 Yufeng Shang
Yufeng Shang Fuling Zhou
Fuling Zhou- Department of Hematology, Zhongnan Hospital of Wuhan University, Wuhan, China
Recently, due to the application of hematopoietic stem cell transplantation and small molecule inhibitor, the survival of acute leukemia is prolonged. However, the 5 year survival rate remains low due to a high incidence of relapse. Immunotherapy is expected to improve the prognosis of patients with relapsed or refractory hematological malignancies because it does not rely on the cytotoxic mechanisms of conventional therapy. In this paper, the advances of immunotherapy in acute leukemia are reviewed from the aspects of Antibody including Unconjugated antibodies, Antibody-drug conjugate and Bispecific antibody, Chimeric Antigen Receptor (CARs), Immune checkpoint, Natural killer cells. The immunological features, mechanisms and limitation in clinic will be described.
Introduction
Acute leukemia is a kind of hematological malignancy with high mortality and poor prognosis and it requires a complex and highly diversified treatment because of its wide prognostic heterogeneity. Although traditional treatments such as chemotherapy and hematopoietic stem cell transplantation make patients obtain complete remission, patients will eventually develop relapse and resistance, leading to disease progression. The average cure rate was only 35% in adult acute lymphoblastic leukemia (ALL) patients and 40% in aged ≤65 years acute myeloid leukemia (AML) patients relying on traditional treatment (1, 2). Allogenic hematopoietic stem cell transplantation (allo-HSCT) increases the cure rate of ALL to about 50%, and offers significant relapse-free survival and overall survival benefits for intermediate- and poor-risk AML, and also cures a proportion of patients with refractory/relapsed AML, but transplantation related mortality (TRM) has reached 20–30% (1, 3–6). In fact, allo-HSCT is also a form of immunotherapy through graft-vs.-leukemia (GvL) effects by the adoptively transferred donor T-cells, which is seen as the first successful clinical application of immunotherapy. Based on GvL effects, the non-myeloablative allogeneic stem cell transplantation has been developed, thus reducing TRM (7). Some anti-AML drugs such azacytidine and sorafenib have shown to promote GvL effects without increasing the risk of graft-vs.-host disease (GVHD) by regulating immune cells (8–10). Besides, donor lymphocyte infusions (DLI) as a form of pure immunotherapy not only could prevent recurrence after transplantation but also have cured a number of acute leukemia patients (11). Therefore, immune related intervention plays a key role in the treatment of acute leukemia.
In recent years, the emergence of immunotherapy has brought hope to hematological malignancies. Here, we reviewed the research progress of immunotherapy in acute leukemia. Immunotherapy mainly includes targeting AML/ALL cell surface antigen (such as CD33, CD123, CLL-1 on AML and CD20, CD19, CD52 on ALL), relieving T/NK cell immunosuppression (such as PD1/PD-L1, CTLA4), using immunopotentiator (such as OX40 agonist) and adoptive cell therapy (such as CART), etc. This study focused on the naked monoclonal antibodies (mAbs), antibody-drug conjugate (ADC), bispecific T cell engager (BiTE), chimeric antigen receptor (CAR) T/NK cell, immune checkpoint inhibitor/immune agonist, etc. An overview of immunotherapy targets for AML and ALL is shown in Figure 1.
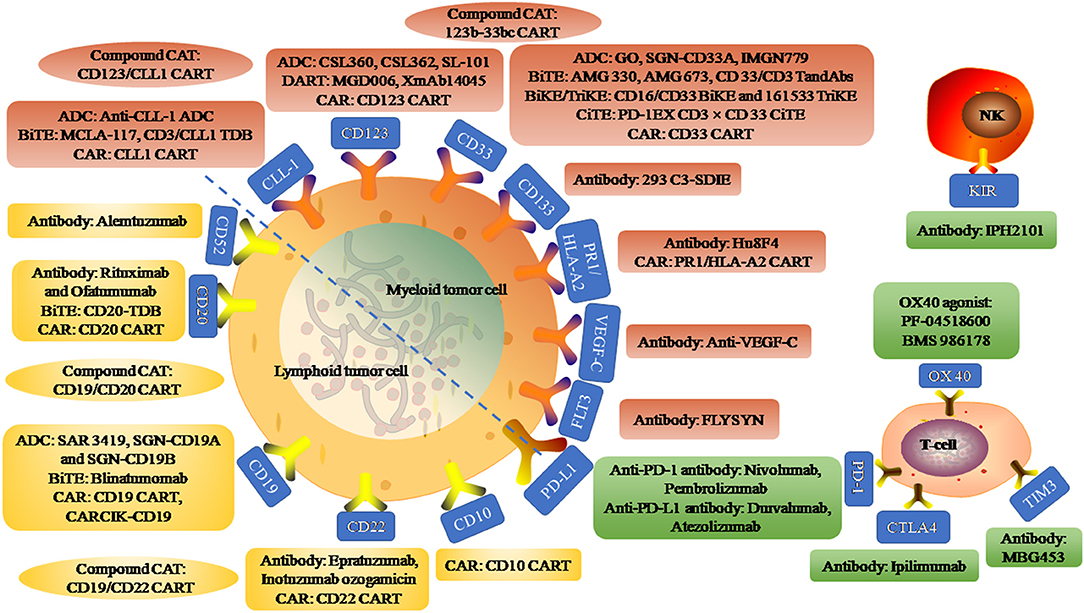
Figure 1. An overview of immunotherapy targets for acute myeloid leukemia and acute lymphoblastic leukemia. Myeloid tumor cell surface antigen targets include CLL-1, CD123, CD33, CD133, PR1/HLA-A2, VEGF-C, and FLT3. Anti-CLL-1 ADC, an antibody-drug conjugate (ADC) targeting CLL-1 that binds to pyrrole diazepine dimer. MCLA-117 is a full-length human IgG1 Bi-Specific T-cell Engagers (BiTE) targeting CLL-1 and CD3. CD3/CLL1 TDB, a CD3 T-cell-dependent bispecific (TDB) full-length humanized IgG1 antibody targeting CLL1. CSL360 and CSL362 are recombinant chimeric antibody targeting CD123 and fully humanized anti-CD123 antibody, respectively. SL-101, an CD123 ADC fused to Pseudomonas exotoxin A. MGD006 is a bispecific CD3xCD123 dual-affinity re-targeting (DART) molecule. XmAb14045, a structured anti-CD123 T cell recruitment antibody. GO, SGN-CD33A, and IMGN779 are CD33 ADC binding to N-acetylgamma-erythromycin, pyrrolidone dimer and DNA alkylation activity, respectively. AMG 330 is a CD33/CD3 BiTE and AMG 673 is the second CD33/CD3 BiTE. CD33/CD3 TandAbs is a directional tandem tetravalent bispecific antibody. CD16/CD33 BiKE is a Bi-Specific Killer Engagers (BiKE) targeting CD16 and CD33, and 161533 TriKE is a Tri-Specific Killer Engagers (TiKE) including a modified IL-15 crosslinking agent between CD 16 and CD 33 single-chain Fv fragments. PD-1EX with CD3xCD33 BiTE, fused the PD-1 extracellular domain (PD-1EX) with CD3xCD 33 BiTE. Hu8F4, a T-cell receptor-like monoclonal antibody targeting PR1/HLA-A2. PR1/HLA-A2 CART, PR1/HLA-A2-specific CART (h8F4-CAR-T cells), containing the scFv of h8F4 fused to CD3 zeta chain through the co-stimulatory domain of CD28. Anti-VEGF-C, an antibody targeting vascular endothelial growth factor C to reduce expansion and enhance differentiation. FLYSYN and 293 C3-SDIE are antibodies targeting FLT3 and CD133. Compound CART include 123b-33bc CART simultaneously targeting CD123 and CD33, and CD123/CLL1 CART simultaneously targeting CD123 and CLL1. Lymphoid tumor cell surface antigen targets include CD52, CD20, CD19, CD22, CD10. Rituximab and Ofatumumab are antibodies targeting CD20. CD20-TDB, is a CD3 TDB full-length humanized immunoglobulin G1 molecule targeting CD20. SAR 3419, SGN-CD19A, and SGN-CD19B are CD 19 ADC that binds to Maytansin, Monoinositol F and Pyrrolidine diazepine, respectively. Blinatumomab, a BiTE with dual specificity for CD19 and CD3. CARCIK-CD19, a cytokine-induced killer (CIK) cells transfected with the transposon CD19CAR. Epratuzumab and Inotuzumab ozogamicin are unconjugated human antibody targeting CD22 and humanized anti-CD 22 ADC, respectively. Alemtuzumab, an anti-CD52 antibody. Compound CART include CD19/CD20 CART simultaneously targeting CD19 and CD20, and CD19/CD22 CART simultaneously targeting CD19 and CD22. Immune targets include inhibitory receptors programmed cell death protein 1 (PD1), cytotoxic T-lymphocyte antigen 4 (CTLA4), T-cell immunoglobulin and mucin-domain containing-3 (TIM3), and stimulatory receptors OX40 on T-cell subsets and their ligands (PD-L1, PD-L2, B7, and OX40L) on AML blasts. IPH2101, anti antibody targeting KIR on NK cell surface.
Unconjugated Antibodies/Antibody-Drug Conjugate (ADC)
Monoclonal antibody binding to leukemia target antigen can lead to direct apoptosis, complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) (12). Potential targets for the treatment of AML include CD33, CD45, CD96, CD123, CD135, C-type lectin-like molecule-1 (CLL1), and FLT3 and so on, among of which CD33 and CD123 also expressed on hematopoietic stem cells (HSCs) while CLL1 absent on megakaryocytic progenitor cells and CD34+/CD38-HSCs (13). The potential targets for ALL include CD19, CD20, CD22, and CD52 etc. (12, 14–16). Antibody therapy include unconjugated antibodies and ADC. ADC can deliver a more potent cytotoxic payload specifically targeting leukemia cells accompanying the conjugated monoclonal antibody if a surface marker to internalize upon binding, and maintain lower systemic concentrations to enhance the effectiveness of induction chemotherapy.
Anti-CD33 Antibody
CD33 is a transmembrane cell surface receptor and a member of the sialic acid-binding immunoglobulin (Ig)-like lectins, containing two conserved tyrosine-based inhibitory signaling motifs for recruitment and activation of the tyrosine phosphatases SHP-1 and SHP-2 or suppressor of cytokine signaling 3 (17). CD33 widely expressed on AML blasts about 90%, and on normal multi-potent myeloid precursor cells, unipotent myeloid colony-forming cells, and maturing myeloid cells, monocytes, peripheral granulocytes and so on (18).
Gemtuzumab Ozogamicin (GO)
GO is the first recombinant humanized ADC targeting CD33, which is linked to N-acetyl-γ-carramycin delivering a DNA-damaging calicheamicin derivative. In 2000, FDA approved GO for recurrent CD 33 + AML aged 60 years or older (19). But in the confirmed III phase clinical study of SWOG S 0106, the addition of GO to induction therapy or after consolidation did not improve the CR rate, DFS or OS, and the mortality rate of GO group increased, so GO automatically withdrew from the market (20). In subsequent studies, it was confirmed that adding GO benefits OS in patients with good cytogenetics and reduces the risk of recurrence (21, 22). Although GO has shown a narrow therapeutic window in early clinical studies, recent reports have proved that the improved dose regimen of GO combined with induced chemotherapy was safe and provided significant survival value for AML patients. Cathy C Zhang has shown that low dose GO combined with induced chemotherapy was able to remove minimal residual diseases more effectively, including LICs, thus achieving a more lasting remission effect and improving the survival of model mice (23). In the randomized phase III EORTC-GIMEMA AML-19 study, for the elderly patients who were not suitable for intensive chemotherapy in the initial treatment of AML, the use of low dose GO was better than the best support therapy in improving OS (4.9 months vs. 3.6 months; HR = 0.69; 95% CI, 0.53~0.90; p = 0.005). The side effects in the two groups were similar and the toxicity was controllable (24). The randomized phase III ALFA 0701 trial showed that adding reduced or segmented doses of GO to standard first-line chemotherapy significantly improved the event-free survival rate in primary AML patients (13.6 months vs. 8.8 months; HR = 0.68; 95% CI, 0.51~0.91) (25). Based on the above two studies, on September 1, 2017, FDA approved GO for the treatment of CD 33 positive adult AML (26). The expression of CD 33 is considered to be a predictor of the efficacy of GO in adult AML (27). The clinical study of stage III in Children's Oncology Group Trial AAML0531 showed that CD 33 splicing polymorphism determined the response of GO to primary AML, especially for CC genotype patients, to conclude that the recurrence rate of GO group was significantly lower than that of non-GO group (26% vs. 49%, p < 0.001) (28). In order to improve the therapeutic response, GO combined with epigenetics therapy is also being studied. For example, in clinical trials of GO combined with histone deacetylase inhibitor Vorinostat and DNA methyltransferase I inhibitor Azacitidine in elderly patients with relapsed or refractory (r/r)AML phase I/II, the ORR was as high as 41.9% in patients receiving maximum tolerated dose (29).
