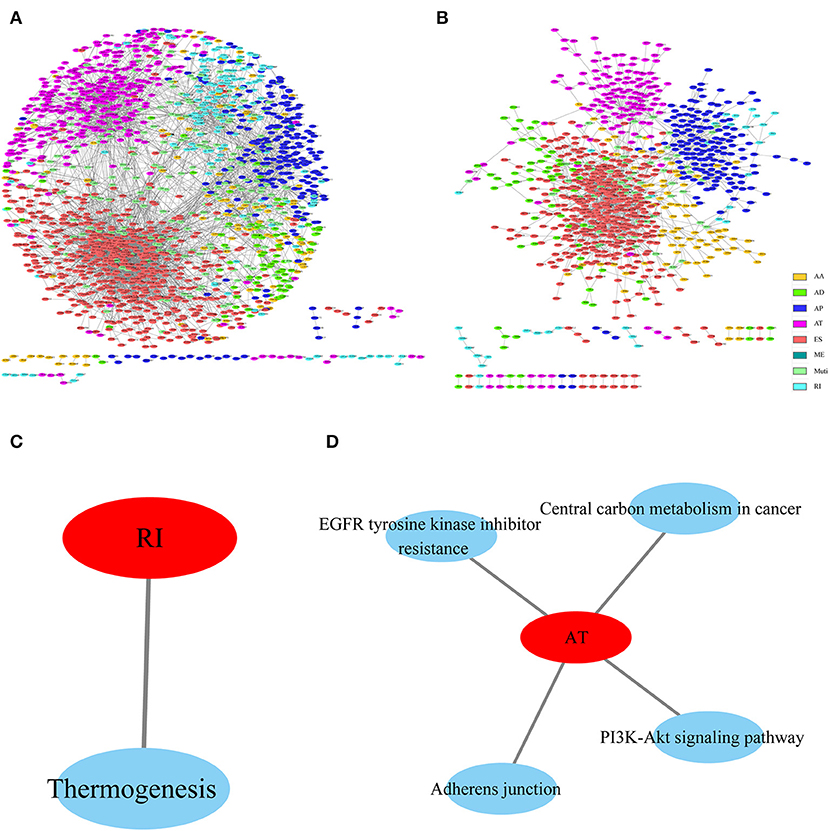
Figure 4. Interaction network of genes with OS (A)- and recurrence (B)-associated AS events for HNSCC, created by Cytoscape. KEGG enrichment analysis of genes with OS (C)- and recurrence (D)-associated AS events.
To observe the relationships between gene expression and prognosis in AS events that were notably correlated with prognosis, TCGA RNA-Seq expression profile data were employed for univariate survival analysis on each gene. Finally, it was discovered that among the 2,410 overall survival-related AS genes, the expression of 399 genes was related to overall survival; whereas, among the 1,457 recurrence-related AS genes, the expression of 194 genes was significantly correlated with recurrence. Furthermore, correlation analysis was performed between these 399 overall-survival genes and the corresponding AS events using the Pearson correlation coefficient. A total of 221 genes (55.39%) were found to be significantly correlated with AS (P < 0.05), indicating that the AS events of most genes were associated with their expression. Besides, of the 194 recurrence-related genes, 89 were markedly correlated with AS (45.88%), demonstrating that the AS events of most genes were markedly associated with their expression.
Genes with a Pearson correlation coefficient between gene expression and AS events of >0.1 or <-0.1 were selected, comprising 199 survival-related genes and 46 recurrence-related ones. Of the 199 overall survival-related genes, 14 were related to recurrence (CCDC23, TTC39A, CTBS, CD44, TAF1D, CCDC84, DHRS12, IFT20, TRABD2A, SPATS2L, CPNE1, NPHP3, PLS3, and LAMP2). Further multivariate Cox analysis was performed on these 14 genes, and it was found that four genes were related to overall survival (CDDC23, NPHP3, PLS3, and IFT20), while six were related to recurrence (NPHP3, TRABD2A, IFT20, TTC39A, CD44, and PLS3). Finally, seven intersecting genes related to both survival and recurrence were selected; their correlations with transcriptome level are shown in Figure 5. It can be observed from the figure that five genes were negatively correlated. Of these, NPHP3 mutation would lead to puberty nephropathy, retinal degeneration, and liver cirrhosis (39), IFT20 has been reported to be related to lung cancer (40), CD44 has been used as a cancer diagnostic marker (41), and PLS3 has also been used as a biomarker to monitor disease progression (42), indicating that most of these genes played important roles in cancer. Besides, to construct prognosis-predicting indexes that were suitable for HNSCC patients and to facilitate clinical practice, these seven feature genes were used to construct a multivariate survival model so as to observe the classification of prognosis by these seven feature genes at AS event and expression profile levels. As displayed in Figure 6, these seven genes presented favorable prognosis classification effects in both datasets, with a large AUC, indicating that these seven genes might serve as prognosis markers of HNSCC.
