 Katsutoshi Sato
Katsutoshi Sato Takashi Shimokawa
Takashi Shimokawa Takashi Imai
Takashi Imai- 1Division of Hematology and Medical Oncology, Icahn School of Medicine at Mount Sinai, The Tisch Cancer Institute, New York, NY, United States
- 2Department of Accelerator and Medical Physics, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Sciences and Technology, Chiba, Japan
- 3Medical Databank, Department of Radiation Medicine, QST Hospital, National Institutes for Quantum and Radiological Science and Technology, Chiba, Japan
In recent years, advanced radiation therapy techniques, including stereotactic body radiotherapy and carbon–ion radiotherapy, have progressed to such an extent that certain types of cancer can be treated with radiotherapy alone. The therapeutic outcomes are particularly promising for early stage lung cancer, with results matching those of surgical resection. Nevertheless, patients may still experience local tumor recurrence, which might be exacerbated by the acquisition of radioresistance after primary radiotherapy. Notwithstanding the risk of tumors acquiring radioresistance, secondary radiotherapy is increasingly used to treat recurrent tumors. In this context, it appears essential to comprehend the radiobiological effects of repeated photon and particle irradiation and their underlying cellular and molecular mechanisms in order to achieve the most favorable therapeutic outcome. However, to date, the mechanisms of acquisition of radioresistance in cancer cells have mainly been studied after repeated in vitro X-ray irradiation. By contrast, other critical aspects of radioresistance remain mostly unexplored, including the response to carbon-ion irradiation of X-ray radioresistant cancer cells, the mechanisms of acquisition of carbon-ion resistance, and the consequences of repeated in vivo X-ray or carbon-ion irradiation. In this review, we discuss the underlying mechanisms of acquisition of X-ray and carbon-ion resistance in cancer cells, as well as the phenotypic differences between X-ray and carbon-ion-resistant cancer cells, the biological implications of repeated in vivo X-ray or carbon-ion irradiation, and the main open questions in the field.
Introduction
The previous decade has seen significant developments in the techniques used in radiotherapy, and advanced radiotherapy has become increasingly adopted. Among advanced radiotherapy techniques, stereotactic body radiation therapy (SBRT) relies on a small irradiation field to precisely deliver high doses of radiation, typically above 10 Gy per fraction, to local tumors. SBRT has been applied to the treatment of various cancers, including lung (1), liver (2), and prostate cancer (3). The therapeutic outcomes of SBRT are particularly promising for early stage lung cancers, with local control rates exceeding 80% (1, 4) and clinical outcomes comparable to those of surgical resection (5, 6).
In addition to SBRT, particle-beam therapy, such as carbon-ion (C-ion) radiotherapy (CIRT), has demonstrated excellent therapeutic outcomes in various types of cancer (7). CIRT has both physical and biological advantages compared with X-ray therapy. With CIRT, tumors are irradiated with C-ions of relativistic energy, which means that C-ions penetrate the body with lower ionization that significantly increases toward the end of the beam path. This radiophysical feature is commonly referred to as the “Bragg-peak” and contributes to increasing the radiation dose delivered to the tumor while minimizing the radiation dose delivered to the skin and surrounding healthy tissues. Furthermore, CIRT has a relative biological effectiveness, which is defined as the ratio of a dose of radiation to the dose of X-ray radiation producing the same biological effects that is >2. Another important feature of CIRT is its effectiveness against conventional X-ray radiotherapy resistant cancers, including melanoma (8) and bone and soft-tissue sarcoma (9–11). Furthermore, CIRT reportedly works as an alternative ablative treatment for early stage lung cancer, in particular for elderly and inoperable patients (12).
Nevertheless, recent studies show that local recurrence can still occur after advanced radiotherapy. For example, an incidence of local recurrence ranging from 4.9 to 19% in patients who received SBRT for lung cancer treatment was reportedly dependent on treatment regimen, tumor stage, and follow-up periods (13–18). Furthermore, 23.3% of patients who received CIRT for the treatment of stage I non-small cell lung cancer also exhibited local recurrence (19).
In cases of tumor recurrence after primary radiotherapy, patients can rarely be treated again with the same radiation regimen, because the tumor might acquire radioresistance, and it is possible that healthy surrounding tissues will not tolerate additional irradiation. Nevertheless, recent studies report that SBRT and CIRT can be used for re-irradiation of recurrent tumors, taking into account both the dose tolerance of healthy tissues and location of the recurrent tumor (20–23). However, several issues related to re-irradiation with SBRT or CIRT still need to be considered. First, only a few studies have reported the clinical outcomes of repeated irradiation, and second, the characteristics of recurrent tumor after primary radiotherapy are largely unknown. In this review, we focus on the biological aspects of acquired X-ray and C-ion resistance in cancer cells and discuss the differences between the consequences of in vitro and in vivo repeated irradiation, the possible mechanisms of acquired resistance in cancer cells, and issues that must be addressed in this research field.
Acquisition of Photon Radioresistance in vitro
Radioresistance acquisition in cancer cells and its underlying mechanisms have been mainly studied using radioresistant cell lines established through repeated in vitro photon (e.g., X-ray or γ-ray) irradiation. Because conventional radiotherapy usually relies on a total dose of ~60 Gy applied in 2-Gy fractions, many studies adopted similar radiation regimens in order to establish radioresistant cell lines (Table 1) (24–41). Importantly, most of these studies showed that the survival of repeatedly irradiated cells was significantly higher than that of the parental cells, which indicated that in vitro, various cell lines could acquire radioresistance following multiple rounds of X-ray irradiation.
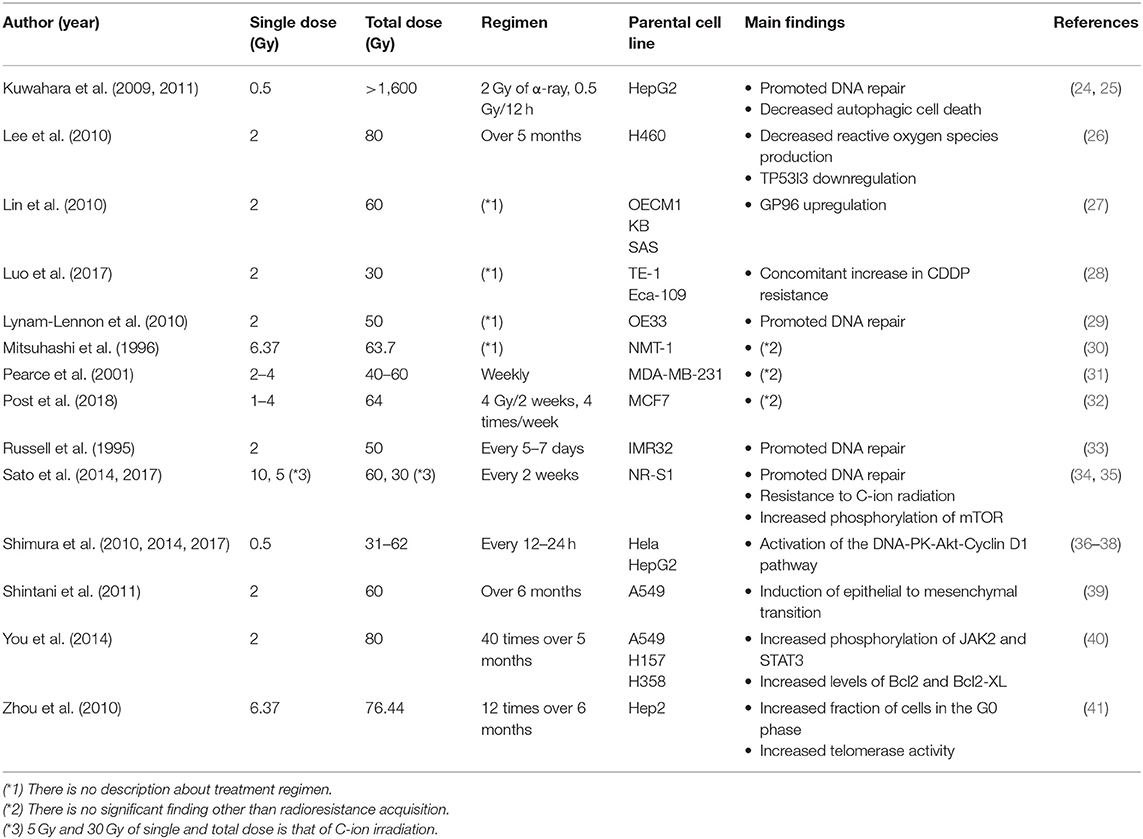
Table 1. Repeated photon irradiation regimen for the establishment of radioresistant cancer cells.
