 Haitang Yang
Haitang Yang Shun-Qing Liang
Shun-Qing Liang Ralph A. Schmid1
Ralph A. Schmid1 Ren-Wang Peng
Ren-Wang Peng- 1Department of General Thoracic Surgery, Department of BioMedical Research, Inselspital, Bern University Hospital, University of Bern, Bern, Switzerland
- 2University of Massachusetts Medical School, Worcester, MA, United States
In non-small cell lung cancer (NSCLC), the most frequent oncogenic mutation in western countries is KRAS, for which, however, there remains no clinically approved targeted therapies. Recent progress on high biological heterogeneity including diverse KRAS point mutations, varying dependence on mutant KRAS, wide spectrum of other co-occurring genetic alterations, as well as distinct cellular status across the epithelial-to-mesenchymal transition (EMT), has not only deepened our understanding about the pathobiology of KRAS-mutant NSCLC but also brought about unprecedented new hopes for precision treatment of patients. In this review, we provide an update on the most recent advances in KRAS-mutant lung cancer, with a focus on mechanistic insights into tumor heterogeneity, the potential clinic implications and new therapies on horizons tailored for KRAS-mutant lung cancer.
Introduction
Lung cancer is the most common cancer with high lethality (1). Carcinogenic Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation is the most common gain-of-function alteration, accounting for ~30% of lung adenocarcinomas in western countries and about 10% of Asian lung adenocarcinomas (2).
As a membrane-bound small GTPase, KRAS switches between the active GTP-bound and inactive GDP-bound status, which is regulated by guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs), respectively (3). The intrinsic GTPase activity of RAS is rather low, but in the presence of GAPs, such as neurofibromin 1 (NF1), its hydrolytic activity can be increased by several orders of magnitude. Reactivation of GDP-bound RAS is mediated by GEFs, such as son of sevenless homolog 1 (SOS1), which promotes the release of bound GDP, and then cellular GTP will replace GDP to bind to RAS. Carcinogenic mutations impair the ability of KRAS to hydrolyze GTP and are thought to lock the oncoprotein in a constitutively active state by activating KRAS downstream signaling cascades, leading to uncontrolled cell proliferation and survival. In patients with lung cancer harboring KRAS mutations, the most mutations occur in codon 12, whereas mutations in codons 13 and 61 are less frequent (4).
In lung cancer, considerable progress in developing molecularly-driven therapeutics has been made in the past decades, mainly including targeted therapies against oncogenic drivers, such as EGFR, HER2, EML4-ALK, MET, ROS1, and BRAF mutations, and immunotherapies in non-oncogene-driven lung cancer, such as PD1 and PDL1 alterations (5, 6). However, for KRAS-mutant lung cancer, the treatment options are still limited, and chemotherapies remain the first-line recommendation.
In this review, we update the recent clinically relevant aspects of the pathobiology of KRAS-mutant non-small cell lung cancer (NSCLC), mainly focusing on tumor heterogeneity, therapeutic implications, and new treatment opportunities.
Heterogeneity in KRAS-Mutant Lung Cancer
Diverse Point Mutations in KRAS
In lung cancer, KRAS mutations occur primarily in adenocarcinoma, whereas they are only rarely seen in squamous cell carcinoma (Figure 1). Diverse point mutations exist within KRAS, the majority of which affect codon 12 of the protein in NSCLC (Figure 1), leading to amino acid substitutions that impair the intrinsic hydrolytic activity and render the KRAS oncoprotein constitutively active.
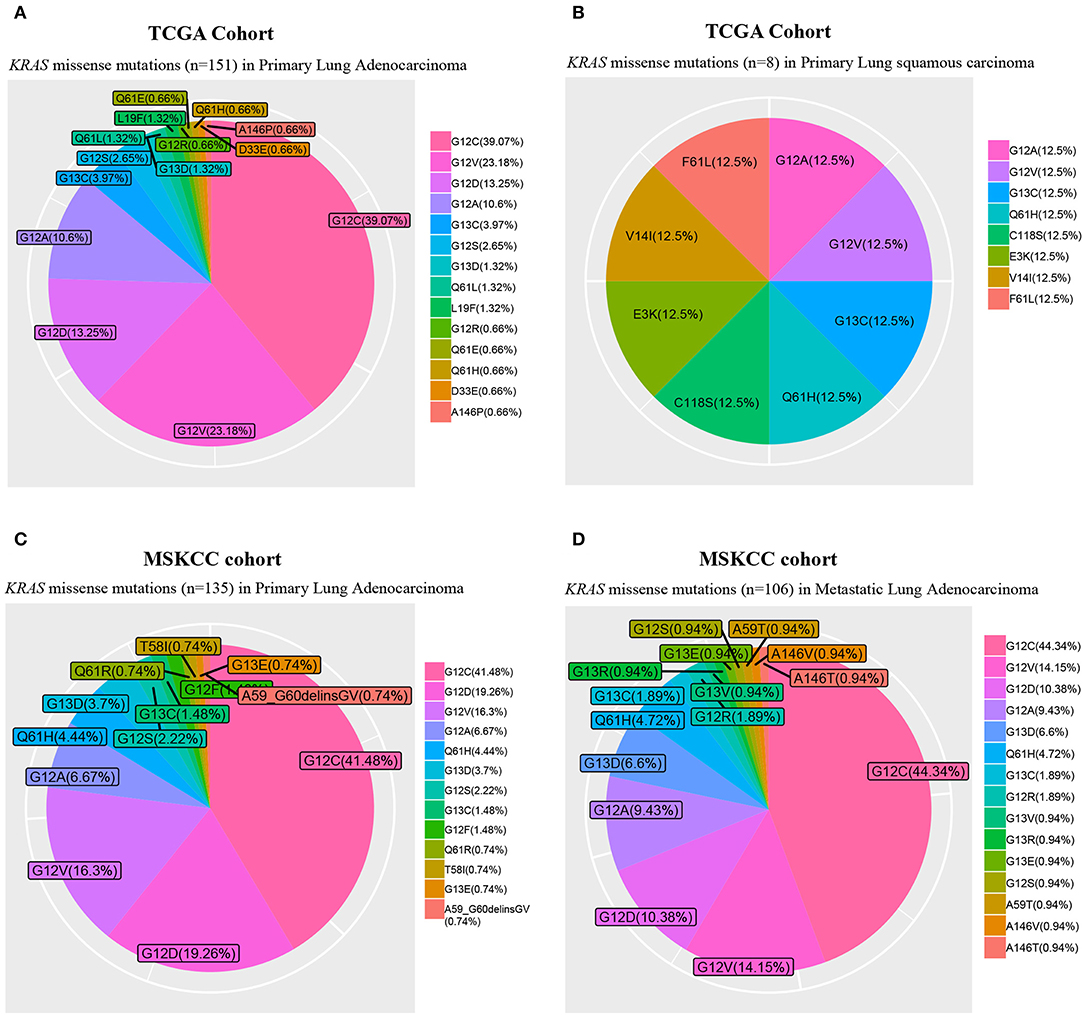
Figure 1. Frequency of KRAS missense mutations. The Cancer Genome Atlas (TCGA) primary lung adenocarcinoma (n = 489; A) and squamous cell carcinoma (n = 492; B) cohorts, and Memorial Sloan Kettering Cancer Center (MSKCC) primary (n = 471; C) and metastatic lung adenocarcinoma (n = 444; D) cohorts.
In lung cancer, the presence of KRAS amino acid substitution influences patients' prognosis and is negatively associated with patient response to targeted therapy (7, 8) and chemotherapy (9–11). Molecular modeling studies showed that different conformations imposed by distinct KRAS oncogene substitutions could lead to altered association with downstream signaling transducers (12). Specifically, compared to wild-type KRAS, the mutant KRASG12C or KRASG12V is less dependent on AKT, which, however, is more intimately engaged by other mutant KRAS proteins.
Mutant KRAS with different amino acid substitutions may also associate with distinct biological behavior (13) and can lead to different clinical outcomes (14–16). In KRAS-mutant lung cancer, tumors carrying KRASG12C exhibited higher ERK1/2 phosphorylation than those with KRASG12D (17). In supporting this observation, studies with genetically engineered mouse model showed that KrasG12C tumors were significantly more sensitive to MEK inhibitor than the KrasG12D ones and that MEK inhibition significantly increased chemotherapeutic efficacy and progression-free survival (PFS) of KRASG12C mice.