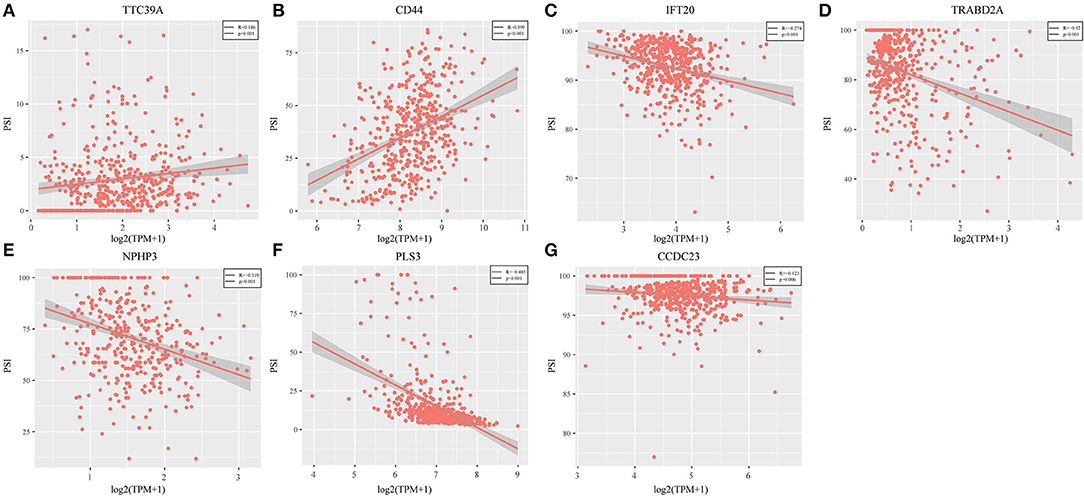
Figure 5. Seven intersected genes related to survival and recurrence were selected. It could be observed TTC39A (A) and CD44 (B) were positively correlated. But IFT20 (C), TRABD2A (D), NPHP3 (E), PLS3 (F), and CCDC23 (G) were negatively correlated (P < 0.05).
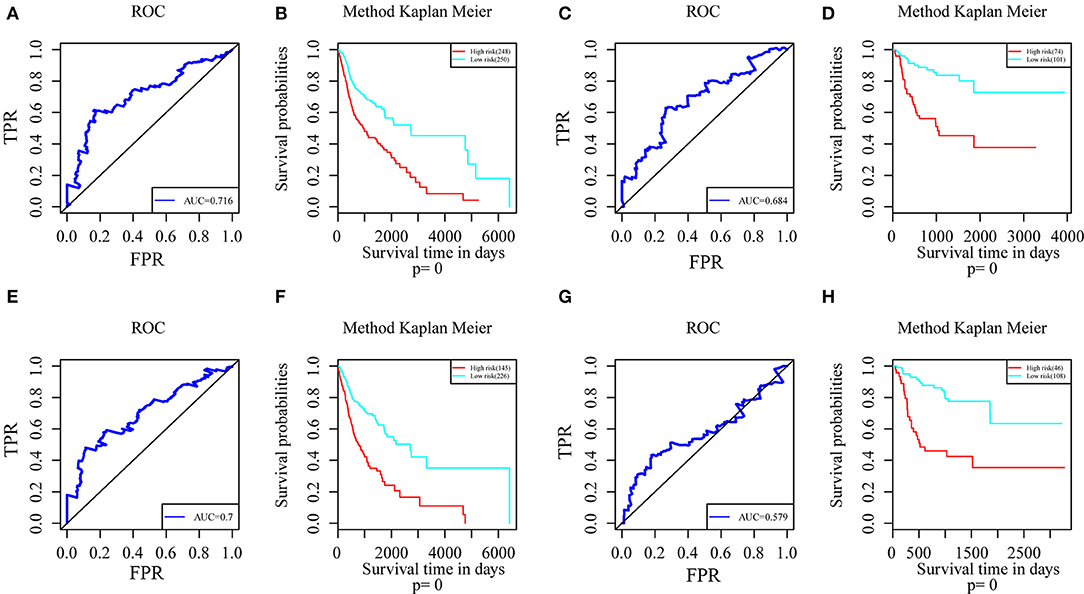
Figure 6. ROC curves and Kaplan-Meier plots of prognostic predictors for seven potential feature genes in HNSCC patients. (A) ROC curves with overall survival AUCs of prognostic predictors plotted for transcriptional events in HNSCC. (B) Kaplan-Meier curves with overall survival of prognostic predictor plotted for transcriptional events in HNSCC. (C) ROC curves with overall survival AUCs of prognostic predictors plotted for AS events in HNSCC. (D) Kaplan-Meier curves with overall survival of prognostic predictor plotted for AS events in HNSCC. (E) ROC curves with recurrence AUCs of prognostic predictors plotted for AS events in HNSCC. (F) Kaplan-Meier curves with recurrence of prognostic predictor plotted for AS events in HNSCC. (G) ROC curves with recurrence AUCs of prognostic predictors plotted for AS events in HNSCC. (H) Kaplan-Meier curves with recurrence of prognostic predictor plotted for AS events in HNSCC.
Cancer results from genetic and epigenetic changes that interfere with basic mechanisms of the normal cell life cycle, such as DNA repair, replication control, and Cell death (43, 44). In the past decade, the relationship between AS and human diseases has been gradually revealed. Changes in the proportions of splicing isomers of certain key proteins are involved in the occurrence and development of cellular case processes. In the development of disease, although synonymous mutation does not cause a change in the encoded protein sequence, it may change the splicing enhancer or silencer of the exons or introns, thereby affecting the splicing process. Similarly, the RNA terminator appears early due to some mutations. Not only does the protein encoded by this RNA have no function, it may also directly affect the insight process of other normal RNAs and cause diseases. Mutations in cis-acting components cause splicing-related diseases such as Parkinson's frontotemporal dementia (FTDP), muscular dystrophy (MD), and TAU disease represented by myoatrophy. Mutations that occur in trans-acting elements can modulate proteins that control the process of insight and cause disease. If DNA cannot be repaired correctly and effectively after replication or damage, it will lead to gene mutations. Gene mutations occurring at regulatory points will affect the transcriptome even