 Xi Chen1,2†
Xi Chen1,2† Zhijie Xu3†Shuangshuang Zeng1,2
Zhijie Xu3†Shuangshuang Zeng1,2 Xiang Wang1,2
Xiang Wang1,2 Wanli Liu1,2
Wanli Liu1,2 Long Qian1,2
Long Qian1,2 Jie Wei1,2
Jie Wei1,2 Xue Yang1,2Qiuying Shen1,2
Xue Yang1,2Qiuying Shen1,2 Zhicheng Gong1,2
Zhicheng Gong1,2 Yuanliang Yan1,2*
Yuanliang Yan1,2*- 1Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Pathology, Xiangya Hospital, Central South University, Changsha, China
Nafamostat mesylate (NM), a synthetic serine protease inhibitor first placed on the market by Japan Tobacco in 1986, has been approved to treat inflammatory-related diseases, such as pancreatitis. Recently, an increasing number of studies have highlighted the promising effects of NM in inhibiting cancer progression. Alone or in combination treatments, studies have shown that NM attenuates various malignant tumors, including pancreatic, colorectal, gastric, gallbladder, and hepatocellular cancers. In this review, based on several activating pathways, including the canonical Nuclear factor-κB (NF-κB) signaling pathway, tumor necrosis factor receptor-1 (TNFR1) signaling pathway, and tumorigenesis-related tryptase secreted by mast cells, we summarize the anticancer properties of NM in existing studies both in vitro and in vivo. In addition, the efficacy and side effects of NM in cancer patients are summarized in detail. To further clarify NM's antitumor activities, clinical trials devoted to validating the clinical applications and underlying mechanisms are needed in the future.
Introduction
Cancer is a worldwide public health problem and has been the major cause of death in recent years (1). Current effective modalities for curing cancers include radiation, surgery, and drugs. Among these therapeutic approaches, chemotherapy is indispensable for tumor therapy and has prolonged the lives of many patients over the past decades. However, treatment failure and side effects are common in chemotherapy, which adversely influence both patient survival and quality of life. Thus, there is a huge demand for powerful chemotherapeutic agents that reduce side effects and provide increased survival benefits for cancer patients. Recently, increasing studies have indicated that protease inhibitors suppress tumor growth and progression (2, 3). In particular, several protease inhibitors, such as bortezomib, carfilzomib, and ixazomib, have been approved by the United States Food and Drug Administration (FDA) for clinical use in the treatment of multiple myeloma (4). All of these findings suggest that protease inhibitors have broad prospects for development as anticancer agents.
Nafamostat mesylate (NM), also known as FUT-175 and 6′-amidino-2-naphthyl-4 -guanidinobenzoate dihydrochloride, is a broad-spectrum serine protease inhibitor synthesized by Fujii et al. (5). Five years later, a company called Japan Tobacco brought it to market. NM is usually used to treat pancreatitis, disseminated intravascular coagulation (DIC), and systemic inflammatory response syndrome via suppression of thrombin, plasmin, kallikrein, trypsin, and Cl esterase in the complement system, as well as factors VIIa, Xa, and XIIa in the coagulation cascade (6–8). A growing contingency of researchers have focused on the potential anticancer effects of NM. As early as 1992, one study confirmed that NM inhibits liver metastasis at concentrations of 10−6–10−7 M in colon adenocarcinoma, which first uncovered its putative effects in cancer therapy. Subsequently, NM has been proven to downregulate the expression of both matrix metalloproteinase−2 (MMP-2) and−9, vascular endothelial growth factor (VEGF) and transforming growth factor beta 1 (TGF-β1), secreted by tumor cells in head and neck squamous cell carcinoma, and NM inhibits angiogenesis and tumor invasion (9). Furthermore, Mander et al. reported the NM significantly inhibits proliferation, migration, and invasion in triple-negative breast cancer, which was identified both in vitro and in vivo (10). Moreover, NM reverses immune resistance induced by interferon-gamma (IFN-ɤ) as a method of increasing programmed cell death ligand-1 (PD-L1) expression in lung and pancreatic cancer (11). Currently, exploration of the antitumor effects of NM are in full swing.
In this report, we concentrate on existing evidence regarding the antitumor activity of NM and discuss the potential mechanisms for NM targeting in cancer. In addition, to evaluate the possibility of NM use in future clinical applications, the effectiveness and adverse effects of NM are also discussed.
Mechanism of NM Anticancer Effects
So far, multiple studies have uncovered the potent anticancer abilities of NM. It is evident that NM inhibits cancer cells proliferation, adhesion and invasion, and suppresses tumor growth in animal models. Furthermore, NM initiates apoptosis both in vitro and in vivo (12–17). In addition, NM has the capacity to provide improvements in sensitivity of the tumor to conventional clinical treatments (15–25). Furthermore, as a synthetic serine protease inhibitor, interest is growing in the use of NM against tumor progression induced by MC-derived tryptase. Tumor cell proliferation and angiogenesis stimulated by tryptase was reversed by NM (26, 27). Herein, to investigate how NM exerts these anticancer effects, we discuss the mechanism for NM targeting (Figure 1).
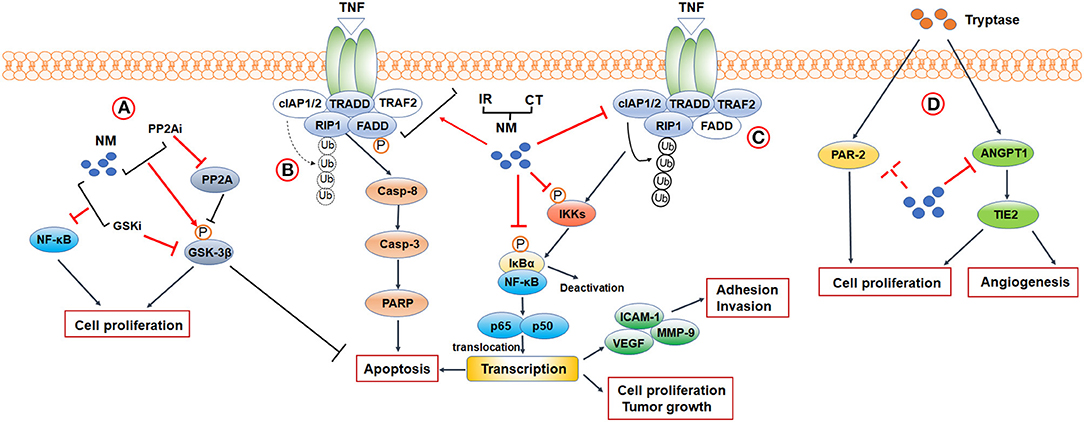
Figure 1. Mechanisms and biological functions of NM in cancer research. (A) PPAR2-GSK3β signaling. (B,C) A crosstalk between NF-κB signaling and apoptosis-related signaling. When TRAF2 is absent (indicated by blank color), c-IAP1/2 is no longer recruited, and RIP1 is not ubiquitinated (indicated by broken arrows). Non-ubiquitinated RIP1 induces caspase-8 dependent apoptosis. Activation of TNFR1 leads to the recruitment of TRADD, TRAF2, c-IAP1/2, and RIP1 to the TNFR1 complex. cIAP1/2 modifies RIP1 with polyubiquitin chains, leading to the activation of canonical NF-κB signaling. (D) Tryptase-mediated PAR-2 and ANGPT1/TIE2 signaling. CT, Chemotherapy; IR, Ionizing Radiation 3; PP2Ai, PP2A inhibitor; GSKi, GSK inhibitor.