Cellular Processes Involved in the Acquisition of Radioresistance Following Repeated Photon Irradiation
The mechanisms of acquisition of radioresistance in cancer cells have been associated with a variety of biological processes (Table 1). However, in many cases, the acquisition of radioresistance can be reasonably explained by the induction of epithelial-to-mesenchymal transition (EMT), which is defined as phenotypic and molecular alterations that result in the loss of epithelial-cell characteristics and the gain of mesenchymal-cell characteristics. As cancer cells undergo EMT, epithelial markers, such as E-cadherin, ZO-1, and cytokeratin, are downregulated, whereas mesenchymal markers, such as N-cadherin, vimentin, snail, and twist, are upregulated, and in some cases, morphological changes lead to the appearance of spindle-shaped cells (42). The most prominent characteristics acquired by cancer cells after EMT are migratory and invasive properties, conferring them a significant metastatic potential, and resistance to chemotherapeutic drugs and ionizing radiation. Indeed, several studies report that EMT in cancer cells, which was defined by reduced E-cadherin protein levels, increased N-cadherin protein levels, and enhanced migration potential, could be induced by single X-ray irradiation (43, 44). Furthermore, Shintani et al. (39) showed that repeated X-ray irradiation of A549 cells (2 Gy weekly for >6 months) induced significant radioresistance and typical EMT (i.e., decreased E-cadherin and increased N-cadherin mRNA and protein levels). Collectively, these data support the notion that EMT induction following X-ray irradiation is a contributing factor in the acquisition of radioresistance.
Another factor involved in the acquisition of radioresistance following repeated X-ray irradiation is an enrichment in cancer stem cells (CSCs), which are known to exhibit higher DNA-repair potential (45) and resistance to reactive oxygen species-induced cytotoxicity (46, 47). CSCs are also often found to be in the G0 phase (48), a quiescent state outside the normal cell cycle and associated with reduced cell proliferation. All of these characteristics are recognized for their role in cellular radioresistance. Additionally, CSCs are important in radioresistance acquisition because of their impact on the heterogeneity of the cell population within a tumor. Indeed, in the hierarchy model, CSCs produce a more differentiated non-CSC progeny exhibiting significant cell-proliferation potential but lacking stem cell properties. Notwithstanding their reduced proliferation rate as compared with their non-CSC progeny, CSCs can self-renew, and maintaining their stemness. Notably, the non-CSC population displays higher radiosensitivity than the CSC population. Consequently, radiation treatment can increase the relative abundance of CSCs in the tumor, which promotes asymmetric cell proliferation and, therefore, an enrichment in CSCs.
Lagadec et al. (49) reported enrichment in CD44high/CD24low breast CSCs following repeated X-ray irradiation of human breast cancer MCF7 and T47D cells, with irradiated cells displaying increased sphere-formation potential. Furthermore, they found that repeated irradiations led CSCs in the G0 phase to reenter the cell cycle, thereby promoting their proliferation, whereas the non-CSC population underwent apoptosis according to the increased fraction of cells in the sub-G1 phase (49). Additionally, Ghisolfi et al. (50) showed that single X-ray irradiation of cancer cells with a dose of 2 Gy to 10 Gy increased the expression of the pluripotency markers OCT3/4 and SOX2 and promoted the enrichment of a CSC subpopulation. Moreover, Mani et al. (51) established a link between EMT and CSCs by demonstrating that TGF-β-induced EMT generated a subpopulation with CSC properties, including characteristic CSC markers, such as CD44high/CD24low and elevated sphere- and mammosphere-formation potential.
To the best of our knowledge, a definitive mechanism responsible for the induction of CSCs remains unclear; however, DNA damage or chromosomal aberration can enhance CSC induction along with increased oncogene activity. Liang et al. (52) showed that DNA damage from UV irradiation and the chromosomal aberrations induced by Mad2 overexpression also increased by Myc and SOX2 expression in human nasopharyngeal carcinoma CNE cell lines and promoted cell dye-exclusion, colony formation, and sphere-formation capacities. These data suggest that the accumulation of DNA damage by repeated X-ray irradiation induces not only EMT but also enrichment of CSCs with increasing oncogenic activity, whereas secondary induction of a CSC subpopulation by EMT (known as cancer plasticity) further contributes to the development of radioresistance.
Molecular Processes Involved in the Acquisition of Radioresistance Following Repeated Photon Irradiation
We and others have independently reported that repeated X-ray irradiation can result in enhanced DNA-repair capacity (24, 29, 33, 34). In our study, the mouse squamous cell carcinoma NR-S1 cell line was irradiated with a total dose of 60 Gy of X-ray radiation applied in 10-Gy fractions in order to establish the X60 radioresistant cancer cell line (Figure 1). Notably, the D10 value (i.e., the radiation dose required to decrease the survival to 10% of the non-irradiated condition) and cell survival after 10 Gy of X-ray radiation were 1.6- and 3.8-fold higher, respectively, for X60 cells than for parental NR-S1 cells (34). Furthermore, 24 h after exposure to 10 Gy X-ray radiation, the number of S139 phosphorylated-H2AX (γ-H2AX) foci, a marker of DNA double-strand breaks (DSBs), was 2.5-fold lower in X60 cells than in NR-S1 cells, indicating that DSBs were repaired more efficiently in X60 cells than in NR-S1 cells (34). Indeed, the collected results of numerous studies (Table 1) further demonstrate that enhanced DNA-repair capacity is a common feature of radioresistant cancer cells arising from repeated X-ray irradiation.
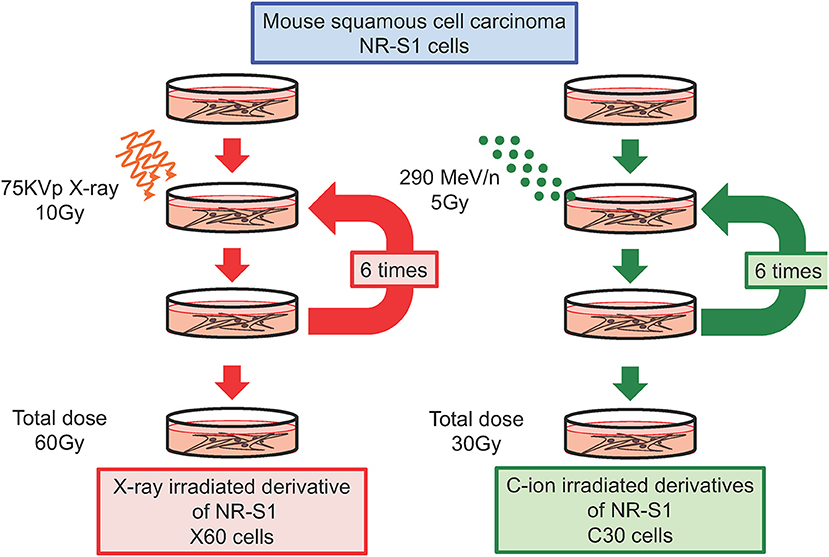
Figure 1. Diagram describing the establishment of radioresistant cancer cells through repeated X-ray or C-ion irradiation. Mouse squamous cell carcinoma NR-S1 cells were irradiated six times at 2-week intervals with 10 Gy of X-ray radiation (left) or 5 Gy of C-ion radiation (left). The radioresistant derivative cell lines exposed to total doses of 60 Gy of X-ray radiation and 30 Gy of C-ion radiation were denoted as X60 and C30 cells, respectively (34, 35).
