 Xiangjun Tang1,2,3†
Xiangjun Tang1,2,3† Shenqi Zhang1†Rui Fu2Li Zhang2Kuanming Huang2Hao Peng2
Shenqi Zhang1†Rui Fu2Li Zhang2Kuanming Huang2Hao Peng2 Longjun Dai2,4*
Longjun Dai2,4* Qianxue Chen1*
Qianxue Chen1*- 1Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, China
- 2Department of Neurosurgery, Taihe Hospital, Hubei University of Medicine, Shiyan, China
- 3Department of Neurosurgery, Affiliated Hospital of Xi'an Jiaotong, University Health Science Center, Xi'an, China
- 4Department of Surgery, University of British Columbia, Vancouver, BC, Canada
The treatment of glioblastoma has been a big challenge for decades in the oncological field mainly owing to its unique biological characteristics, such as high heterogeneity, diffusing invasiveness, and capacity to resist conventional therapies. The mRNA-based therapeutic modality holds many superior features, including easy manipulation, rapid and transient expression, and adaptive convertibility without mutagenesis, which are suitable for dealing with glioblastoma's complexity and variability. Synthetic anticancer mRNAs carried by various vehicles act as the ultimate attackers of the tumor across biological barriers. In this modality, specifically targeted glioblastoma treatment can be guaranteed by adding targeting molecules at certain levels. The choice of mRNA-bearing vehicle and administration method is a fully patient-tailored selection. This review covers the advantages and possible limitations of mRNA-based gene therapy, the in vitro synthesis of mRNA, the feasible methods for synthetic mRNA delivery and clinical therapeutic prospects of mRNA-based gene therapy for glioblastoma.
Background
Glioblastoma multiforme (GBM) is the most common and aggressive primary brain tumor with inferior prognosis. GBM is thought to arise from the neuroglial stem or progenitor cells and has been defined by WHO as a grade IV glioma (1, 2). It is recurrent in almost all patients (3). GBM affects 3–4 people out of every 100,000 per year, with a sustained and highly significant incidence rise across all ages (4, 5). The outcomes for treating GBM retain gloomy, though surgical techniques and adjuvant therapies have progressively developed for decades. GBM remains a virtually incurable disease, resulting in a death rate of greater than 95% within 5 years of diagnosis (6). The main reasons that GBM is challenging to treat relate both the restriction of surgical resection and the resistance to irradiation and chemotherapy (7). Not only the drugs were prevented to enter into gliomas' cells by blood-brain barrier (BBB) and brain-tumor barrier (BTB) in the brain, but also the complexity of tumor composition and diffusing invasiveness have hindered the better effective treatment for over three decades (8, 9). There is an urgent need for advancement in treatment strategies to improve outcomes for GBM patients. Since Wolff et al. pioneered the concept of nucleic acid based therapy, reporting functional protein expression in target organs after the direct injection of plasmid DNA or mRNA (10), gene therapy has held a great potential to provide a viable alternative to conventional treatments toward effectively overcoming cancer progression in GBM (11).
Till now, four main types of vectors have been widely used in gene-related therapy, including plasmids, viral vectors, cosmids, and artificial chromosomes. Viral vectors used for gene therapy are associated with severe safety issues (12, 13). Meanwhile, low transfection efficiency and the potential to induce mutations limited the development of non-viral DNA vectors (14). Recent research reported that novel stabilized mRNA constructs have become more attractive alternatives to the most commonly used DNA-based plasmid (pDNA) (15, 16), mainly due to their ease of manipulation and safety in clinical applications. This review discusses the advantages and possible limitations of mRNA-based gene therapy, the feasible methods for mRNA-based gene delivery and clinical therapeutic prospects of mRNA-based gene therapy for glioblastoma.
The Beneficial Aspects of mRNA-based Gene Transfer
Synthetic mRNA has emerged as an efficient gene transfection tool, and a wide range of therapeutic applications have been developed (17). Prolonged intracellular persistence of mRNA is a basic prerequisite for synthetic mRNA to be effectively used in gene therapy. mRNA-based gene have significantly enhanced translational efficience of foreign mRNA in host cells, mainly owe to the discovery of 5′mRNA anti-reverse cap analogs (ARCA), the insertion of additional untranslated regions, and poly(A) tails (15, 18–20). To summarize, compared with pDNA delivery in gene therapy, mRNA-based gene treatment has more significant virtues: (i) pDNA is translated into the nucleus and mRNA is translated in the cytoplasm directly. The mRNA transfection is efficient even in quiescent cells, which is obviously different from pDNA transfection; (ii) the risk of insertional mutagenesis can be ignored by the nature of RNA. Hence, mRNA has a significant security compared to DNA in gene therapy for clinical applications; (iii) the immunogenic reaction of toll-like receptor-activated mRNA is weaker than the unmethylated CpG motifs of DNA recognized by TLR9; (iv) the mRNA transfection into host cells can be much easier because its construct is far smaller than pDNA. Furthermore, mRNA gene therapy circumvents the need for selecting a specific promoter, and thus the transfection process is relatively efficient and facile; (v) protein translation takes place almost immediately after mRNA transfection because of it's functionality in the cytoplasm without the need to enter into the nucleus (15, 21–24). Mainly because of the unstable structure and ubiquitous presence of RNase, the biggest concern about mRNA-based gene vehicle has been its stability and durability during its application. Table 1 lists the comparisons between mRNA- and DNA-based gene carriers as regards to practical applications.
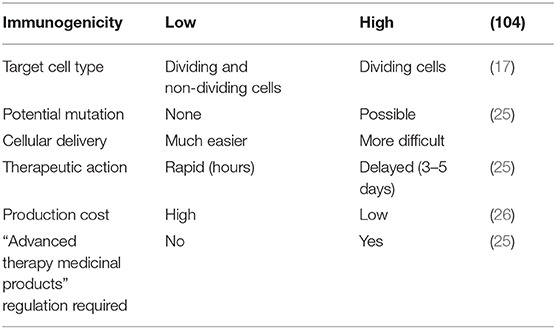
Table 1. Comparisons between mRNA- and DNA-based gene carriers.