further, resulting in differential proteins that activate or inhibit key functions, leading to infinite proliferation. In all steps of gene expression, AS may provide the greatest potential for molecular diversity and controlled regulation in cells (45). Various molecular complexes composed of RBP, structural RNA, and other protein factors bind to pre-mRNA at various locations (the RNA binding motif) and mediate (46). AS occurs through the expression of complexes on the pre-mRNA regulatory sequence or changes in the gene sequence. AS is not only an important mechanism in the normal cell cycle but is also a key mechanism for the occurrence of gene mutations leading to various pathologies, including tumors. Therefore, identification and analysis of AS are important to advance our understanding of tumor biology.
Head and neck cancers include tumors in head and neck tissues or organs except for the eye, brain, ear, thyroid, and esophagus, and over 90% of head and neck cancers are squamous cell carcinomas (47). The world has witnessed annual totals of about 600,000 new head and neck tumor cases and over 300,000 deaths (48). Head and neck cancer ranks top among systemic tumors in terms of the diversities of its primary site and pathological type. Unfortunately, the survival rate of head and neck cancer patients is low due to the lack of effective risk assessment and means of early diagnosis (49). Head and neck cancer is a highly complicated and heterogeneous disease. Tumor heterogeneity has mediated multiple clinical subtypes of head and neck cancer, which possesses different sensitivity and prognosis to chemotherapeutics and targeted preparations (50). The investigation and clinical application of biomarkers is the key to prognosis assessment, molecular classification, grade determination, recurrence assessment, and early selection of appropriate therapeutics and means. According to the literature, the abnormal splicing level of the GSN gene is remarkably higher in tumor tissues than in para-carcinoma tissues and regulates the proliferation process of the HNSCC cell line (51). Meanwhile, the oncogene DOCK5 variant plays a critical role in the human papilloma virus (HPV)-negative HNSCC and is involved in tumor proliferation, migration, and invasion (52). Moreover, it is also reported that the AS of the CD44 (53), LAMA3, DST (54), and ESRP genes has similar effects. Taken together, these results confirm that AS plays an important part in HNSCC genesis and development, which prompted us to systemically analyze the AS time in HNSCC clinical samples and dig out the potential prognosis markers and therapeutic targets.