As part of the investigation of the molecular mechanisms underlying the acquisition of radioresistance, several groups have highlighted a relationship between DNA repair and pro-survival signaling pathways, such as the Akt and mechanistic target of rapamycin (mTOR) pathways. Shimura et al. (36) suggested a potential molecular mechanism for the acquisition of radioresistance induced by repeated X-ray irradiation, showing that cyclin D1 expression and Akt phosphorylation levels were increased in X-ray-resistant derivatives of HeLa and HepG2 cells established following repeated irradiation. These radioresistant cancer cell lines also displayed constitutively elevated levels of DSBs, as measured by H2AX and ataxia telangiectasia mutated (ATM) phosphorylation, relative to those in parental cell lines. Strikingly, downregulation of cyclin D1 in radioresistant HeLa and HepG2 derivatives decreased H2AX-, ATM-, and Akt-phosphorylation levels, as well as cell survival, after further X-ray irradiation. Therefore, they proposed that repeated X-ray irradiation triggered cyclin D1 overexpression and forced cell cycle progression, which in turn caused further DNA damage and led to the activation of both Akt signaling and DNA-dependent protein kinase activity, a central component in the non-homologous end joining DSB-repair pathway. Eventually, these signals promoted further cyclin D1 overexpression as part of a positive-feedback loop that likely resulted in the acquisition of radioresistance (36, 38, 53, 54).
In addition to Akt signaling, mTOR signaling has been associated with the acquisition of X-ray resistance in cancer cells. Chang et al. (55) established radioresistant derivatives of PC-3, DU145, and LNCaP cells following repeated X-ray irradiation with a dose of 2 Gy/day for 5 consecutive days (55) and showed that these radioresistant cancer cell lines exhibited both mesenchymal and CSC phenotypic traits. Interestingly, they found that radioresistant cells treated with BEZ235, a specific inhibitor of the phosphoinositide 3 (PI3) and mTOR kinases, displayed decreased expression of mesenchymal (N-cadherin, vimentin, and snail) and CSC (OCT3/4, SOX2, and CD44) markers, increased expression of the epithelial marker E-cadherin, and reduced cell survival. They further showed that BEZ235, the PI3-kinase inhibitor BKM120, and the mTOR-kinase inhibitor rapamycin, suppressed the expression of DNA-repair proteins induced by X-ray irradiation, including Ku80, BCRA1, and BRCA2 (56). Moreover, we independently demonstrated that mTOR signaling was enhanced in X60 radioresistant cancer cells as compared with parental NR-S1 cells, whereas rapamycin treatment decreased their radioresistant phenotype (35). Importantly, rapamycin also suppresses both non-homologous end joining and homologous recombination (HR) DSB-repair pathways (57). Collectively, these results indicate that activation of the pro-survival Akt and mTOR signaling pathways can eventually increase the DNA-repair capacity of repeatedly irradiated cancer cells and thereby promote the acquisition of radioresistance.
Acquisition of C-ion Radioresistance in vitro
Acquisition of C-Ion Resistance Following Repeated X-Ray Irradiation
In clinical practice, CIRT is an effective treatment for locally recurrent tumors after primary radiotherapy, likely because cell killing by C-ion radiation is independent of various cellular or tumor characteristics, including p53 status (58), cell cycle phase (59, 60), and hypoxia (61–63). Although these findings suggest that C-ion radiation should be effective against radioresistant cancer cells arising from repeated X-ray irradiation, no available experimental data supported this hypothesis. Therefore, in a recent study, we determined whether C-ion radiation could efficiently kill X60 radioresistant cancer cells. Contrary to our expectations, we found that compared with parental NR-S1 cells, X60 cells exhibited significant levels of resistance against C-ion radiation (34). Furthermore, 24 h after C-ion irradiation, the number of γ-H2AX foci was 2.5-fold lower in X60 cells than in NR-S1 cells. These observations indicated that repeated X-ray irradiation of cancer cells with a relatively high dose of 10 Gy per fraction could induce not only X-ray resistance but also C-ion resistance (34). We believe that further investigations using such radioresistant cells will likely lead to the discovery of novel mechanisms contributing to C-ion resistance in cancer cells.
To gain further insight into the underlying mechanisms of radioresistance in X60 cells, in a recent study, we compared several biological and morphological traits of X60 cells and parental NR-S1 cells, including cell shape and size, number of heterochromatin domains in the nucleus, and DNA content (34). Additionally, we analyzed the correlation between these factors and X-ray or C-ion resistance. Interestingly, we found that the number of heterochromatin domains was strongly correlated with both X-ray and C-ion resistance, which suggested that heterochromatin components or the dynamics of heterochromatin were also involved in the acquisition of radioresistance.
Indeed, previous studies show that heterochromatin proteins, such as HP1α (64) and CAF1 (65), are directly involved in DNA repair, and recent studies report that DNA damage in heterochromatin domains is mainly repaired by the HR machinery (66, 67). The damaged heterochromatin first move to the periphery of the heterochromatin domain to prevent abnormal recombination or deleterious expansion at satellite or repetitive DNA sequences, after which Rad51, the core component of the HR machinery, accumulates at DNA-damage sites located at the periphery of the heterochromatin domain (66, 67). Although these studies were conducted in Drosophila and yeast cells, Jakob et al. (68) observed similar dynamics of damaged heterochromatin and DNA-repair following heavy ion-beam irradiation of mammalian cells, finding that immediately after irradiation, DSBs were formed in the heterochromatin along the uranium ion-beam track. Within 30 min, the DSB sites relocated to the periphery of the heterochromatin domain, and replication protein A, a marker of DNA-end resection during HR repair, accumulated at these DSB sites (68). In agreement with these findings, our preliminary results showed that 1 h after X-ray or C-ion irradiation, X60 cells displayed an increased number of Rad51 foci as compared with NR-S1 cells (Figure 2). Because Rad51 is a central factor in the HR machinery, Rad51 foci are commonly considered to represent sites of ongoing DSB repair by HR.
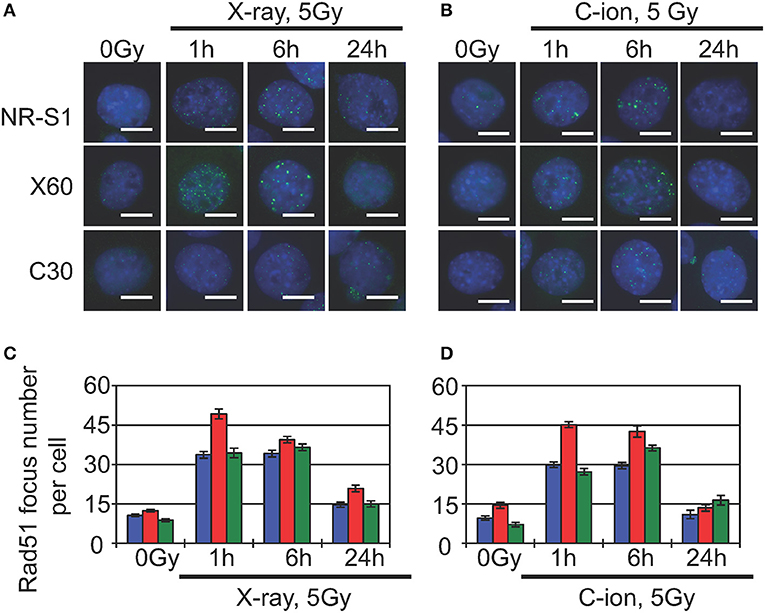
Figure 2. Analysis of Rad51-foci formation in parental NR-S1 cells and radioresistant X60 and C30 cells. (A,B) Representative immunofluorescence images of NR-S1, X60, and C30 cells irradiated with 10 Gy of X-ray radiation (A) or 5 Gy of C-ion radiation (B), as previously described (51, 56). At the indicated time after irradiation, cells were fixed and labeled with anti-Rad51 antibodies (green) using standard procedures (34, 35). Nuclei were counterstained by Hoechest33342 (blue). Scale bars, 10 μm. (C,D) Histograms showing the average number of Rad51 foci per cell following X-ray (C) and C-ion (D) irradiation of NR-S1 (blue), X60 (red), and C30 (green) cells. Data represent the mean ± standard deviation.