Taken together, different amino acid substitutions in oncogenic KRAS lead to heterogeneity in biological behaviors of the mutant protein, implying the need of genotype-specific analysis to identify clinically relevant subgroups of patients that may ultimately influence treatment decisions. It also should be taken into account for different downstream signaling pathways to be inhibited for patients with tumors carrying different KRAS amino acid substitutions.
KRAS Dependence Score and EMT
The concept of KRAS dependence or independence was proposed based on the observations that in both patients and cell lines, tumors frequently exhibit unexplained intrinsic resistance to KRAS-targeted therapy, by either inhibitors or genetic ablations. Mutant KRAS has been considered as an oncogenic driver. However, whether it is indispensable in each tumor carrying this oncogene is not clear. Early evidence suggested that not all KRAS-mutant tumor cells are dependent on KRAS (18, 19), and that some KRAS-mutant cancer cells, including lung (20) and pancreatic cancer cells (21, 22), can survive in the absence of the KRAS oncogene. These observations provide additional layers of evidence that make targeting KRAS-mutant tumors more complex.
Oncogenic KRAS can activate various downstream effector pathways, and the best characterized are phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) (23–25). Oncogenic KRAS signaling proceeded by different downstream effectors may lead to phenotypic variance in cancer, but to what extent the downstream effectors contribute to the oncogenic phenotype is not fully understood. Recently, Yuan et al. designed a combinatorial siRNA-based approach to functionally discern the link between KRAS downstream effectors and phenotypic variation in a large panel of cancer cell lines, and identified two major subtypes within KRAS-mutant cancers based on the dependence on KRAS or RSK (Ribosomal Protein S6 Kinase A1) (25). Interestingly, besides the distinct morphologies and effector landscapes, the two subtypes also differ in metabolic status with therapeutically tractable vulnerabilities. The heterogeneity in effector signaling pathways across KRAS-mutant cells presents a significant challenge to identify universal synthetic partners lethal to mutant KRAS.
It is well-documented that the epithelial-to-mesenchymal transition (EMT) process is closely related to therapy resistance. Interestingly, KRAS-mutant cancer cells dependent on or addicted to KRAS oncogene are more associated with an epithelial phenotype, whereas those independent of KRAS adopt a mesenchymal phenotype (18). Importantly, KRAS-mutant cancer cells differing in EMT status vary in their responses to MEK inhibitors (26), as EMT rewires the expression of receptor tyrosine kinases (RTKs), a consequence of differential feedback activation of the MAPK pathway following MEK inhibition. In epithelial-like cancer cells, ERBB3 is preferentially activated by feedback signaling, which reactivates MEK and AKT signaling. In mesenchymal-like KRAS-mutant cancer cells, reactivation of MEK and AKT was dominantly driven by FGFR1. Signaling transduced by FGFR is normally suppressed by the sprouty proteins (SPRY4), but MEK inhibition represses the negative regulation of SPRY4. In line with this, another independent study using short hairpin (sh) RNA screen had similar findings in KRAS-mutant lung and pancreatic cancer cells (27). These findings provide a strong therapeutic rationale to treat epithelial KRAS-mutant lung cancer (high epithelial markers) with clinically available ERBB and MEK inhibitors, and mesenchymal-like KRAS-mutant lung cancers (high FGFR1) by combined therapy with FGFR and MEK inhibitors.
The association of tumor response to MEK inhibitor therapy to EMT status of cancer cells was further investigated by a more recent study (28). Peng et al. identified an inverse correlation between MAPK signaling dependency and a zinc finger E-box binding homeobox 1 (ZEB1)–mediated EMT in patient samples harboring KRAS, BRAF, or NRAS mutations. Mechanistic results indicated that MAPK dependency is dictated by the functional interplay between scaffold protein interleukin-17 receptor D (IL17RD) and ZEB1. Mechanistically, in mesenchymal-like KRAS-mutant lung cancer cells, ZEB1 directly represses IL17RD to mediate the resistance to MEK inhibitors. Based on this, ZEB1 suppression by miR-200 expression or histone deacetylase inhibitor (mocetinostat) re-sensitized mesenchymal cells to MEK inhibition and markedly reduced in vivo tumor growth. This study provided the mechanistic support for combinatorial treatment (MEK plus histone deacetylase inhibitors) for KRAS-mutant lung cancer, and, again, highlighted the importance of stratification of epithelial and mesenchymal subsets in decision-making for treating KRAS-mutant lung cancer.
Genetic Alterations Co-occurring With KRAS Mutations
Heterogeneity in KRAS-mutant tumors also arises from co-occurring alterations of other genes, e.g., TP53, CDKN2A/2B, STK11, and KEAP1 (Figure 2). Compelling evidence showed that co-occurring genomic changes could profoundly affect biological behaviors (29–31), clinical outcomes (32), and therapeutic vulnerabilities of KRAS-mutant cancers (33, 34).
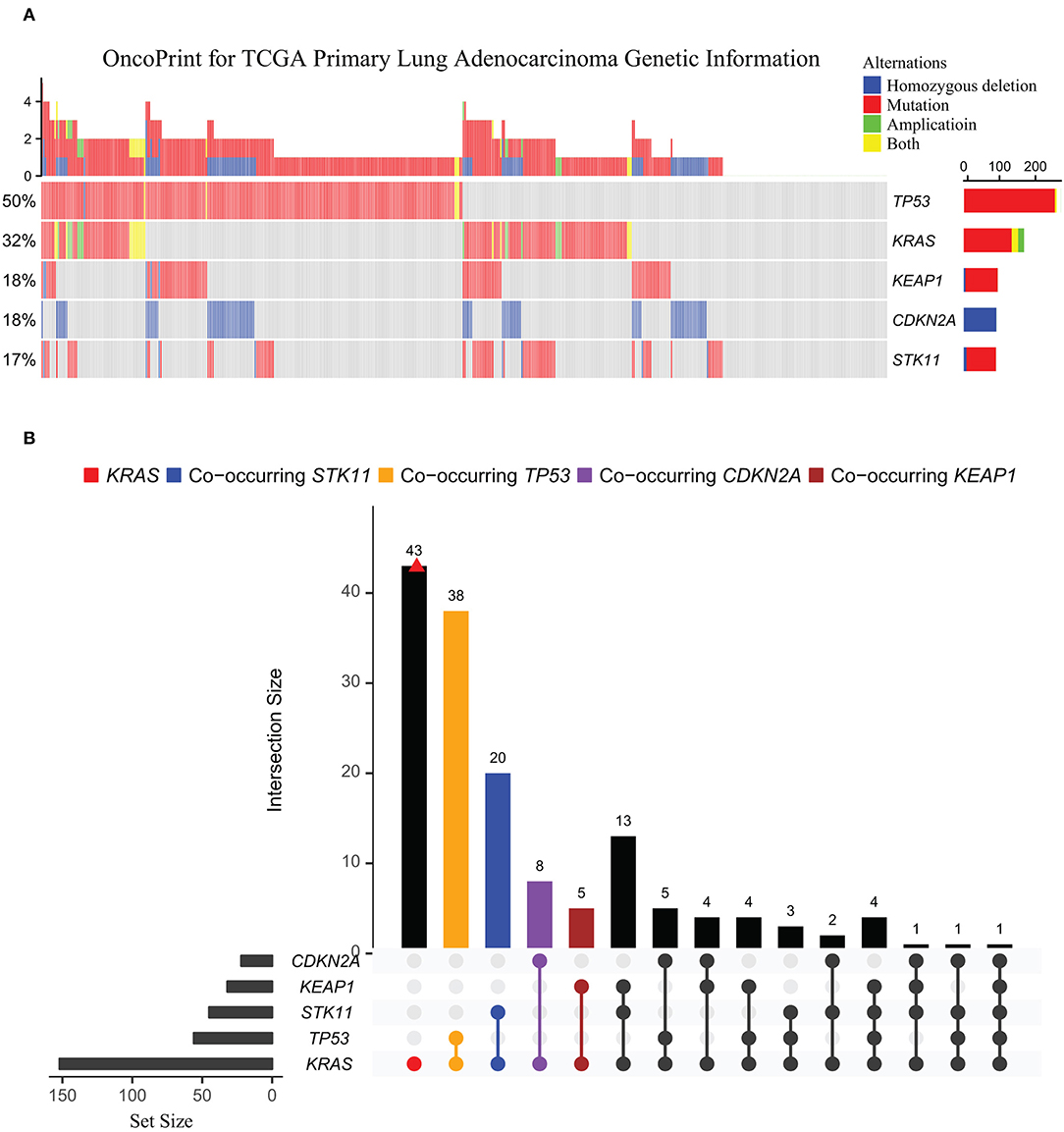
Figure 2. Oncoprint for common genetic alterations co-occurring with KRAS in TCGA lung adenocarcinoma cohort. (A) The genetic landscape of KRAS, TP53, KEAP1, CDKN2A, and STK11 in TCGA primary lung adenocarcinoma cohort (n = 489). (B) UpSetR plot visualizing the intersections of other mutations co-occurring with KRAS-mutant across TCGA lung adenocarcinomas. The intersection points are indicated by different colors: CDKN2A (purple), KEAP1 (brown), STK11 (blue), and TP53 (orange). Intersections among the co-occurring genes were connected with a line, with frequency of each co-occurring mutation type shown on the bar plot. Set metadata is shown to the left (charts).