In vitro Synthesization of Target Gene-bearing mRNA
The structure of synthetic mRNA is the same as the structure of natural mRNA, containing a cap, 5′ and 3′ UTRs and a poly(A)-tail and the encoded gene of interest. Regardless of the application, the vital factor is bioavailability of the synthetic mRNA. In recent years, the cap structure of the eukaryotic mRNAs naturally occurring at the 5′end has been studied on the therapeutic use of mRNAs (22, 27, 28). The cap is involved in mRNA's maturation, nuclear export, initiation of translation, and their turnover through interacting with highly specialized cap-binding proteins (29, 30). Due to the existence of NTPs and a regular cap such as m7GpppGNpN, polymerase-mediated transcripts are highly capped in a reverse orientation (i.e., Gpppm7GpNpN) up to one-third to one-half of total transcripts (28, 31). Such reverse-capped transcripts significantly reduce the translational efficiency of mRNA. However, 3′-O-methyl, 3′-H, or 2′-O-methyl modified anti-reverse cap analogs (ARCAs) of the m7Guo can achieve 100% correct orientation, thereby resulting in higher translational efficiency of synthetic mRNAs (18, 32–34). It can also improve resistance to enzymatic degradation (35). ARCA is now widely used in in vitro synthesization of mRNA. 3′ UTR is another key regulator of intracellular kinetics of an mRNA molecule (36). The length of the 3′ UTR is a critical factor since the longer of the mRNAs 3′ UTRs the shorter of the half-life, meanwhile mRNAs with shorter 3′ UTRs are less efficiently translated (37, 38). Human globin 3′ UTRs are now being commonly used in mRNA synthesization, mainly based on the distinctive feature of human erythrocytes (17). Practically, the human 5′ UTR with Kozak sequence, standardized 3′ UTR sequence and ARCA cap analog are all commercially available. The presence and length of the 3′-poly(A)-tail in mRNA also have great importance for efficient translation and stability (39). Different administration route may result in diverse average half-life of protein production from transfected modified mRNA, it ranges from 50 h in vitro to 7–30 h in vivo (40). The majority of mRNA decay are began with deadenylation of the poly(A)-tail total to ~10 nucleotides (41, 42), so a poly (T120) sequence was always introduced in the Tail PCR process in our previous studies (16, 20). The Figure 1 shows the flow chart of mRNA synthesis in vitro.
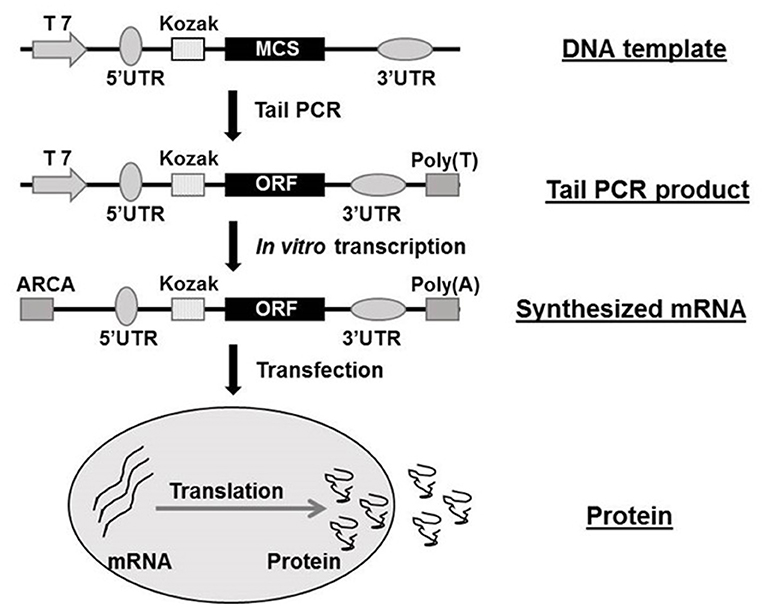
Figure 1. Flow chat of mRNA synthesization in vitro. ARCA, anti-reverse cap analog; MCS, multiple cloning site; ORF, open reading frame; PCR, polymerase chain reaction; UTR, untranslated region. The flow chat was reproduced from Guo et al. (16).
Delivery of Synthesized mRNA
Synthetic mRNAs have to be located in the cytoplasm of targeted cells to be translated into corresponding proteins using the host cell's machinery. As Kowalski et al. indicated, the delivery of exogenous synthetic mRNA is quite complex and requiring overcome numerous obstacles in extra- and intra-cellular to reach the cytoplasm exogenous synthetic mRNA delivery process is quite multifaceted and faces multiple extra- and intra-cellular barriers to reach the cytoplasm (43). Under most circumstances, the cell membrane is a major and formidable barrier for synthetic mRNA's intracellular delivery. The characteristics of mRNA and lipid bilayer components of the cell membrane are essential factors when we design an ideal method to deliver synthetic mRNAs to targeted cells. In general, the delivery of mRNA in intracellular is not as easy as small oligonucleotides, in part because of its larger size (300–5,000 kDa, up to 15 kb) and poly-anionic feature, and it needs encapsulation into nanoparticle (43–45). The cell membrane is primarily made up of a lipid bilayer composed of zwitterionic and negatively charged phospholipids (43). The polar heads of the phospholipids point toward the aqueous environment and the hydrophobic tails form a hydrophobic core (43, 46). The ion channels and ion pumps mounted in the lipid bilayer maintain a negative potential (−40 to −80 mV) across the cell membrane, forming an electro-barrier for highly negatively charged mRNA molecules (47). Thus, appropriate supplementary measures are required to facilitate synthetic mRNA crossing the cell membrane. It is amazingly stable and sustained long time if the mRNA transferred into the cytoplasm (22). Since the relatively small size and single chain structure of mRNA, majority of the available delivery tools were shown to work better with mRNA which were well used for studying in plasmid DNA delivery (48, 49). After this, several mRNA delivery methods that are being utilized in preclinical and clinical studies are explicated and related pros and cons are also discussed.
Naked mRNA Delivery
The naked mRNA spontaneous uptake by cells is not satisfied, even though the transient expression was demonstrated in some studies (10, 50, 51). Some clinical trials using intratumoral injection of naked mRNA to encode tumor-associated antigens into patients with advanced melanoma and with liver cancer are currently being undertaken (43). Naked mRNAs can be passively transferred into cells of interest by electroporation, which is used on a certain type of cells in preclinical studies. When cells are treated with short high-field electric pulses, the difference of voltage along the cell membrane can cause temporary perforation, allowing mRNA molecules passively pass through the hole and spread into the cell. The electroporation-mediated delivery efficiency can be influenced by the electrical field, ionic strength of the medium, the cell type and other membrane permeability-related factors (22). Therefore, the electroporation conditions must be carefully balanced between the high uptake of synthetic mRNA and the high percentage of healthy cells. The earliest successful electroporation mRNA transfections were conducted on human hematopoietic cells and dendritic cells, which showed superior to spontaneous uptake and lipofection (48, 52). Later on, this method was also applied in other types of cells, such as stem cells and lymphocytes (53, 54). More recently, the high throughput electroporators were also successfully used to electroporate large volumes of cells (55, 56). Theoretically, electroporation and electroporator can be used in a variety of cells without special reagents, but expensive equipment is needed and more cells and mRNAs are required for each transfection. Because of the relatively high cell mortality rate, the electroporation conditions for each cell type need to be optimized to achieve the best results.