Furthermore, several studies report that HR repair can remove complex DNA lesions, including clustered DNA damage (69) and DNA-protein crosslinks (70, 71). Given that X60 cells exhibit increased numbers of heterochromatin domains and radiation-induced Rad51 foci as compared with NR-S1 cells (Figure 2), it appears conceivable that enhanced HR-repair capacity and heterochromatin dynamics contribute to X-ray and C-ion resistance in X60 cells. Whether such a mechanism of acquisition of radioresistance could be specific to X60 cells or shared by other radioresistant cancer cell lines remains to be explored. Nevertheless, future studies focusing on further our understanding of the underlying mechanisms of DSB repair in heterochromatin domains could, therefore, represent a significant breakthrough toward elucidating the mechanisms of acquisition of radioresistance in cancer cells and identifying novel therapeutic strategies for the treatment of radioresistant tumors.
Acquisition of C-Ion Resistance Following Repeated C-Ion Irradiation
To date, only a limited number of studies have investigated the acquisition of C-ion resistance, and it remains unclear whether repeated C-ion irradiation can lead to radioresistance in cancer cells. To address this question, in a recent study, we irradiated NR-S1 cells with a total dose of 30 Gy of C-ion radiation applied in 5-Gy fractions in order to establish a C30 radioresistant cancer cell line (Figure 1). This C-ion irradiation regimen is biologically equivalent to the X-ray irradiation regimen used to establish the X60 cell line, because NR-S1 cells exhibit comparable survival following exposure to 5 Gy and 10 Gy of C-ion and X-ray radiation, respectively. Interestingly, when we assessed the X-ray and C-ion radiation sensitivity of C30 cells using a colony formation assay, we found that C30 cells displayed moderate resistance to C-ion radiation but not to X-ray radiation (35).
Although it remains unclear why repeated X-ray irradiation but not repeated C-ion irradiation conferred significant C-ion-resistance to NR-S1 derivative-cells, we believe that enrichment in CSCs might be a contributing factor. Indeed, it is widely recognized that CSCs are more resistant to X-ray radiation and anticancer drugs than more differentiated cancer cells. Furthermore, as noted, several studies report that repeated X-ray irradiation can increase the CSC fraction within a cancer cell population (49, 50, 72–74). Conversely, Cui et al. (75) showed that C-ion radiation could efficiently kill CSCs both in vitro and in vivo. Therefore, it is conceivable that repeated X-ray irradiation but not repeated C-ion irradiation could contribute to enriching a radioresistant subpopulation with CSC-like characteristics. However, there is one element in contradiction to this hypothesis. Because most CSCs are in a G0 quiescent state (45), the non-CSC subpopulation might proliferate faster; therefore, if the primary factor in the acquisition of X-ray and C-ion resistance is enrichment in CSCs, the level of radioresistance of a growing X60 cell population, for example, could gradually decrease over time, which was not observed (34, 35).
Collectively, these findings indicate that CSC enrichment and other mechanisms, such as genetic alterations and mutations (76), might jointly contribute to the acquisition of both X-ray and C-ion resistance in cancer cells. Although further investigations are required to elucidate these mechanisms, the current body of evidence suggests that C-ion irradiation does not induce radioresistance and can be used for the treatment of locally recurrent tumors arising after primary CIRT.
Effects of in vivo Repeated Photon or C-ion Irradiation
Numerous studies are focused on translating results obtained with in vitro models of radioresistant cancer cells into clinical practice. Therefore, it appears essential to determine whether phenotypic changes resulting from repeated photon or particle irradiation also occur in vivo. However, to the best of our knowledge, the effects of in vivo repeated photon or particle irradiation on the acquisition of radioresistance in cancer cells has not been reported.
To address this question and examine whether the characteristics of repeatedly irradiated tumors differ from those of parental tumors, we recently established in vivo models of regrown irradiated tumors (77). To this end, NR-S1-derived tumors engrafted into C3H/He mice were irradiated with single doses of γ-ray (30 Gy) or C-ion (15 Gy) radiation, which have comparable effects on tumor growth. Two weeks after irradiation, we harvested the irradiated tumors and transplanted them into healthy mice, and 2 weeks later, the regrown tumors were irradiated again, with this irradiation/regrowth/transplant process repeated six times in total. The resulting repeatedly irradiated tumors were exposed to total doses of 180 Gy of γ-ray radiation and 90 Gy of C-ion radiation and denoted as G180 and C90 in vivo regrown tumor models, respectively (Figure 3). We then examined differences in tumor-growth potential, spontaneous metastasis from the primary site to the lung surface, tumor-grafted mouse survival, and radiosensitivity between non-irradiated NR-S1-derived tumors and G180 and C90 tumors.
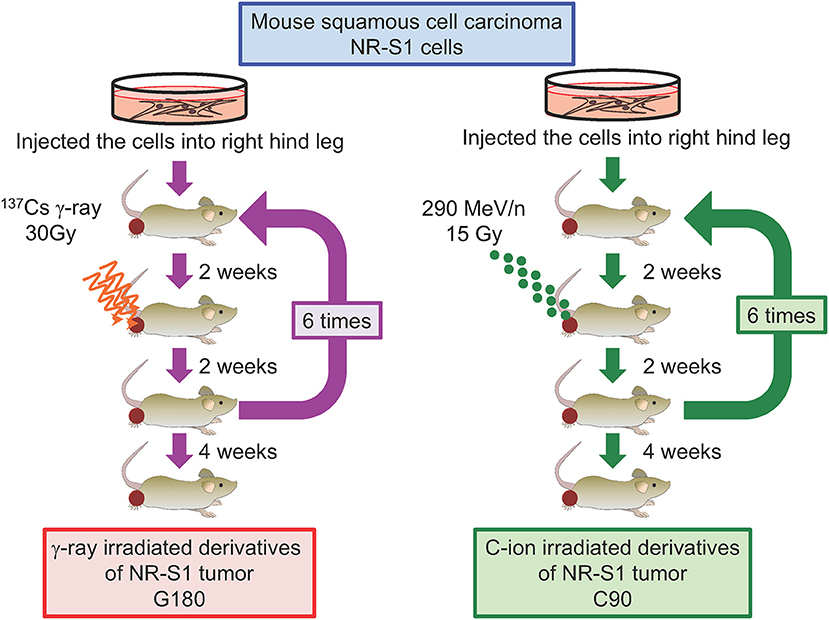
Figure 3. Diagram describing the establishment of regrown tumor models through repeated γ-ray or C-ion irradiation. Mouse squamous cell carcinoma NR-S1 cells were injected into the right hind leg of healthy C3H/He mice. Upon reaching ~10 mm in diameter, tumors derived from NR-S1 cells were irradiated with 30 Gy of γ-ray radiation or 15 Gy of C-ion radiation and allowed to regrow for 2 weeks before transplant into the right hind leg of healthy mice. The irradiation/regrow/transplant procedure was performed six times in total, resulting in regrown tumors exposed to total doses of 180 Gy of γ-ray radiation and 90 Gy of C-ion radiation, respectively. The tumors were finally harvested for analysis 4 weeks after the final irradiation (77).
Notably, G180 tumors displayed drastically increased tumor-growth rates and metastatic potential compared with those of non-irradiated tumors, and mice grafted with G180 tumors displayed significantly shorter survival than those grafted with non-irradiated tumors. By contrast, the characteristics of the C90 tumors remained comparable to those of non-irradiated tumors. Importantly, X-ray and C-ion irradiation of G180 and C90 tumors did not affect the relative tumor-growth rates, spontaneous lung metastasis, and survival of tumor-grafted mice as compared with non-irradiated tumors. Furthermore, colony formation assays performed using cells isolated from non-irradiated, G180, and C90 tumors showed that they all added similar sensitivity to X-ray and C-ion radiation (77). Moreover, compared with non-irradiated and C90 tumors, G180 tumors harbored numerous microvessels and expressed genes associated with angiogenesis and metastasis, including VEGFA, HIF1A, FN1, MMP2, MMP9, PAI1, and PLAU. Together, these data indicated that, contrary to repeated in vitro irradiation, repeated in vivo γ-ray and C-ion irradiation did not lead to the acquisition of radioresistance in regrown tumors. However, repeated photon irradiation but not particle irradiation appeared to enhance tumor growth and metastasis, resulting in an increased aggressiveness of regrown tumors.