NF-κB Signaling
Nuclear factor-κB (NF-κB) is an inducible transcription factor comprising 5 family members, designated as NF-κB1/p50, NF-κB2/p52, RELA/p65, RELB, and c-REL, which bind to consensus DNA sequences at promoter regions as heterodimers and homodimers to activate target genes (28). So far, two major signaling pathways are considered to mediate NF-κB activation: the canonical and non-canonical NF-κB signaling pathways. In the canonical pathway, NF-κb activation occurs via degradation of the inhibitor of κB (IκB) family, consisting of characteristic members IκBα and several structurally related proteins, to which cytoplasmic NF-κB binds in a resting state. After stimulation by some stress or infection factor, the IκB kinase (IKK) complex, composed of catalytic (IKKα and IKKβ) and regulatory (IKKγ) subunits, is activated, subsequently leading to phosphorylation of IκBs. Phosphorylated IκBs further undergo ubiquitylation and proteasome-mediated degradation, promoting release and translocation of NF-κB dimers to regulate gene transcription (29). The entire canonical pathway induces activation of NF-κB heterodimers, including p50 and p65 or p50 and c-Rel, displaying rapid and transient characteristics. In contrast, activation of the non-canonical pathway generates p53 and RELB at a slow and persistent speed with involvement of only IKKα (30). Based on these two pathways, NF-κB activation mediates a wide variety of human disease, particularly cancers.
NF-κB is frequently hyperactivated in several cancers, and its subunits have crucial roles in tumor proliferation, migration and resistance to radiotherapy and chemotherapy. Although the canonical NF-κB signaling pathway has been extensively studied in all kinds of cancers, there still new breakthroughs occur in this field. For instance, in breast and lung cancer, inflammatory cytokines, such as IFN-α and tumor necrosis factor (TNF), activate signal transducer and activator of transcription 1 (STAT1) and NF-κB/p65 in brain metastatic cells in response to stimulation by carcinoma–astrocyte gap junctions composed of protocadherin 7 and connexin 43, leading to tumor growth and chemoresistance (31). The relationship between the upstream and downstream inflammatory cytokines and NF-κB has been intensively studied, and this research revealed a novel role of NF-κB in carcinoma–astrocyte interaction models. Grinberg-Bleyer et al. reported that only deficiency of c-Rel in NF-κB subunits could suppress the generation and maintenance of activated regulatory T cells for impeding immune tolerance. Mice lacking c-REL experienced delayed melanoma growth and enhanced antiprogrammed cell death-1 (PD-1) immunotherapy responses (32). Since p65 containing NF-κB complexes are primarily responsible for cellular activation responses, targeting only c-Rel may prevent undesirable side effects. In addition, investigations have focused on the interplay between malignancy and the non-canonical pathway in recent years. In human glioblastoma cells, transcription of the C250T mutant telomerase (TERT) promoter was reactivated by p52 cooperating with the E-twenty-six family of transcription factors, promoting further regulation of TERT transcription and tumorigenicity (33). Accordingly, targeting critical NF-κB subunits or dysfunction of upstream pathways leading to NF-κB activation may afford cancer patients considerable treatment prospects.
Recently, NM has been shown to potently inhibit canonical NF-κB pathways and further suppress the aggressive behavior of various cancer, such as pancreatic cancer (12–14), colorectal (15), gastric (16), and hepatocellular carcinoma (17). In the study of Uwagawa et al. NM was found to inhibit IκBα phosphorylation and NF-κB DNA-binding activity in a dose-dependent and time-dependent manner in pancreatic cancer cells (12). Another study showed that the activity of intercellular adhesion molecule-1 (ICAM-1), VEGF, and MMP-9, the downstream transcription target genes of NF-κB (34–36), were stifled owing to NM-induced suppression of NF-κB activity, inhibiting pancreatic cancer cell adhesion, anoikis, and invasion (13). Moreover, NM could significantly decreased NF-κB activation which reducing tumor growth, neovascularization and prolonged survival by apoptosis modulation in pancreatic in vivo models (13, 14). Researches over the past decade have demonstrated that NM inhibited proliferation and induced apoptosis by IκBα/NF-κB/p65 nuclear translocation and caspase families regulation in colorectal, gastric, and hepatocellular carcinoma cells (15–17). In in vivo model of colorectal, hepatocellular and gastric cancers, NM have confirmed effective antineoplastic properties with reduced diameters, volumes, and weights of tumors as a single agent (15–17).
Furthermore, aberrant activation of NF-κB signaling has been proven as one potential mechanism underlying chemotherapy and radiotherapy failure. Intriguingly, a function of NM for enhancing the sensitivity of chemotherapy and radiotherapy though inhibition of NF-κB activation was reported in pancreatic, gastric, gallbladder and hepatocellular cancer (Tables 1, 2). Previous researches have reported that the combination of NM and chemotherapeutics agents, such as oxaliplatin, gemcitabine, paclitaxel, and nab-paclitaxel could increase sensitivity of single-agent and combination chemotherapy in pancreatic cancer. The chemo-sensitization abilities of NM were mainly reported by downregulation of IκBα phosphorylation and generating synergistic cytotoxicity, which inhibited NF-κB activation. Previous researches have reported that the combination of NM and chemotherapeutics agents, such as oxaliplatin, gemcitabine, paclitaxel, and nab-paclitaxel could increase sensitivity of single-agent and combination chemotherapy in pancreatic cancer. The chemo-sensitization abilities of NM were mainly reported by downregulation of IκBα phosphorylation and generating synergistic cytotoxicity, which inhibited NF-κB activation (18–22). In vivo studies have also demonstrated that NM sensitizes cells to the chemotherapy and further inhibits pancreatic tumor growth (18–22). Similar studies have revealed that NM inhibited NF-κB activity in hepatocellular carcinoma, gallbladder and gastric cancers both in vitro and in vivo. Specifically, after treatment of NM combined with gemcitabine in gallbladder cancer (25), with paclitaxel in gastric cancer (16) or with TNF-α in hepatocellular carcinoma (17), depression of NF-κB activation was stimulated. Another study also confirmed that the combination of NM sensitized oxaliplatin-induced NF-κB/p65 activation via suppressing phosphorylated IκBα and IKKα/β in colorectal cancer, resulting in reduced cell proliferation, increased apoptosis in vitro and decreased tumor growth in vivo (15). NM was also shown to act as a radiosensitizer via inhibition of NF-κB in colorectal (24) and pancreatic cancer (23). Of note, except for dysfunction of NF-κB, dysregulation of Mdm2 results in increasing p53 expression in response to NM treatment combined with 5 Gy ionizing radiation (IR) in pancreatic cancer (23). A number of studies indicate that both NF-κB and p53 are activated in response to DNA damage by ionizing radiation and modulate each other's activities (37).