Lipoplex- and Polyplex-Mediated Transfection
Another method for synthetic mRNA delivery is the complexation of mRNA nucleic acids with cationic lipids or polymers forming lipoplexes and polyplexes through spontaneous charge interactions (57). The carrier materials bind nucleic acids and protect their cargo from degradation by forming tight particles containing PH-sensitive molecules that escape from the endosome and enter the cytoplasm after endocytosis (22, 58). Furthermore, these carriers can be functionalized for specific cell or tissue delivery of synthetic mRNA by modifying carrier's formulation (59). Now, lipoplex- and polyplex-mediated mRNA transfection is being commonly used in various preclinical studies. However, because cationic liposomes have relatively high cytotoxicity and may interfere with cell metabolism in different cells, cationic lipid-related carriers have been mainly limited to in vitro studies. In recent years, more complex mRNA vectors have been created, such as PH-reactive polymer nanoparticles, which can also be systematically delivered in vivo (60).
Inorganic Nanoparticle-Mediated Delivery
Although current mRNA delivery technologies are mainly concentrated on cationic polymers and liposomes, inorganic nanoparticles have also been developed. In 2009, Zohra et al. for the first time introduced that carbonate apatite inorganic nanoparticles bond with cationic liposomes of DOTAP (N-[1-(2,3-Dioleoloxy) propyl]-N, N, N-trimethyl ammonium chloride) could successfully generate high transfection efficiency of luciferase mRNA in both mitotic and non-mitotic cells (61). As an additional advantage, inorganic carbonate apatite combining with DOTAP could facilitate DOTAP-mediated mRNA expression (62). So, inorganic nanoparticle holds a promising potential to be widely used for synthetic mRNA delivery.
Polypeptide-Mediated Delivery
As another type of synthetic vehicles, precisely designed polypeptides are also used to deliver mRNA to the cell cytoplasm (63). Amphiphilic cationic feature of the polypeptide mainly determines the mRNA delivery function. As shown in Mastrobattista et al.'s recent study, the GALA peptide functionalized the target mRNA polyplexes (PPx-GALA) in dendritic cells (DCs), and the cellular uptake of mRNA that PPx-GALA complex is 18 times higher than lipofectamine without causing cytotoxicity (64). The conjugation of precisely designed peptide to mRNA polyplexes not only promotes the mRNA expression but also plays a significant role in targeting the specific type of cells or tissue.
Virus-Mediated Delivery
Synthetic mRNA can also be delivered into cells of interest by viral particles, which is different from conventional transfection. Such gene delivery-related viral infection requires cloning the target gene into a specific virus system and packaging specific cells to obtain the “modified” virus. Alphavirus, Sendai virus, and retrovirus have been utilized for mRNA delivery (65, 66). Retrovirus-mediated mRNA transfection can be delivered to the cytoplasm as a direct translation template for interest proteins, but the vector needs to be altered to prevent reverse translation (22). The advantage is that the infection efficiency is particularly high, especially some of the primary cells and living cells which are difficult to transfect. In addition, mRNA can avoid being degraded outside the cell by retroviral particle and the viral envelope may be modified for synthetic mRNA-transfer (67). Generally speaking, the virus system is time-consuming, expensive and complex, and may be potentially dangerous if it is improperly operated.
Cell- Mediated Delivery
Cell-mediated gene therapy has been extensively studied for decades. A variety of types of cells have been involved in this field, including hematopoietic stem cell (HSC) (68), mesenchymal stem cell (MSC) (69), dendritic cell (DC) (70), macrophage (71), natural killer (NK) cell (72), and chimeric antigen receptor T (CAR-T) cell (73). Anti-CD19 CAR-T cell products, the first FDA approved gene therapy for patients with pre-B cell acute lymphoblastic leukemia or B-cell lymphomas, have revolutionized anti-cancer therapy, giving a new treatment option for patients who have difficulty to receive standard treatment (74). DNA-based engineering strategies have been used in the majority of cell-mediated gene therapies. However, the advantage of mRNA transfection is that the rate and duration of target gene expression can be well-managed by adjusting the quantity of mRNA, which may avoid some DNA-engineered cell-induced adverse events, such as cytokine release syndrome following the infusion of CAR-expressing T cells. Recently, as an alternative modality, mRNA has been utilized for cell-mediated gene delivery. The studied cell type includes DC (75), NK cell (72), T cell (76), HSC (77), and MSC (16, 78, 79). The most notable advantage of using cell-mediated mRNA delivery lies in vehicle cell's homing capacity. The target mRNAs can be transfected into vehicle cells by preferred delivery method, such as electroporation, viral or non-viral delivery. As illustrated in Figure 2, at the target site, mRNA-carried message can be transferred to recipient cells through several pathways: (1) cell-to-cell contact, such as DCs and CAR-T cell; (2) encoded protein produced in the vehicle cells, in the case of secreting protein or proteins containing transacting activator of transcription (TAT) segment; (3) parent cell-derived exosomes containing both mRNA and protein. Several clinical trials have been reported to use autologous DCs loaded with mRNA as a treatment of various cancers (22). In our recent work, the therapeutic efficacy of anticancer gene-engineered MSCs was also demonstrated in glioblastoma animal model (16).