An integrative study of genomics, transcriptomics, and proteomics in early-stage and chemo-refractory KRAS-mutant lung adenocarcinomas identified three major subsets defined by co-occurring genetic alterations in STK11/LKB1 (KL subgroup), TP53 (KP subgroup), and CDKN2A/B (KC subgroup) (29). The three subgroups differ in biological properties and therapeutic vulnerabilities, with KC tumors associated with suppressed mTORC1 signaling and KL tumors with lower expression of immune markers (e.g., PD-L1) if KEAP1 co-mutated, while higher levels of somatic mutations, inflammatory and immune checkpoint markers, and prolonged relapse-free survival were observed in KP tumors. Further, KL cells exhibited heightened vulnerability to HSP90 inhibition. This work argued that genomic alterations co-occurring with mutant KRAS stratify lung adenocarcinomas and define pathobiological properties and therapeutic vulnerabilities.
A more recent study of a large patient cohort (n = 330) with advanced KRAS-mutant lung cancer identified co-mutated KEAP1 as an independent prognostic marker for poorer survival [HR = 1.96; P < 0.001] and as being associated with less response to chemotherapy [HR = 1.64; P = 0.03] and immune therapy [HR = 3.54; P = 0.003] (30). Another study showed that presence of co-mutated Trp53 reduces sensitivity to combined treatment with MEK inhibitor and chemotherapy in KrasG12C-driven murine lung cancer, which supports further clinical investigations of the combination therapy for patients with lung cancer harboring KRASG12C and wild-type p53 (17). Finally, yet importantly, STK11/LKB1 alterations have been described as a major driver of primary resistance to PD-1 blockade in KRAS-mutant lung adenocarcinoma (31, 35).
Supporting this, a recent study (36) showed that among 377 non-squamous NSCLC patients treated with platinum-doublet chemotherapy (carboplatin or cisplatin and pemetrexed) plus pembrolizumab (anti-PD1), the therapy response was significantly associated with the genetic status of STK11. Specifically, patients with genomic alterations of STK11 (N = 102) were associated with significantly shorter PFS (4.8 vs. 7.2 months, HR 1.5, 95% CI 1.1–2.0; P = 0.0063) and shorter overall survival (10.6 vs. 16.7 months, HR 1.58, 95% CI 1.09–2.27; P = 0.0083) compared with patients without STK11 alteration (N = 275). Also, the objective response rate (RR) was significantly different between the two groups (32.6 vs. 44.7%, P = 0.049). More importantly, the addition of pembrolizumab to platinum-doublet chemotherapy did not significantly improve PFS (4.8 vs. 4.3 months, HR 1.13, 95% CI 0.83–1.54, P = 0.75) or overall survival (10.6 vs. 10.3 months, HR 1.03, 95% CI 0.71–1.49, P = 0.79) compared to the chemotherapy alone (36). This study defines a subgroup of patients with STK11 alterations who do not benefit from immunotherapy, indicating the importance of cancer genetic information for stratification of patients who would benefit from immune checkpoint blockade. Apparently, co-occurring alterations further increase the heterogeneous complexity, which may explain inconsistent outcomes of clinical trials with KRAS-mutant lung cancers.
New Horizons for Treating KRAS-Mutant Lung Cancer
Refocusing on Direct Targeting of KRAS
For decades, KRAS was considered undruggable due to its high affinity for GTP and the lack of a clear binding pocket. Enormous attempts and efforts had been made, but all failed to identify compounds that could effectively and directly target mutant RAS. Since then, there has been little advance. However, with new technologies in drug development and novel mechanistic insights into RAS biology, attention has been refocused on the approach that directly interferes with the function of RAS oncoproteins, with more effort given to find the way to target mutant alleles specifically.
Earlier studies have identified small molecules selectively recognizing and irreversibly inactivating one specific KRAS-mutant allele harboring a G12C amino acid substitution (37, 38). A breakthrough of direct RAS targeting was finally made by Ostrem et al., who, by using a novel screening technology called tethering, developed a new strategy to target mutant KRASG12C specifically without affecting the wild-type protein (37). This work also suggested that the previous perception of mutant KRAS was persistently locked in its active GTP-bound state might not be true.
Later on, Lim et al. reported the synthesis of a GDP analog, SML-8-73-1, which contains an electrophilic chloroacetamide attached to the β-phosphate. This analog can covalently modify cysteine 12 of KRasG12C and, as a result, it competes with GTP and GDP for active site binding in a cellular context (38). Despite the pioneering development of the KRASG12C-specific inhibitors, follow-up studies indicated that these initial compounds showed only limited potency (39, 40). In a search for more effective compounds or analogs, ARS853 was developed (40), which selectively reduced KRAS-GTP levels by more than 90% and increased the in vitro hydrolytic reaction rate by 600-fold compared to the initial compound used in Ostrem et al. (37). At the micromolar range, ARS853 potently suppressed MAPK and PI3K-AKT signaling. Thus, KRASG12C mutant protein is in a dynamically rather than a statically active state and targeting the inactive, GDP-bound form of KRAS is a realistic and promising anti-RAS therapeutic. These striking findings were recently translated into in vivo studies, in which a new covalent KRASG12C-specific inhibitor, ARS-1620, showed rapid and durable tumor regression in mice (41).
These studies prompt a revisit to target KRAS oncoproteins directly. Recent discoveries have enabled further development and investigation of more compounds of this family in clinical trials (Table 1). Encouraging phase I clinical trial data of AMG510 (Amgen, clinical trial information: NCT03600883) in 32 patients with KRASG12C mutation (14 with NSCLC, 19 with colorectal cancer, and 2 with appendix cancer) were just released in ASCO 2019. Five of 10 evaluable patients with NSCLC had a partial response, and four had stable disease, in total achieving a disease control rate of 90% (9/10). Additionally, 13 of 18 evaluable patients with colorectal cancer experienced stable disease. Twenty-six patients were still under study, and nine discontinued. Importantly, the treatment was well-tolerated, with primarily grade 1 events (68%). Two grade 3 treatment-related AEs were reported (anemia and diarrhea). No grade 4 or more severe treatment-related adverse effects were reported. MRTX849 is another potent, highly selective, and orally available small-molecule inhibitor of KRASG12C (42). MRTX849 shows broad-spectrum anti-tumor activity in a panel of patient- and cell-derived in vivo tumor models with KRAS G12C-substitution, with complete tumor regression observed in a subset of these models.
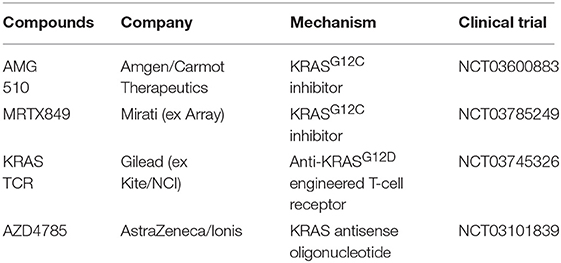
Table 1. Ongoing clinical trials involving direct targeting of KRAS.
Different from the inhibitors that directly target mutant KRAS, AZD4785 is a KRAS antisense oligonucleotide that targets the KRAS gene irrespective of its mutation status (43). Despite AZD4785 being safe and well-tolerated, the first phase I trial failed, which might be due to the fact that AZD4785 targets both mutant and wild-type KRAS mRNA for degradation.
Tran and colleagues described a case of a patient with metastatic colorectal cancer treated with autologous T cells specific for mutant KRASG12D, which was restricted to the major histocompatibility complex class I allele HLA-C*08:02 (44). Despite the rarity of HLA-C*08:02, this study demonstrated the promise of T-cell–based immunotherapy for targeting KRASG12D and HLA-C*08:02. Further evaluation in more patients is warranted.