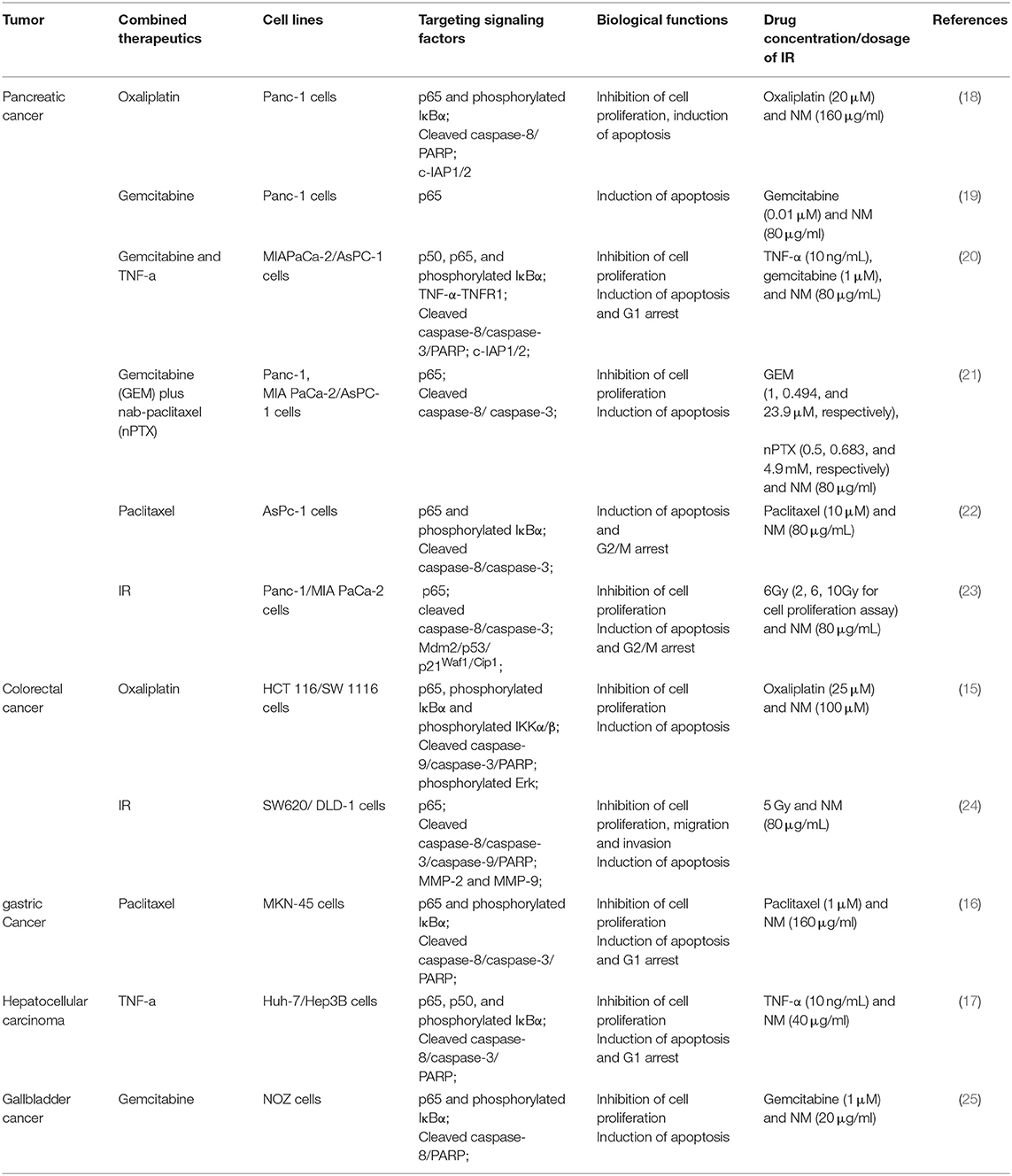
Table 1. The anti-tumor activities of combination of NM and chemotherapy/radiotherapy in vitro.
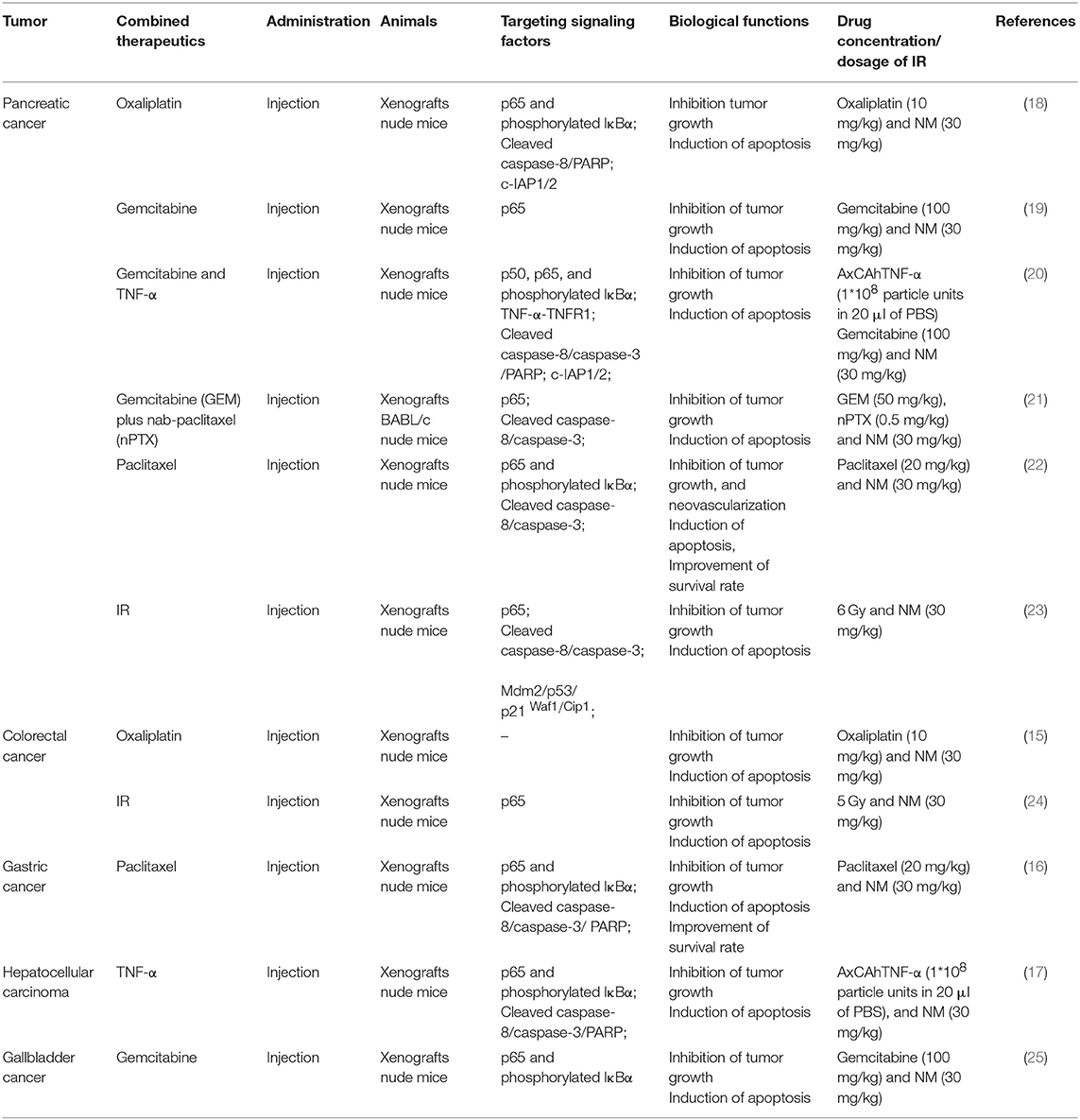
Table 2. The anti-tumor activities of combination of NM and chemotherapy/radiotherapy in vivo.