SGN-CD33A
An ADC targeting CD33 conjugated to a highly potent, synthetic DNA cross-linking pyrrolobenzodiazepine (PBD) dimer via a protease-cleavable linker leading to cell death. SGN-CD33A overcome some limitations of GO, such as the non-uniform conjugation of the toxin with the antibody, the drug's relatively slow internalization kinetics, and toxin extrusion via drug transporters (30). In combination with hypomethylating agents (HMAs) treating AML patients, the remission rate was significantly increased, but the hematological toxicity was increased at the same time (31). SGN-CD33A has been reported to cause liver damage, especially sinusoidal syndrome, which has been terminated by FDA. Dose-adjusted SGN-CD33A can reduce adverse reactions. Phase I clinical studies are under way to assess its toxicity and efficacy (32).
IMGN779
Another preclinical studies of anti-CD-33 antibodies, a conjugate with DNA alkylation activity, possessing good antitumor effects in mouse model and AML cell lines, whose cytotoxic activity involved DNA damage, cell-cycle arrest, and apoptosis (33). In order to improve the specificity of target cells for AML, alternative target antigens, such as CD25, FLT3 in the early clinical stage, are being studied (34, 35).
Anti-CD123 Antibody
Interleukin-3 (IL-3) receptor α (CD123) is not only constitutively expressed on normal committed hematopoietic progenitor cells, but also highly expressed in AML blasts, including leukemic stem cells (LSCs) (36, 37). IL-3 is an activated T cell product that bridges innate and adaptive immunity and contributes to several immunopathologies.
CSL360
CSL 360 is a recombinant chimeric immunoglobulin G 1 anti CD123 monoclonal antibody, which can recognize the CD123(+)/CD131(–) phenotype expressed by LSCs exhibiting anti leukemia activity by neutralizing IL-3 to prevent IL-3 binding to its receptor (38, 39). In vitro, blockade of CD123 in AML cell lines abolishes proliferation induced by IL-3 (39, 40). Stage I clinical trials proved its safety, but it did not show definite anti-leukemia activity in most patients with AML (38). CSL360 modified with diethylenetriaminepentaacetic acid for complexing 111In and 13-mer nuclear translocation sequence peptides to enable nuclear importation in LSCs for Auger electron radioimmunotherapy of AML was studied by Leyton et al. (41).
CSL362
The second generation humanized, affinity-matured and Fc-engineered anti CD123 monoclonal antibody, induces stronger ADCC by high affinity to CD16 of NK cells (42, 43). Several studies have proved that it has good antitumor activity in mice and can prolong the survival time of mice (42, 44). Xie et al. reported a case of leukemia cell resistant to autologous ADCC. After allogeneic hematopoietic stem cell transplantation, CSL362 combined with donor-derived NKs effectively dissolved these cells, suggesting that CSL362 can be used as a promising therapy for chemotherapy and transplantation (45). Another antibody SL-101, which is a novel antibody-conjugate comprising an anti-CD123 single-chain Fv fused to Pseudomonas exotoxin A, is also in the preclinical study (46).
Anti-CLL-1 Antibody
CLL-1 is a type II transmembrane glycoprotein of myeloid differentiation antigen, which is highly expressed in AML blasts and LSCs (13, 47). Unlike other targets such as CD33 and CD123, CLL-1 is not expressed on normal HSCs, which can ensure hematopoietic recovery, therefore provide a potential therapeutic target for AML treatment (48).
CLL-1 ADC
CLL-1 ADC is an anti CLL-1 antibody that contains a highly potent PBD dimer DNA alkylating and cross-linking agent, conjugated through a novel self-immolative disulfide linker, which is very effective in eliminating xenografts in mice or monkey leukemia cells and has little toxicity. It is expected to be an effective and safe treatment for AML (49).
Other antibodies against AML include Hu8F4 (T-cell receptor-like monoclonal antibody binding PR1/HLA-A2 complex on the surface of AML cells), VEGF-C (Vascular Endothelial Growth Factor-C) antibody (50–53).
Selected trials of antibody directed therapy for AML has been completed or is ongoing as shown in Table 1.
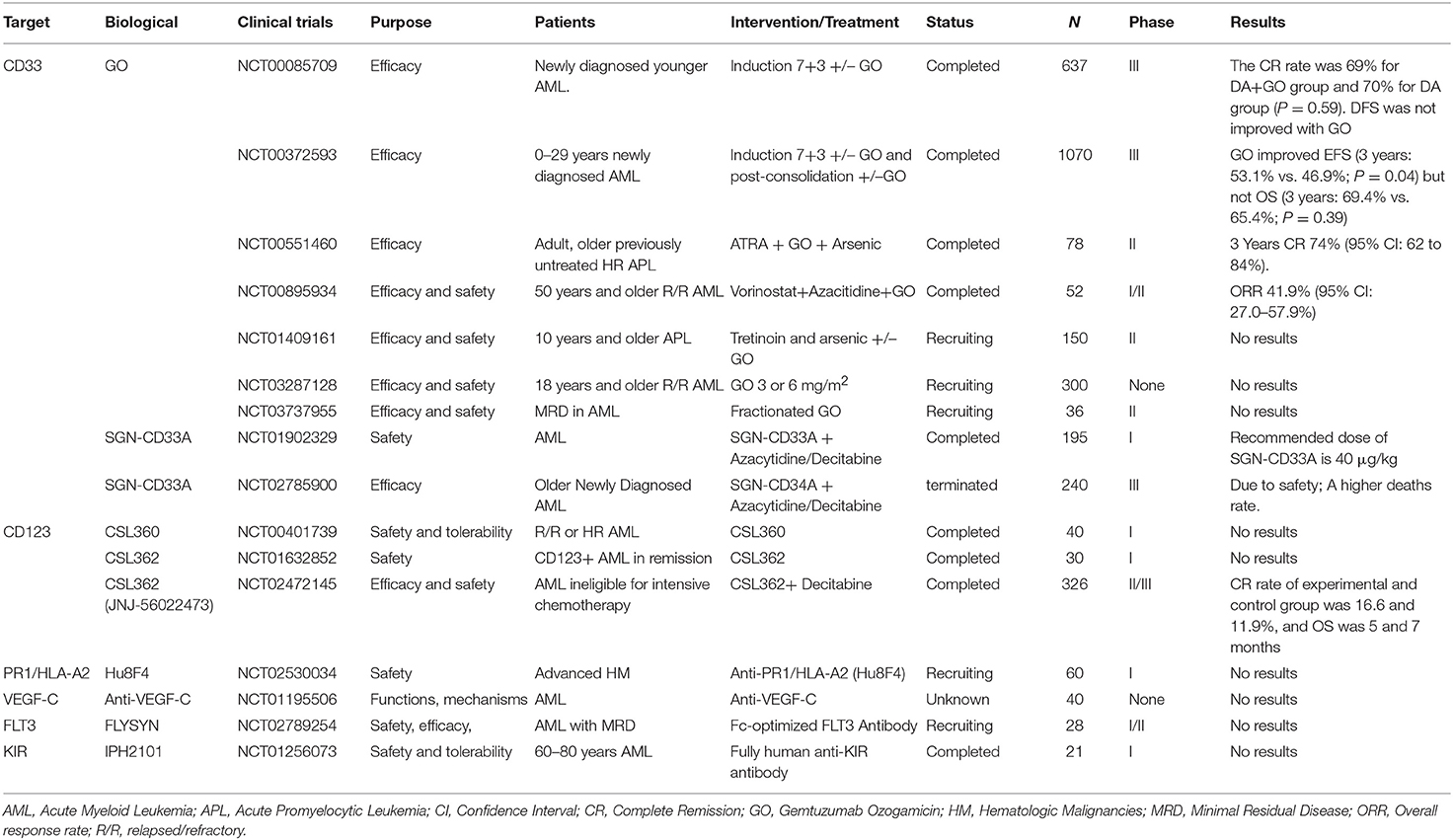
Table 1. Selected completed or ongoing trials of antibody directed therapy for AML.
Anti-CD20 Antibody
CD20, a non-glycosylated transmembrane phosphoproteinis, is a B-lineage specific antigen expressed on both normal and malignant cells during nearly all stages of B-cell differentiation, but not expressed on hematopoietic stem cells, pro-B cells and plasma cells (54). It has an important role in cell cycle progression, differentiation, and modulation of apoptosis pathways (55).
Rituximab
A chimeric antibody targeting CD20 containing a murine variable region and a human Fc region. In the study of Thomas et al. (56) to investigate the impact of addition of rituximab to the modified hyper-CVAD regimen, Hyper-CVAD combined with Rituximab could improve the outcome of young CD20 positive Ph(–) B-ALL patients, with the complete remission duration 70% vs. 38% in 3 years, OS 75% vs. 47% in 3 years, but it did not improve the survival of elderly patients (56). In a randomized Graall-R 2005 Study to evaluate the addition of rituximab to a conventional chemotherapy backbone in 209 patients ages 18 to 59 years with newly diagnosed CD20-positive Ph-negative B-cell ALL, results showed that the cumulative incidence of relapse (CIR) was decreased by Rituximab (2 years CIR 18% vs. 30.5%, p = 0.02), but OS was not longer (2 year OS, 71% vs. 64%; p = 0.095). Rituximab enhances the efficacy of chemotherapy without additive toxicity, but the improvement in the treatment of ALL is modest (57–59).
Ofatumumab
A second generation anti CD20 monoclonal antibody that binds to a site different than Rituximab. Ofatumumab is more effective than Rituximab in inducing cytotoxic mediated by ADCC and CDC. In 55 patients with newly diagnosed ALL and 4 patients with CR who underwent a median of 8 cycles of Ofatumumab, the results showed that with the exception of 1 patient, other patients (98%) achieved CR after the first cycle, and 53 patients (93%) obtained MRD negative, and 3 year CRD and OS were 78 and 68%, respectively. The OS of patients with CD20 < 20% and ≥20% in 3 years were 82 and 64%, respectively (p = 0.96) (60). It was confirmed to be safe and highly effective in patients with CD 20 positive ALL.
Anti-CD22 Antibody
CD22 is expressed in early stages of B-cell development until terminal differentiation except for plasma cells and expressed in most (>90%) patients with B-cell ALL (61). When CD22 binds to the monoclonal antibody, antibody/antigen complex will be rapid internalization leading to antitumor effect (62).
Epratuzumab
An unconjugated human monoclonal antibody that binds to the third extracellular domain of CD22. The combination of CD 22 with Epratuzumab results in the direct phosphorylation of upstream inhibition receptors of BCR signaling (63). The tolerance of Epratuzumab combined with reinduction chemotherapy is good, but compared with chemotherapy alone, the rate of CR2 is not significantly improved. Although increasing the times of Epratuzumab, MRD still has no significant improvement (64). In the SWOG S0910 study, for adult r/r pre-B ALL, the total effective rate (CR + PR) of the adult r/ r pre-B ALL was 52% when Epratuzumab combined with clofarabine and cytarabine, significantly higher than the previous efficacy of clofarabine/cytarabine alone (17%) (65). In the multicenter prospective phase II study of Hyper-CVAD combined with Epratuzumab for young r/r CD22 pre-B ALL, almost half of the patients were MRD negative. It is expected to improve the survival of patients if it is used in first-line therapy especially before allogeneic hematopoietic stem cell transplantation because of Epratuzumab's ability to reduce MRD levels (66).