These findings could suggest that repeated irradiation affects the tumor microenvironment rather than the tumor itself or its CSC subpopulation. Although our investigations could not determine whether repeated photon irradiation enriched the CSC subpopulation in regrown tumors, our data showed that G180 tumor cells in suspension culture had a significantly higher sphere-formation potential than their non-irradiated and C90 counterparts (77). Because CSCs are characterized by significant resistance to cytotoxic agents, including radiation and anticancer drugs, we believe that G180 tumors are likely enriched in tumor-initiating cells (TICs) that differ from a typical CSC subpopulation. To date, such phenomena have not been reported, and further studies will be required to determine which cells among cancer and stromal cells are mostly affected by repeated in vivo irradiation and what mechanisms lead to increased aggressiveness in repeatedly irradiated tumors.
Crucially, these findings also demonstrated that repeated C-ion irradiation was far less prone to induce acquisition of radioresistance and enhance tumor aggressiveness, as assessed by tumor growth, metastatic potential, and prognosis of tumor-grafted mice. Although it remains necessary to ascertain why C-ion radiation effectively suppressed tumor aggressiveness and TIC or CSC subpopulations, we believe that the accumulated evidence supports CIRT as a promising treatment for local recurrent tumors.
Relationship Between the 4Rs of Radiotherapy and Radioresistance Acquisition
Tumor shrinkage by fractionated radiotherapy has been explained by the “4Rs” of radiotherapy, where each “R” represents “Repair,” “Repopulation,” “Redistribution,” and “Reoxygenation” (78). “Repair” denotes the difference in cell survival of tumor cells and normal tissue between single or fractionated irradiation at the same radiation dose (79) and is basically measured by colony formation assay, followed by calculation of α and β values using a linear-quadratic model to quantify the radiosensitivity of each cell (80). “Repopulation” describes regeneration of normal tissue, such as skin and mucosal tissue (81). This concept relies on experimental results showing that the recovery of skin and mucosal tissue occurs faster than regrowth of gross tumor mass, and that each fractionation regime of radiotherapy can be determined based on these differences. Because α and β values and difference in recovery between tumor and normal tissue are used for treatment planning of radiotherapy, they are recognized as important therapeutic components. “Redistribution” indicates synchronization of the cell cycle in cells exposed to radiation. The tumor harbors multiple cell types exhibiting various cell cycle phases (82). Upon treatment of the tumor with radiotherapy, the relatively radiosensitive cell fractions, such as those in the G2/M phase, will die first, whereas the relatively radioresistant cell fractions, such as those in the G1 and S phases, will survive. However, cells surviving the first round of irradiation will enter a radiosensitive phase of the cell cycle during subsequent rounds and eventually will subsequently be efficiently killed. “Reoxygenation” describes changes in well-oxygenated areas of an irradiated tumor (83). Partial oxygen pressure in the peripheral tumor is higher than that in the center, because nutrition and oxygen at the periphery is well-supplied by tumor blood vessels (78). The partial oxygen pressure enhances the cell-killing effect, because radiation-induced reactive oxygen species, such as OH radical, initiated DNA breakage (84). Therefore, well-oxygenated areas of a tumor (i.e., the periphery) are killed first, followed by vascularization of the central tumor along with tumor shrinkage. Repetition of this process enhances the efficacy of radiotherapy. The 4Rs reasonably describe the process of tumor shrinkage during radiotherapy and are useful for recognizing tumor and environmental conditions the determine radioresistance or radiosensitivity.
On the other hand, our previous results suggest the possibility that use of the 4Rs might not be appropriate for planning secondary radiotherapy. At the very least, using the same definitions as those used to determine primary radiotherapy might not be suitable for secondary radiotherapy. If the tumor targeted for secondary radiotherapy has acquired radioresistance via EMT, the total dose required to control the tumor should be increased. This suggests that the α and β values of the tumor cells and the dose fractionation used to prevent normal-tissue complications should be changed. Therefore, this suggests that “Repair” and “Repopulation” should be properly adjusted in the planning of secondary radiotherapy. In cases where primary radiotherapy fails to control tumor growth, followed by tumor regrowth within the irradiation field, this suggests that radioresistant cancer cells, such as CSCs, likely exist in the target area. If these tumors are treated with another round of irradiation, “Reoxygenation” might not be suitable for interpreting tumor radiosensitivity, because the CSCs might be in a quiescent state and capable of surviving within the hypoxic area. In addition to the induction of the radioresistant cancer cells, our data showed that repeated photon irradiation in vivo promoted acquisition of a more aggressive phenotype in the tumors. These characteristic changes do not fit the classical 4Rs of radiotherapy. Although our results were obtained by experiments using mouse cancer cell lines rather that human specimens, and the results in vitro did not match those obtained in vivo, they indicated that other hallmarks are required to interpret possible radioresistant or aggressive fractions in target tumors for planning secondary radiotherapy targeting regrown tumors. Given that hypoxic areas are primary niches of CSCs (85), and the tumor vasculature clearly changes after irradiation (86), imaging techniques used to identify hypoxic areas and well-vascularized areas in target tumors will likely be useful for planning secondary radiotherapy. Indeed, drugs targeting hypoxic areas have been developed (87, 88), and tumor blood vessels can be visualized by contrast-enhanced magnetic resonance imaging (89).
What are the Main Open Questions in the Field?
Although numerous studies show that repeated X-ray irradiation can lead to increased radioresistance in various cancer cells, the primary source of radioresistant cancer cells remains elusive. Therefore, it is imperative to determine whether the selection of inherently radioresistant cells or emergence of radioresistant cells due to de novo genetic alterations is the primary cause of the acquisition of radioresistance in order to prevent the appearance of radioresistance and improve patient care. With the recent development of genetic barcoding techniques (90–93), we can now label a large number of cells within a given population. Combined with high-throughput DNA sequencing, genetic barcoding could be used to track and identify the type(s) of cells that can survive and proliferate following repeated irradiation.
Although extensive efforts have been made to investigate the mechanisms of acquisition of radioresistance in cancer cells using in vitro models, the radiobiological effects of repeated in vivo irradiation remain poorly understood. Tumors are complex ecosystems comprising various cell types, including cancer, stromal, and immune cells. Furthermore, the tumor environment is partly heterogeneous, with hypoxic or nutrient-deprived areas. In this regard, our in vivo data suggest that changes in the tumor microenvironment, including angiogenesis, might be critical for the prognosis of mice bearing regrown tumors after repeated irradiation.
In addition to the limited amount of data available concerning the effects of repeated in vivo irradiation, the differences between photon and particle irradiation remain largely unknown. Indeed, photon and particle radiation have distinct physical properties, and their resulting biological effects might be very different. For example, particle radiation produces a high density of reactive oxygen species and clustered DNA damage along the particle-beam track (94). Nevertheless, there is, to date, no sensible theory linking the physical characteristics of radiation to their biological effects, including high relative biological effectiveness in cell-survival assays and suppression of metastasis both in vitro and in vivo.
Answers to these questions would definitely promote the understand of why repeated C-ion irradiation does not appear to induce significant radioresistance in cancer cells, whereas repeated X-ray irradiation leads to significant resistance to both X-ray and C-ion radiation in NR-S1 cells. The identification of potential targets to enhance or elicit radiosensitization could facilitate the development of novel therapeutic strategies for the treatment of radioresistant tumors and recurrent tumors after primary radiotherapy.