Besides these classical strategies, some novel molecular inhibiters were confirmed to improve the antitumor effects of NM through pathways related to NF-κB signaling. Haruki et al. found that addition of glycogen synthase kinase-3 (GSK-3) inhibitor to NM significantly decreased the NF-κB/p65 activation and inhibited the cell proliferation in pancreatic cancers, though using GSK-3 inhibitor alone did not alleviate NF-κB activation. They also demonstrated that GSK-3β, one of the GSK-3 isomers dephosphorylated by protein phosphatase 2 (PPA2), was phosphorylated and acted as an inactive form after treating by NM. In that case, they further confirmed that NM combined with PP2A inhibitor significantly up-regulated phosphorylation of GSK-3β and promoted cellular apoptosis comparing to NM or PP2A alone, suggesting NM may improve the therapeutic outcomes of pancreatic cancer (38). Taken together, NM, alone or in combination with other anticancer treatments, plays a powerful role as a candidate intervention in NF-κB signaling for cancer therapy.
TNFR1 Signaling
Apoptosis is a cellular suicide program that plays a pivotal role in the repression of cancer development. Among various mechanisms that contribute to apoptosis, the tumor necrosis factor receptor-1 (TNFR1) signaling pathway has emerged as one of the key mediators. TNFR1 is ubiquitously expressed on almost all cells of the human body and is the primary mediator stimulated by soluble TNF. When TNF binds and activates TNFR1, both pro-apoptosis pathways and anti-apoptosis pathways are affected, such as canonical NF-κB signaling based on subsequent formation of complexes. On the one hand, a pro-apoptotic cascade reaction consisting of multiple proteins, such as Fas-associated death domain (FADD), TNF receptor-associated death domain (TRADD), and the receptor-interacting protein kinase 1 (RIP1), occurs and forms caspase-8. All of these proteins are designated as complex II and further generate caspase-3 and polyadenosine ribose polymerase (PARP) to induce apoptosis (39, 40). Nevertheless, this cascade can be curbed by several factors, such as cellular inhibitor of apoptosis 1 and 2 (c-IAP1/2) (41), an NF-κB–regulated gene for suppressing apoptosis and promoting cell survival. On the other hand, the TNF-TNFR1 complex also recruits a series of proteins, including TNF receptor associated factor 2 (TRAF2), TRADD, c-IAP1/2, and RIP1. Among them, c-IAP1/2 is recruited by TRAF2 and further modifies RIP1 with polyubiquitin chains. This complex, known as complex I, leads to the activation of canonical NF-κB signaling pathways (39, 40). Therefore, targeting TNFR1-induced pro-apoptotic pathways to counteract the role of canonical NF-κB signaling pathways in stimulation of antiapoptotic transcriptional programs may represent a pivotal approach for cancer therapy.
The NM-induced apoptosis was identified in cultured cell and animal models, and demonstrated in human cancers such as pancreatic, colorectal, gastric, hepatocellular, and gallbladder cancers (12–18, 20–25). Multiple sources of data support that NM accelerates apoptosis in dual ways that not only prevent activation of NF-κB-regulated antiapoptotic process but also act on initiation of signaling molecules in TNFR1-induced apoptosis. Uwagawa et al. demonstrated that NM upregulates expression of TNFR1 in a dose-dependent manner and elevates phosphorylation of FADD on Ser-194 in a time-dependent manner. Based on activation of these two mediators and inhibition of NF-κB activation, expression of caspase-8 was upregulated to facilitate apoptosis (12). Furthermore, numerous studies have reported that NM plus chemotherapeutic agents or IR triggers expression of cleaved caspase-8, caspase-3, caspase-9, and PARP compared to using chemotherapy or IR alone in pancreatic, colorectal, gastric, gallbladder, and hepatocellular cancer, closely followed by downregulation of various forms of NF-κB (15, 16, 18, 20–22, 24, 25). Specifically, NM inhibits oxaliplatin-induced and TNF-α- and gemcitabine-induce c-IAP1/2 activity in pancreatic cancer (18, 20). These findings suggest that NM potentiates antitumor effects via inhibition of c-IAP1/2 expression in a positive feedback loop where restraining NF-κB activation negatively regulates c-IAP1/2 expression, which in turn represses activation of NF-κB and promotes apoptosis. Thus, an important consideration in anticancer fields is induction of downstream apoptotic pathways by NM.
Inhibitory Effects Against Tryptase
Tryptase, a trypsin-like serine-proteinases with a molecular weight of 134 kDa, is the most abundant secretion of mast cells (MCs) (42). Tryptase contains a hydrolyzing peptide that binds to the carboxyl terminus of basic residues and a tetrameric structure consisting of non-covalently linked subunits, with an adequately active form stored in MCs (43). Two types of tryptase are expressed on human MCs, alpha and beta. Tryptase-α is the major circulating isoform, and tryptase-β is the major form stored in secretory granules (42). Normally, tryptase acts as an indicator to provide information about the distribution and activation status of MCs, so levels of serum tryptase may reflect disease states, such as allergy reaction, mastocytosis, and other inflammatory reactions (44, 45). Nevertheless, there is increasing evidence to support the view that tryptase is a vital mediator in biological pathways, including tissue remodeling and carcinogenesis (46).
Currently, an enormous number of studies have uncovered the association between MC-released tryptase and tumor progression, especially for neovascularization. For instance, a positive linear correlation was demonstrated between serum mast cell densities positive for tryptase (MCDPT) and microvascular density and endothelial area in patients undergoing surgery in colorectal, breast and gastric cancers, indicating that serum tryptase levels may represent a novel surrogate angiogenic marker in cancer patients (27, 47, 48). Involvement of tryptase in the degradation of extracellular matrix and the release of angiogenic factors, such as VEGF receptor and fibroblast growth factor-2, has been clearly demonstrated (49). Based on these findings, tryptase itself or the process that tryptase acts on might become potential drug targets for cancer therapy.
The pharmacological property of NM makes this compound an interesting candidate for new anticancer agent by inhibiting tryptase in colon and pancreatic cancer. Yoshii et al. identified that in colon carcinoma, NM blocks cell proliferation induced by treatment of tryptase or protease-activated receptor-2 (PAR-2) in a concentration-dependent fashion (27). PAR-2 is stimulated by tryptase (47) and mediates proliferative effects via phosphorylation of mitogen-activated protein kinase in colon carcinoma (27). In addition, PAR-2 upregulates MMP expression and plasminogen activator, leading to degradation of the extracellular matrix (48) and has become a hallmark for poor prognosis in combination with MCDPT. It is unclear whether NM directly inhibits expression of PAR-2 to exert its antiproliferative effects. Similar inhibitory effects of NM on tryptase-induced cell proliferation were observed in pancreatic cancer by Guo et al. They also demonstrated that NM possessed the ability to reverse tube formation caused by tryptase via inhibiting the expression of angiogenesis-related genes, angiopoietin-1 (ANGPT1) and TIE2 (26). It is well-known that ANGPT1 targets the TIE2 receptor as an agonist to regulate vascular maturation and stabilization (50). To date, investigations of NM inhibitory effects against tryptase for anti-cancer therapy are relatively scarce and limited to in vitro studies. Although application of NM is mentioned as a possible research direction, studies on the mechanism of targeting tryptase cancer therapy are in their infancy. More studies are needed both in vitro and in vivo in the future.