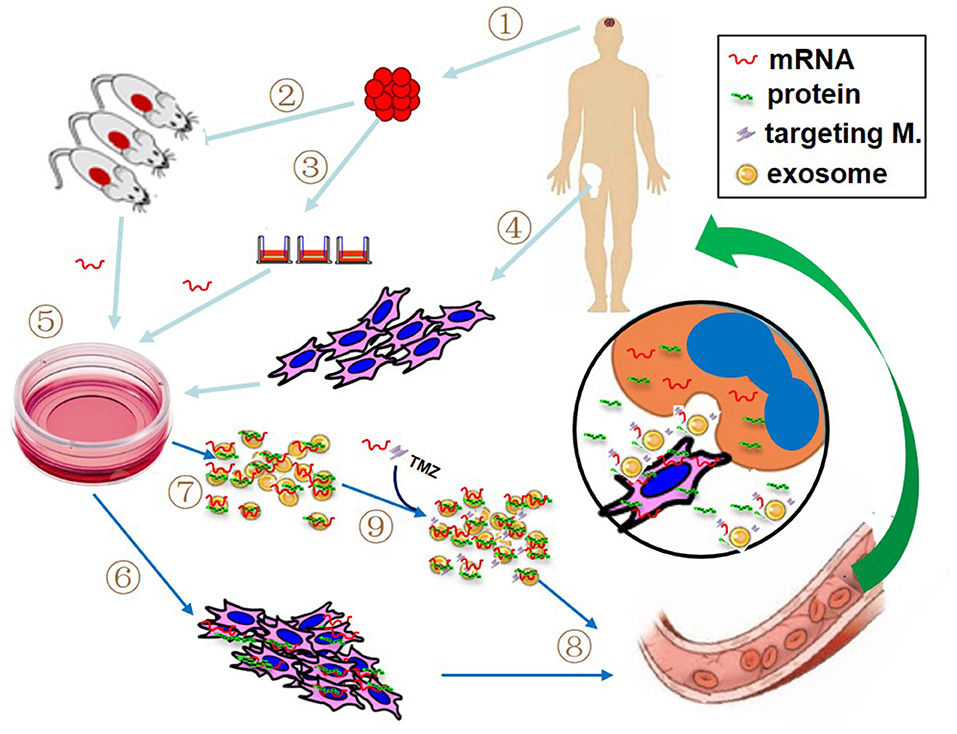
Figure 2. Proposed patient-tailored glioblastoma treatment with mRNA-based therapeutic modality. If practical, the target mRNA is predetermined using resected tumor tissue (1) with patient-derived xenografted animal model (2) or real-time detection (3). Meanwhile, patient's own vehicle cells (like bone marrow-derived mesenchymal stem cells, MSCs) are prepared (4). The synthetic target mRNA is delivered into cultured vehicle cells by preferred transfection method (5) and the target mRNA-bearing cells (6) and associated exosomes (7) are collected for administration (8). If necessary, additional mRNA, specific targeting molecules and/or chemotherapeutics such as temozolomide (TMZ) can be directly loaded into the isolated exosomes prior to the administration (9). The insert picture illustrates the possible action modes of mRNA in the tumor site, including mRNA product (i.e., protein) from both vehicle cell and recipient cell, cell-to-cell contact, and exosome-mediated attack molecules.
Exosome-Mediated Delivery
As the key mediators for intercellular communications, exosomes play an important part in mRNA delivery. Exosomes are nanoscale membrane vesicles mainly composed of circular double-layer lipid membranes and intracapsular contents (80). During the biogenesis of exosomes, the vesicular membrane is formed through two steps of reverse invagination of the cellular plasma membrane, resulting in vesical membrane outside-facing-out. The biological significance of this property is 2-fold. Firstly, this membrane orientation is a necessary prerequisite for the application of exosomes to targeted cancer therapy due to the targeting molecules from mother cells are also exist in the exosomes (81). Secondly, the content of vesicles is closely connected with plasma membrane reverse invagination. Theoretically, anything in the cell cytoplasm can be inwrapped in exosomes (82, 83), including synthetic mRNA from transfected parent cells. Therefore, the delivered synthetic mRNA could exist inside of exosomes in two forms, mRNA and/or relevant protein (Figure 2). Furthermore, synthetic mRNA and chemotherapeutic drugs can also be directly loaded into isolated exosomes through conventional transfection technology (84). Isolated exosomes can be passively transmitted throughout the body, but their ability to target distribution depends primarily on the surface-derived targeting molecules form parent cells (84). In the recipient cells, exosomes are primarily absorbed into the cell by endocytosis, membrane fusion, or receptor-mediated internalization (85). Due to the cell-free nature and biological characteristics, exosomes replace cell-based modality (like CAR-T) and directly attack tumor cells. In regards to cancer therapy, exosomes have following advantages. First, exosomes can be used as “off-the-shelf” reagents so that the doses can be controlled according to the therapeutic condition. Second, their nanoscale size provides the possibility for solid tumor therapy such as GBM. Third, the combined and/or alternate use of cell-based and cell-free platforms will enhance the application value for cell-based cancer therapy (86).
The Therapeutic Prospects of mRNA-based Gene Therapy for Glioblastoma
The reason why GBM is difficult to treat rests with its special biological characteristics, such as located in the brain and defended by both BBB and BTB, high heterogeneity, diffusing invasiveness, and capacity to resist conventional therapies. As mechanical barriers, BBB and BTB protect GBM from certain therapeutic agents. The high heterogeneity of GBM probably means that it cannot be well-cured by any single particular drug, especially biomarker-related agents. Its invasive growth property determines the highest recurrence rate after surgical resection, and its high capacity of drug resistance suggests that it cannot be efficiently treated by the sustained use of any specific drug. Taken together, an ideal anti-GBM strategy should adapt all challenges and meet the following requirements: (1) Anticancer actions are specifically confined to the tumor site; (2) personalized treatment plan according to the sensitivity of anticancer drugs predetermined; (3) multiple anticancer mechanisms can work simultaneously; and (4) the anticancer agents can be adaptively replaced. Of course, because GBM grows in a restricted intracranial environment, if practical, surgical resection is always the first treatment upon diagnosis, otherwise, the patients may die from some GBM-related complications, such as cerebral hernia, and we may not have the opportunity to provide any therapeutic intervention.
The Use of mRNA-Based Anticancer Gene Products as the Ultimate Attackers of the Tumor Has Special Superiority for GBM Treatment
As described earlier, nano-scaled target mRNA-bearing lipoplexes, polyplexes, liposomes and exosomes are all able to cross BBB and BTB in the brain (87, 88), overcoming the natural barriers. The rapid and transient expression nature of synthetic mRNA, which has been considered the biggest weakness of using mRNA for gene therapy, is just suitable for dealing with GBM's complexity and variability through its adaptive convertibility. It is also because of its short-term high-level expression, mRNA-based gene therapy has been intensively and almost exclusively focused on cancer treatment (89, 90). The selection of anti-oncogene depends mainly on the specific situation of each patient. If possible, predetermination should be performed using in vitro real-time detection of tumor cells or patient-derived xenografted animal model immediately after surgical resection (91, 92).