Concluding Remarks
To date, few reports have been published describing the acquisition of radioresistance in repeatedly irradiated tumor cells, particularly after particle irradiation. A series of experiments using models of tumors repeatedly irradiated with either photon or particle radiation show that repeated in vitro irradiation with relatively high doses of X-ray radiation can induce significant resistance to both X-ray and C-ion radiation. By contrast, repeated in vitro irradiation with relative biological effectiveness doses of C-ion radiation does not contribute to the acquisition of X-ray or C-ion resistance in tumor cells. Somewhat surprisingly, repeated X-ray or C-ion irradiation of in vivo regrown tumor models does not increase their radioresistance; however, repeated photon irradiation but not C-ion irradiation increased tumor aggressiveness. Because the evidence was limited to a single tumor cell type, further studies are required to conclusively determine the effects of repeated irradiation on the acquisition of radioresistance in tumors.
Author Contributions
KS contributed to the design and wrote the manuscript. TS and TI rewrote and made edits. TI conducted the final review.
Funding
This work was supported by JPSP KAKENHI Grant Nos. 15K15467 and 24591857, and by the Center of Innovation Program (COI stream from JST) and P-CREATE grant from AMED, and was performed as a research project with heavy ions at NIRS-HIMAC (No. 15J183).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank their colleagues in ex-Research Center for Charged Particle Therapy at the National Institute of Radiological Sciences. This research was supported in part by the Research Project Heavy Ions at NIRS-HIMAC. We also thank Editage (www.editage.com) for English language editing.
References
1. Ohnishi H, Shirato H, Nagata Y, Hiraolka M, Fujino M, Gomi K, et al. Hypofractinated stereoptactic radiotherapy (HypoFXSRT) for stage I non-small cell lung cancer: updated results of 257 patients in a Japnese multi-institutional study. J Thorac Oncol. (2007) 2:S94–100. doi: 10.1097/JTO.0b013e318074de34
2. Sanuki N, Takeda A, Oku Y, Mizuno T, Aoki Y, Eriguchi T, et al. Stereotactic body radiotherapy for small hepatocellular carcinoma: a retrospective outcome analysis in 185 patients. Acta Oncol. (2014) 53:399–404. doi: 10.3109/0284186X.2013.820342
3. Freeman DE, King CR. Stereotactic body radiotherapy for low-risk prostate cancer: five-year outcomes. Radio Oncol. (2011) 6:3. doi: 10.1186/1748-717X-6-3
4. Shioyama Y, Onishi H, Takayama K, Matsuo Y, Takeda A, Yamashita H, et al. Clinical outcomes of stereotactic body radiotherapy for patients with stage I small-cell lung cancer: analysis of a subset of the Japanese radiological society multi-institutional SBRT study group database. Technol Cancer Res Treat. (2018). 17:1533033818783904. doi: 10.1177/1533033818783904
5. Kastelijn EA, El Sharouni SY, Hofman FN, Van Putte BP, Monninkhof EM, Van Vulpen M, et al. Clinical outcomes in early-stage NSCLC treated with stereotactic body radiotherapy versus surgical resection. Anticancer Res. (2015) 35:5607–14.
6. Ackerson BG, Tong BC, Hong JC, Gu L, Chino J, Trotter JW, et al. Stereotactic body radiation therapy versus sublobar resection for stage I NSCLC. Lung Cancer. (2018) 125:185–91. doi: 10.1016/j.lungcan.2018.09.020
7. Kamada T, Tsujii H, Blakely EA, Debus J, De Neve W, Durante M, et al. Carbon ion radiotherapy in Japan: an assessment of 20 years of clinical experience. Lancet Oncol. (2015). 16:e93–e100. doi: 10.1016/S1470-2045(14)70412-7
8. Tsuji H, Ishikawa H, Yanagi T, Hirasawa N, Kamada T, Mizoe JE. Carbon-ion radiotherapy for locally advanced or unfavorably located choroidal melanoma: a Phase I/II dose-escalation study. Int J Radiat Oncol Biol Phys. (2007) 67:857–62. doi: 10.1016/j.ijrobp.2006.09.022
9. Imai R, Kamada T, Tsuji H, Yanagi T, Baba M, Miyamoto T, et al. Carbon ion radiotherapy for unresectable sacral chordomas. Clin Cancer Res. (2004) 10:5741–6. doi: 10.1158/1078-0432.CCR-04-0301
10. Jingu K, Tsujii H, Mizoe JE, Hasegawa A, Bessho H, Takagi R, et al. Carbon ion radiation therapy improves the prognosis of unresectable adult bone and soft-tissue sarcoma of the head and neck. Int J Radiat Oncol Biol Phys. (2012) 82:2125–31. doi: 10.1016/j.ijrobp.2010.08.043
11. Serizawa I, Kagei K, Kamada T, Imai R, Sugahara S, Okada T, et al. Carbon ion radiotherapy for unresectable retroperitoneal sarcomas. Int J Radiat Oncol Biol Phys. (2009). 75:1105–10. doi: 10.1016/j.ijrobp.2008.12.019
12. Takahashi W, Nakajima M, Yamamoto N, Tsujii H, Kamada T, Tsujii H. Carbon ion radiotherapy in a hypofractionation regimen for stage I non-small-cell lung cancer. J Radiat Res. (2014) 55:i26–7. doi: 10.1093/jrr/rrt216
13. Spratt DE, Wu AJ, Adeseye V, Din SU, Shaikh F, Woo KM, et al. Recurrence patterns and second primary lung cancers after stereotactic body radiation therapy for early-stage non-small-cell lung cancer: implications for surveillance. Clin Lung Cancer. (2016) 17:177–83.e2. doi: 10.1016/j.cllc.2015.09.006
14. Giuliani ME, Hope A, Mangona V, Guckenberger M, Mantel F, Peulen H, et al. Predictors and patterns of regional recurrence following lung SBRT: a report from the elekta lung research group. Clin Lung Cancer. (2017) 18:162–8. doi: 10.1016/j.cllc.2016.10.006
15. Hobbs CJ, Ko SJ, Paryani NN, Accurso JM, Olivier KR, Garces Y, et al. Stereotactic body radiotherapy for medically inoperable stage I-II non-small cell lung cancer: the Mayo clinic experience. Mayo Clin Proc Innov Qual Outcomes. (2017) 2:40–8. doi: 10.1016/j.mayocpiqo.2017.11.001
16. Samson P, Rehman S, Juloori A, DeWees T, Roach M, Bradley J, et al. Local control for clinical stage I non-small cell lung cancer treated with 5-fraction stereotactic body radiation therapy is not associated with treatment schedule. Pract Radiat Oncol. (2018) 8:404–13. doi: 10.1016/j.prro.2018.04.004
17. von Reibnitz D, Shaikh F, Wu AJ, Treharne GC, Dick-Godfrey R, Foster A, et al. Stereotactic body radiation therapy (SBRT) improves local control and overall survival compared to conventionally fractionated radiation for stage I non-small cell lung cancer (NSCLC). Acta Oncol. (2018) 57:1567–73. doi: 10.1080/0284186X.2018.1481292
18. Shintani T, Matsuo Y, Iizuka Y, Mitsuyoshi T, Mizowaki T. A Retrospective long-term follow-up study of stereotactic body radiation therapy for non-small cell lung cancer from a single institution: incidence of late local recurrence. Int J Radiat Oncol Biol Phys. (2018) 100:1228–36. doi: 10.1016/j.ijrobp.2018.01.050
19. Koto M, Miyamoto T, Yamamoto N, Nishimura H, Yamada S, Tsujii H. Local control and recurrence of stage I non-small cell lung cancer after carbon ion radiotherapy. Radiother Oncol. (2004) 71:147–56. doi: 10.1016/j.radonc.2004.02.007
20. Høyer M. Re-irradiation with stereotactic body radiation therapy (SBRT). Chin Clin Oncol. (2017) 6 (Suppl. 2):S15. doi: 10.21037/cco.2017.07.01
21. Ogawa Y, Shibamoto Y, Hashizume C, Kondo T, Iwata H, Tomita N, et al. Repeat stereotactic body radiotherapy (SBRT) for local recurrence of non-small cell lung cancer and lung metastasis after first SBRT. Radiat Oncol. (2018) 13:136. doi: 10.1186/s13014-018-1080-4