Safety and Efficacy
NM is a synthetic low-molecular-weight serine protease inhibitor clinically used for acute pancreatitis and applied as an anticoagulant during extracorporeal circulation supportive treatment with a short half-life time (~23 min) (51). Evidence illustrating its clinical use for pancreatitis is suggested in animal models, illustrating that NM decreases the mortality of rats in an experimental acute pancreatitis model in a dose-dependent manner (Infusion at doses of 0.5–50 mg/kg/min) (6). In aspects of anticoagulation, a retrospective study indicated that NM could substitute for heparin in application of extracorporeal membrane oxygenation (ECMO), a novel rescue measure for circulatory and/or respiratory failure. Results showed that NM reduces anticoagulation values of patient to safe levels with an infused rate of 0.2–0.5 mg/kg per hour (52). In patients with acute kidney injury who received continuous renal replacement (CRRT) therapy, a single-center randomized study was performed and found that mean filter lifespan, a representation of treatment efficacy, was significantly longer in patients receiving NM than in those without NM, and no adverse events associated with NM administration were noted (53). For hematological malignancies patients diagnosed as DIC, the DIC resolution rates are 40.3 and 56.3% on days 7 and 14 after treatment of NM (0.06–0.20 mg/kg/day) (7). While compared to protease inhibitor gabexate mesylate (GM), there was no significant difference of DIC resolution rates between these two inhibitors. With respect to cancer therapy, three studies have been conducted to explore the possibility of NM's clinical application in pancreatic tumors treated with a combination of NM and gemcitabine chemotherapy. A phase I study was performed with 12 patients enrolled, and the regimen contained administration of gemcitabine at a fixed dose of 1,000 mg/m2 for 30 min on days 1, 8, and 15 of each 28-day cycle, accompanied by administration of NM starting via a port-catheter system for 24 h before infusion of gemcitabine. The starting dose of NM was 2.4 mg/kg with increments of 1.2 mg/kg until 4.8 mg/kg, and no patients experienced dose-limiting toxic effects at any level of incremental dose. In that case, the recommended dose of NM in combination with full-dose gemcitabine is 4.8 mg/kg (54). Next, based on the recommended dosage in a phase I trial, a single-arm phase II study occurred in a single center to evaluate treatment efficacy of 35 patients with unresectable and metastatic pancreatic cancer. The overall response rate was 17.1%, and 25% of patients who required opioids for cancer-related pain decreased their intake. From this evidence, the regimen revealed an effective improvement compared to standard chemotherapy with gemcitabine (55). Finally, in a retrospective single-center study, jaundice, ascites, high lymphocyte count, and high serum CA19-9 levels were investigated as poor prognostic factors for overall survival in patients with unresectable pancreatic cancer (56).
As an anticoagulant, the risk of bleeding is one of the most common adverse effects (>5%) associated with NM use. A retrospective study found that NM (500 mg was mixed with 5% dextrose and 250 mL water at an infusion rate of 20 mg/h) significantly increased bleeding complications (16.4%) in patients receiving ECMO compared to patients receiving heparin therapy (7.1%) (57). However, in 101 patients who received CRRT, use of NM tended to be correlated with decreased incidence of bleeding complications compared to the use of unfractionated heparin (6.6 vs. 16%) (58). Furthermore, headache (2.2%), nausea (1.3%), and fever (0.9%) were observed in a prospective, observational study including 832 patients with leukocytapheresis (59). Kim et al. reported a case of rare adverse reaction, anaphylactic shock, caused by NM in Korea after a 10-min infusion of NM for hemodialysis (60). In hematological malignancies patients with DIC, a retrospective study found that adverse events, such as hyperkalemia and hyponatremia, were more commonly observed in patients receiving NM (0.06–0.20 mg/kg/day) compared to GM (7). These findings indicate hints for exploration of adverse effects in cancer therapy. In unresectable pancreatic cancer, in vivo studies found that known adverse effects of NM, such as hyperkalemia, hyponatremia, and hepatopathy, were comparable between mice treated with NM at a dosage of 30 μg/g and PBS three times a week for 6 weeks (14). Moreover, another in vivo study of pancreatic cancer showed no significant difference in liver toxicity between combined injection of TNF-α and NM compared to injection of each compound alone (17). In a single-arm, single center, phase II trial, leukopenia and neutropenia primarily appeared in pancreatic cancer patients who received NM (4.8 mg/kg continuous regional arterial infusion) with gemcitabine (1,000 mg/m2 intravenously) on days 1, 8, and 15 (55). Thus, the superiority of NM in reducing adverse reactions is obvious in current clinical therapy, but randomized controlled studies are still necessary to detect adverse reactions and their rates of occurrence. In light of the above detailed findings, NM exhibits low toxicity and advantages based on combination therapy with traditional chemotherapeutic agents, suggesting that combined treatment of NM and chemotherapy may represent a novel promising strategy for cancer.
Discussion
The Potential Immune Therapy of NM
To date, there are several contractional points regarding immunomodulatory functions of NM. By reducing of granzyme activity and cytotoxic T lymphocytes cytolysis, NM suppressed local C5a/C3a production and attenuated T cell auto-reactivity in experimental autoimmune encephalomyelitis (61, 62). In stroke rat models, NM decreased the generation of proinflammatory factors TNF-α, interleukin-1β (IL-1β), inducible nitric oxide synthase and cyclo-oxygenase-2, and elicited the expression of anti-inflammatory mediators CD206, TGF-β, IL-10, and IL-4 (63). On the contrary, it was identified that NM play a role in immune activity enhancement. NM effectively increased the activity of T lymphocytes and natural killer cells in hepatic resection patients (64). Also, NM induced CD8 T-cell proliferation, and played a role as a co-adjuvant for peptide vaccination (65). As mentioned above, the major anti-cancer mechanism of NM was the down-regulation of TNF-α-induced NF-κB activation. Fujiwara et al. have demonstrated that NM could act as an immunotherapy sensitizer combined with TNF-α and gemcitabine in hepatocellular carcinoma and pancreatic cancer (17, 20, 21). Also, NM were demonstrated affected anti-angiogenic activity by MCs-derived tryptase in colon carcinoma (26, 27). In all, the existing evidence suggests that NM is inclined to show immune activation activity in antitumor therapy.
Although the role of NM in inflammatory-related pathway has been reported, the direct evidence of NM antitumor effects in immune cells is still not clarified. PD-L1 is a crucial factor in inhibition of T cell-mediated responses. The previous study has uncovered that NM reversed IFN-γ-induced up-regulation of PD-L1 in lung cancer and pancreatic cancer cells (Figure 2) (66). The pivotal immune escape factor human leukocyte antigen-ABC (HLA-ABC) may help differentiate benign from malignant indeterminate pulmonary lesions (67). The study of Homma et al. was confirmed that NM influenced IFN-γ-induced HLA-ABC up-regulation in lung cancer and pancreatic cancer (11), suggesting that NM may improve the treatment of immune resistant cancer. The inhibited effects of NM are associated with PD-L1-mediated immune evasion pathways including PAR-1, STAT1, STAT3, protein kinase B (Akt), interferon regulatory factors-1, extracellular signal-regulated kinase signals, but not NF-κB pathway. More interesting, inhibition of IFN-γ-induced PD-L1 upregulation was not found in above researches (68–72). Hence, the underlying mechanisms of how NM exerted its inhibition remains to be explored. More studies are warranted to figure out the correlation between NM and inflammatory factors and investigate the inhibition effects of NM in human tumors.