Whether direct inhibition of KRAS with these new compounds is sufficient remains a question, given the presence of KRAS independence in tumor cells harboring KRAS mutations. Concurrent inhibition of collateral dependencies may be required to potentiate the effectiveness of those compounds.
Reinforcing MEK Inhibitors
To date, most efforts to treat cancers with RAS mutations have focused on targeting downstream effectors of mutant RAS, such as RAF, MEK, or PI3K, each of which is druggable. Although, as described above, different KRAS mutations show a preference for activating different downstream signaling, hyperactivation of the mitogen-activated protein kinase (MAPK) pathway is generally recognized as a key feature in KRAS-driven lung cancer cells. One reason is that the G12C substitution (44%), the most common subtype in KRAS-mutant lung cancer, shows more prominent engagement with MAPK signaling. Supporting these findings, we performed pooled drug sensitivity analysis based on publicly available dataset in Genomics of Drug Sensitivity in Cancer, which revealed that, compared with KRAS-wild-type lung cancer cells, KRAS-mutant lung cancer cells are exclusively more sensitive to various MEK inhibitors rather than those targeting other oncogenic pathways (Figure 3). This explains why MEK inhibitors have been the most widely investigated, typically as a combination therapy, despite the presence of multiple inhibitors that are being explored to target different KRAS-activated pathways.
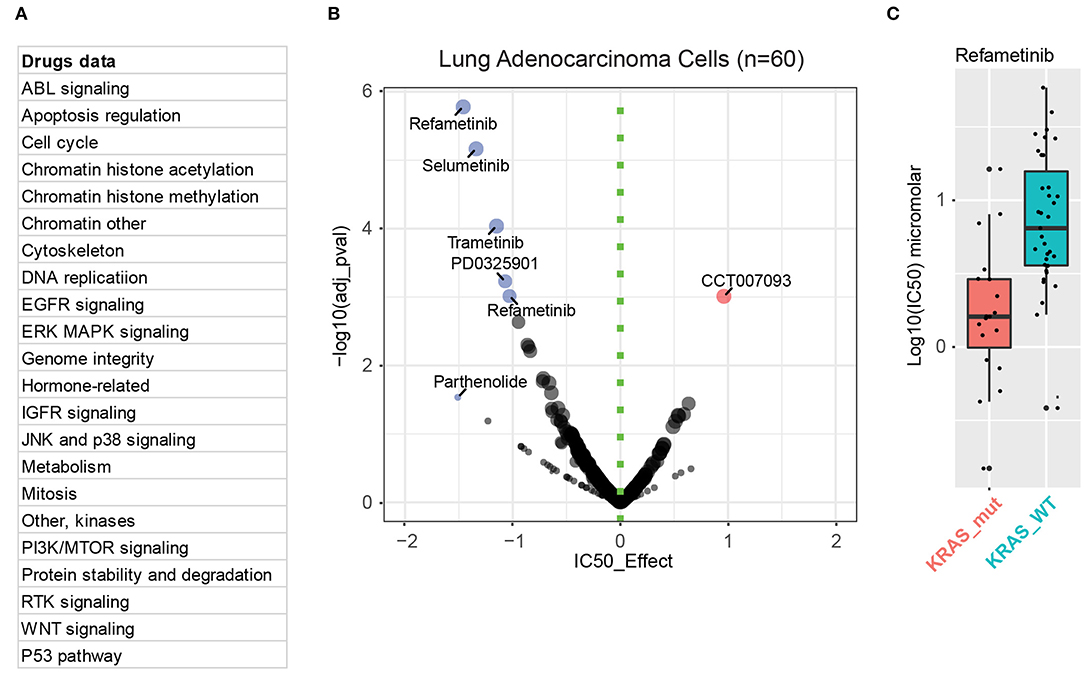
Figure 3. MEK mediates the key downstream effect in KRAS-mutant lung cancer cells. (A) Drug information incorporated in the Genomics of Drug Sensitivity in Cancer database. (B) Integrative analysis of drug sensitivity data of KRAS-mutant (n = 20) lung adenocarcinoma cells compared to KRAS-wild-type (WT; n = 40) ones. In the volcano plot, the x-axis indicates the IC50 effect, with effect < 0 representing KRAS-mutation sensitive inhibitors (in blue) compared with KRAS-WT ones. The color intensity and the circle size are proportional to significance value (the y-axis). (C) Sensitivity analysis of KRAS-mutant (in red) and WT (in blue) cells to one of the MEK inhibitors, refametinib.
Rethinking Combination Treatment With Chemotherapy
In the clinic, conventional chemotherapy is widely used to treat patients with KRAS-mutant NSCLC, although chemotherapy plus immune checkpoint blockade has been recently approved as the first-line regimen for NSCLC, including patients with KRAS mutations (45, 46). Clinical trials evaluating the efficacy of selumetinib, a potent MEK inhibitor, to potentiate chemotherapy, have recently been conducted.
A survival benefit of selumetinib plus docetaxel in comparison with docetaxel alone was demonstrated in a phase II clinical trial (47). Specifically, the primary endpoint of the study—median overall survival—was 9.4 months for the combination (selumetinib plus docetaxel) compared with 5.2 months for the control (placebo and docetaxel), although this difference was not statistically significant [hazard ratio (HR) for death 0·80, 80% CI 0·56–1·14; one-sided p = 0·21]. The median PFS was significantly improved in patients receiving selumetinib (5.3 vs. 2.1 months; HR for progression 0·58, 80% CI 0·42–0·79; one-sided p = 0·014) as was the response rate (37% vs. 0%; p < 0·0001). Subsequently, a subgroup analysis demonstrated that patients harboring G12V and G12C KRAS mutations appeared to experience higher RR and longer PFS for the combination arm, which was recently confirmed in preclinical mouse model (17).
Following this encouraging result, a further phase III clinical trial in KRAS-mutant lung cancer was conducted, which, however, failed to reproduce the significant benefit in patients treated with the combination compared with docetaxel alone (48). In this larger cohort trial, median PFS was 3.9 months with the combination group and 2.8 months with the control group (difference, 1.1 months; HR, 0.93 [95% CI, 0.77–1.12]; P = 0.44). Median overall survival was 8.7 and 7.9 months, respectively (difference, 0.9 months; HR, 1.05 [95% CI, 0.85–1.30]; P = 0.64). There is a marginally significant objective RR (20.1% in combination group vs. 13.7% in the control group; odds ratio, 1.61 [95% CI, 1.00–2.62]; P = 0.05). Whereas, the inconsistency with previous phase II trial results was unclear, a multitude of possible mechanisms, such as the aforementioned genomic (diverse point mutations and co-occurring alterations) and phenotypic (different EMT status) heterogeneity within the recruited patients, might be anticipated. In addition, chemotherapeutics used for combination treatment may need to be reconsidered in future studies, given that distinct amino acid substitutions of KRAS oncoproteins differed in their response to the commonly-used chemotherapy agents (9, 11). Nonetheless, these results did not rule out the effectiveness of this combination therapy in a subgroup of patients with KRAS mutations and deserved more detailed analysis, which might provide mechanistic information that facilitates patient stratification and prediction of potential responders.
Trametinib is another selective and potent MEK inhibitor that has been clinically approved for BRAF mutant cancers (mainly melanoma). Like selumetinib, the efficacy of trametinib, alone or in combination with docetaxel, has been evaluated in KRAS-mutant NSCLC. In a phase II trial, trametinib as a monotherapy showed RR and PFS similar to docetaxel in previously treated KRAS-mutant NSCLC (49). Another phase II study with KRAS-mutant NSCLC (n = 54, including 19 with G12C, 9 with G12D, 9 with G12A) documented a trend toward worse PFS (HR = 1.86, p = 0.06) and survival (HR = 1.80, p = 0.14) in G12C patients compared to non-G12C patients (50). Trametinib plus docetaxel had a RR of 33% and median survival of 11.1 months in patients with recurrent KRAS-mutant NSCLC. These results suggest that clinical responses to combined trametinib and docetaxel may differ between G12C and non-G12C patients.
Rewiring SHP2 Activities
SHP2 is a non-receptor protein tyrosine phosphatase, encoded by the PTPN11 gene that is ubiquitously expressed. SHP2 is involved in signal transduction downstream of multiple growth factors, cytokine, and integrin receptors and, not surprisingly, functions as an essential player in oncogenesis (51, 52). Upon the activation of RTKs, the SH2 domain of SHP2 binds to the phosphorylated tyrosine residues and various substrates, such as RTKs, scaffolds and adaptor proteins, which enables SHP2 in its active state for enzymatical removal of phosphates (dephosphorylation) from the substrates.