The Highly Specific Targeting Capability of mRNA-Based Modality Is Essential to Achieve the Most Effective Treatment of Cancer With Limited Side Effects
In this mRNA-based therapeutic modality, the GBM targeting can be achieved at least in three levels. (1) Cell level. The tumor-homing property has been verified in several types of cells, including NK cells, DCs, and MSCs (72, 93, 94). In addition to their ability of tumor-directed migration and incorporation, these types of cells are easy acquisition and fast ex vivo expansion. More importantly, they are feasible for autologous transplantation. These cells are also able to cross BBB especially under brain tumorous condition (95, 96). The targeting capacity of these cells can be further enhanced by transfecting specific targeting molecules (e.g., CAR-T). The therapeutic efficacy of GBM by cell-mediated and mRNA-based modality has heterogeneity in preclinical studies and a number of them are currently in different phases of clinical trials (16, 20, 96). (2) Exosome level. As aforementioned, anticancer mRNA-bearing exosomes are able to target tumor cells directed by the targeting molecules in their membrane, which originate from their parent cells. Furthermore, additional targeting molecules can be loaded into exosomes after their isolation. Several GBM-specific targeting peptides have been precisely investigated. These peptides act on a different part of the tumor cell through different mechanisms (97). (3) Molecular-level. Under certain circumstances, the ultimate attack molecules kill tumor cells in a tumor-specific manner. For example, tumor necrosis factor-related apoptosis inducing ligand (TRAIL) induces apoptosis specifically in tumor cells through binding with death receptors. This target specificity is determined by the differential expression of death receptors in tumor cells (98).
The Administration Route of mRNA Is a Patient-Tailored Selection
The selection of delivery routes of synthetic mRNA to GBM patients depends on the general evaluation of various parameters, including patient's glioma grade, stage, surgical, and chemotherapy history, as well as the available forms of synthetic mRNA carriers. In general, intravenous infusion is the safest and most practical administration method for various forms of synthetic mRNA. Other options include local injection, cerebrospinal fluid infusion, interventional infusion and administration through the nasopharyngeal pathway. However, it is worth noting that the alternate use and sometimes a combination of different mRNA content and different delivery method could be significantly beneficial for the patients with GBM. In our previous clinical trial, the combination of local application of MSCs during surgical operation and intravenous infusion of MSCs after surgery achieved an ideal outcome (99).
Although no clinical trials of mRNA-based GBM therapy have yet been completed, some are underway. A phase II randomized, blinded trial of CMV RNA-pulsed dendritic cells with tetanus-diphtheria toxoid vaccine in patients with newly-diagnosed glioblastoma is ongoing1. The mRNA-based therapy has not been widely adopted in treating GBM, but it had made great progress in other diseases. A phase 1 clinical trial showed that rabies vaccination based on mRNA encoding was safe and could produce functional antibodies (100). Martin Sebastian et al. reported a phase I/II a study of mRNA-based cancer immunotherapy on non-small cell lung cancer. The patients of this trial were well-tolerated and the antigen-specific immune responses were detected in 63% of assessable patients after five injections (101). The therapeutic prospects of mRNA-based gene therapy for GBM could proliferate when noted obstacles are overcome, such as the targeted delivery in vivo, rapid degradation, and short half-life. We also have to address the repetitive application and sufficient delivery efficiency in the brain.
Conclusions
The dissatisfactory outcome of GBM treatment has retained for decades. The reason why GBM is difficult to treat rests with its special biological characteristics such as high heterogeneity, diffusing invasiveness, and capacity to resist conventional therapies, as well as the existence of biological barriers, e.g., BBB and BTB. Compared with conventional DNA-based strategy, synthetic mRNA-mediated therapeutic modality holds many superior features including easy manipulation, rapid and transient expression, and adaptive convertibility without mutagenesis, which is suitable for dealing with glioblastoma's complexity and variability. Synthetic anticancer mRNAs carried by various vehicles, such as organic or inorganic nanoparticle-encapsulated complexes, patient-derived DCs or MSCs and their corresponding exosomes, act as the ultimate attackers of the tumor across biological barriers. In this mRNA-based therapeutic modality, specifically targeted GBM treatment can be guaranteed by adding targeting molecules at certain levels. The choice of mRNA-bearing vehicle and administration method is a fully patient-tailored selection. To date, mRNA has been used in preclinical clinical trials such as cancer immunotherapy, infectious disease control and regenerative medicine (24, 102, 103), but, anticancer treatment is the most developed application for mRNA (89, 104). The mRNA-based therapeutic modality holds a great promising potential to be efficiently utilized for the patients with GBM.
Ethics Statement
Ethical approval was waived since we used only publicly available data and materials in this study.
Author Contributions
XT, LZ, and QC designed this research. RF, KH, and HP collected relevant literatures. XT and LD wrote and revised the manuscript. SZ helped to correct the language. All authors have read and approved the final manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (No. 81702482) and Natural Science Foundation of Hubei Province of China (No. 2017CFB562).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
mRNA, messenger Ribonucleic Acid; GBM, Glioblastoma multiforme; WHO, World Health Organization; BBB, blood-brain barrier; BTB, brain-tumor barrier; pDNA, DNA-based plasmid; ARCA, anti-reverse cap analogs; PCR, Polymerase Chain Reaction; DOTAP, N-[1-(2,3-dioleoloxy)propyl]-N,N,N-trimethyl ammonium chloride; DCs, dendritic cells; HSC, hematopoietic stem cell; MSC, mesenchymal stem cell; NK, natural killer; CAR-T, chimeric antigen receptor T; TRAIL, tumor necrosis factor-related apoptosis inducing ligand.
Footnotes
References
1. Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Primers. (2015) 1:15017. doi: 10.1038/nrdp.2015.17
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
3. Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci USA. (2000) 97:6242–4. doi: 10.1073/pnas.97.12.6242
4. Philips A, Henshaw DL, Lamburn G, O'Carroll MJ. Brain tumours: rise in glioblastoma multiforme incidence in England 1995-2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health. (2018) 2018:7910754. doi: 10.1155/2018/7910754
5. Walker EV, Davis FG, CBTR founding affiliates. Malignant primary brain and other central nervous system tumors diagnosed in Canada from 2009 to 2013. Neuro Oncol. (2019) 21:360–9. doi: 10.1093/neuonc/noy195
6. Jhanwar-Uniyal M, Labagnara M, Friedman M, Kwasnicki A, Murali R. Glioblastoma: molecular pathways, stem cells and therapeutic targets. Cancers. (2015) 7:538–55. doi: 10.3390/cancers7020538
7. Tomiyama A, Ichimura K. Signal transduction pathways and resistance to targeted therapies in glioma. Semin Cancer Biol. (2019) 58:118–29. doi: 10.1016/j.semcancer.2019.01.004
8. Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. (2011) 13:e17. doi: 10.1017/S1462399411001888
9. Westphal M, Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci. (2011) 12:495–508. doi: 10.1038/nrn3060
10. Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. (1990) 247(4949 Pt 1):1465–8.