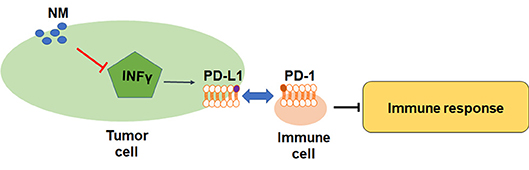
Figure 2. Mechanisms and biological functions of NM in immune response.
A Brief Comparison Between NM and Other Protease Inhibitors
To further evaluate availability of NM for cancer therapy, herein, we briefly compared the function and clinical values between NM and other protease inhibitors. A series of synthetic serine protease inhibitors such as GM and bortezomib have been used for clinical application. Up to now, GM has been therapeutically used for DIC and acute pancreatitis in Asian countries (73). The antitumor effects of NM have been analyzed (74–76) and compared to GM in post-endoscopic retrograde cholangiopancreatography pancreatitis (PEP), wound healing and DIC (7, 77, 78). Similar to NM, GM exerted significant antitumorigenic effects on suppression of TNF-α-induced NF-κB expression and promotion of caspase-3, 7 activity in human pancreatic cancer cells (74). In addition, in vitro and in vivo studies reported that GM attenuated MMP activities and reduced cell growth, invasion and angiogenesis in colon cancer (75). These effects are also identified in a series of colorectal cancer cells with KRAS, BRAF and PIK3CA mutations (76). Since these genetic mutations have been emerged as predictive biomarkers for patients who might fail to EGFR-targeted therapy (79), these preliminary findings indicate that GM may represent a promising therapeutic agent for metastatic colorectal cancer. However, pharmacological mechanisms and clinical studies of GM on cancer therapy are still limited. Furthermore, the in vitro experiment demonstrated that NM could more effectively suppress the pancreatic protease activities than GM up to 10–100 times. A meta-analysis demonstrated the decreased risk of PEP was correlated with NM, while not associated with GM (77). The wound healing ability of NM was also identified to be more efficient than GM in rat models (78). However, the association between GM and NM were not observed in hematological malignancies-induced DIC. There were no significant differences in the DIC resolution rates between GM and NM groups, but the frequency of adverse effects was relatively higher in NM groups (7). Based on these discoveries, the comparable effects of NM and GM suggested the more potential usage of NM on cancer treatment, and further studies are still needed to validate safety and efficacy of NM in clinical trials.
The protease inhibitor bortezomib is the first drug approved by FDA for multiple myeloma treatment. Compared to bortezomib, NM showed the analogous inhibitory effects on NF-κB activation and IFN-γ-induced PD-L1 expression (66). Moreover, the preclinical and clinical data identified that NM could sensitize solid tumors to chemotherapy or classical chemo-radiotherapy (15–24, 37, 80). Nevertheless, FDA-approved proteases inhibitors such as bortezomib were observed mainly distributing in blood and/or bone marrow instead of solid tumors (3). Considering the limitations of current protease inhibitors, NM may apply for the treatment of solid tumor as a new aspect. In summary, these evidences indicate that prospects of NM for boarder clinical application are desirable even if currently only used in Asian countries.
The Potential Clinical Uses of NM
So far, NM has been approved for treating diseases of digestive and hematological systems such as pancreatitis and DIC, and studies have confirmed potentiality clinical use of NM in various diseases. Herein, we summarized potential pharmacological action of NM in non-tumor diseases distributed in nervous, circulatory, respiratory system, and infectious. In in vivo models of nervous system, for example cerebral ischemia, transient middle cerebral artery occlusion rats indicated the blood-brain barrier (BBB) protective function of NM through inhibition of thrombin. The symptom such as neuronal damage, brain infarcts, brain oedema, and motor dysfunction caused by impaired BBB could be reduced after NM treatment (81, 82). Moreover, NM regulated cardiovascular functions through increasing nitric oxide generation via the Akt/eNOS signaling pathway, which indicating that NM might serve as a safeguard for preventing cardiovascular diseases (83). Asthma is one of the serious respiratory diseases all over the world. In rat models with asthma, the treatment of NM showed a decreased eosinophil and neutrophil infiltration, and decreased levels of inflammatory factors such as IL-5, IL-6, IL-13, and IL-17 in bronchoalveolar lavage fluid. Also, the nuclear NF-κB activity reduced in lung tissues (84). It indicated the applicable function of NM in respiratory systems. As for antimicrobial activity of NM, Inman et al. found that NM exerts a dose-dependent inhibition of chlamydial infection. The in vitro and in vivo models showed that NM could effectively minimized pathological features of chlamydial-induced arthritis, including inflammatory infiltration and joint damage (85). Additionally, NM could inhibit middle east respiratory syndrome coronavirus infections (86) and the ebola virus disease in in vitro studies (87). Obviously, it can be concluded that the diverse anti-inflammatory and NF-κB modulating abilities of NM may provide hopeful clinical applications for future pharmaceutical development.
The Limitations of Research Progress
Currently, a detailed mechanism for how NM fosters inhibition of IκBα phosphorylation in malignant cancer cells remains elusive. Which molecules are regulated by NM in tryptase-mediated angiogenesis also remains unknown. At this point, the modulatory effects of NM on tumor growth are distinct, particularly when combined with chemo- and radiotherapy. Additional clinical studies are essential to identify the pathological significance of NM in cancer onset and to compare therapeutic responses between conventional therapy alone and in combination with NM. Furthermore, the immune involvements of NM are worthy to deeply dig either in immunoadjuvant therapy or directly in immunocyte sensitization.
Conclusions
NM is a serine protease inhibitor associated with inhibition of tumor progression in various tumors. Herein, we summarized the pharmacological mechanisms and evaluated the clinical application of NM for cancer therapy. Current research demonstrates that NM blocks canonical NF-κB signaling, targets TNFR1-stimulated cleavage of caspase families, and the tryptase of mast cells to improve therapeutic outcome. As a potential novel anti-cancer agent, NM has been validated to ameliorate cancer therapy resistance and avoid immune resistance. Existing clinical data showed relatively preferable efficacy and safety in NM treatment. Compared with other proteases inhibitors, NM has comparable antitumor activities and advantages in human pancreatic and colorectal cancer. All above evidence highlight the superiority of NM. Nevertheless, the question remains whether NM could be used in treatment for cancer patients, and additional preclinical and clinical studies are essential to be further conducted.