Previous studies demonstrated that the adaptive reactivation of MAPK signaling in the presence of a MEK inhibitor was mediated by the loss of MAPK-dependent negative feedback loops and the consequent induction of RTKs signaling (26, 53). Recent studies in KRAS-mutant lung cancer (54, 55) and pancreatic adenocarcinoma (55), KRAS-amplified gastric carcinoma (56), and multiple other cancer models expressing mutant or wild-type KRAS (57, 58) revealed that the anti-tumor effect of MEK inhibitor treatment could be dramatically potentiated by concurrent SHP2 inhibition.
Specifically, targeting of MEK alone is frequently hampered by adaptive resistance, which is complex and context-dependent, and can involve activation of various RTKs, including ERBB family, AXL, PDGFR-α, or FGFR1 (26, 53, 59–62). Strikingly, SHP2, the key integrator of RTK-RAS signaling, was necessary for various contexts upon MEK blockade and required to re-establish MAPK signaling. Strong synergy was observed when SHP2 and MEK were simultaneously targeted, resulting in sustained inhibition of tumor growth in different cancer models.
These studies provided compelling evidence supporting further investigations of combining SHP2 and MEK inhibitors for patients with KRAS-mutant cancer. Fitting this tendency, the recent development of potent allosteric SHP2 inhibitors strengthens the interest in targeting SHP2 in cancer (63). A clinical trial (NCT03114319) investigating TNO155, an SHP2 inhibitor, in patients with K-/N-/H-RAS, BRAF, or PTPN11 mutant tumors is ongoing.
Resurging Autophagy Inhibition
Potential therapeutic interventions to inhibit autophagy have been extensively studied in cancer. Tumor cells depend on macroautophagy to cope with oncogene-induced metabolic stress. Notably, in human cancer cell lines or tumors bearing KRAS mutations, high levels of basal autophagy were observed, making inhibition of autophagy therapeutically actionable in KRAS-driven tumors (64, 65).
Three simultaneously published studies signal a resurgence of interest to inhibit autophagy in KRAS-driven cancer (66–68). These studies indicated that upon the inhibition of the MAPK pathway, KRAS-mutant tumors depend on autophagy for survival and that, as a result, blocking this protective mechanism by concomitant inhibition of autophagy and MEK or ERK kinases is likely to be therapeutically beneficial in patients with KRAS-mutant pancreatic ductal adenocarcinoma, NRAS-mutant melanoma, and BRAF-mutant colorectal cancer (66, 67). More importantly, Kinsey et al. initiated off-label treatment for a patient with metastatic PDAC with trametinib and hydroxychloroquine, both of which have been clinically approved for other indications. They observed a striking disease response with a 50% reduction in tumor burden without toxicity (67).
Based on the intriguing findings, further clinical investigations are required to determine the benefits of combined MEK and autophagy for patients with activating mutations in the RAS–RAF–MEK–ERK pathway.
Rewiring KRAS Activation
In normal cases, KRAS is activated in response to signaling from upstream RTKs. However, oncogenic KRAS mutations “lock” the protein in a constitutively active state, activating KRAS-dependent signaling in a RTKs-independent pattern. In line with this, clinical trials confirmed that patients with KRAS-mutant cancers generally have a poor response to the first generation of EGFR inhibitors, such as erlotinib and gefitinib, and the presence of KRAS mutations is used as a biomarker to exclude patients for EGFR inhibitor therapy (7, 8, 69).
However, recent studies have challenged this paradigm, which instead demonstrated that the activation of ERBB signaling was required for KRASG12D-driven lung tumorigenesis in preclinical mice models and that pan-ERBB inhibition other than EGFR inhibition alone was strikingly effective to inhibit KRAS-mutant tumor growth and progression (61, 62).
In humans, the ERBB family contains HER1 (EGFR, ERBB1), HER2 (NEU, ERBB2), HER3 (ERBB3), and HER (ERBB4). Previous studies showed that ERBB3 activation was associated with resistance to MEK inhibition in KRAS-mutant NSCLC cells (26, 59). In a recent study by Kruspig et al. (61), the authors showed that multiple ERBB ligands (e.g., Areg, Ereg, Nrg3, Nrg4, and Hbegf) and receptors (for example, Erbb2 and Erbb3) were highly expressed in a KRASG12D mouse model. Neratinib, a multi-ERBB inhibitor (70, 71), almost completely suppressed the emergence of tumors. In sharp contrast, erlotinib failed to reproduce the same effect. Further mechanistic analyses of seemingly contradictory results revealed that ERBB activity establishes a feed-forward loop to amplify signaling through the core RAS-ERK cascade to sustain survival and proliferation in KRAS-mutant NSCLC. Indeed, pan-ERBB inhibition enhanced the potency of MEK inhibition in vitro and in vivo.
Similarly, an independent study by Moll et al. demonstrated a requirement for ERBB signaling to support the progression of KRASG12D-driven lung cancer (62). In this study, an independent pan-ERBB inhibitor, afatinib, was used. Genetic mouse models revealed that EGFR deletion attenuates mutant KRAS activity and transiently reduces tumor growth. However, EGFR inhibition initiated a rapid resistance mechanism involving non-EGFR ERBB family members, which triggered a tumor escape mechanism. This provided an explanation, at least to some extent, for the poor unresponsiveness of KRAS-mutant lung cancer patients to the first-generation TKIs targeting EGFR alone. More importantly, afatinib blocked compensatory ERBB2 and ERBB3 activation, whereas erlotinib and gefitinib did not. Together, these studies suggested the therapeutic potential of pan-ERBB inhibitors in KRAS-driven tumors.
Notably, both studies revealed a requirement for simultaneous inhibition of multiple ERBB while targeting a single member of the family was not effective. These preclinical studies provide new insights into KRAS-driven tumorigenesis and bring new hope for KRAS-mutant lung cancer patients. Further trials, such as combined MEK with pan-ERBB inhibitors, are highly needed to determine the translational significance of the pan-ERBB inhibition strategy for patients, which can be easily and quickly conducted given that both inhibitors have been clinically approved (72, 73).
Re-examining Downstream Partnership: CRAF (RAF1) but Not A-RAF or B-RAF
KRAS oncogenes signal through a cascade of downstream effectors, among which the most important one is the RAF/MEK/ERK cascade. The direct RAS downstream effectors within the RAF/MEK/ERK pathway are the RAF kinases, including A-, B-, and C-RAF. However, it is not well-known how these individual RAF kinases contribute to KRAS-mutant tumor initiation and development.
Recent studies showed that C-RAF rather than B-RAF plays a crucial role in mediating KRAS oncogenic signaling (74, 75). Targeting of C-Raf rather than of B-Raf kinase could recapitulate the effect of Kras ablation and effectively inhibit tumor development without inducing significant toxicities in mouse models of Kras/Trp53-mutant lung adenocarcinoma. This work suggested that distinct RAF kinases likely play different roles in mediating KRAS oncogenic signaling.
In a more recent study, ablation of B- or C-RAF was concomitant with KrasG12V induction (76). C-Raf ablation completely prevented Kras-driven NSCLC without inducing deleterious effects, which, however, was not the case with B-Raf ablation, indicating that B-Raf is dispensable for Kras oncogenic signaling. Moreover, ablation of C-Raf did not affect Mek or Erk phosphorylation, suggesting that C-Raf-mediated Kras signaling is independent of the MAPK cascade. Further, the same group showed that combined inhibition of C-Raf and Egfr induced complete regression of pancreatic ductal adenocarcinomas in Kras/Trp53-driven GEM models and PDXs without apparent toxicity (77). Together, these studies provided compelling evidence that C-Raf, but not B-Raf or A-Raf, may mediate the oncogenic signaling in KRAS-driven cancer. More importantly, the therapeutic effect observed by ablation of C-Raf was likely due to disrupting the interaction of the C-Raf protein with other partners, such as BCL2, ASK1, MST2, and ROKα (76), whereas not via modulating MAPK cascade that is also essential for normal homeostasis. This might explain, to some extent, why the elimination of C-Raf did not induce systemic toxicity, in contrast to MEK inhibitors.
Notably, the therapeutic effect achieved by C-Raf ablation could not be reproduced by three C-Raf inhibitors that are designed to block the kinase activity other than the protein expression, confirming that the non-kinase activity of C-RAF instead of the conventional MAPK cascade is critical for the ability of KRAS-dependent oncogenic transformation. The striking finding of these studies, which is at odds with the currently ongoing efforts to develop C-RAF kinase inhibitors, implies, instead, the need for strategies to block C-RAF kinase-independent activities or induce its degradation.