Inotuzumab Ozogamicin (InO)
A humanized anti-CD 22 ADC combined with calicheamicin, a cytotoxic antibiotic agent (67). After InO binds CD22 then internalizes itself rapidly, the conjugated calicheamicin was delivered intracellularly causing double-strand DNA cleavage and cell apoptosis. In a phase III clinical trial involving 326 r/r ALL patients to compare with InO and standard care, patients were randomly divided into InO group and standard intensive chemotherapy group (67). The complete remission rate in InO group was significantly higher than that in standard treatment group (80.7% [95% CI, 72.1–87.7] vs. 29.4% [95% CI, 21.0–38.8], p < 0.001). The MRD negative rate in InO group was significantly increased (78.4% vs. 28.1% P < 0.001). PFS in InO group was prolonged (median PFS: 5.0 months [95% CI, 3.7~5.6], 1.8 months [95% CI, 1.5~2.2]), and median OS was 7.7 months (95% CI, 6.0–9.2) vs. 6.7 months (95% CI, 4.9~8.3). InO significantly improved the remission rate in patients with higher CD22 levels (≥90%) and lower CD22 levels (<90%). It is worth noting that InO has a good response rate to both high and low tumor loads, while Blinatumomab has a relatively good response rate to patients with low tumor load.
Anti-CD52 Antibody
CD52 is a glycoprotein linked to the cell membrane by a phosphatidylinositol glycan linkage, and is expressed on the surface of nearly all normal and malignant B-lymphocytes and T-lymphocytes, monocytes, and macrophages, but not on plasma cells and HSCs (68). It was reported CD52 played a role in cell lysis via ADCC, complement-mediated cell lysis, and possibly apoptosis (69).
Alemtuzumab
Alemtuzumab is a recombinant DNA-derived humanized monoclonal antibody that targets CD52. Binding to CD52 induces cell lysis through complement activation and ADCC (70). Single drug Alemtuzumab has limited effect on relapsed or refractory acute leukemia. In phase I study of CALGB, the use of Alemtuzumab after remission can eradicate MRD, but it is also associated with viral infection (71). Studies have shown that low-dose Alemtuzumab before transplant may efficiently prevent severe acute and chronic GVHD by its T-cell depleting effect (72, 73).
Anti-CD19 Antibody
CD19 appears at early stages and persists through all stages of B-cell maturation and is homogeneously expressed on malignant cells. It is one of the important membrane antigens involved in the activation and proliferation of B cells.
SAR 3419
A humanized anti-CD 19 ADC that binds to a derivative of the potent microtubule-acting cytotoxic agent, Maytansin, which induces cell cycle arrest and apoptosis (74). In the study of xenotransplantation of CD19 preB ALL and mixed lineage leukemia (MLL) in mice, SAR3419 delayed the progression of the disease (75). However, in the multicenter, single-arm phase II clinical trial in patients with r/r ALL, only 4 of the 17 evaluable patients responded (ORR was 25.5%) and duration of response was only 1.9 months (76). Because of its unsatisfactory therapeutic response, the clinical trial was terminated earlier suggesting that SAR3419 monotherapy is not ideal for r/r ALL.
SGN-CD19A
A humanized ADC targeting CD 19 linked to microtubule interfering agent monomethyla-uristatin F, which inhibits microtubule assembly, triggers cell-cycle arrest of G2-M phase growth and induces cell apoptosis. In the study of exploring SGN-CD19A safety in the treatment of r/r B-ALL and Lymphoma, the phase I clinical trial study showed good tolerance to SGN-CD19A. The objective response rate was 30% in 33 patients who could be evaluated (NCT01786096).
SGN-CD19B
A highly active ADC proved by preclinical studies, targeting CD 19 based on Pyrrolidine diazepine, which releases DNA crosslinkers instead of microtubules inhibitors. In vivo experiments on monkeys, SGN-CD19B effectively cleaned CD20 B lymphocytes from peripheral blood and lymphoid tissues. Phase I clinical trials are under way to explore the therapeutic potential of SGN-CD19 in r/r B-NHL.
Selected trials of antibody directed therapy for ALL has been completed or is ongoing as shown in Table 2.
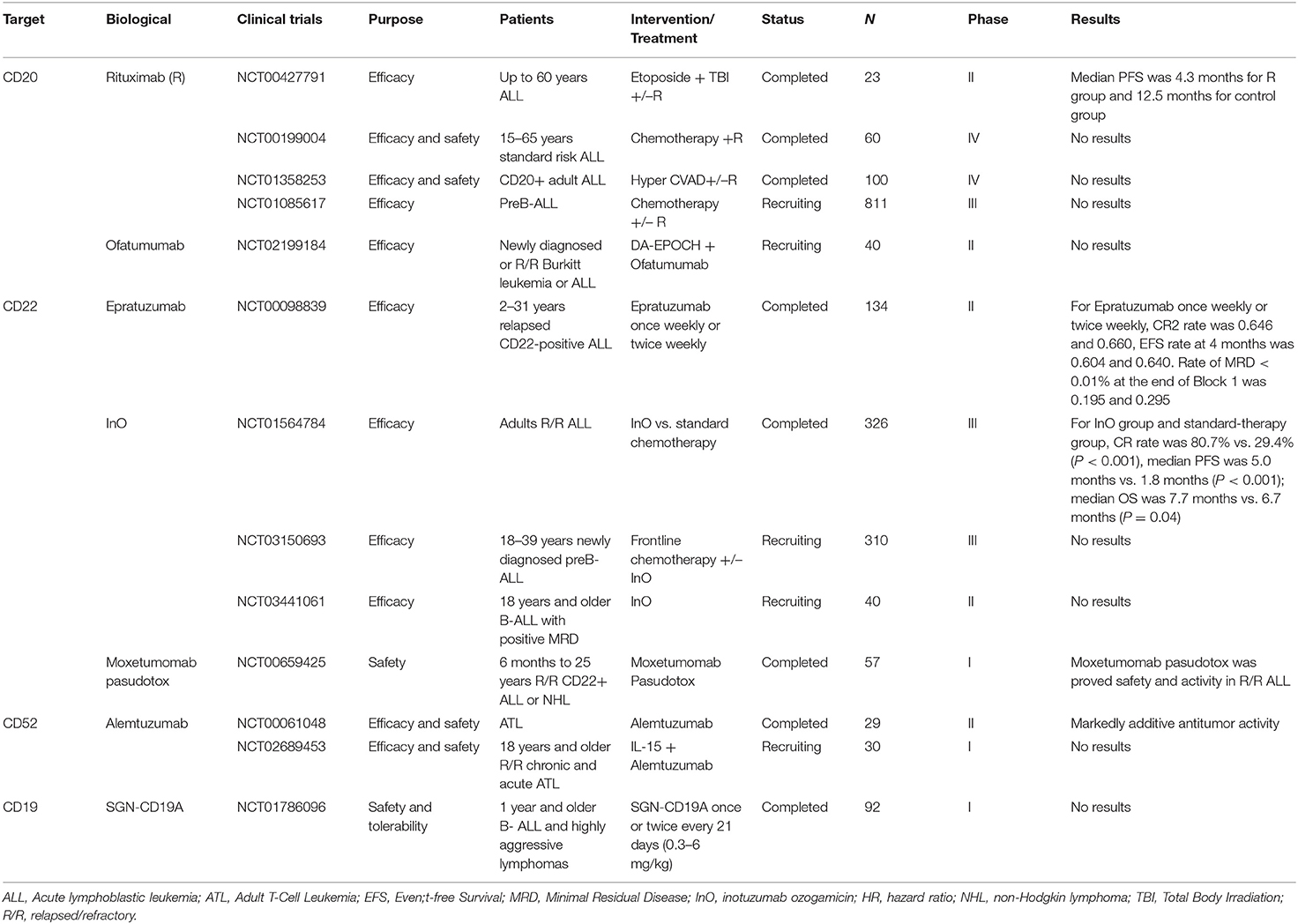
Table 2. Selected completed or ongoing trials of antibody directed therapy for ALL.
Bispecific Antibody/Trispecific Antibody
Bi- and trispecific antibodies, are single chain variable fragment (Scfv) consisting of at least two different specific antibodies one for tumor-associated surface antigens and the other for surface antigens on effector cells, such as CD3 ε on T cells or CD16 on NK cells. Through the double specificity of BiTE, the tumor cells were combined with T effector cells in HLA-independent manner.
CD3/CD33 Bispecific T-Cell Engagers (BiTE)
AMG 330
CD3/CD33 BiTE antibody is dual specificity for CD3 and the sialic acid-binding lectin CD33. The cytotoxicity of AMG 330 to AML cells can be mediated not only by T cells, but also by killing CD33 MDSCs (77). In the presence of AML cells, AMG 330 specifically induced expression of CD69 and CD25 as well as release of IFN-γ, TNF, interleukin (IL)-2, IL-10, and IL-6 (78). AMG 330 can overcome some limitations of CD 33 targeted drugs. It neither regulates the expression of CD 33 nor is affected by the activity of ABC transporter. The density of target antigen, the dose of antibody and the ratio of E/T are the key factors to determine the effect of BiTE (79). The results showed that T cell ligands could regulate AMG 330, and inhibitory ligands PD-L1 and PD-L2 decreased the cytotoxicity of AMG 330, while the activated ligands CD 80 and CD 86 enhanced the cytotoxicity of AMG 330. The synergistic therapy with the regulation of T cell receptor signal can further enhance the effect of this targeted therapy (80, 81). The expression of CD 33 and splicing polymorphism are also associated with the cytotoxicity of AMG 330 in vitro (82). A phase I clinical study of AMG 330 for r/r AML is ongoing (NCT02520427).
AMG 673
An extended half-life BiTE. At present, there is no pre-clinical data report on AMG 673, but clinical research on r/r AML is being carried out (NCT03224819).
CD33/CD3 TandAbs
CD 33/CD3 directional tandem tetravalent bispecific antibody (TandAbs). These antibodies provide two binding sites for each antigen to maintain the affinity of a bivalent antibody. Its molecular weight exceeds the renal clearance threshold, so it has a longer half-life than the smaller antibody structure. This TandAbs can induce strong, dose-dependent cell lysis in CD 33 AML cells. This effect is regulated by the ratio of effector cells to target cells, and the existence of T cells is strictly required. The activation, proliferation and maximal AML cell lysis of T cells were related to the high affinity of CD 33 and CD3. High affinity TandAbs has a wide range of activity in vitro in primary or r/r AML patients. Delayed and inhibited tumor growth was observed in HL-60 xenotransplantation model of immunodeficient mice. CD 33/CD3 TandAbs is the potential of a new approach to the treatment of AML (83).
CD16/CD33 Bispecific Killer Engagers (BiKE)/Bispecific Killer Engagers (TriKE)
CD16/CD33 BiKE
CD16/CD33 BiKE was also named “1633” which can induce the cytotoxicity of NK cells to human AML cells by eliminating MDSCs and reverse the MDSC mediated immunosuppression of NK cells (84, 85). When incubated with AML cells, CD16/CD33 BiKE is able to specifically trigger the cytotoxicity and cytokine release of NK cells. CD16/CD33 BiKE can overcome the autoinhibitory signal and effectively induce the inhibitory effect of NK cell effector on AML. In vitro, CD16/CD33 BiKE ± ADAM 17 inhibitor enhances the activation and specificity of NK cells to CD 33 (+) AML, and may be used for recurrent AML or post-transplant anti-leukemia therapy in the future (85).