11. Caffery B, Lee JS, Alexander-Bryant AA. Vectors for glioblastoma gene therapy: viral & non-viral delivery strategies. Nanomaterials. (2019) 9:E105. doi: 10.3390/nano9010105
12. Rosenecker J, Huth S, Rudolph C. Gene therapy for cystic fibrosis lung disease: current status and future perspectives. Curr Opin Mol Ther. (2006) 8:439–45.
13. Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. (2010) 363:355–64. doi: 10.1056/NEJMoa1000164
14. Sanders N, Rudolph C, Braeckmans K, De Smedt SC, Demeester J. Extracellular barriers in respiratory gene therapy. Adv Drug Deliv Rev. (2009) 61:115–27. doi: 10.1016/j.addr.2008.09.011
15. Leonhardt C, Schwake G, Stogbauer TR, Rappl S, Kuhr JT, Ligon TS, et al. Single-cell mRNA transfection studies: delivery, kinetics and statistics by numbers. Nanomedicine. (2013) 10:679–88. doi: 10.1016/j.nano.2013.11.008
16. Guo XR, Yang ZS, Tang XJ, Zou DD, Gui H, Wang XL, et al. The application of mRNA-based gene transfer in mesenchymal stem cell-mediated cytotoxicity of glioma cells. Oncotarget. (2016) 7:55529–42. doi: 10.18632/oncotarget.10835
17. Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3' UTRs identified by cellular library screening. Mol Ther. (2018) 27:824–36. doi: 10.1016/j.ymthe.2018.12.011
18. Stepinski J, Waddell C, Stolarski R, Darzynkiewicz E, Rhoads RE. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3'-O-methyl)GpppG and 7-methyl (3'-deoxy)GpppG. RNA. (2001) 7:1486–95.
19. Jemielity J, Stepinski J, Jaremko M, Haber D, Stolarski R, Rhoads RE, et al. Synthesis of novel mRNA 5' cap-analogues: dinucleoside P1, P3-tri-, P1, P4-tetra-, and P1, P5-pentaphosphates. Nucleosides Nucleotides Nucleic Acids. (2003) 22:691–4. doi: 10.1081/NCN-120022611
20. Guo XR, Hu QY, Yuan YH, Tang XJ, Yang ZS, Zou DD, et al. PTEN-mRNA engineered mesenchymal stem cell-mediated cytotoxic effects on U251 glioma cells. Oncol Lett. (2016) 11:2733–40. doi: 10.3892/ol.2016.4297
21. Bangel-Ruland N, Tomczak K, Fernandez Fernandez E, Leier G, Leciejewski B, Rudolph C, et al. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy. J Gene Med. (2013) 15:414–26. doi: 10.1002/jgm.2748
22. Vallazza B, Petri S, Poleganov MA, Eberle F, Kuhn AN, Sahin U. Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdiscip Rev RNA. (2015) 6:471–99. doi: 10.1002/wrna.1288
23. Tavernier G, Andries O, Demeester J, Sanders NN, De Smedt SC, Rejman J. mRNA as gene therapeutic: how to control protein expression. J Control Release. (2011) 150:238–47. doi: 10.1016/j.jconrel.2010.10.020
24. Meng Z, O'Keeffe-Ahern J, Lyu J, Pierucci L, Zhou D, Wang W. A new developing class of gene delivery: messenger RNA-based therapeutics. Biomater Sci. (2017) 5:2381–92. doi: 10.1039/c7bm00712d
25. Berraondo P, Martini PGV, Avila MA, Fontanellas A. Messenger RNA therapy for rare genetic metabolic diseases. Gut. (2019) 68:1323–30. doi: 10.1136/gutjnl-2019-318269
26. Li B, Zhang X, Dong Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. (2019) 11:e1530. doi: 10.1002/wnan.1530
27. Grudzien-Nogalska E, Kowalska J, Su W, Kuhn AN, Slepenkov SV, Darzynkiewicz E, et al. Synthetic mRNAs with superior translation and stability properties. Methods Mol Biol. (2013) 969:55–72. doi: 10.1007/978-1-62703-260-5_4
28. Rhoads RE. Synthetic mRNA: production, introduction into cells, and physiological consequences. Methods Mol Biol. (2016) 1428:3–27. doi: 10.1007/978-1-4939-3625-0_1
29. Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA. (2011) 2:277–98. doi: 10.1002/wrna.52
30. Furuichi Y. Discovery of m(7)G-cap in eukaryotic mRNAs. Proc Jpn Acad Ser B Phys Biol Sci. (2015) 91:394–409. doi: 10.2183/pjab.91.394
31. Pasquinelli AE, Dahlberg JE, Lund E. Reverse 5' caps in RNAs made in vitro by phage RNA polymerases. RNA. (1995) 1:957–67.
32. Kocmik I, Piecyk K, Rudzinska M, Niedzwiecka A, Darzynkiewicz E, Grzela R, et al. Modified ARCA analogs providing enhanced translational properties of capped mRNAs. Cell Cycle. (2018) 17:1624–36. doi: 10.1080/15384101.2018.1486164
33. Jemielity J, Fowler T, Zuberek J, Stepinski J, Lewdorowicz M, Niedzwiecka A, et al. Novel “anti-reverse” cap analogs with superior translational properties. RNA. (2003) 9:1108–22. doi: 10.1261/rna.5430403
34. Grudzien-Nogalska E, Stepinski J, Jemielity J, Zuberek J, Stolarski R, Rhoads RE, et al. Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol. (2007) 431:203–27. doi: 10.1016/S0076-6879(07)31011-2
35. Wojtczak BA, Sikorski PJ, Fac-Dabrowska K, Nowicka A, Warminski M, Kubacka D, et al. 5'-Phosphorothiolate dinucleotide cap analogues: reagents for messenger RNA modification and potent small-molecular inhibitors of decapping enzymes. J Am Chem Soc. (2018) 140:5987–99. doi: 10.1021/jacs.8b02597
36. Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene. (2001) 265:11–23. doi: 10.1016/s0378-1119(01)00350-x
37. Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. (2011) 473:337–42. doi: 10.1038/nature10098
38. Tanguay RL, Gallie DR. Translational efficiency is regulated by the length of the 3' untranslated region. Mol Cell Biol. (1996) 16:146–56.