Revitalizing Chemotherapy
Currently, the platinum-based chemotherapy is still widely used for patients with KRAS-mutant lung cancer. However, the efficacy of chemotherapy is very limited, and durable response is generally short. Considerable efforts have been made to potentiate the efficacy of chemotherapy in KRAS-mutant cancer. Unfortunately, a recent phase III study has once again frustrated this attempt, which showed no additional survival benefit from combined MEK inhibitors compared to docetaxel alone (48).
Oncogenic KRAS signaling also involves PI3K-AKT-mTOR, via the interaction with the catalytic subunits of PI3K (78–80). Blocking RAS-mediated PI3K activation has also shown to inhibit the progression of KRAS-driven tumors. However, high toxicities of targeting PI3K, AKT or mTOR, in combination with MEK inhibitors, have prevented their approval for use in human patients (81–83).
We recently found that activation of mTOR signaling mediates a key resistance mechanism to chemotherapy in KRAS-mutant lung cancer (84). We observed exclusively hyperactivated mTOR signaling in lung cancer patient samples with KRAS-mutations but not in those carrying wild-type KRAS. Combined clinically approved mTOR inhibitor and chemotherapy showed a strong synergism in inhibiting proliferation of cancer cells harboring KRAS-mutation specifically. Additionally, the efficacy of this combination treatment correlates with the magnitude of mTOR activity induced by chemotherapy alone. Our results pinpoint a rational and readily translatable strategy that combines mTOR inhibitors with standard chemotherapy to treat KRAS-mutant lung cancer.
A recently published study provided novel hints to potentiate platinum-based chemotherapy in multiple cancer types (85). Jin et al. used multi-step kinome screens and identified MAST1, an AGC serine/threonine protein, as a key mediator of cisplatin resistance. The mechanistic study showed that MAST1 expression was increased in resistant cells and functioned as a MAP3K (MAPK kinase kinase), thereby activating MEK1 in cisplatin-resistant cells. Knockdown of MAST1 re-sensitized the resistant cells to cisplatin in vitro and in vivo. Further investigations showed that cisplatin directly binds to cysteine 142 site of MEK1, restricting its access to C-RAF that typically phosphorylates MEK1. In this case, MAST1 took over C-RAF to re-activate MAPK cascade in cisplatin-resistant cells, and inhibition of MAST1 led to decreased MEK1 phosphorylation, explaining the effectiveness of targeting MAST1 in overcoming cisplatin resistance. More interestingly, MAST1 expression was shown to be specific to cisplatin rather than other similar agents (for instance, 5-fluorouracil) that interfere with DNA replication. This finding may be particularly relevant in the oncogenic RAS/BRAF setting, which mainly activates downstream MAPK signaling.
In a recent study (86), we reported that pemetrexed-resistant KRAS-mutant lung cancer cells assume a mesenchymal phenotype and cross-resist MEK inhibitors. Mechanistically, acquisition of resistance enables KRAS-mutant lung cancer cells to bypass canonical KRAS effectors but entail hyperactive AXL/eIF4E, increased protein turnover in the endoplasmic reticulum (ER), and adaptive activation of an ER stress-relief unfolded protein response survival pathway whose integrity is maintained by HSP90. In line with these mechanistic findings, HSP90 inhibitors synergistically enhance antitumor effects of pemetrexed and MEK inhibitors in multiple in vitro and in vivo models, validating a rational combination strategy to treat KRAS-mutant lung cancer.
Reactivating Anti-tumor Immunity
Immune surveillance is generally dormant in cancer via dysregulation of immune checkpoints, such as the upregulation of the immunosuppressive protein PD-L1 for the evasion of the host immune system. Considerably convincing evidence has shown the importance of immune checkpoint blockade for treating various cancers (87). However, the therapeutic efficacy varies individually due to the high heterogeneity of tumors and the lack of reliable biomarkers to stratify the patients. Currently, limited biomarkers, such as PD-L1 expression (88) and tumor mutation load (TMB) (89–91), are clinically used to predict the immunotherapy benefit.
Interestingly, several preclinical studies suggested that tumors harboring KRAS mutations might be associated with a vulnerability to immunotherapies, in particular those with concomitant TP53 mutations (29, 92, 93). Mechanistic studies indicated that oncogenic KRAS could stabilize PD-L1 mRNA through post-transcriptional modifications of the AU-rich element binding protein tristetraprolin (TTP) (94). Specifically, KRAS-MEK signaling contributes to phosphorylation and inhibition of TTP through the kinase MK2. In the same study, a high correlation between MAPK activation and elevated PD-L1 expression was observed in KRAS-mutant human lung and colorectal tumors.
A landmark trial (KEYNOTE-024) demonstrated that pembrolizumab was superior to chemotherapy in advanced NSCLC (potentially including patients with KRAS-mutations), among which more than 50% had high PD-L1 expression (95). Follow-up studies of the KEYNOTE-024 cohort revealed continuous survival benefit in patients treated with pembrolizumab as first-line monotherapy compared to those treated with chemotherapy (96).
Importantly, two remarkable phase III trials (KEYNOTE-189 and KEYNOTE-407) demonstrated that addition of pembrolizumab (Keytruda, anti-PD1) could significantly prolong the survival of NSCLC patients (45, 97). The promising results from these studies have led to the approval of pembrolizumab in combination with chemotherapy (carboplatin-pemetrexed) for metastatic, non-squamous NSCLC, and the approval of pembrolizumab in combination with chemotherapy (carboplatin and Taxol) for patients with squamous lung carcinoma, excluding those carrying EGFR or ALK mutations. Although patients with KRAS-mutant NSCLC were potentially included in the two studies, specific efficacy of the combination therapy on KRAS-mutant NSCLC remains to be investigated.
A recent systematic review and meta-analysis study, which integrated multiple randomized clinical trials, showed that immune checkpoint inhibitors significantly prolonged overall survival in the KRAS-mutant subgroup (HR, 0.65; 95% CI, 0.44–0.97; P = 0.03) but not in the KRAS wild-type one (HR, 0.86; 95% CI, 0.67–1.11; P = 0.24; interaction, P = 0.24) (98). Another meta-analysis study that incorporated 509 patients (138 of KRAS-mutant and 371 with KRAS-wild-type NSCLC) showed that, compared to docetaxel chemotherapy, immune checkpoint inhibitors improved overall survival in patients with previously-treated KRAS-mutant NSCLC (HR = 0.64 [95% confidence interval, 0.43–0.96], P = 0.03) (99), but not in patients with wild-type KRAS (HR = 0.88 [95% confidence interval, 0.68–1.13], P = 0.30). These results indicate that KRAS mutational status is a potential biomarker for survival benefits to immune checkpoint inhibitors. However, two other studies with advanced non-squamous NSCLC reported that the efficacy of immune checkpoint inhibitors is independent of KRAS-mutant status (100, 101). Thus, further studies with a stratification of KRAS genetic status are still needed.
Although, preclinical studies using immune-competent mouse models verified the promising efficacy of checkpoint blockade in the Kras-mutant setting (92, 102, 103), a majority of these studies relied on mouse models with a single genetic background, which limits the power to assess the potential influence of other co-occurring mutations (e.g., STK11 alterations). Co-occurring genetic mutations, which can lead to differential downstream effectors engaged by mutant KRAS, have been reported to significantly affect the tumor immune signatures (25, 29, 31, 35) and responses to immunotherapy (30). This might explain why a pooled analysis of patient response data does not consistently support the association between benefits of immunotherapy and KRAS mutations (98–101).
Conclusions
Targeting KRAS has represented a tremendous unmet clinical need. Nevertheless, the challenge of clinical treatment of KRAS-mutant cancers seems not to be insurmountable. Now, a new wave of attempts is motivated to target KRAS directly, which has long been considered undruggable. A striking response has been achieved with AMG510 in patients with KRASG12C. New treatment strategies based on a deeper understanding of the pathobiology of oncogenic KRAS, such as abolishing C-RAF, blocking the universal rewiring of SHP2, and the protective autophagy in response to MEK inhibitors are highly promising with preliminary success in human patients. Conventional approaches, such as combined chemotherapy and mTOR inhibitors, as well as combined cisplatin and MAST1 inhibitors, are also encouraging but require further investigations in patients. The great success of immunotherapy has been witnessed in the treatment of patients with various tumors, but more evidence is required in cancer patients with KRAS mutations.