At present, few complete gene studies on HNSCC AS have been published. A study aimed at identifying AS events (ASE) in human papillomavirus (HPV)-negative HNSCC demonstrated that the analysis of ASE in HPV-negative HNSCC identified multiple changes that may be associated with carcinogenesis, including carcinogenic DOCK5 variants (52). A recent study conducting a genome-wide analysis of AS events using RNA-seq data from the TCGA program in a sample of 464 HNSC patients revealed new AS events associated with carcinogenesis and the immune microenvironment (55). Likewise, another study systematically analyzed the RNA binding protein (RBP) gene mutations, copy number, and gene expression pattern, and changes of AS in these tumors, and AS sequence enrichment and the motif of the change in cancer RBP expression (56). However, screening studies related to survival and recurrence have not been widely studied in HNSCC through the selection of characteristic genes and the establishment of prognostic models.
This paper systemically analyzed 555 HNSCC samples from the TCGA database and mined the relationships between AS events and prognosis. Meanwhile, the key genes affecting the prognosis of HNSCC were also analyzed and mined based on the genome-wide selective splicing events, and the HNSCC samples were classified into high or low risk using the prognosis model constructed based on the gene expression profiles and AS events. Our results verified that AS events can serve as prognosis-predicting factors and potential therapeutic targets under a large sample size, a finding that contributes to the more accurate guidance of clinical treatment and judgment of prognosis. As a consequence, seven potential feature genes were mined, namely NPHP3, TRABD2A, IFT20, TTC39A, CD44, PLS3, and CDDC23. Additionally, multivariate survival model analysis suggested that these seven feature genes could classify the prognosis of HNSCC well at the AS time and gene expression levels.
Finally, despite the limited ability to detect individual events, we demonstrated that associated AS events can be used to construct high-performance prognostic indicators for HNSCC risk stratification, which is promising in clinical practice. In addition, we found excellent splicing-related networks. These results will be most valuable for deciphering the functional contribution of RNA splicing in HNSCC tumorigenesis. In-depth analysis of RNA splicing patterns may indeed reveal new cancer drivers and provide insights into the mechanisms of these pathways.
The datasets generated for this study can be found in the TCGA.
Informed consent was obtained from all the patients before surgery, and all experiments were conducted by following the bioethics rules issued by the Research Ethics Committee of Central South University, Changsha, China.
YT and XZ designed research. ZL and XC analyzed data. JL and TW statistical data. CC and GLin in functional analysis. GC and YL wrote and modified the paper. MW, GZ, and GLiu prepared all the figures. JW modified the language in the revisions. All authors have read and approved the final manuscript.
The Natural Science Foundation of Fujian Province (no. 2015J01512). The Middle- and Young-Aged Key Personnel Projects of Fujian Provincial Health and Family Planning Commission, China (no. 2015-ZQN-JC-39). The Medical Innovation Project of Fujian Provincial Health and Family Planning Commission, China. The Science and Technology Projects of Quanzhou Science and Technology Bureau, Quanzhou, Fujian, China (no. 2014Z22). The Natural Science Youth Founds of Jiangxi Province (no. 2016BAB215249). The Natural Science Foundation of China (nos. 81660294, 81602389, 81472696, 81202128, 81272974).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank members of their laboratory and their collaborators for their research work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00087/full#supplementary-material
AS, Alternative splicing; OS, Overall survival; HNSCC, head and neck squamous cell carcinoma; ES, Exon Skip; ME, Mutually Exclusive Exons; RI, Retained Intron; AP, Alternate Promoter; AT, Alternate Terminator; AD, Alternate Donor site; AA, Alternate Acceptor site; AUC, areas under the curve; HR, hazard ratio; TPR, Ture Positive Rate; FPR, False Positive Rate.