39. Sergeeva OV, Koteliansky VE, Zatsepin TS. mRNA-based therapeutics - Advances and perspectives. Biochemistry. (2016) 81:709–22. doi: 10.1134/S0006297916070075
40. Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. (2015) 217:345–51. doi: 10.1016/j.jconrel.2015.08.007
41. Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. (2007) 8:113–26. doi: 10.1038/nrm2104
42. Chen CY, Shyu AB. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA. (2011) 2:167–83. doi: 10.1002/wrna.40
43. Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. (2019) 27:710–28. doi: 10.1016/j.ymthe.2019.02.012
44. Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. (2015) 15:7300–6. doi: 10.1021/acs.nanolett.5b02497
45. Fenton OS, Kauffman KJ, McClellan RL, Appel EA, Dorkin JR, Tibbitt MW, et al. Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Adv Mater. (2016) 28:2939–43. doi: 10.1002/adma.201505822
46. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. (2018) 19:281–96. doi: 10.1038/nrm.2017.138
47. Flewelling RF, Hubbell WL. The membrane dipole potential in a total membrane potential model. Applications to hydrophobic ion interactions with membranes. Biophys J. (1986) 49:541–52. doi: 10.1016/S0006-3495(86)83664-5
48. Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, et al. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. (2001) 98:49–56. doi: 10.1182/blood.v98.1.49
49. Zou S, Scarfo K, Nantz MH, Hecker JG. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int J Pharm. (2010) 389:232–43. doi: 10.1016/j.ijpharm.2010.01.019
50. Diken M, Kreiter S, Selmi A, Britten CM, Huber C, Tureci O, et al. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. (2011) 18:702–8. doi: 10.1038/gt.2011.17
51. Probst J, Weide B, Scheel B, Pichler BJ, Hoerr I, Rammensee HG, et al. Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. (2007) 14:1175–80. doi: 10.1038/sj.gt.3302964
52. Kalady MF, Onaitis MW, Padilla KM, Emani S, Tyler DS, Pruitt SK. Enhanced dendritic cell antigen presentation in RNA-based immunotherapy. J Surg Res. (2002) 105:17–24. doi: 10.1006/jsre.2002.6435
53. Smits E, Ponsaerts P, Lenjou M, Nijs G, Van Bockstaele DR, Berneman ZN, et al. RNA-based gene transfer for adult stem cells and T cells. Leukemia. (2004) 18:1898–902. doi: 10.1038/sj.leu.2403463
54. Rabinovich PM, Komarovskaya ME, Wrzesinski SH, Alderman JL, Budak-Alpdogan T, Karpikov A, et al. Chimeric receptor mRNA transfection as a tool to generate antineoplastic lymphocytes. Hum Gene Ther. (2009) 20:51–61. doi: 10.1089/hum.2008.068
55. Choi Y, Yuen C, Maiti SN, Olivares S, Gibbons H, Huls H, et al. A high throughput microelectroporation device to introduce a chimeric antigen receptor to redirect the specificity of human T cells. Biomed Microdevices. (2010) 12:855–63. doi: 10.1007/s10544-010-9440-3
56. Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. (2010) 17:147–54. doi: 10.1038/cgt.2009.61
57. Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. (2006) 116:255–64. doi: 10.1016/j.jconrel.2006.06.024
58. Midoux P, Pichon C, Yaouanc JJ, Jaffres PA. Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol. (2009) 157:166–78. doi: 10.1111/j.1476-5381.2009.00288.x
59. Pichon C, Midoux P. Mannosylated and histidylated LPR technology for vaccination with tumor antigen mRNA. Methods Mol Biol. (2013) 969:247–74. doi: 10.1007/978-1-62703-260-5_16
60. Su X, Fricke J, Kavanagh DG, Irvine DJ. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol Pharm. (2011) 8:774–87. doi: 10.1021/mp100390w
61. Zohra FT, Chowdhury EH, Nagaoka M, Akaike T. High performance mRNA transfection through carbonate apatite-cationic liposome conjugates. Biomaterials. (2009) 30:4006–13. doi: 10.1016/j.biomaterials.2009.02.050
62. Zohra FT, Chowdhury EH, Nagaoka M, Akaike T. Drastic effect of nanoapatite particles on liposome-mediated mRNA delivery to mammalian cells. Anal Biochem. (2005) 345:164–6. doi: 10.1016/j.ab.2005.06.031
63. Ardejani MS, Orner BP. Materials science. Obey the peptide assembly rules. Science. (2013) 340:561–2. doi: 10.1126/science.1237708
64. Lou B, De Koker S, Lau CYJ, Hennink WE, Mastrobattista E. mRNA polyplexes with post-conjugated GALA peptides efficiently target, transfect, and activate antigen presenting cells. Bioconjug Chem. (2019). 30:461–75. doi: 10.1021/acs.bioconjchem.8b00524
66. Galla M, Schambach A, Baum C. Retrovirus-based mRNA transfer for transient cell manipulation. Methods Mol Biol. (2013) 969:139–61. doi: 10.1007/978-1-62703-260-5_10
67. Munch RC, Muhlebach MD, Schaser T, Kneissl S, Jost C, Pluckthun A, et al. DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther. (2011) 19:686–93. doi: 10.1038/mt.2010.298
68. Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. (2011) 12:301–15. doi: 10.1038/nrg2985
69. Bianco P. “Mesenchymal” stem cells. Annu Rev Cell Dev Biol. (2014) 30:677–704. doi: 10.1146/annurev-cellbio-100913-013132
70. Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, Jacobs JF, et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res. (2011) 17:5725–35. doi: 10.1158/1078-0432.CCR-11-1261
71. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. (2010) 327:656–61. doi: 10.1126/science.1178331
72. Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. (2016) 7:105. doi: 10.3389/fimmu.2016.00105
73. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. (2012) 12:265–77. doi: 10.1038/nrc3258
74. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. (2019) 16:372–85. doi: 10.1038/s41571-019-0184-6
75. Fornaguera C, Guerra-Rebollo M, Angel Lazaro M, Castells-Sala C, Meca-Cortes O, Ramos-Perez V, et al. mRNA delivery system for targeting antigen-presenting cells in vivo. Adv Healthc Mater. (2018) 7:e1800335. doi: 10.1002/adhm.201800335