Given the presence of a variety of potent and specific chemicals, treating KRAS-mutant lung cancer remains a significant challenge, implying that the problem might be mechanism-related rather than the efficacy of targeting itself. A critical point is a high heterogeneity within KRAS-mutant tumors. To maximize the patient benefit, it cannot be more important than molecularly guided stratification on top of KRAS mutations.
Author Contributions
HY and S-QL wrote the manuscript. RS reviewed the manuscript. R-WP outlined and revised the manuscript.
Funding
This study was funded by grants from the Swiss Cancer League, the Cancer League of the Canton of Bern, and China Scholarship Council.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
2. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. (2013) 24:2371–6. doi: 10.1093/annonc/mdt205
3. Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. (2005) 1756:81–2. doi: 10.1016/j.bbcan.2005.10.001
4. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. (2009) 6:201–5. doi: 10.1513/pats.200809-107LC
5. Cortinovis D, Abbate M, Bidoli P, Capici S, Canova S. Targeted therapies and immunotherapy in non-small-cell lung cancer. Ecancermedicalscience. (2016) 10:648. doi: 10.3332/ecancer.2016.648
6. Nagano T, Tachihara M, Nishimura Y. Molecular mechanisms and targeted therapies including immunotherapy for non-small cell lung cancer. Curr Cancer Drug Targets. (2018) 19:595–630. doi: 10.2174/1568009619666181210114559
7. Rulli E, Marabese M, Torri V, Farina G, Veronese S, Bettini A, et al. Value of KRAS as prognostic or predictive marker in NSCLC: results from the TAILOR trial. Ann Oncol. (2015) 26:2079–84. doi: 10.1093/annonc/mdv318
8. Papadimitrakopoulou V, Lee JJ, Wistuba II, Tsao AS, Fossella FV, Kalhor N, et al. The BATTLE-2 study: a biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol. (2016) 34:3638–47. doi: 10.1200/JCO.2015.66.0084
9. Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. (2011) 22:235–7. doi: 10.1093/annonc/mdq680
10. Park S, Kim JY, Lee SH, Suh B, Keam B, Kim TM, et al. KRAS G12C mutation as a poor prognostic marker of pemetrexed treatment in non-small cell lung cancer. Korean J Intern Med. (2017) 32:514–22. doi: 10.3904/kjim.2015.299
11. Renaud S, Guerrera F, Seitlinger J, Reeb J, Voegeli AC, Legrain M, et al. KRAS-specific amino acid substitutions are associated with different responses to chemotherapy in advanced non-small-cell lung cancer. Clin Lung Cancer. (2018) 19:e919–e931. doi: 10.1016/j.cllc.2018.08.005
12. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. (2012) 104:228–39. doi: 10.1093/jnci/djr523
13. Renaud S, Seitlinger J, Falcoz PE, Schaeffer M, Voegeli AC, Legrain M, et al. Specific KRAS amino acid substitutions and EGFR mutations predict site-specific recurrence and metastasis following non-small-cell lung cancer surgery. Br J Cancer. (2016) 115:346–53. doi: 10.1038/bjc.2016.182
14. Yu HA, Sima CS, Shen R, Kass S, Gainor J, Shaw A, et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J Thorac Oncol. (2015) 10:431–7. doi: 10.1097/JTO.0000000000000432
15. Fu XH, Chen ZT, Wang WH, Fan XJ, Huang Y, Wu XB, et al. KRAS G12V mutation is an adverse prognostic factor of Chinese gastric cancer patients. J Cancer. (2019) 10:821–8. doi: 10.7150/jca.27899
16. Wiesweg M, Kasper S, Worm K, Herold T, Reis H, Sara L, et al. Impact of RAS mutation subtype on clinical outcome-a cross-entity comparison of patients with advanced non-small cell lung cancer and colorectal cancer. Oncogene. (2019) 38:2953–66. doi: 10.1038/s41388-018-0634-0
17. Li S, Liu S, Deng J, Akbay EA, Hai J, Ambrogio C, et al. Assessing therapeutic efficacy of MEK inhibition in a KRAS(G12C)-driven mouse model of lung cancer. Clin Cancer Res. (2018) 24:4854–64. doi: 10.1158/1078-0432.CCR-17-3438
18. Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. (2009) 15:489–500. doi: 10.1016/j.ccr.2009.03.022
19. Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T, et al. Identification of mutant K-Ras-dependent phenotypes using a panel of isogenic cell lines. J Biol Chem. (2013) 288:2403–13. doi: 10.1074/jbc.M112.394130
20. Mou H, Moore J, Malonia SK, Li Y, Ozata DM, Hough S, et al. Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis. Proc Natl Acad Sci USA. (2017) 114:3648–53. doi: 10.1073/pnas.1620861114
21. Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. (2014) 158:185–97. doi: 10.1016/j.cell.2014.06.003
22. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. (2014) 514:628–32. doi: 10.1038/nature13611
23. McCormick F. KRAS as a therapeutic target. Clin Cancer Res. (2015) 21:1797–801. doi: 10.1158/1078-0432.CCR-14-2662
24. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
25. Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, et al. Differential effector engagement by oncogenic KRAS. Cell Rep. (2018) 22:1889–902. doi: 10.1016/j.celrep.2018.01.051
26. Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, et al. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS-mutant lung cancer. Cancer Discov. (2016) 6:754–69. doi: 10.1158/2159-8290.CD-15-1377
27. Manchado E, Weissmueller S, Morris JP IV, Chen CC, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. (2016) 534:647–51. doi: 10.1038/nature18600
28. Peng DH, Kundu ST, Fradette JJ, Diao L, Tong P, Byers LA, et al. ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism. Sci Transl Med. (2019) 11:eaaq1238. doi: 10.1126/scitranslmed.aaq1238
29. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. (2015) 5:860–77. doi: 10.1158/2159-8290.CD-14-1236
30. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res. (2018) 24:334–40. doi: 10.1158/1078-0432.CCR-17-1841
31. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
32. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol. (2019) 14:606–16. doi: 10.1016/j.jtho.2018.12.013
33. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. (2017) 23:1362–8. doi: 10.1038/nm.4407
34. Wang X, Min S, Liu H, Wu N, Liu X, Wang T, et al. Nf1 loss promotes Kras-driven lung adenocarcinoma and results in Psat1-mediated glutamate dependence. EMBO Mol Med. (2019) 11:e9856. doi: 10.15252/emmm.201809856
35. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. (2018) 8:822–35. doi: 10.1158/2159-8290.CD-18-0099
36. Skoulidis F, Arbour KC, Hellmann MD, Patil PD, Marmarelis ME, Awad MM, et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J Clin Oncol. (2019) 37(15 Suppl.):102. doi: 10.1200/JCO.2019.37.15_suppl.102
37. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. (2013) 503:548–51. doi: 10.1038/nature12796
38. Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. (2014) 53:199–204. doi: 10.1002/anie.201307387
39. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. (2016) 351:604–8. doi: 10.1126/science.aad6204
40. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. (2016) 6:316–29. doi: 10.1158/2159-8290.CD-15-1105
41. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. (2018) 172 578–589.e517. doi: 10.1016/j.cell.2018.01.006
42. Papadopoulos KP, Ou S-HI, Johnson ML, Christensen J, Velastegui K, Potvin D, et al. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J Clin Oncol. (2019) 37(15 Suppl.):TPS3161. doi: 10.1200/JCO.2019.37.15_suppl.TPS3161
43. Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. (2017) 9:eaal5253. doi: 10.1126/scitranslmed.aal5253
44. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
45. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
46. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
47. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. (2013) 14:38–47. doi: 10.1016/S1470-2045(12)70489-8
48. Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. (2017) 317:1844–53. doi: 10.1001/jama.2017.3438
49. Blumenschein GR Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol. (2015) 26:894–901. doi: 10.1093/annonc/mdv072
50. Gadgeel SM, Miao J, Riess JW, Mack PC, Gerstner GJ, Burns TF, et al. S1507: phase II study of docetaxel and trametinib in patients with G12C or non-G12C KRAS mutation positive (+) recurrent non-small cell lung cancer (NSCLC). J Clin Oncol. (2019) 37:9021. doi: 10.1200/JCO.2019.37.15_suppl.9021
51. Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. (2007) 109:862–7. doi: 10.1182/blood-2006-07-028829
52. Matozaki T, Murata Y, Saito Y, Okazawa H, Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci. (2009) 100:1786–93. doi: 10.1111/j.1349-7006.2009.01257.x