161533 TriKE
Although “1633” has anti-tumor effects, it lacks the ability to induce proliferation of NK cells. IL-15 plays an important role in the proliferation and activation of NK cells. Therefore, a Trike, including a modified IL-15 crosslinking agent between CD 16 and CD 33 single-chain Fv fragments (ScFvs), named “161533” was developed, this structure is capable of inducing NK cells proliferation and survival while allowing CD33-specific cytotoxic activity (86). Clinical trial of CD16/IL-15/CD33 Trikes in the treatment of high-risk heme malignancies is currently underway (NCT03214666).
CD3/CD123 Dual-Affinity Re-Targeting (DART)
MGD006
CD3CD123-DART
DART is composed of two antigen-binding specific (A+B) heavy chains and light chain variable regions on two independent polypeptide chains (VLA-VHB-VLB-VHA), and is stabilized by another C-terminal bridge (87). MGD006 is a bispecific molecule that recognizes CD3 and CD123 membrane proteins, redirecting T cells to kill CD123-expressing cells, which can induce the activation and expansion of T cells and mediate the rapid clearance of AML (88). It kills AML cells and primary AML cells in a dose-dependent manner, and the safety and effectiveness are also verified in monkey (89, 90). Phase I clinical studies have been conducted on the safety of MGD006 in the treatment of r/r AML or intermediate-2/high-risk MDS (NCT02152956).
XmAb14045
A uniquely structured anti-CD123 T cell recruitment antibody developed by Xencor. XmAb technology ensures structural stability and prolongs serum half-life by retaining inactive FC fractions. The safety and toxicity of XmAb14045 were evaluated in patients with CD123+ hematologic malignancies in phase 1 I clinical study (NCT02730312).
CD3/CLL1 BiTE
CLL-1 is highly expressed in AML blasts and LSCs. Unlike other targets such as CD33 and CD123, CLL-1 is not expressed on normal HSCs, which can ensure hematopoietic recovery (48). The double-specific CLL-1/CD3 antibody (MCLA-117), a full-length human IgG1 bispecific antibody, lacking Fc effector function and containing a common light chain, was developed. MCLA-117 is able to induce target antigen-specific cytotoxicity of allogeneic or autologous T cells to primary AML cells at low E:T ratio. Phase I clinical study incorporating all subtypes of adult r/r AML and newly diagnosed, high-risk cytogenetic elderly AML to verify the safety, toxicity and efficacy of MCLA-117(NCT03038230). Other studies on CLL1 BiTE, such as CD3/CLL1 TDB (T cell-dependent bispecific), a full length human IgG1 bispecific antibody engineered for improved pharmacokinetic and altered Fc-mediated functions could extended half-lives, have shown that it has good PK properties and antitumor activity, and has little effect on the non-target of HSC (48).
CD3/CD19 BiTE
Blinatumomab (AMG103)
A BiTE immunotherapy with dual specificity for CD19 and CD3. Blinatumomab simultaneously binds CD3+ cytotoxic T cells and CD19+ B cells and redirects T cells to lyse malignant cells (91). First of all, the phase I clinical study of MRD positive B-ALL treated with Blinatumomab was carried out. The results showed that Blinatumomab had a good response to B-ALL regardless of MRD after hemotherapy (92). A subsequent phase II clinical study of r/r preB-ALL indicated that Blinatumomab significantly increased ORR and OS. After Blinatumomab 2 cycles, ORR was 69% and the median survival time was 9.8 months (93). In a multi-center, single-arm, phase II clinical study to observe the safety and efficacy of Blinatumomab in the treatment of adult r/r preB-ALL, the adverse reactions were mainly granulocytopenia fever and controlled neurotoxicity. The CR rate was 32% [95% CI, 26%~40%] and the median remission time was 6.7 months. About 31% (95% CI, 25%~39%) of the patients had MRD response (94). Based on the above research, FDA approved the application of Blinatumomab to Ph(–) r/r preB-ALL on December 3, 2014. There are many clinical studies on Blinatumomab, including the phase III clinical study of Blinatumomab for the first/second recurrence of ALL, the phase II clinical trial for recurrent Ph + ALL and the first-line treatment for MRD + ALL. In a study of 405 preB-ALL patients who received a large number of previous pretreatments, patients were randomly divided into single drug Blinatumomab group and standard chemotherapy group. The results showed that the median survival time of Blinatumomab group was significantly better than that of chemotherapy group (7.7 months vs. 4.0 months) (95). In the adult r/r preB ALL treated with Blinatumomab, the OS of the patients with MRD reaction was significantly longer than that of the patients without MRD reaction, and the T cell amplification was found to be larger in the long-term survivors. It suggests that the long-term survival of the patients may be related to the MRD reaction and the high expansion of T cells (96).
CD3/CD20 BiTE
CD20-TDB
B cells targeting anti-CD20/CD3 T cell dependent bispecific antibody (CD20-TDB), is a full-length humanized immunoglobulin G1 molecule. In monkeys, CD20-TDB at a single dose of 1 mg/kg can effectively kill B cells in peripheral blood and lymphoid tissues and exhibit pharmacokinetic characteristics similar to those of conventional monoclonal antibodies (97).
CiTE
One mechanism of limiting BiTE activity may be T cell incompetence and exhaustion driven by PD-1/PD-L1 and so on. Blocking the PD-1/PD-L1 pathway can increase the cytotoxicity of CD33/CD3 BiTE to leukemia cells (81). Herrmann et al. fused the PD-1 extracellular domain (PD-1EX) with CD3 × CD 33 BiTE to form CiTE. Studies have shown that CiTE with PD-1EX domain increases the specificity. Early clinical trials are under way. If preliminary trials prove to be free of severe immune-related adverse events, this could be an important step forward (98, 99).
Selected trials of bispecific or Tri-Specific T cell or killer Engagers antibody directed therapy for AML and ALL has been completed or is ongoing as shown in Table 3.
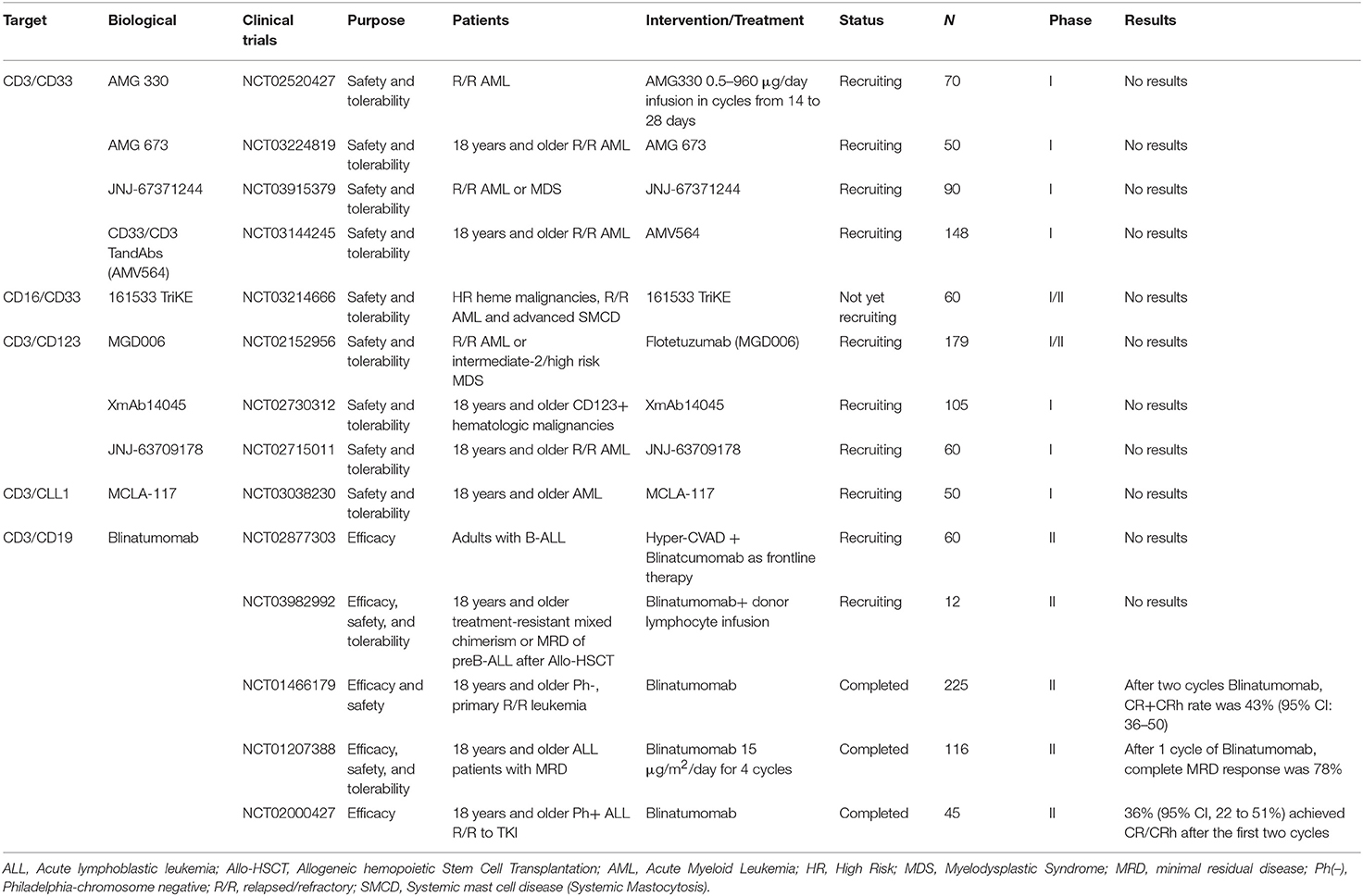
Table 3. Selected completed or ongoing trials of bispecific or Tri-Specific T cell or killer Engagers antibody directed therapy for AML and ALL.
Chimeric Antigen Receptor (CARs)
CARs are genetically engineered cell membrane binding receptors which can activate T cells by linking from the extracellular antigen binding region to the intracellular signal domain via the spacer, whose effect on target antigen is independent of MHC (100). Genetic material encoding the chimeric antigen receptor is transferred into the patient's T cells using transfection, gamma retroviral or lentiviral recombinant vectors, or a transposon system (101–104). The advantages and limitations of each approach have not been fully elucidated recently. CARs contain an extracellular tumor antigen-recognizing scFv linked to an intracellular signaling component comprised of T-cell receptor (TCR) primary domain and often encompassing additional co-stimulatory endodomains (e.g., CD28, CD137 (4-1BB), CD134 (OX40), CD27, ICOS, etc.) (105). CAR transmits a signal by its ligand to the intracellular T cells through a signaling domain, typically the CD3-Zeta chain. The incorporation of co-stimulatory molecules can augment the effects of zeta chain signaling and hence enhance T cell proliferation and persistence (106). According to co-stimulators in signal transduction domain, CAR could be divided into four generations. The first-generation CAR T cells lacked co-stimulatory molecules and had little anti tumor activity (107). The second-generation CARs contain one co-stimulatory signaling domain CD28 or 4-1BB and the third-generation CARs contain two co-stimulatory signaling domains, with the first consisting of a CD28 or 4-1BB domain and the second provided by other molecules, such as OX40, CD28, or 4-1BB (105). Whether third generation CARs benefit more than second generation CARs remains unknown (108). Recent preclinical results indicate that CD137-based co-stimulatory domains are better than those based on CD28 at preventing T cell exhaustion (109). In recent years, the fourth-generation “armored CAR” T cells are engineered to additionally express cytokines or co-stimulatory ligands, aimed at enhancing expansion and longevity of the CAR T cells (110). Optimization of CARs design is an effective way to improve anti-tumor effect.
CD33 CART
CD33 CART
Wang et al. demonstrated that CD33 CAR-T cells exhibited potent antileukemic efficacy, and CAR-T cells with 4-1BB co-stimulatory ligands performed better in antileukemic function than with CD28. Further analysis showed that CD33 4-1BBz.CAR-T cells had an increased central memory compartment and showed resistance to exhaustion (111). In NSG mice model of AML xenotransplantation, a novel second-generation anti-CD33 CAR that incorporates a 4-1BB-CD3ζ signaling tail, CD 33 CART can significantly reduce tumor load and prolong survival time (112). But it also causes toxic effects on normal hematopoietic cells. In order to reduce the toxicity, SS Kenderian et al. designed a transient expression of mRNA anti-CD33 CAR (113). Recently published in Cell, using genetic engineering to inactivate the CD33 gene of HSCs can avoid the toxic effect of CD33 CART on bone marrow, thus making it possible for CD33 CART to treat AML (114).