22. Shirai K, Ohno T, Saitoh JI, Okamoto M, Katoh H, Murata K, et al. Prospective study of isolated recurrent tumor re-irradiation with carbon-ion beams. Front Oncol. (2019) 9:181. doi: 10.3389/fonc.2019.00181
23. Milano MT, Kong FS, Movsas B. Stereotactic body radiotherapy as salvage treatment for recurrence of non-small cell lung cancer after prior surgery or radiotherapy. Transl Lung Cancer Res. (2019) 8:78–87. doi: 10.21037/tlcr.2018.08.15
24. Kuwahara Y, Li L, Baba T, Nakagawa H, Shimura T, Yamamoto Y, et al. Clinically relevant radioresistant cells efficiently repair DNA double-strand breaks induced by X-rays. Cancer Sci. (2009) 100:747–52. doi: 10.1111/j.1349-7006.2009.01082.x
25. Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar MH, Fukumoto M, Shimura T, et al. Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. (2011) 2:e177. doi: 10.1038/cddis.2011.56
26. Lee YS, Oh JH, Yoon S, Kwon MS, Song CW, Kim KH, et al. Differential gene expression profiles of radioresistant non-small-cell lung cancer cell lines established by fractionated irradiation: tumor protein p53-inducible protein 3 confers sensitivity to ionizing radiation. Int J Radiat Oncol Biol Phys. (2010) 77:858–66. doi: 10.1016/j.ijrobp.2009.12.076
27. Lin TY, Chang JT, Wang HM, Chan SH, Chiu CC, Lin CY, et al. Proteomics of the radioresistant phenotype in head-and-neck cancer: Gp96 as a novel prediction marker and sensitizing target for radiotherapy. Int J Radiat Oncol Biol Phys. (2010) 78:246–56. doi: 10.1016/j.ijrobp.2010.03.002
28. Luo J, Wang W, Tang Y, Zhou D, Gao Y, Zhang Q, et al. mRNA and methylation profiling of radioresistant esophageal cancer cells: the involvement of Sall2 in acquired aggressive phenotypes. J Cancer. (2017) 8:646–56. doi: 10.7150/jca.15652
29. Lynam-Lennon N, Reynolds JV, Pidgeon GP, Lysaght J, Marignol L, Maher SG. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal adenocarcinoma. Radiat Res. (2010) 174:703–11. doi: 10.1667/RR2295.1
30. Mitsuhashi N, Takahashi T, Sakurai H, Nozaki M, Akimoto T, Hasegawa M, et al. A radioresistant variant cell line, NMT-1R, isolated from a radiosensitive rat yolk sac tumour cell line, NMT-1: differences of early radiation-induced morphological changes, especially apoptosis. Int J Radiat Biol. (1996) 69:329–36. doi: 10.1080/095530096145887
31. Pearce AG, Segura TM, Rintala AC, Rintala-Maki ND, Lee H. The generation and characterization of a radiation-resistant model system to study radioresistance in human breast cancer cells. Radiat Res. (2001) 156:739–50. doi: 10.1667/0033-7587(2001)156[0739:TGACOA]2.0.CO;2
32. Post AEM, Smid M, Nagelkerke A, Martens JWM, Bussink J, Sweep FCGJ, et al. Interferon-stimulated genes are involved in cross-resistance to radiotherapy in tamoxifen-resistant breast cancer. Clin Cancer Res. (2018) 24:3397–408. doi: 10.1158/1078-0432.CCR-17-2551
33. Russell J, Wheldon TE, Stanton P. A radioresistant variant derived from a human neuroblastoma cell line is less prone to radiation-induced apoptosis. Cancer Res. (1995) 55:4915–4921.
34. Sato K, Imai T, Okayasu R, Shimokawa T. Heterochromatin domain number correlates with X-ray and carbon-ion radiation resistance in cancer cells. Radiat Res. (2014) 182:408–19. doi: 10.1667/RR13492.1
35. Sato K, Azuma R, Imai T, Shimokawa T. Enhancement of mTOR signaling contributes to acquired X-ray and C-ion resistance in mouse squamous carcinoma cell line. Cancer Sci. (2017) 108:2004–10. doi: 10.1111/cas.13323
36. Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y, et al. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene. (2010) 29:4826–37. doi: 10.1038/onc.2010.238
37. Shimura T, Noma N, Sano Y, Ochiai Y, Oikawa T, Fukumoto M, et al. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother Oncol. (2014) 112:302–7. doi: 10.1016/j.radonc.2014.07.015
38. Shimura T. Targeting the AKT/cyclin D1 pathway to overcome intrinsic and acquired radioresistance of tumors for effective radiotherapy. Int J Radiat Biol. (2017) 93:381–5. doi: 10.1080/09553002.2016.1257832
39. Shintani Y, Okimura A, Sato K, Nakagiri T, Kadota Y, Inoue M, et al. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann Thorac Surg. (2011) 92:1794–804. doi: 10.1016/j.athoracsur.2011.07.032
40. You S, Li R, Park D, Xie M, Sica GL, Cao Y, et al. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol Cancer Ther. (2014) 13:606–16. doi: 10.1158/1535-7163.MCT-13-0608
41. Zhou FX, Xiong J, Luo ZG, Dai J, Yu H J, Liao ZK, et al. cDNA expression analysis of a human radiosensitive-radioresistant cell line model identifies telomere function as a hallmark of radioresistance. Radiat Res. (2010) 174:550–7. doi: 10.1667/RR1657.1
42. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. (2012) 2:442–54. doi: 10.1038/nrc822
43. Jung JW, Hwang SY, Hwang JS, Oh ES, Park S, Han IO. Ionising radiation induces changes associated with epithelial-mesenchymal transdifferentiation and increased cell motility of A549 lung epithelial cells. Eur J Cancer. (2007) 43:1214–24. doi: 10.1016/j.ejca.2007.01.034
44. Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, et al. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys. (2011) 81:1530–7. doi: 10.1016/j.ijrobp.2011.06.1956
45. Maugeri-Saccà M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. (2012) 11:1627–36. doi: 10.1158/1535-7163.MCT-11-1040
46. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. (2009) 458:780–3. doi: 10.1038/nature07733
47. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. (2011) 19:387–400. doi: 10.1016/j.ccr.2011.01.038
48. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. (2013) 14:329–40. doi: 10.1038/nrm3591
49. Lagadec C, Vlashi E, Della Donna L, Meng Y, Dekmezian C, Kim K, et al. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. (2010) 12:R13. doi: 10.1186/bcr2479
50. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS ONE. (2012) 7:e43628. doi: 10.1371/journal.pone.0043628
51. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
52. Liang Y, Zhong Z, Huang Y, Deng W, Cao J, Tsao G, et al. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem. (2010) 285:4931–40. doi: 10.1074/jbc.M109.048397
53. Shimura T. Acquired radioresistance of cancer and the AKT/GSK3b/cyclin D1 overexpression cycle. J Radiat Res. (2011) 52:539–44. doi: 10.1269/jrr.11098
54. Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle. (2013) 12:773–82. doi: 10.4161/cc.23719
55. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. (2013) 4:e875. doi: 10.1038/cddis.2013.407
56. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. (2014) 5: e1437. doi: 10.1038/cddis.2014.415
57. Chen H, Ma Z, Vanderwaal RP, Feng Z, Gonzalez-Suarez I, Wang S, et al. The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiat Res. (2011) 175:214–24. doi: 10.1667/RR2323.1
58. Takahashi A, Matsumoto H, Yuki K, Yasumoto J, Kajiwara A, Aoki M, et al. High-LET radiation enhanced apoptosis but not necrosis regardless of p53 status. Int J Radiat Oncol Biol Phys. (2004) 60:591–7. doi: 10.1016/j.ijrobp.2004.05.062
59. Schlaff CD, Krauze A, Belard A, O'Connell JJ, Camphausen KA. Bringing the heavy: carbon ion therapy in the radiobiological and clinical context. Radiat Oncol. (2014) 9:88. doi: 10.1186/1748-717X-9-88