Author Contributions
YY, XC and ZX wrote this review article. XC, ZX, SZ, XW, WL, LQ, JW, XY, QS and ZG performed administrative and technical support. YY designed the study and contributed to manuscript preparation. All authors reviewed and approved the final version of the manuscript.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities of Central South University (2019zzts800), the National Natural Science Foundation of China (No. 81803035, 81572946, 81703036), the Natural Science Foundation of Hunan Province (2019JJ50932), and the Youth Fund of Xiangya Hospital (No. 2017Q17).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Elsevier's English Language Editing Service for assistance with language editing, and we thank all members of the Center for Molecular Medicine for their critical comments.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. (2017) 67:7–30. doi: 10.3322/caac.21387
2. Eatemadi A, Aiyelabegan HT, Negahdari B, Mazlomi MA, Daraee H, Daraee N, et al. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed Pharmacother. (2017) 86:221–31. doi: 10.1016/j.biopha.2016.12.021
3. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. (2017) 14:417–33. doi: 10.1038/nrclinonc.2016.206
4. Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. (2017) 36:561–84. doi: 10.1007/s10555-017-9707-8
5. Fujii S, Hitomi Y. New synthetic inhibitors of C1r, C1 esterase, thrombin, plasmin, kallikrein and trypsin. Biochim Biophys Acta. (1981) 661:342–5. doi: 10.1016/0005-2744(81)90023-1
6. Iwaki M, Ino Y, Motoyoshi A, Ozeki M, Sato T, Kurumi M, et al. Pharmacological studies of FUT-175, nafamostat mesilate. V. Effects on the pancreatic enzymes and experimental acute pancreatitis in rats. Jpn J Pharmacol. (1986) 41:155–62. doi: 10.1254/jjp.41.155
7. Minakata D, Fujiwara SI, Ikeda T, Kawaguchi SI, Toda Y, Ito S, et al. Comparison of gabexate mesilate and nafamostat mesilate for disseminated intravascular coagulation associated with hematological malignancies. Int J Hematol. (2019) 109:141–6. doi: 10.1007/s12185-018-02567-w
8. Ohtake Y, Hirasawa H, Sugai T, Oda S, Shiga H, Matsuda K, et al. Nafamostat mesylate as anticoagulant in continuous hemofiltration and continuous hemodiafiltration. Contrib Nephrol. (1991) 93:215–7. doi: 10.1159/000420222
9. Yamashita Y, Ishiguro Y, Sano D, Kimura M, Fujita K, Yoshida T, et al. Antitumor effects of Nafamostat mesilate on head and neck squamous cell carcinoma. Auris Nasus Larynx. (2007) 34:487–91. doi: 10.1016/j.anl.2006.12.002
10. Mander S, You DJ, Park S, Kim DH, Yong HJ, Kim DS, et al. Nafamostat mesilate negatively regulates the metastasis of triple-negative breast cancer cells. Arch Pharm Res. (2018) 41:229–42. doi: 10.1007/s12272-017-0996-9
11. Homma S, Hayashi K, Yoshida K, Sagawa Y, Kamata Y, Ito M. Nafamostat mesilate, a serine protease inhibitor, suppresses interferon-gamma-induced up-regulation of programmed cell death ligand 1 in human cancer cells. Int Immunopharmacol. (2018) 54:39–45. doi: 10.1016/j.intimp.2017.10.016
12. Uwagawa T, Li Z, Chang Z, Xia Q, Peng B, Sclabas GM, et al. Mechanisms of synthetic serine protease inhibitor (FUT-175)-mediated cell death. Cancer. (2007) 109:2142–53. doi: 10.1002/cncr.22658
13. Fujiwara Y, Furukawa K, Haruki K, Shimada Y, Iida T, Shiba H, et al. Nafamostat mesilate can prevent adhesion, invasion and peritoneal dissemination of pancreatic cancer thorough nuclear factor kappa-B inhibition. J Hepatobiliary Pancreat Sci. (2011) 18:731–9. doi: 10.1007/s00534-011-0390-9
14. Furukawa K, Iida T, Shiba H, Fujiwara Y, Uwagawa T, Shimada Y, et al. Anti-tumor effect by inhibition of NF-kappaB activation using nafamostat mesilate for pancreatic cancer in a mouse model. Oncol Rep. (2010) 24:843–50. doi: 10.3892/or.2010.843
15. Lu YX, Ju HQ, Wang F, Chen LZ, Wu QN, Sheng H, et al. Inhibition of the NF-kappaB pathway by nafamostat mesilate suppresses colorectal cancer growth and metastasis. Cancer Lett. (2016) 380:87–97. doi: 10.1016/j.canlet.2016.06.014
16. Haruki K, Shiba H, Fujiwara Y, Furukawa K, Iwase R, Uwagawa T, et al. Inhibition of nuclear factor-kappaB enhances the antitumor effect of paclitaxel against gastric cancer with peritoneal dissemination in mice. Dig Dis Sci. (2013) 58:123–31. doi: 10.1007/s10620-012-2311-4
17. Haruki K, Shiba H, Fujiwara Y, Furukawa K, Iwase R, Uwagawa T, et al. Inhibition of nuclear factor-kappaB enhances the antitumor effect of tumor necrosis factor-alpha gene therapy for hepatocellular carcinoma in mice. Surgery. (2013) 154:468–78. doi: 10.1016/j.surg.2013.05.037
18. Gocho T, Uwagawa T, Furukawa K, Haruki K, Fujiwara Y, Iwase R, et al. Combination chemotherapy of serine protease inhibitor nafamostat mesilate with oxaliplatin targeting NF-kappaB activation for pancreatic cancer. Cancer Lett. (2013) 333:89–95. doi: 10.1016/j.canlet.2013.01.019
19. Uwagawa T, Chiao PJ, Gocho T, Hirohara S, Misawa T, Yanaga K. Combination chemotherapy of nafamostat mesilate with gemcitabine for pancreatic cancer targeting NF-kappaB activation. Anticancer Res. (2009) 29:3173–8. Available online at: http://ar.iiarjournals.org/content/29/8/3173.long
20. Fujiwara Y, Shiba H, Iwase R, Haruki K, Furukawa K, Uwagawa T, et al. Inhibition of nuclear factor kappa-B enhances the antitumor effect of combination treatment with tumor necrosis factor-alpha gene therapy and gemcitabine for pancreatic cancer in mice. J Am Coll Surg. (2013) 216:320–32 e3. doi: 10.1016/j.jamcollsurg.2012.09.016
21. Horiuchi T, Uwagawa T, Shirai Y, Saito N, Iwase R, Haruki K, et al. New treatment strategy with nuclear factor-kappaB inhibitor for pancreatic cancer. J Surg Res. (2016) 206:1–8. doi: 10.1016/j.jss.2016.06.047
22. Fujiwara Y, Furukawa K, Shimada Y, Iida T, Shiba H, Uwagawa T, et al. Combination paclitaxel and inhibitor of nuclear factor kappaB activation improves therapeutic outcome for model mice with peritoneal dissemination of pancreatic cancer. Pancreas. (2011) 40:600–7. doi: 10.1097/MPA.0b013e31820b9257
23. Shirai Y, Shiba H, Iwase R, Haruki K, Fujiwara Y, Furukawa K, et al. Dual inhibition of nuclear factor kappa-B and Mdm2 enhance the antitumor effect of radiation therapy for pancreatic cancer. Cancer Lett. (2016) 370:177–84. doi: 10.1016/j.canlet.2015.10.034
24. Sugano H, Shirai Y, Horiuchi T, Saito N, Shimada Y, Eto K, et al. Nafamostat mesilate enhances the radiosensitivity and reduces the radiation-induced invasive ability of colorectal cancer cells. Cancers. (2018) 10:E386. doi: 10.3390/cancers10100386