CD 123 CART
CD 123 CART and CD 123-CAR cytokines induced killer cells (in which PBMC is activated by interferon γ and IL-2) have become a potential therapy for AML (115, 116). It is worth noting that CD 123-CART on normal hematopoietic function is smaller than that of CD33 CART. Modification of anti-CD123 scFv with VH and VL chains of different monoclonal antibodies can reduce the bone marrow toxicity of AML mice (117). The first clinical trial of CART-123 for r/r AML was conducted in our country in 2016 (NCT02937103), and there are other two trials ongoing (NCT03556982)/ (NCT03672851). Stage I clinical trials of CD123-CART for recurrent AML after Allo-HSCT are also underway (NCT03114670).
CLL1 CART
CLL1 CART
CLL-1 CART cells generated by Zhou using the scFv region of the mAb against CLL-1 coupled to the co-stimulatory domains of CD28, 4-1BB, and the CD3-ζ chain could specifically dissolve CLL-1 cells and primary AML cells in vitro (118). Anti-tumor activity was also observed in AML mice and the survival time of mice was prolonged. Importantly, CLL-1 CART can specifically clear CLL-1 myeloid progenitor cells and mature myeloid cells, whereas normal HSCs are unaffected by their lack of CLL-1 expression (118, 119).
Other potential target antigens for AML include CD44v6 (120), FR β (121), NKG2D (122), and PR1/HLA-A2 (53), and so on.
CD19 CART
CD19 CART
The CR rate of r/r ALL treated with CART19 was as high as 70–90%, and a sustained remission was observed without additional treatment (123–125). In order to improve the efficacy, the main focus is on the design of CAR. Phase I trials of 19–28z CART in 16 patients with r/r ALL showed a total CR rate of 88%, enabling most patients to transition to allo-HSCT (126). The safety and long-term results of 53 adult patients with relapsed ALL treated with 19–28z CART were evaluated. Fourteen cases developed severe cytokine release syndrome (26%) and 1 case died. Eighty three percentage of the patients achieved complete remission. Median follow-up was 29 months (range 1–65 months), median EFS was 6.1 months (95% CI, 5.0–11.5 months) and median OS was 12.9 months (95% CI, 8.7–23.4 months). In patients with low tumor load, the median OS was 20.1 months, and the incidence of cytokine release syndrome and neurotoxic events was significantly lower than that in patients with high tumor load (127). The third generation CART19 1928zT2 designed by Li Peng also achieved a good effect in the treatment of relapsed ALL (128). Besides, 4-1BB co-stimulatory signal is also engineered with the use of lentiviral-vector technology for gene transfer and permanent T-cell modification, named CTL019. After 30 patients with r/r ALL received CTL019 treatment, 27 cases (90%) received CR. The 6 months EFS was 67% (95% CI, 51~88) and OS was 78% (95% CI, 65~95) (61). Another study of 29 patients with r/r B-ALL treated with CD 19 4-1BB co-stimulated CART cells concluded the 86% MRD negative rate (129). FDA has granted CTL 019 breakthrough treatment status.
Compound CART
The compound of CART: 123b-33bc CAR T, targeting CD 123 and CD 33 at the same time, not only eliminate the tumor load of AML, but also prevent the recurrence caused by the escape of antigen or the persistence of LSC. It has strong antitumor activity in vivo (130). With regard to CD123/CLL1 CART, phase II clinical trials plan to recruit 20 patients with r/r AML younger than 70 years old to verify their safety and toxicity.
According to research, it shows that PD-1 inhibitor can regulate the CART cell responses, suggesting that the PD-1 pathway may be critical for determining the T-cell immunotherapy response of CAR-modified cells. The combination of PD-1 inhibitor and CART is except to improve the treatment response and remission time.
Selected trials of CAR cell therapy for AML and ALL has been completed or is ongoing as shown in Table 4.
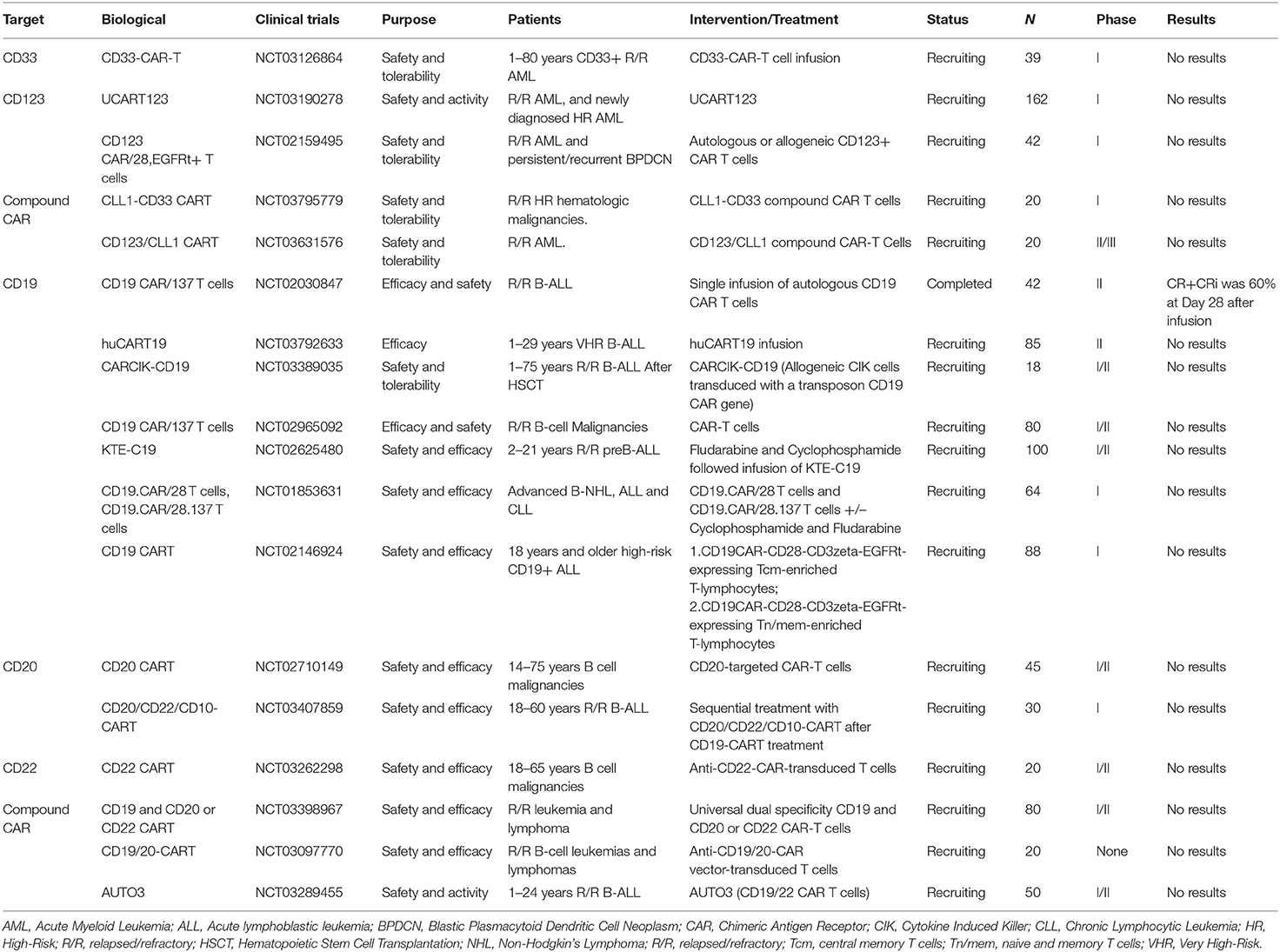
Table 4. Selected completed or ongoing trials of CAR cell therapy for AML and ALL.
Immune Checkpoint Inhibitor and Immune Agonist
The two main immune checkpoint blocking pathways in clinical studies are cytotoxic T lymphocyte antigen (CTLA-4) and programmed cell death pathway. Programmed cell death pathways include programmed cell death protein 1 (PD-1) and its ligands, programmed death ligands 1 (PD-L1) and 2 (PD-L2). Although most clinical studies of blocking PD-1 and CTLA-4 with humanized monoclonal antibodies are directed at solid tumors and lymphoma, PD-1 and CTLA-4 also play a role in leukemia, GVL and GVHD (131). Blocking CTLA-4 and blocking PD-1/PD-L1 pathway with anti PD-L1 antibody enhanced the anti-leukemia immune response in mice (131, 132). Combined blocking of PD1/PD-L1 and Tim-3/galectin-9 may help to prevent CD8 (+) T cell failure in patients with advanced AML and other malignant hematological diseases (133). Studies have shown that epigenetic drugs can regulate the expression of immunological checkpoint molecules on TIL and tumor cells (134), such as Azacytidine upregulating the expression of PD-1 and PD-L1 in MDS/AML. The up-regulation of these genes may be related to the emergence of Azacytidine resistance and the decrease of survival time (135). To improve response rates, epigenetic drugs combined with PD-1/PDL-1 inhibitors are also being tested in clinical trials (NCT02397720, NCT02530463). The timing of immune checkpoint blocking may be more important for the occurrence of hematological malignancies, especially leukemia, because the tumor itself often destroys the host's immune function. Due to high tumor burden and proliferation rate in acute leukemia, the disease may develop before checkpoint antibodies (especially when used alone) have sufficient time to activate the immune response. Therefore, when there is minimal residual disease and a complete immune system, the application of immune checkpoint inhibitors may achieve the best results. A number of clinical trials are currently underway to use checkpoint antibodies as a single drug or in combination with standard chemotherapy regimens to treat newly diagnosed and relapsed leukemia patients.
OX40, a member of the tumor necrosis factor receptor family, is an activating receptor expressed on the surface of activated CD4+ T cells and CD8+ T cells. OX40 signaling activates downstream NF-κB and PI3K pathways, whose sustained activation prolongs T cell survival and promotes the activation and proliferation of T cells. OX40 also inhibits the differentiation and activity of T-reg cell and improves immunosuppressive effects thereby to reinforce antitumor immunity. Besides, OX40L signal transduction promotes NK cells activation, cytokines production, cytotoxicity and the like (136–138). OX40/OX40L has received widespread attention as a potential target for immune activation. Clinical trial of OX40 antibody to treat leukemia is ongoing (NCT03390296, NCT03410901). However, OX40 is expressed on leukemic blasts in a substantial percentage of patients with AML, and so after stimulation with OX40 agonist, OX40 can mediate proliferation and release of cytokines that act as growth and survival factors for the leukemic cells (136). Therefore, the therapeutic effect of OX40 antibody on leukemia needs further verification.
Selected trials of Immune Checkpoint Inhibitor therapy for AML and ALL has been completed or is ongoing as shown in Table 5.