1. Hung YH, Liu SA, Wang CC, Wang CP, Jiang RS, Wu SH. Treatment outcomes of unknown primary squamous cell carcinoma of the head and neck. PLoS ONE. (2018) 13:e0205365. doi: 10.1371/journal.pone.0205365
2. Chan JYK, Zhen G, Agrawal N. The role of tumor DNA as a diagnostic tool for head and neck squamous cell carcinoma. Sem Cancer Biol. (2019) 55:1–7. doi: 10.1016/j.semcancer.2018.07.008
3. Sannigrahi MK, Sharma R, Panda NK, Khullar M. Role of non-coding RNAs in head and neck squamous cell carcinoma: a narrative review. Oral Dis. (2018) 24:1417–27. doi: 10.1111/odi.12782
4. Talmi YP, Takes RP, Alon EE, Nixon IJ, Lopez F, de Bree R, et al. Prognostic value of lymph node ratio in head and neck squamous cell carcinoma. Head Neck. (2018) 40:1082–90. doi: 10.1002/hed.25080
5. Manohar PM, Sapir E, Bellile E, Swiecicki PL, Pearson AT, Prince ME, et al. Capecitabine after surgical salvage in recurrent squamous cell carcinoma of head and neck. Otolaryngol Head Neck Surg. (2017) 157:995–7. doi: 10.1177/0194599817722948
6. Grossberg AJ, Mohamed ASR, Elhalawani H, Bennett WC, Smith KE, Nolan TS, et al. Imaging and clinical data archive for head and neck squamous cell carcinoma patients treated with radiotherapy. Scient Data. (2018) 5:180173. doi: 10.1038/sdata.2018.173
7. Karabajakian A, Gau M, Reverdy T, Neidhardt EM, Fayette J. Induction chemotherapy in head and neck squamous cell carcinoma: a question of belief. Cancers. (2018) 11:15. doi: 10.3390/cancers11010015
8. Hamilton SN, Tran E, Berthelet E, Wu J, Olson R. Early (90-day) mortality after radical radiotherapy for head and neck squamous cell carcinoma: a population-based analysis. Head Neck. (2018) 40:2432–40. doi: 10.1002/hed.25352
9. Tomczak K, Czerwinska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. (2015) 19:A68–77. doi: 10.5114/wo.2014.47136
10. Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. (2018) 34:211–24.e6. doi: 10.1016/j.ccell.2018.07.001
11. Wei L, Jin Z, Yang S, Xu Y, Zhu Y, Ji Y. TCGA-assembler 2: software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics. (2018) 34:1615–7. doi: 10.1093/bioinformatics/btx812
12. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. (2010) 463:457–63. doi: 10.1038/nature08909
13. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. (2008) 456:470–6. doi: 10.1038/nature07509
14. Salton M, Misteli T. Small molecule modulators of Pre-mRNA splicing in cancer therapy. Trends Mol Med. (2016) 22:28–37. doi: 10.1016/j.molmed.2015.11.005
15. Li Y, Sun N, Lu Z, Sun S, Huang J, Chen Z, et al. Prognostic alternative mRNA splicing signature in non-small cell lung cancer. Cancer Lett. (2017) 393:40–51. doi: 10.1016/j.canlet.2017.02.016
16. Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, et al. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative pre-mRNA splicing. Mol Cell Biol. (2015) 35:3071–82. doi: 10.1128/MCB.00202-15
17. Su C-H, Dhananjaya D, Tarn WY. Alternative splicing in neurogenesis and brain development. Front Mol Biosci. (2018) 5:12. doi: 10.3389/fmolb.2018.00012
18. Van Alstyne M, Simon CM, Sardi SP, Shihabuddin LS, Mentis GZ, Pellizzoni L. Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. (2018) 32:1045–59. doi: 10.1101/gad.316059.118
19. Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell. (2002) 2:9–15. doi: 10.1016/S1535-6108(02)00091-0
20. Martinez-Montiel N, Rosas-Murrieta NH, Anaya Ruiz M, Monjaraz-Guzman E, Martinez-Contreras R. Alternative splicing as a target for cancer treatment. Int J Mol Sci. (2018) 19:545. doi: 10.3390/ijms19020545
21. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. (2014) 33:5311–8. doi: 10.1038/onc.2013.533