60. Okayasu R. Repair of DNA damage induced by accelerated heavy ions-A mini review. Int J Cancer. (2012) 130:991–1000. doi: 10.1002/ijc.26445
61. Hirayama R, Uzawa A, Takase N, Matsumoto Y, Noguchi M, Koda K, et al. Evaluation of SCCVII tumor cell survival in clamped and non-clamped solid tumors exposed to carbon-ion beams in comparison to X-rays. Mutat Res. (2013) 756:146–51. doi: 10.1016/j.mrgentox.2013.05.008
62. Tinganelli W, Ma NY, Von Neubeck C, Maier A, Schicker C, Kraft-Weyrather W, et al. Influence of acute hypoxia and radiation quality on cell survival. J Radiat Res. (2013) 54 (Suppl. 1):i23–30. doi: 10.1093/jrr/rrt065
63. Tinganelli W, Durante M, Hirayama R, Krämer M, Maier A, Kraft-Weyrather W, et al. Kill-painting of hypoxic tumours in charged particle therapy. Sci Rep. (2015) 5:17016. doi: 10.1038/srep17016
64. Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. (2009) 185:577–86. doi: 10.1083/jcb.200810035
65. Baldeyron C, Soria G, Roche D, Cook AJ, Almouzni G. HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J Cell Biol. (2011) 193:81–95. doi: 10.1083/jcb.201101030
66. Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell. (2011) 144:732–44. doi: 10.1016/j.cell.2011.02.012
67. Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, et al. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol. (2015) 17:1401–11. doi: 10.1038/ncb3258
68. Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, et al. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. (2011) 39:6489–99. doi: 10.1093/nar/gkr230
69. Hada M, Georgakilas AG. Formation of clustered DNA damage after high-LET irradiation: a review. J Radiat Res. (2008) 49:203–10. doi: 10.1269/jrr.07123
70. Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, Salem AM. Repair and biochemical effects of DNA-protein crosslinks. Mutat Res. (2011) 711:113–22. doi: 10.1016/j.mrfmmm.2010.12.007
71. Culard F, Bouffard S, Charlier M. High-LET irradiation of a DNA- binding protein: protein-protein and DNA-protein crosslinks. Radiat Res. (2005) 164:774–80. doi: 10.1667/RR3456.1
72. López J, Poitevin A, Mendoza-Martínez V, Pérez-Plasencia C, García-Carrancá A. Cancer-initiating cells derived from established cervical cell lines exhibit stem-cell markers and increased radioresistance. BMC Cancer. (2012) 12:48. doi: 10.1186/1471-2407-12-48
73. Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. (2014) 110:538–45. doi: 10.1016/j.radonc.2013.10.040
74. Sun L, Moritake T, Zheng YW, Suzuki K, Gerelchuluun A, Hong Z, et al. In vitro stemness characterization of radio-resistant clones isolated from a medulloblastoma cell line ONS-76. J Radiat Res. (2013) 54:61–9. doi: 10.1093/jrr/rrs078
75. Cui X, Oonishi K, Tsujii H, Yasuda T, Matsumoto Y, Furusawa Y, et al. Effects of carbon ion beam on putative colon cancer stem cells and its comparison with X-rays. Cancer Res. (2011) 71:3676–87. doi: 10.1158/0008-5472.CAN-10-2926
76. Durante M, Bedford JS, Chen DJ, Conrad S, Cornforth MN, Natarajan AT, et al. From DNA damage to chromosome aberrations: joining the break. Mutat Res. (2013) 756:5–13. doi: 10.1016/j.mrgentox.2013.05.014
77. Sato K, Nitta N, Aoki I, Imai T, Shimokawa T. Repeated photon and C-ion irradiations in vivo have different impact on alteration of tumor characteristics. Sci Rep. (2018) 8:1458. doi: 10.1038/s41598-018-19422-x
78. Withers HR. The four R's of radiotherapy. In: Lett JT, Adler H, editors. Advances in Radiation Biology, Vol 5. Cambridge, MA: Academic Press, Inc. (1975). p. 241–71.
79. Elkind MM. Fractional dose radiotherapy and its relationship to survival curve shape. Cancer Treat Rev. (1976) 3:1–15. doi: 10.1016/S0305-7372(76)80014-X
80. Brenner DJ. The linear-quadratic model is an appropriate methodology for determining isoeffective doses at large doses per fraction. Semin Radiat Oncol. (2008) 18:234–9. doi: 10.1016/j.semradonc.2008.04.004
81. EJ. Hall, AJ. Giaccia editors. Time, dose, and fractionation in radiotherapy. In: Radiobiology for Radiologists. Philadelphia, PA: Lippincottt Williams & Wilkins (2011). p. 391–418.
82. Withers HR. Cell cycle redistribution as a factor in multifraction irradiation. Radiology. (1975) 114:199–202. doi: 10.1148/114.1.199
83. van Putten LM, Kallman RF. Oxygenation status of a transplantable tumor during fractionated radiation therapy. J Natl Cancer Inst. (1968) 40:441–5. doi: 10.1097/00007890-196808000-00017
84. Hirayama R, Ito A, Noguchi M, Matsumoto Y, Uzawa A, Kobashi G, et al. OH radicals from the indirect actions of X-rays induce cell lethality and mediate the majority of the oxygen enhancement effect. Radiat Res. (2013) 180:514–23. doi: 10.1667/RR13368.1
85. Peitzsch C, Perrin R, Hill RP, Dubrovska A, Kurth I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int J Radiat Biol. (2014) 90:636–52. doi: 10.3109/09553002.2014.916841
86. Crokart N, Jordan BF, Baudelet C, Ansiaux R, Sonveaux P, Grégoire V, et al. Early reoxygenation in tumors after irradiation: determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys. (2005) 63:901–10. doi: 10.1016/j.ijrobp.2005.02.038
87. Stieb S, Eleftheriou A, Warnock G, Guckenberger M, Riesterer O. Longitudinal PET imaging of tumor hypoxia during the course of radiotherapy. Eur J Nucl Med Mol Imaging. (2018) 45:2201–17. doi: 10.1007/s00259-018-4116-y
88. Hirata K, Yamaguchi S, Shiga T, Kuge Y, Tamaki N. The Roles of hypoxia imaging using 18F-fluoromisonidazole positron emission tomography in glioma treatment. J Clin Med. (2019) 24:E1088. doi: 10.3390/jcm8081088
89. Nitta N, Takakusagi Y, Kokuryo D, Shibata S, Tomita A, Higashi T, et al. Intratumoral evaluation of 3D microvasculature and nanoparticle distribution using a gadolinium-dendron modified nano-liposomal contrast agent with magnetic resonance micro-imaging. Nanomedicine. (2018) 14:1315–24. doi: 10.1016/j.nano.2018.03.006
90. Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. (2015). 21:440–8. doi: 10.1038/nm.3841
91. Kalhor R, Mali P, Church GM. Rapidly evolving homing CRISPR barcodes. Nat Methods. (2017) 14:195–200. doi: 10.1038/nmeth.4108
92. Guernet A, Mungamuri SK, Cartier D, Sachidanandam R, Jayaprakash A, Adriouch S, et al. CRISPR-barcoding for intratumor genetic heterogeneity modeling and functional analysis of oncogenic driver mutations. Mol Cell. (2016) 63:526–38. doi: 10.1016/j.molcel.2016.06.017
93. Nguyen LV, Pellacani D, Lefort S, Kannan N, Osako T, Makarem M, et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature. (2015) 528: 267–71. doi: 10.1038/nature15742
Keywords: cancer, radioresistance, acquisition, X-ray radiation, carbon-ion radiation, repeated irradiation, DNA repair, aggressiveness
Citation: Sato K, Shimokawa T and Imai T (2019) Difference in Acquired Radioresistance Induction Between Repeated Photon and Particle Irradiation. Front. Oncol. 9:1213. doi: 10.3389/fonc.2019.01213
Received: 14 August 2019; Accepted: 23 October 2019;
Published: 12 November 2019.
Edited by:
Ira I. Skvortsova, Innsbruck Medical University, AustriaReviewed by:
Zhongxing Liao, University of Texas MD Anderson Cancer Center, United StatesMichael W. Epperly, University of Pittsburgh, United States
Copyright © 2019 Sato, Shimokawa and Imai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takashi Imai, aW1haS50YWthc2hpQHFzdC5nby5qcA==