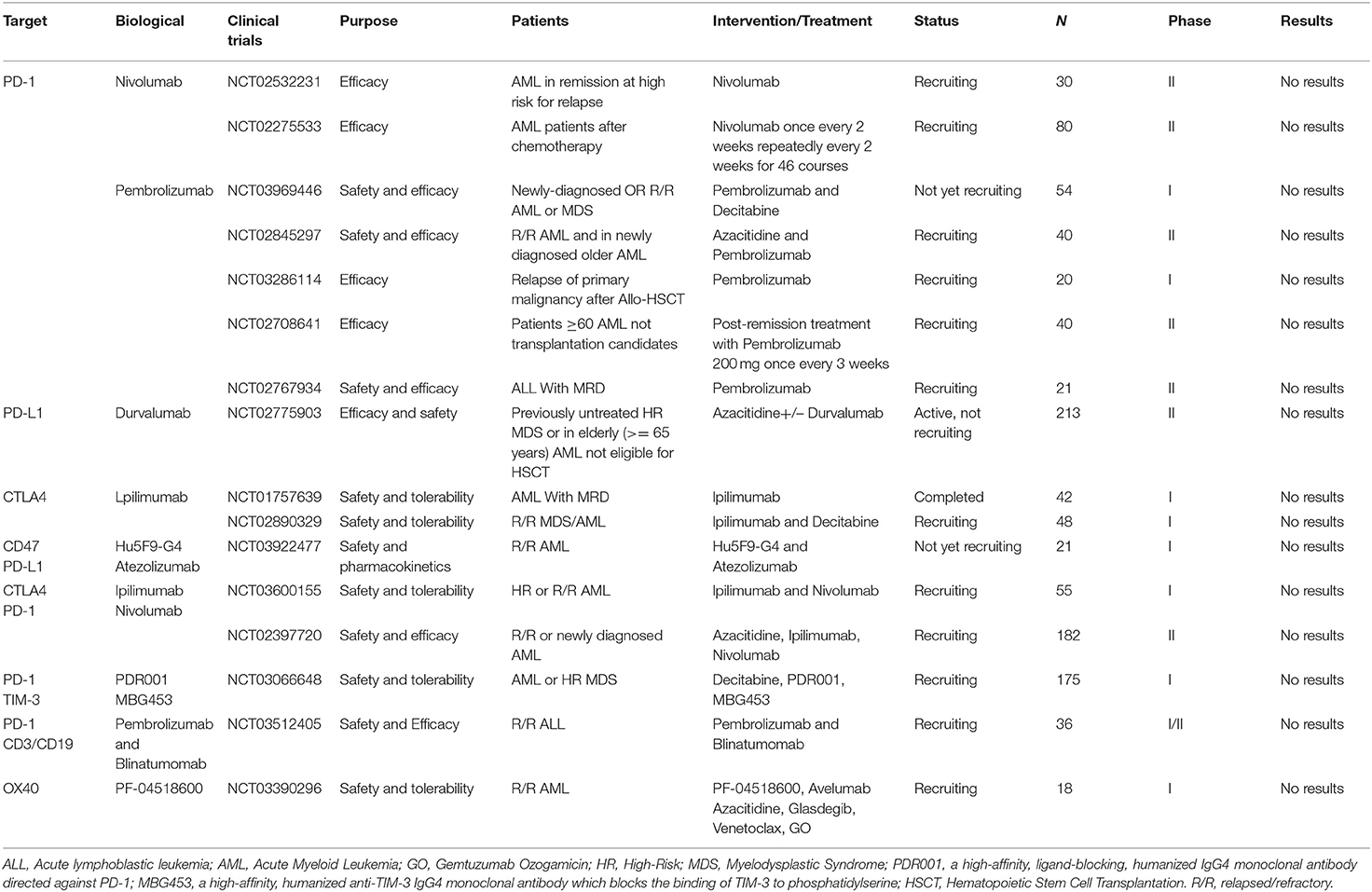
Table 5. Selected completed or ongoing trials of Immune Checkpoint Inhibitor therapy for AML and ALL.
NK Cells
NK cells can exert direct cytotoxic activity without antigen-specific initiation and activate other immune cells to promote anti-leukemia immune response (139). The activity of NK cells in patients with acute leukemia is affected by a number of mechanisms, including reduced expression of activated receptors and decreased cytokine secretion (140, 141). In addition, the low expression of NK ligands in leukemia cells and the production of soluble immunosuppressive factor are also the reasons of impaired NK cells activity (142, 143). NK cell-based tumor immunotherapy strategies include anti KIR antibodies, cytokines (including IL-15 and IL-2), adoptive HLA- haploidentical NK cells and use of NK cell lines (139, 144–146). Other strategies include reducing Treg cells and using bispecific antibodies, such as CD16 on NK cells and leukemia cell-specific antigens, which significantly enhance the cytotoxicity mediated by antibody dependent cells, one of the main mechanisms for eliminating leukemic cells.
IL-2 diphtheria toxin fusion protein (IL2DT) can enhance the clearance of AML by haplotype NK cells. After 7 days of NK cells adoptive infusion, the depletion of Treg and the persistence of NK cells predicted a good clinical response. IL2DT could reduce the host Treg cells and then improve the efficacy of haploid NK cells in the treatment of AML (147). Cytokine induced memory like NK cells enhance its cytotoxicity to myeloid leukemia, and this effect is independent of KIR, which has been confirmed in NSG transplanted tumor model mice (148). Recent studies have shown that ATO enhances the cytotoxicity of NK cells, and the mechanism may be that ATO can change the receptor and ligand structure of NK cells (149). Samudio et al. used oncolytic virus HSV to activate peripheral blood mononuclear cells to lyse leukemia cells. This activation depends, at least in part, on TLR-2, and acts primarily by activating NK cells (150). Fc-optimized CD133 antibody (293 C3-SDIE) significantly enhanced the activation, degranulation and lysis of primary CD133-positive AML cells by allogeneic and autologous NK cells, thereby promoting the clearance of AML cells by NK cells (151). Williams et al. found that CD16 NK-92 cells combined with an antibody against leukemia stem cell antigen could improve the survival of AML model mice (44).
Selected trials of NK cells therapy and other immunotherapies for AML and ALL has been completed or is ongoing as shown in Table 6.
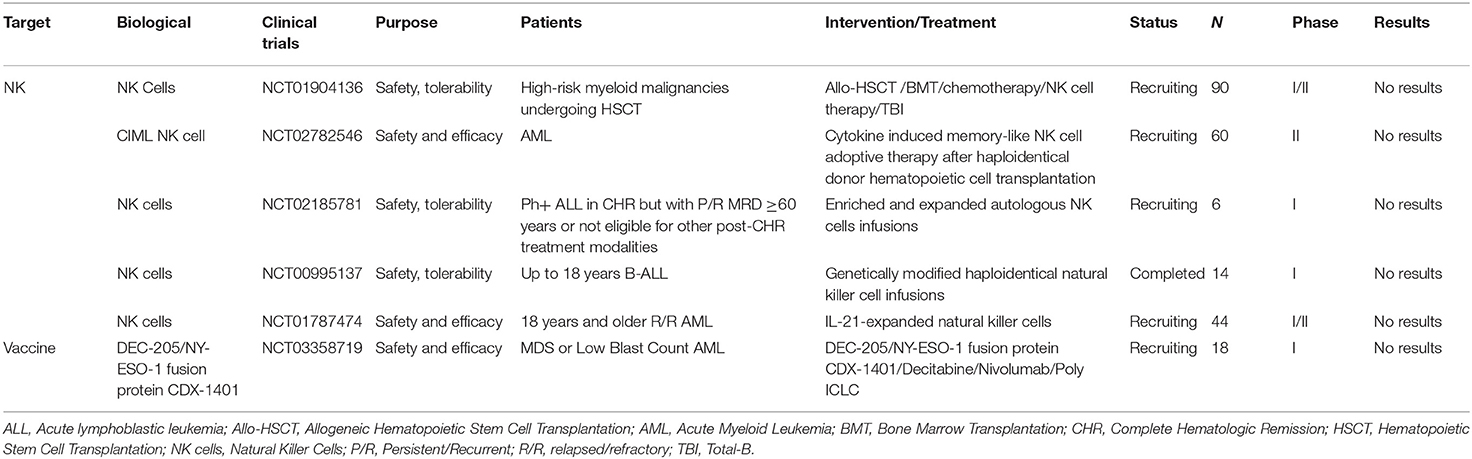
Table 6. Selected completed or ongoing trials of NK cells therapy and other immunotherapies for AML and ALL.
Conclusions
The use of immunotherapy in the treatment of acute leukemia has greatly improved the choice of treatment. It not only provides an opportunity for hematopoietic stem cell transplantation, but also increases the MRD response rate, reduces the MRD level, and further improves the remission time and survival time. Combined use of multiple immunotherapies, such as checkpoint inhibitors combined with other immunotherapy, can improve efficacy and duration of response. Although immunotherapy exhibits a good prospect, its toxicity deserves attention. The side effects of immunotherapy on normal HSCs are urgent problems to be solved, and more precise targets need to be studied to reduce the damage to normal cells.
Author Contributions
FZ and YS designed the study. YS collected data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China under Grant [number: 81770179].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. (2011) 29:532–43. doi: 10.1200/JCO.2010.30.1382
2. Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. (1999) 341:1051–62. doi: 10.1056/NEJM199909303411407
3. Goldstone AH, Richards SM, Lazarus HM, Tallman MS, Buck G, Fielding AK, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. (2008) 111:1827–33. doi: 10.1182/blood-2007-10-116582
4. Raff T, Gokbuget N, Luschen S, Reutzel R, Ritgen M, Irmer S, et al. Molecular relapse in adult standard-risk ALL patients detected by prospective MRD monitoring during and after maintenance treatment: data from the GMALL 06/99 and 07/03 trials. Blood. (2007) 109:910–5. doi: 10.1182/blood-2006-07-037093
5. Baron F, Labopin M, Ruggeri A, Ehninger G, Bonifazi F, Stelljes M, et al. Umbilical cord blood versus unrelated donor transplantation in adults with primary refractory or relapsed acute myeloid leukemia: a report from Eurocord, the Acute Leukemia Working Party and the Cord Blood Committee of the Cellular Therapy and Immunobiology Working Party of the EBMT. Blood Cancer J. (2019) 9:46. doi: 10.1038/s41408-019-0204-x
6. Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. (2009) 301:2349–61. doi: 10.1001/jama.2009.813
7. Baron F, Maris MB, Sandmaier BM, Storer BE, Sorror M, Diaconescu R, et al. Graft-versus-tumor effects after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. J Clin Oncol. (2005) 23:1993–2003. doi: 10.1200/JCO.2005.08.136
8. Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML). Blood. (2012) 119:3361–9. doi: 10.1182/blood-2011-09-377044
9. Ehx G, Fransolet G, de Leval L, D'Hondt S, Lucas S, Hannon M, et al. Azacytidine prevents experimental xenogeneic graft-versus-host disease without abrogating graft-versus-leukemia effects. Oncoimmunology. (2017) 6:e1314425. doi: 10.1080/2162402X.2017.1314425
10. Mathew NR, Baumgartner F, Braun L, O'Sullivan D, Thomas S, Waterhouse M, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. (2018) 24:282–91. doi: 10.1038/nm.4484
11. Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. (2004) 103:767–76. doi: 10.1182/blood-2003-02-0342
12. Kantarjian H, Thomas D, Wayne AS, O'Brien S. Monoclonal antibody-based therapies: a new dawn in the treatment of acute lymphoblastic leukemia. J Clin Oncol. (2012) 30:3876–83. doi: 10.1200/JCO.2012.41.6768
13. Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. (2004) 64:8443–50. doi: 10.1158/0008-5472.CAN-04-1659
14. Buckley SA, Walter RB. Update on antigen-specific immunotherapy of acute myeloid leukemia. Curr Hematol Malig Rep. (2015) 10:65–75. doi: 10.1007/s11899-015-0250-9
15. Gasiorowski RE, Clark GJ, Bradstock K, Hart DN. Antibody therapy for acute myeloid leukaemia. Br J Haematol. (2014) 164:481–95. doi: 10.1111/bjh.12691
16. Ai J, Advani A. Current status of antibody therapy in ALL. Br J Haematol. (2015) 168:471–80. doi: 10.1111/bjh.13205
17. Macauley MS, Crocker PR, Paulson JC. Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol. (2014) 14:653–66. doi: 10.1038/nri3737
18. Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. (2012) 119:6198–208. doi: 10.1182/blood-2011-11-325050
19. Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. (2001) 7:1490–6.
20. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. (2013) 121:4854–60. doi: 10.1182/blood-2013-01-466706
21. Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. (2014) 15:986–96. doi: 10.1016/S1470-2045(14)70281-5
22. Loke J, Khan JN, Wilson JS, Craddock C, Wheatley K. Mylotarg has potent anti-leukaemic effect: a systematic review and meta-analysis of anti-CD33 antibody treatment in acute myeloid leukaemia. Ann Hematol. (2015) 94:361–73. doi: 10.1007/s00277-014-2218-6
23. Zhang CC, Yan Z, Pascual B, Jackson-Fisher A, Huang DS, Zong Q, et al. Gemtuzumab Ozogamicin (GO) inclusion to induction chemotherapy eliminates leukemic initiating cells and significantly improves survival in mouse models of acute myeloid leukemia. Neoplasia. (2018) 20:1–11. doi: 10.1016/j.neo.2017.10.008
24. Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. (2016) 34:972–9. doi: 10.1200/JCO.2015.64.0060
25. Lambert J, Pautas C, Terre C, Raffoux E, Turlure P, Caillot D, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase 3 ALFA-0701 trial. Haematologica. (2018) 104:113–19. doi: 10.3324/haematol.2018.188888
26. Jen EY, Ko CW, Lee JE, Del Valle PL, Aydanian A, Jewell C, et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res. (2018) 24:3242–6. doi: 10.1158/1078-0432.CCR-17-3179
27. Khan N, Hills RK, Virgo P, Couzens S, Clark N, Gilkes A, et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia. (2017) 31:1059–68. doi: 10.1038/leu.2016.309
28. Lamba JK, Chauhan L, Shin M, Loken MR, Pollard JA, Wang YC, et al. CD33 splicing polymorphism determines gemtuzumab ozogamicin response in de novo acute myeloid leukemia: report from randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol. (2017) 35:2674–82. doi: 10.1200/JCO.2016.71.2513
29. Walter RB, Medeiros BC, Gardner KM, Orlowski KF, Gallegos L, Scott BL, et al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a phase I/II study. Haematologica. (2014) 99:54–9. doi: 10.3324/haematol.2013.096545
30. Minagawa K, Jamil MO, Al-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor t-cells for acute myeloid leukemia. PLoS ONE. (2016) 11:e0166891. doi: 10.1371/journal.pone.0166891
31. Fathi AT, Erba HP, Lancet JE, Stein EM, Ravandi F, Faderl S, et al. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood. (2018) 132:1125–33. doi: 10.1182/blood-2018-03-841171
32. Stein EM, Walter RB, Erba HP, Fathi AT, Advani AS, Lancet JE, et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood. (2018) 131:387–96. doi: 10.1182/blood-2017-06-789800
33. Kovtun Y, Noordhuis P, Whiteman KR, Watkins K, Jones GE, Harvey L, et al. IMGN779, a novel CD33-targeting antibody-drug conjugate with DNA-alkylating activity, exhibits potent antitumor activity in models of AML. Mol Cancer Ther. (2018) 17:1271–9. doi: 10.1158/1535-7163.MCT-17-1077
34. Flynn MJ, Zammarchi F, Tyrer PC, Akarca AU, Janghra N, Britten CE, et al. ADCT-301, a Pyrrolobenzodiazepine (PBD) dimer-containing Antibody-Drug Conjugate (ADC) targeting CD25-expressing hematological malignancies. Mol Cancer Ther. (2016) 15:2709–21. doi: 10.1158/1535-7163.MCT-16-0233
35. Snyder JT, Malinao MC, Dugal-Tessier J, Atkinson JE, Anand BS, Okada A, et al. Metabolism of an oxime-linked antibody drug conjugate, AGS62P1, and characterization of its identified metabolite. Mol Pharm. (2018) 15:2384–90. doi: 10.1021/acs.molpharmaceut.8b00225
36. Testa U, Riccioni R, Diverio D, Rossini A, Lo Coco F, Peschle C. Interleukin-3 receptor in acute leukemia. Leukemia. (2004) 18:219–26. doi: 10.1038/sj.leu.2403224
37. Vergez F, Green AS, Tamburini J, Sarry JE, Gaillard B, Cornillet-Lefebvre P, et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. (2011) 96:1792–8. doi: 10.3324/haematol.2011.047894
38. He SZ, Busfield S, Ritchie DS, Hertzberg MS, Durrant S, Lewis ID, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma. (2015) 56:1406–15. doi: 10.3109/10428194.2014.956316
39. Sun Q, Woodcock JM, Rapoport A, Stomski FC, Korpelainen EI, Bagley CJ, et al. Monoclonal antibody 7G3 recognizes the N-terminal domain of the human interleukin-3 (IL-3) receptor alpha-chain and functions as a specific IL-3 receptor antagonist. Blood. (1996) 87:83–92.
40. Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. (2009) 5:31–42. doi: 10.1016/j.stem.2009.04.018
41. Leyton JV, Williams B, Gao C, Keating A, Minden M, Reilly RM. MicroSPECT/CT imaging of primary human AML engrafted into the bone marrow and spleen of NOD/SCID mice using 111In-DTPA-NLS-CSL360 radioimmunoconjugates recognizing the CD123+/CD131- epitope expressed by leukemia stem cells. Leuk Res. (2014) 38:1367–73. doi: 10.1016/j.leukres.2014.09.005
42. Lee EM, Yee D, Busfield SJ, McManus JF, Cummings N, Vairo G, et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica. (2015) 100:914–26. doi: 10.3324/haematol.2014.113092
43. Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. (2014) 28:2213–21. doi: 10.1038/leu.2014.128
44. Williams BA, Wang XH, Leyton JV, Maghera S, Deif B, Reilly RM, et al. CD16(+)NK-92 and anti-CD123 monoclonal antibody prolongs survival in primary human acute myeloid leukemia xenografted mice. Haematologica. (2018) 103:1720–9. doi: 10.3324/haematol.2017.187385
45. Xie LH, Biondo M, Busfield SJ, Arruda A, Yang X, Vairo G, et al. CD123 target validation and preclinical evaluation of ADCC activity of anti-CD123 antibody CSL362 in combination with NKs from AML patients in remission. Blood Cancer J. (2017) 7:e567. doi: 10.1038/bcj.2017.52
46. Han L, Jorgensen JL, Brooks C, Shi C, Zhang Q, Nogueras Gonzalez GM, et al. Antileukemia efficacy and mechanisms of action of SL-101, a novel anti-CD123 antibody conjugate, in acute myeloid leukemia. Clin Cancer Res. (2017) 23:3385–95. doi: 10.1158/1078-0432.CCR-16-1904
47. van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. (2007) 110:2659–66. doi: 10.1182/blood-2007-03-083048
48. Leong SR, Sukumaran S, Hristopoulos M, Totpal K, Stainton S, Lu E, et al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. (2017) 129:609–18. doi: 10.1182/blood-2016-08-735365
49. Zheng B, Yu SF, Del Rosario G, Leong SR, Lee GY, Vij R, et al. An anti-CLL-1 antibody-drug conjugate for the treatment of acute myeloid leukemia. Clin Cancer Res. (2018) 25:1358–68. doi: 10.1158/1078-0432.CCR-18-0333
50. Sergeeva A, He H, Ruisaard K, St John L, Alatrash G, Clise-Dwyer K, et al. Activity of 8F4, a T-cell receptor-like anti-PR1/HLA-A2 antibody, against primary human AML in vivo. Leukemia. (2016) 30:1475–84. doi: 10.1038/leu.2016.57
51. Kampen KR, Scherpen FJG, Mahmud H, Ter Elst A, Mulder AB, Guryev V, et al. VEGFC antibody therapy drives differentiation of AML. Cancer Res. (2018) 78:5940–8. doi: 10.1158/0008-5472.CAN-18-0250
52. Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood. (2011) 117:4262–72. doi: 10.1182/blood-2010-07-299248
53. Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. (2016) 18:985–94. doi: 10.1016/j.jcyt.2016.05.001
54. Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. (1981) 67:134–40. doi: 10.1172/JCI110005
55. Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, et al. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res. (2008) 14:1561–70. doi: 10.1158/1078-0432.CCR-07-1254
56. Thomas DA, O'Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, Wierda W, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. (2010) 28:3880–9. doi: 10.1200/JCO.2009.26.9456
57. Desjonqueres A, Chevallier P, Thomas X, Huguet F, Leguay T, Bernard M, et al. Acute lymphoblastic leukemia relapsing after first-line pediatric-inspired therapy: a retrospective GRAALL study. Blood Cancer J. (2016) 6:e504. doi: 10.1038/bcj.2016.111
58. Levato L, Molica S. Rituximab in the management of acute lymphoblastic leukemia. Expert Opin Biol Ther. (2018) 18:221–6. doi: 10.1080/14712598.2018.1425389
59. Maury S, Chevret S, Thomas X, Heim D, Leguay T, Huguet F, et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med. (2016) 375:1044–53. doi: 10.1056/NEJMoa1605085
60. Wei G, Wang J, Huang H, Zhao Y. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J Hematol Oncol. (2017) 10:150. doi: 10.1186/s13045-017-0516-x
61. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
62. Carnahan J, Wang P, Kendall R, Chen C, Hu S, Boone T, et al. Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties. Clin Cancer Res. (2003) 9:3982S−90S.
63. Lumb S, Fleischer SJ, Wiedemann A, Daridon C, Maloney A, Shock A, et al. Engagement of CD22 on B cells with the monoclonal antibody epratuzumab stimulates the phosphorylation of upstream inhibitory signals of the B cell receptor. J Cell Commun Signal. (2016) 10:143–51. doi: 10.1007/s12079-016-0322-1
64. Raetz EA, Cairo MS, Borowitz MJ, Lu X, Devidas M, Reid JM, et al. Re-induction chemoimmunotherapy with epratuzumab in relapsed acute lymphoblastic leukemia (ALL): phase II results from Children's Oncology Group (COG) study ADVL04P2. Pediatr Blood Cancer. (2015) 62:1171–5. doi: 10.1002/pbc.25454
65. Advani AS, McDonough S, Coutre S, Wood B, Radich J, Mims M, et al. SWOG S0910: a phase 2 trial of clofarabine/cytarabine/epratuzumab for relapsed/refractory acute lymphocytic leukaemia. Br J Haematol. (2014) 165:504–9. doi: 10.1111/bjh.12778
66. Chevallier P, Chantepie S, Huguet F, Raffoux E, Thomas X, Leguay T, et al. Hyper-CVAD + epratuzumab as a salvage regimen for younger patients with relapsed/refractory CD22-positive precursor B-cell acute lymphocytic leukemia. Haematologica. (2017) 102:e184–6. doi: 10.3324/haematol.2016.159905
67. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. (2016) 375:740–53. doi: 10.1056/NEJMoa1509277
68. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology. (2009) 128:260–70. doi: 10.1111/j.1365-2567.2009.03115.x
69. Frampton JE, Wagstaff AJ. Alemtuzumab. Drugs. (2003) 63:1229–43; discussion 1245–6. doi: 10.2165/00003495-200363120-00003
70. Golay J, Manganini M, Rambaldi A, Introna M. Effect of alemtuzumab on neoplastic B cells. Haematologica. (2004) 89:1476–83.