22. Singh R, Gupta SC, Peng WX, Zhou N, Pochampally R, Atfi A, et al. Regulation of alternative splicing of Bcl-x by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. (2016) 7:e2262. doi: 10.1038/cddis.2016.168
23. Hamdollah Zadeh MA, Amin EM, Hoareau-Aveilla C, Domingo E, Symonds KE, Ye X, et al. Alternative splicing of TIA-1 in human colon cancer regulates VEGF isoform expression, angiogenesis, tumour growth and bevacizumab resistance. Mol Oncol. (2015) 9:167–78. doi: 10.1016/j.molonc.2014.07.017
24. Marzese DM, Manughian-Peter AO, Orozco JIJ, Hoon DSB. Alternative splicing and cancer metastasis: prognostic and therapeutic applications. Clin Exp Metast. (2018) 35:393–402. doi: 10.1007/s10585-018-9905-y
25. Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. (2005) 20:881–90. doi: 10.1016/j.molcel.2005.10.026
26. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Disc. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
27. Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. (2006) 66:283–9. doi: 10.1158/0008-5472.CAN-05-2749
28. Zhu J, Chen Z, Yong L. Systematic profiling of alternative splicing signature reveals prognostic predictor for ovarian cancer. Gynecol Oncol. (2018) 148:368–74. doi: 10.1016/j.ygyno.2017.11.028
29. Bjorklund SS, Panda A, Kumar S, Seiler M, Robinson D, Gheeya J, et al. Widespread alternative exon usage in clinically distinct subtypes of Invasive Ductal Carcinoma. Scient Rep. (2017) 7:5568. doi: 10.1038/s41598-017-05537-0
30. Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. (2017) 32:204–20.e15. doi: 10.1016/j.ccell.2017.07.003
31. Marcelino Meliso F, Hubert CG, Favoretto Galante PA, Penalva LO. RNA processing as an alternative route to attack glioblastoma. Hum Genet. (2017) 136:1129–41. doi: 10.1007/s00439-017-1819-2
32. Ryan M, Wong WC, Brown R, Akbani R, Su X, Broom B, et al. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. (2016) 44:D1018–22. doi: 10.1093/nar/gkv1288
33. Cen WN, Pang JS, Huang JC, Hou JY, Bao WG, He RQ, et al. The expression and biological information analysis of miR-375-3p in head and neck squamous cell carcinoma based on 1825 samples from GEO, TCGA, and peer-reviewed publications. Pathol Res Practice. (2018) 214:1835–47. doi: 10.1016/j.prp.2018.09.010
34. Tugizimana F, Steenkamp PA, Piater LA, Dubery IA. A conversation on data mining strategies in LC-MS untargeted metabolomics: pre-processing and pre-treatment steps. Metabolites. (2016) 6:40. doi: 10.3390/metabo6040040
35. Ronneberg TA, Freeland SJ, Landweber LF. Genview and gencode: a pair of programs to test theories of genetic code evolution. Bioinformatics. (2001) 17:280–1. doi: 10.1093/bioinformatics/17.3.280
36. Ryan MC, Cleland J, Kim R, Wong WC, Weinstein JN. SpliceSeq: a resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts. Bioinformatics. (2012) 28:2385–7. doi: 10.1093/bioinformatics/bts452
37. O'Quigley J, Moreau T. Cox's regression model: computing a goodness of fit statistic. Comp Methods Programs Biomed. (1986) 22:253–6. doi: 10.1016/0169-2607(86)90001-5
38. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. (2017) 45:D362–d8. doi: 10.1093/nar/gkw937
39. Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. (2003) 34:455–9. doi: 10.1038/ng1216
40. Dong YZ, Meng XM, Li GS. Long non-coding RNA SNHG15 indicates poor prognosis of non-small cell lung cancer and promotes cell proliferation and invasion. Europ Rev Med Pharmacol Sci. (2018) 22:2671–9. doi: 10.26355/eurrev_201805_14963
41. Matsumura Y, Tarin D. Significance of CD44 gene products for cancer diagnosis and disease evaluation. Lancet. (1992) 340:1053–8. doi: 10.1016/0140-6736(92)93077-Z