53. Caunt CJ, Sale MJ, Smith PD, Cook SJ. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. (2015) 15:577–92. doi: 10.1038/nrc4000
54. Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. (2018) 24:961–7. doi: 10.1038/s41591-018-0023-9
55. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. (2018) 24:954–60. doi: 10.1038/s41591-018-0024-8
56. Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. (2018) 24:968–77. doi: 10.1038/s41591-018-0022-x
57. Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. (2018) 8:1237–49. doi: 10.1158/2159-8290.CD-18-0444
58. Lu H, Liu C, Velazquez R, Wang H, Dunkl LM, Kazic-Legueux M, et al. SHP2 inhibition overcomes RTK-mediated pathway re-activation in KRAS mutant tumors treated with MEK inhibitors. Mol Cancer Ther. (2019) 18:1323–34. doi: 10.1158/1535-7163.MCT-18-0852
59. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. (2014) 7:86–93. doi: 10.1016/j.celrep.2014.02.045
60. Boshuizen J, Koopman LA, Krijgsman O, Shahrabi A, van den Heuvel EG, Ligtenberg MA, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat Med. (2018) 24:203–12. doi: 10.1038/nm.4472
61. Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci Transl Med. (2018) 10:eaao2565. doi: 10.1126/scitranslmed.aao2565
62. Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. Afatinib restrains K-RAS-driven lung tumorigenesis. Sci Transl Med. (2018) 10:eaao2301. doi: 10.1126/scitranslmed.aao2301
63. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. (2016) 535:148–52. doi: 10.1038/nature18621
64. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. (2011) 25:460–70. doi: 10.1101/gad.2016311
65. Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci USA. (2016) 113:182–7. doi: 10.1073/pnas.1515617113
66. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. (2019) 25:628–40. doi: 10.1038/s41591-019-0368-8
67. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. (2019) 25:620–7. doi: 10.1038/s41591-019-0367-9
68. Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci USA. (2019) 116:4508–517. doi: 10.1073/pnas.1817494116
69. Cadranel J, Mauguen A, Faller M, Zalcman G, Buisine MP, Westeel V, et al. Impact of systematic EGFR and KRAS mutation evaluation on progression-free survival and overall survival in patients with advanced non-small-cell lung cancer treated by erlotinib in a French prospective cohort (ERMETIC project–part 2). J Thorac Oncol. (2012) 7:1490–502. doi: 10.1097/JTO.0b013e318265b2b5
70. Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. (2010) 28:3076–83. doi: 10.1200/JCO.2009.27.9414
71. Martin M, Holmes FA, Ejlertsen B, Delaloge S, Moy B, Iwata H, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2017) 18:1688–700. doi: 10.1016/S1470-2045(17)30717-9
72. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. (2014) 15:213–22. doi: 10.1016/S1470-2045(13)70604-1
73. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. (2015) 16:141–51. doi: 10.1016/S1470-2045(14)71173-8
74. Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. (2011) 19:652–63. doi: 10.1016/j.ccr.2011.04.002
75. Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov. (2011) 1:128–36. doi: 10.1158/2159-8290.CD-10-0044
76. Sanclemente M, Francoz S, Esteban-Burgos L, Bousquet-Mur E, Djurec M, Lopez-Casas PP, et al. c-RAF ablation induces regression of advanced Kras/Trp53 mutant lung adenocarcinomas by a mechanism independent of MAPK signaling. Cancer Cell. (2018) 33 217–28.e214. doi: 10.1016/j.ccell.2017.12.014
77. Blasco MT, Navas C, Martin-Serrano G, Grana-Castro O, Lechuga CG, Martin-Diaz L, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and C-RAF. Cancer Cell. (2019) 35 573–87.e576. doi: 10.1016/j.ccell.2019.03.002
78. Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. (2011) 2:261–74. doi: 10.1177/1947601911408079
79. Salt MB, Bandyopadhyay S, McCormick F. Epithelial-to-mesenchymal transition rewires the molecular path to PI3K-dependent proliferation. Cancer Discov. (2014) 4:186–99. doi: 10.1158/2159-8290.CD-13-0520
80. Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. (2019) 25:796–807. doi: 10.1158/1078-0432.CCR-18-0368
81. Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. (2015) 75:183–9. doi: 10.1007/s00280-014-2615-5
82. Mita M, Fu S, Piha-Paul SA, Janku F, Mita A, Natale R, et al. Phase I trial of MEK 1/2 inhibitor pimasertib combined with mTOR inhibitor temsirolimus in patients with advanced solid tumors. Invest New Drugs. (2017) 35:616–26. doi: 10.1007/s10637-017-0442-3
83. Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, et al. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. (2018) 119:1471–6. doi: 10.1038/s41416-018-0322-4
84. Liang SQ, Buhrer ED, Berezowska S, Marti TM, Xu D, Froment L, et al. mTOR mediates a mechanism of resistance to chemotherapy and defines a rational combination strategy to treat KRAS-mutant lung cancer. Oncogene. (2019) 38:622–36. doi: 10.1038/s41388-018-0479-6
85. Jin L, Chun J, Pan C, Li D, Lin R, Alesi GN, et al. MAST1 drives cisplatin resistance in human cancers by rewiring cRaf-independent MEK activation. Cancer Cell. (2018) 34 315–30.e317. doi: 10.1016/j.ccell.2018.06.012
86. Yang H, Liang SQ, Xu D, Yang Z, Marti TM, Gao Y, et al. HSP90/AXL/eIF4E-regulated unfolded protein response as an acquired vulnerability in drug-resistant KRAS-mutant lung cancer. Oncogenesis. (2019) 8:45. doi: 10.1038/s41389-019-0158-7
87. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
88. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
89. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
90. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. (2018) 36:633–41. doi: 10.1200/JCO.2017.75.3384
91. Wang Z, Duan J, Cai S, Han M, Dong H, Zhao J, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. (2019) 5:696–702. doi: 10.1001/jamaoncol.2018.7098
92. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2017) 23:3012–24. doi: 10.1158/1078-0432.CCR-16-2554
93. Owada Y, Muto S, Takagi H, Inoue T, Watanabe Y, Yamaura T, et al. Correlation between mutation burden of tumor and immunological/clinical parameters in considering biomarkers of immune checkpoint inhibitors for non-small cell lung cancer (NSCLC). (2017) 35(15 Suppl.):e23184. doi: 10.1200/JCO.2017.35.15_suppl.e23184
94. Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. (2017) 47 1083–99.e1086. doi: 10.1016/j.immuni.2017.11.016
95. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
96. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. (2019) 37:537–46. doi: 10.1200/JCO.18.00149
97. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
98. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. (2018) 4:210–6. doi: 10.1001/jamaoncol.2017.4427
99. Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: a meta-analysis and review. Oncotarget. (2017) 8:48248–52. doi: 10.18632/oncotarget.17594
100. Jeanson A, Tomasini P, Souquet-Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC). J Thorac Oncol. (2019) 14:1095–101. doi: 10.1016/j.jtho.2019.01.011
101. Passiglia F, Cappuzzo F, Alabiso O, Bettini AC, Bidoli P, Chiari R, et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br J Cancer. (2019) 120:57–62. doi: 10.1038/s41416-018-0234-3
102. Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK inhibition improves anti-tumor immunity and T cell function in murine Kras mutant lung cancer. Cell Rep. (2019) 27 806–19.e805. doi: 10.1016/j.celrep.2019.03.066
103. Lee JW, Zhang Y, Eoh KJ, Sharma R, Sanmamed MF, Wu J, et al. The combination of MEK inhibitor with immunomodulatory antibodies targeting programmed death 1 and programmed death ligand 1 results in prolonged survival in Kras/p53-driven lung cancer. J Thorac Oncol. (2019) 14:1046–60. doi: 10.1016/j.jtho.2019.02.004
Keywords: lung cancer, KRAS, mitogen-activated protein kinases, heterogeneity, targeted therapy, immunotherapy
Citation: Yang H, Liang S-Q, Schmid RA and Peng R-W (2019) New Horizons in KRAS-Mutant Lung Cancer: Dawn After Darkness. Front. Oncol. 9:953. doi: 10.3389/fonc.2019.00953
Received: 24 July 2019; Accepted: 09 September 2019;
Published: 25 September 2019.
Edited by:
Alessandro Rimessi, University of Ferrara, ItalyReviewed by:
Noriaki Sunaga, Gunma University, JapanTimothy F. Burns, University of Pittsburgh, United States
Copyright © 2019 Yang, Liang, Schmid and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ren-Wang Peng, cmVud2FuZy5wZW5nJiN4MDAwNDA7aW5zZWwuY2g=