71. Tibes R, Keating MJ, Ferrajoli A, Wierda W, Ravandi F, Garcia-Manero G, et al. Activity of alemtuzumab in patients with CD52-positive acute leukemia. Cancer. (2006) 106:2645–51. doi: 10.1002/cncr.21901
72. Spyridonidis A, Liga M, Triantafyllou E, Themeli M, Marangos M, Karakantza M, et al. Pharmacokinetics and clinical activity of very low-dose alemtuzumab in transplantation for acute leukemia. Bone Marrow Transpl. (2011) 46:1363–8. doi: 10.1038/bmt.2010.308
73. Sharma K, Janik JE, O'Mahony D, Stewart D, Pittaluga S, Stetler-Stevenson M, et al. Phase II study of alemtuzumab (CAMPATH-1) in patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Clin Cancer Res. (2017) 23:35–42. doi: 10.1158/1078-0432.CCR-16-1022
74. Blanc V, Bousseau A, Caron A, Carrez C, Lutz RJ, Lambert JM. SAR3419: an anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin Cancer Res. (2011) 17:6448–58. doi: 10.1158/1078-0432.CCR-11-0485
75. Carol H, Szymanska B, Evans K, Boehm I, Houghton PJ, Smith MA, et al. The anti-CD19 antibody-drug conjugate SAR3419 prevents hematolymphoid relapse postinduction therapy in preclinical models of pediatric acute lymphoblastic leukemia. Clin Cancer Res. (2013) 19:1795–805. doi: 10.1158/1078-0432.CCR-12-3613
76. Kantarjian HM, Lioure B, Kim SK, Atallah E, Leguay T, Kelly K, et al. A Phase II study of coltuximab ravtansine (SAR3419) monotherapy in patients with relapsed or refractory acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. (2016) 16:139–45. doi: 10.1016/j.clml.2015.12.004
77. Jitschin R, Saul D, Braun M, Tohumeken S, Volkl S, Kischel R, et al. CD33/CD3-bispecific T-cell engaging (BiTE(R)) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. (2018) 6:116. doi: 10.1186/s40425-018-0432-9
78. Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther. (2014) 13:1549–57. doi: 10.1158/1535-7163.MCT-13-0956
79. Laszlo GS, Gudgeon CJ, Harrington KH, Dell'Aringa J, Newhall KJ, Means GD, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. (2014) 123:554–61. doi: 10.1182/blood-2013-09-527044
80. Laszlo GS, Gudgeon CJ, Harrington KH, Walter RB. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J. (2015) 5:e340. doi: 10.1038/bcj.2015.68
81. Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia. (2016) 30:484–91. doi: 10.1038/leu.2015.214
82. Laszlo GS, Beddoe ME, Godwin CD, Bates OM, Gudgeon CJ, Harrington KH, et al. Relationship between CD33 expression, splicing polymorphism, and in vitro cytotoxicity of gemtuzumab ozogamicin and the CD33/CD3 BiTErAMG 330. Haematologica. (2018) 104:e59–62. doi: 10.3324/haematol.2018.202069
83. Reusch U, Harrington KH, Gudgeon CJ, Fucek I, Ellwanger K, Weichel M, et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin Cancer Res. (2016) 22:5829–38. doi: 10.1158/1078-0432.CCR-16-0350
84. Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. (2014) 123:3016–26. doi: 10.1182/blood-2013-10-533398
85. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. (2013) 19:3844–55. doi: 10.1158/1078-0432.CCR-13-0505
86. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 Trispecific Killer Engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. (2016) 22:3440–50. doi: 10.1158/1078-0432.CCR-15-2710
88. Campagne O, Delmas A, Fouliard S, Chenel M, Chichili GR, Li H, et al. Integrated pharmacokinetic/pharmacodynamic model of a bispecific CD3xCD123 DART molecule in nonhuman primates: evaluation of activity and impact of immunogenicity. Clin Cancer Res. (2018) 24:2631–41. doi: 10.1158/1078-0432.CCR-17-2265
89. Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. (2015) 7:289ra82. doi: 10.1126/scitranslmed.aaa5693
90. Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. (2016) 127:122–31. doi: 10.1182/blood-2014-05-575704
91. Loffler A, Gruen M, Wuchter C, Schriever F, Kufer P, Dreier T, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia. (2003) 17:900–9. doi: 10.1038/sj.leu.2402890
92. Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. (2011) 29:2493–8. doi: 10.1200/JCO.2010.32.7270
93. Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. (2014) 32:4134–40. doi: 10.1200/JCO.2014.56.3247
94. Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. (2015) 16:57–66. doi: 10.1016/S1470-2045(14)71170-2
95. Kantarjian H, Stein A, Gokbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. (2017) 376:836–47. doi: 10.1056/NEJMoa1609783
96. Zugmaier G, Gokbuget N, Klinger M, Viardot A, Stelljes M, Neumann S, et al. Long-term survival and T-cell kinetics in relapsed/refractory ALL patients who achieved MRD response after blinatumomab treatment. Blood. (2015) 126:2578–84. doi: 10.1182/blood-2015-06-649111
97. Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. (2015) 7:287ra70. doi: 10.1126/scitranslmed.aaa4802
98. Rettig MP, DiPersio JF. CiTE antibody for AML. Blood. (2018) 132:2425–7. doi: 10.1182/blood-2018-10-879668
99. Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A, et al. Bifunctional PD-1 x alphaCD3 x alphaCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood. (2018) 132:2484–94. doi: 10.1182/blood-2018-05-849802
100. Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. (2014) 65:333–47. doi: 10.1146/annurev-med-060512-150254
101. Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. (2009) 32:169–80. doi: 10.1097/CJI.0b013e318194a6e8
102. Lee J, Sadelain M, Brentjens R. Retroviral transduction of murine primary T lymphocytes. Methods Mol Biol. (2009) 506:83–96. doi: 10.1007/978-1-59745-409-4_7
103. Quintas-Cardama A, Yeh RK, Hollyman D, Stefanski J, Taylor C, Nikhamin Y, et al. Multifactorial optimization of gammaretroviral gene transfer into human T lymphocytes for clinical application. Hum Gene Ther. (2007) 18:1253–60. doi: 10.1089/hum.2007.088
104. Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. (2008) 16:580–9. doi: 10.1038/sj.mt.6300404
105. Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol. (2016) 13:25–40. doi: 10.1038/nrclinonc.2015.187
106. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mols Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
107. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transpl. (2010) 16:1245–56. doi: 10.1016/j.bbmt.2010.03.014
108. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
109. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
110. Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. (2014) 20:127–33. doi: 10.1097/PPO.0000000000000034
111. Li S, Tao Z, Xu Y, Liu J, An N, Wang Y, et al. CD33-specific chimeric antigen receptor T cells with different co-stimulators showed potent anti-leukemia efficacy and different phenotype. Hum Gene Ther. (2018) 29:626–39. doi: 10.1089/hum.2017.241
112. O'Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. (2015) 100:336–44. doi: 10.3324/haematol.2014.112748
113. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. (2015) 29:1637–47. doi: 10.1038/leu.2015.52
114. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–53 e19. doi: 10.1016/j.cell.2018.05.013
115. Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. (2013) 122:3138–48. doi: 10.1182/blood-2012-12-474056
116. Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. (2013) 161:389–401. doi: 10.1111/bjh.12282
117. Thokala R, Olivares S, Mi T, Maiti S, Deniger D, Huls H, et al. Redirecting specificity of T cells using the Sleeping Beauty System to express chimeric antigen receptors by mix-and-matching of VL and VH domains targeting CD123+ tumors. PLoS ONE. (2016) 11:e0159477. doi: 10.1371/journal.pone.0159477
118. Wang J, Chen S, Xiao W, Li W, Wang L, Yang S, et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J Hematol Oncol. (2018) 11:7. doi: 10.1186/s13045-017-0553-5
119. Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol Ther. (2017) 25:2202–13. doi: 10.1016/j.ymthe.2017.05.024
120. Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. (2013) 122:3461–72. doi: 10.1182/blood-2013-04-493361
121. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRbeta-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. (2016) 30:1355–64. doi: 10.1038/leu.2016.35
122. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
123. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. (2013) 21:2122–9. doi: 10.1038/mt.2013.154
124. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. (2014) 123:2625–35. doi: 10.1182/blood-2013-11-492231
125. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra38. doi: 10.1126/scitranslmed.3005930
126. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
127. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
128. Weng J, Lai P, Qin L, Lai Y, Jiang Z, Luo C, et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J Hematol Oncol. (2018) 11:25. doi: 10.1186/s13045-018-0572-x
129. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
130. Petrov JC, Wada M, Pinz KG, Yan LE, Chen KH, Shuai X, et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. (2018) 32:1317–26. doi: 10.1038/s41375-018-0075-3
131. Fevery S, Billiau AD, Sprangers B, Rutgeerts O, Lenaerts C, Goebels J, et al. CTLA-4 blockade in murine bone marrow chimeras induces a host-derived antileukemic effect without graft-versus-host disease. Leukemia. (2007) 21:1451–9. doi: 10.1038/sj.leu.2404720
132. Koestner W, Hapke M, Herbst J, Klein C, Welte K, Fruehauf J, et al. PD-L1 blockade effectively restores strong graft-versus-leukemia effects without graft-versus-host disease after delayed adoptive transfer of T-cell receptor gene-engineered allogeneic CD8+ T cells. Blood. (2011) 117:1030–41. doi: 10.1182/blood-2010-04-283119
133. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
134. Zhang M, Xiao XQ, Jiang YF, Liang YS, Peng MY, Xu Y, et al. DNA demethylation in PD-1 gene promoter induced by 5-azacytidine activates PD-1 expression on Molt-4 cells. Cell Immunol. (2011) 271:450–4. doi: 10.1016/j.cellimm.2011.08.014
135. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
136. Nuebling T, Schumacher CE, Hofmann M, Hagelstein I, Schmiedel BJ, Maurer S, et al. The immune checkpoint modulator OX40 and its ligand OX40L in NK-cell immunosurveillance and acute myeloid leukemia. Cancer Immunol Res. (2018) 6:209–21. doi: 10.1158/2326-6066.CIR-17-0212
137. Buchan SL, Rogel A, Al-Shamkhani A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood. (2018) 131:39–48. doi: 10.1182/blood-2017-07-741025
138. Biagi E, Dotti G, Yvon E, Lee E, Pule M, Vigouroux S, et al. Molecular transfer of CD40 and OX40 ligands to leukemic human B cells induces expansion of autologous tumor-reactive cytotoxic T lymphocytes. Blood. (2005) 105:2436–42. doi: 10.1182/blood-2004-07-2556
139. Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res. (2014) 20:3390–400. doi: 10.1158/1078-0432.CCR-13-1766
140. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood. (2002) 99:3661–7. doi: 10.1182/blood.V99.10.3661
141. Lion E, Willemen Y, Berneman ZN, Van Tendeloo VF, Smits EL. Natural killer cell immune escape in acute myeloid leukemia. Leukemia. (2012) 26:2019–26. doi: 10.1038/leu.2012.87
142. Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. (2011) 96:1302–9. doi: 10.3324/haematol.2010.039743
143. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. (2002) 419:734–8. doi: 10.1038/nature01112
144. Szczepanski MJ, Szajnik M, Welsh A, Foon KA, Whiteside TL, Boyiadzis M. Interleukin-15 enhances natural killer cell cytotoxicity in patients with acute myeloid leukemia by upregulating the activating NK cell receptors. Cancer Immunol Immunother. (2010) 59:73–9. doi: 10.1007/s00262-009-0724-5
145. Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol. (2014) 26:161–72. doi: 10.1016/j.smim.2014.02.002
146. Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
147. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
148. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
149. Alex AA, Ganesan S, Palani HK, Balasundaram N, David S, Lakshmi KM, et al. Arsenic trioxide enhances the NK cell cytotoxicity against acute promyelocytic leukemia while simultaneously inhibiting its bio-genesis. Front Immunol. (2018) 9:1357. doi: 10.3389/fimmu.2018.01357
150. Samudio I, Rezvani K, Shaim H, Hofs E, Ngom M, Bu L, et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood. (2016) 127:2575-86. doi: 10.1182/blood-2015-04-639088
Keywords: immunotherapy, acute myeloid leukemia, acute lymphoblastic leukemia, antibody-drug conjugate, bispecific antibody, chimeric antigen receptor, immune checkpoint
Citation: Shang Y and Zhou F (2019) Current Advances in Immunotherapy for Acute Leukemia: An Overview of Antibody, Chimeric Antigen Receptor, Immune Checkpoint, and Natural Killer. Front. Oncol. 9:917. doi: 10.3389/fonc.2019.00917
Received: 04 July 2019; Accepted: 03 September 2019;
Published: 19 September 2019.
Edited by:
Onder Alpdogan, Thomas Jefferson University, United StatesReviewed by:
Frederic Baron, University of Liège, BelgiumGuru Prasad Maiti, Oklahoma Medical Research Foundation, United States
Copyright © 2019 Shang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fuling Zhou, emhvdWZ1bGluZ0B3aHUuZWR1LmNu