76. Eggers R, Philippi A, Altmeyer MO, Breinig F, Schmitt MJ. Primary T cells for mRNA-mediated immunotoxin delivery. Gene Ther. (2018) 25:47–53. doi: 10.1038/gt.2017.87
77. Antony JS, Latifi N, Haque A, Lamsfus-Calle A, Daniel-Moreno A, Graeter S, et al. Gene correction of HBB mutations in CD34(+) hematopoietic stem cells using Cas9 mRNA and ssODN donors. Mol Cell Pediatr. (2018) 5:9. doi: 10.1186/s40348-018-0086-1
78. Hamann A, Nguyen A, Pannier AK. Nucleic acid delivery to mesenchymal stem cells: a review of nonviral methods and applications. J Biol Eng. (2019) 13:7. doi: 10.1186/s13036-019-0140-0
79. Toh WS, Lai RC, Zhang B, Lim SK. MSC exosome works through a protein-based mechanism of action. Biochem Soc Trans. (2018) 46:843–53. doi: 10.1042/BST20180079
80. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. (2014) 14:195–208. doi: 10.1038/nri3622
81. Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, et al. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/ζ complex. J Immunol. (2002) 168:3235–41. doi: 10.4049/jimmunol.168.7.3235
82. Kim HS, Choi DY, Yun SJ, Choi SM, Kang JW, Jung JW, et al. Proteomic analysis of microvesicles derived from human mesenchymal stem cells. J Proteome Res. (2012) 11:839–49. doi: 10.1021/pr200682z
83. Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. (2011) 2:282. doi: 10.1038/ncomms1285
84. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
85. Kourembanas S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol. (2015) 77:13–27. doi: 10.1146/annurev-physiol-021014-071641
86. Tang XJ, Sun XY, Huang KM, Zhang L, Yang ZS, Zou DD, et al. Therapeutic potential of CAR-T cell-derived exosomes: a cell-free modality for targeted cancer therapy. Oncotarget. (2015) 6:44179–90. doi: 10.18632/oncotarget.6175
87. El Andaloussi S, Lakhal S, Mager I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. (2013) 65:391–7. doi: 10.1016/j.addr.2012.08.008
88. Wood MJ, O'Loughlin AJ, Samira L. Exosomes and the blood-brain barrier: implications for neurological diseases. Ther Deliv. (2011) 2:1095–9. doi: 10.4155/tde.11.83
89. Van Tendeloo VF, Ponsaerts P, Berneman ZN. mRNA-based gene transfer as a tool for gene and cell therapy. Curr Opin Mol Ther. (2007) 9:423–31.
90. Uzgun S, Nica G, Pfeifer C, Bosinco M, Michaelis K, Lutz JF, et al. PEGylation improves nanoparticle formation and transfection efficiency of messenger RNA. Pharm Res. (2011) 28:2223–32. doi: 10.1007/s11095-011-0464-z
91. Luo J, Guo XR, Tang XJ, Sun XY, Yang ZS, Zhang Y, et al. Intravital biobank and personalized cancer therapy: the correlation with omics. Int J Cancer. (2014) 135:1511–6. doi: 10.1002/ijc.28632
92. Tuveson D, Hanahan D. Translational medicine: cancer lessons from mice to humans. Nature. (2011) 471:316–7. doi: 10.1038/471316a
93. Xie C, Yang Z, Suo Y, Chen Q, Wei D, Weng X, et al. Systemically infused mesenchymal stem cells show different homing profiles in healthy and tumor mouse models. Stem Cells Transl Med. (2017) 6:1120–31. doi: 10.1002/sctm.16-0204
94. Moniri MR, Sun XY, Rayat J, Dai D, Ao Z, He Z, et al. TRAIL-engineered pancreas-derived mesenchymal stem cells: characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. (2012) 19:652–8. doi: 10.1038/cgt.2012.46
95. Conaty P, Sherman LS, Naaldijk Y, Ulrich H, Stolzing A, Rameshwar P. Methods of mesenchymal stem cell homing to the blood-brain barrier. Methods Mol Biol. (2018) 1842:81–91. doi: 10.1007/978-1-4939-8697-2_6
96. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. (2018) 119:1171–81. doi: 10.1038/s41416-018-0258-8
97. Raucher D. Tumor targeting peptides: novel therapeutic strategies in glioblastoma. Curr Opin Pharmacol. (2019) 47:14–9. doi: 10.1016/j.coph.2019.01.006
98. Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat J, Warnock GL. Potential implications of mesenchymal stem cells in cancer therapy. Cancer Lett. (2011) 305:8–20. doi: 10.1016/j.canlet.2011.02.012
99. Zhang ZX, Guan LX, Zhang K, Zhang Q, Dai LJ. A combined procedure to deliver autologous mesenchymal stromal cells to patients with traumatic brain injury. Cytotherapy. (2008) 10:134–9. doi: 10.1080/14653240701883061
100. Alberer M, Gnad-Vogt U, Hong HS, Mehr KT, Backert L, Finak G, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. (2017) 390:1511–20. doi: 10.1016/S0140-6736(17)31665-3
101. Sebastian M, Schroder A, Scheel B, Hong HS, Muth A, von Boehmer L, et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother. (2019) 68:799–812. doi: 10.1007/s00262-019-02315-x
102. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov. (2014) 13:759–80. doi: 10.1038/nrd4278
103. Van Lint S, Renmans D, Broos K, Dewitte H, Lentacker I, Heirman C, et al. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev Vaccines. (2015) 14:235–51. doi: 10.1586/14760584.2015.957685
Keywords: mRNA, gene therapy, glioblastoma, patient-tailored, targeting molecules
Citation: Tang X, Zhang S, Fu R, Zhang L, Huang K, Peng H, Dai L and Chen Q (2019) Therapeutic Prospects of mRNA-Based Gene Therapy for Glioblastoma. Front. Oncol. 9:1208. doi: 10.3389/fonc.2019.01208
Received: 27 August 2019; Accepted: 23 October 2019;
Published: 08 November 2019.
Edited by:
Liam Chen, Johns Hopkins University, United StatesReviewed by:
Han Shen, Westmead Institute for Medical Research, AustraliaPierpaolo Peruzzi, Brigham and Women's Hospital, Harvard Medical School, United States
Copyright © 2019 Tang, Zhang, Fu, Zhang, Huang, Peng, Dai and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Longjun Dai, ljdai@mail.ubc.ca; Qianxue Chen, chenqx666@whu.edu.cn
†These authors have contributed equally to this work