42. Tang N, Gibson H, Germeroth T, Porcu P, Lim HW, Wong HK. T-plastin (PLS3) gene expression differentiates Sezary syndrome from mycosis fungoides and inflammatory skin diseases and can serve as a biomarker to monitor disease progression. Br J Dermatol. (2010) 162:463–6. doi: 10.1111/j.1365-2133.2009.09587.x
43. Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genetics. (2016) 17:284–99. doi: 10.1038/nrg.2016.13
44. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
45. Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. (2014) 15:689–701. doi: 10.1038/nrg3778
46. Siegfried Z, Karni R. The role of alternative splicing in cancer drug resistance. Curr Opin Genet Dev. (2018) 48:16–21. doi: 10.1016/j.gde.2017.10.001
47. Orlandi E, Alfieri S, Simon C, Trama A, Licitra L. Treatment challenges in and outside a network setting: head and neck cancers. Europ J Surg Oncol. (2019) 45:40–5. doi: 10.1016/j.ejso.2018.03.012
48. Maghami E, Koyfman SA, Weiss J. Personalizing postoperative treatment of head and neck cancers. Am Soc Clin Oncol Educ Book. (2018) 38:515–22. doi: 10.1200/EDBK_201087
49. Wierzbicka M, Napierala J. Updated national comprehensive cancer network guidelines for treatment of head and neck cancers 2010–2017. Otolaryngol Pol. (2017) 71:1–6. doi: 10.5604/01.3001.0010.7193
50. Shang W, Zhang Q, Huang Y, Shanti R, Alawi F, Le A, et al. Cellular plasticity-targeted therapy in head and neck cancers. J Dental Res. (2018) 97:654–64. doi: 10.1177/0022034518756351
51. Kelley DZ, Flam EL, Guo T, Danilova LV, Zamuner FT, Bohrson C, et al. Functional characterization of alternatively spliced GSN in head and neck squamous cell carcinoma. Transl Res. (2018) 202:109–19. doi: 10.1016/j.trsl.2018.07.007
52. Liu C, Guo T, Xu G, Sakai A, Ren S, Fukusumi T, et al. Characterization of alternative splicing events in HPV-negative head and neck squamous cell carcinoma identifies an oncogenic DOCK5 variant. Clin Cancer Res. (2018) 24:5123–32. doi: 10.1158/1078-0432.CCR-18-0752
53. Wang SJ, Wong G, de Heer AM, Xia W, Bourguignon LY. CD44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope. (2009) 119:1518–30. doi: 10.1002/lary.20506
54. Li R, Ochs MF, Ahn SM, Hennessey P, Tan M, Soudry E, et al. Expression microarray analysis reveals alternative splicing of LAMA3 and DST genes in head and neck squamous cell carcinoma. PLoS ONE. (2014) 9:e91263. doi: 10.1371/journal.pone.0091263
55. Li ZX, Zheng ZQ, Wei ZH, Zhang LL, Li F, Lin L, et al. Comprehensive characterization of the alternative splicing landscape in head and neck squamous cell carcinoma reveals novel events associated with tumorigenesis and the immune microenvironment. Theranostics. (2019) 9:7648–65. doi: 10.7150/thno.36585
Keywords: head and neck squamous cell carcinoma (HNSCC), TCGA database, prognosis, Alternative Splicing (AS), oncology
Citation: Li Z, Chen X, Wei M, Liu G, Tian Y, Zhang X, Zhu G, Chen C, Liu J, Wang T, Lin G, Wang J, Cai G and Lv Y (2020) Systemic Analysis of RNA Alternative Splicing Signals Related to the Prognosis for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 10:87. doi: 10.3389/fonc.2020.00087
Received: 08 August 2019; Accepted: 16 January 2020;
Published: 07 February 2020.
Edited by:
Wojciech Golusinski, Poznan University of Medical Sciences, PolandReviewed by:
Michal Masternak, University of Central Florida, United StatesCopyright © 2020 Li, Chen, Wei, Liu, Tian, Zhang, Zhu, Chen, Liu, Wang, Lin, Wang, Cai and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gengming Cai, Y2dta2d4QGhvdG1haWwuY29t; Yunxia Lv, ODMzOTQwNDVAcXEuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.