25. Iwase R, Haruki K, Fujiwara Y, Furukawa K, Shiba H, Uwagawa T, et al. Combination chemotherapy of nafamostat mesylate with gemcitabine for gallbladder cancer targeting nuclear factor-kappaB activation. J Surg Res. (2013) 184:605–12. doi: 10.1016/j.jss.2013.06.003
26. Guo X, Zhai L, Xue R, Shi J, Zeng Q, Gao C. Mast cell tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int J Mol Sci. (2016) 17:E834. doi: 10.3390/ijms17060834
27. Yoshii M, Jikuhara A, Mori S, Iwagaki H, Takahashi HK, Nishibori M, et al. Mast cell tryptase stimulates DLD-1 carcinoma through prostaglandin- and MAP kinase-dependent manners. J Pharmacol Sci. (2005) 98:450–8. doi: 10.1254/jphs.FPJ05002X
28. Cildir G, Low KC, Tergaonkar V. Noncanonical NF-kappaB signaling in health and disease. Trends Mol Med. (2016) 22:414–29. doi: 10.1016/j.molmed.2016.03.002
29. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. (2012) 12:121–32. doi: 10.1038/nrc3204
30. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
31. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–8. doi: 10.1038/nature18268
32. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. (2017) 170:1096–108.e13. doi: 10.1016/j.cell.2017.08.004
33. Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. (2015) 17:1327–38. doi: 10.1038/ncb3240
34. Harada M, Morimoto K, Kondo T, Hiramatsu R, Okina Y, Muko R, et al. Quinacrine inhibits ICAM-1 transcription by blocking DNA binding of the NF-kappaB subunit p65 and sensitizes human lung adenocarcinoma A549 cells to TNF-alpha and the fas ligand. Int J Mol Sci. (2017) 18:E2603. doi: 10.3390/ijms18122603
35. Luo M, Hou L, Li J, Shao S, Huang S, Meng D, et al. VEGF/NRP-1axis promotes progression of breast cancer via enhancement of epithelial-mesenchymal transition and activation of NF-kappaB and beta-catenin. Cancer Lett. (2016) 373:1–11. doi: 10.1016/j.canlet.2016.01.010
36. Ning L, Ma H, Jiang Z, Chen L, Li L, Chen Q, et al. Curcumol suppresses breast cancer cell metastasis by inhibiting MMP-9 via JNK1/2 and Akt-dependent NF-kappaB signaling pathways. Integr Cancer Ther. (2016) 15:216–25. doi: 10.1177/1534735416642865
37. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. (2013) 12:86. doi: 10.1186/1476-4598-12-86
38. Haruki K, Shiba H, Shimada Y, Shirai Y, Iwase R, Fujiwara Y, et al. Glycogen synthase kinase-3beta activity plays a key role in the antitumor effect of nafamostat mesilate in pancreatic cancer cells. Ann Gastroenterol Surg. (2018) 2:65–71. doi: 10.1002/ags3.12025
39. Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem Pharmacol. (2016) 116:1–10. doi: 10.1016/j.bcp.2016.03.009
40. Hayden MS, Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Semin Immunol. (2014) 26:253–66. doi: 10.1016/j.smim.2014.05.004
41. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. (2003) 114:181–90. doi: 10.1016/S0092-8674(03)00521-X
42. de Souza Junior DA, Santana AC, da Silva EZ, Oliver C, Jamur MC. The role of mast cell specific chymases and tryptases in tumor angiogenesis. Biomed Res Int. (2015) 2015:142359. doi: 10.1155/2015/142359
43. Ribatti D, Ranieri G. Tryptase, a novel angiogenic factor stored in mast cell granules. Exp Cell Res. (2015) 332:157–62. doi: 10.1016/j.yexcr.2014.11.014
44. Valent P. Mast cell activation syndromes: definition and classification. Allergy. (2013) 68:417–24. doi: 10.1111/all.12126
45. Vitte J. Human mast cell tryptase in biology and medicine. Mol Immunol. (2015) 63:18–24. doi: 10.1016/j.molimm.2014.04.001
46. Ni WW, Cao MD, Huang W, Meng L, Wei JF. Tryptase inhibitors: a patent review. Expert Opin Ther Pat. (2017) 27:919–28. doi: 10.1080/13543776.2017.1322064
47. Qian N, Li X, Wang X, Wu C, Yin L, Zhi X. Tryptase promotes breast cancer angiogenesis through PAR-2 mediated endothelial progenitor cell activation. Oncol Lett. (2018) 16:1513–20. doi: 10.3892/ol.2018.8856
48. Ammendola M, Leporini C, Marech I, Gadaleta CD, Scognamillo G, Sacco R, et al. Targeting mast cells tryptase in tumor microenvironment: a potential antiangiogenetic strategy. Biomed Res Int. (2014) 2014:154702. doi: 10.1155/2014/154702
49. Wroblewski M, Bauer R, Cubas Cordova M, Udonta F, Ben-Batalla I, Legler K, et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat Commun. (2017) 8:269. doi: 10.1038/s41467-017-00327-8
50. Kim J, Park DY, Bae H, Park DY, Kim D, Lee CK, et al. Impaired angiopoietin/Tie2 signaling compromises Schlemm's canal integrity and induces glaucoma. J Clin Invest. (2017) 127:3877–96. doi: 10.1172/JCI94668
51. Keck T, Balcom JH, Antoniu BA, Lewandrowski K, Warshaw AL, Fernandez-del Castillo CF. Regional effects of nafamostat, a novel potent protease and complement inhibitor, on severe necrotizing pancreatitis. Surgery. (2001) 130:175–81. doi: 10.1067/msy.2001.115827
52. Park JH, Her C, Min HK, Kim DK, Park SH, Jang HJ. Nafamostat mesilate as a regional anticoagulant in patients with bleeding complications during extracorporeal membrane oxygenation. Int J Artif Organs. (2015) 38:595–9. doi: 10.5301/ijao.5000451
53. Choi JY, Kang YJ, Jang HM, Jung HY, Cho JH, Park SH, et al. Nafamostat mesilate as an anticoagulant during continuous renal replacement therapy in patients with high bleeding risk: a randomized clinical trial. Medicine. (2015) 94:e2392. doi: 10.1097/MD.0000000000002392
54. Uwagawa T, Misawa T, Sakamoto T, Ito R, Gocho T, Shiba H, et al. A phase I study of full-dose gemcitabine and regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. Ann Oncol. (2009) 20:239–43. doi: 10.1093/annonc/mdn640
55. Uwagawa T, Misawa T, Tsutsui N, Ito R, Gocho T, Hirohara S, et al. Phase II study of gemcitabine in combination with regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. Am J Clin Oncol. (2013) 36:44–8. doi: 10.1097/COC.0b013e31823a53b2
56. Furukawa K, Uwagawa T, Iwase R, Haruki K, Fujiwara Y, Gocho T, et al. Prognostic factors of unresectable pancreatic cancer treated with nafamostat mesilate combined with gemcitabine chemotherapy. Anticancer Res. (2012) 32:5121–6. Available online at: http://ar.iiarjournals.org/content/32/11/5121
57. Lim JY, Kim JB, Choo SJ, Chung CH, Lee JW, Jung SH. Anticoagulation during extracorporeal membrane oxygenation; nafamostat mesilate versus heparin. Ann Thorac Surg. (2016) 102:534–9. doi: 10.1016/j.athoracsur.2016.01.044
58. Makino S, Egi M, Kita H, Miyatake Y, Kubota K, Mizobuchi S. Comparison of nafamostat mesilate and unfractionated heparin as anticoagulants during continuous renal replacement therapy. Int J Artif Organs. (2016) 39:16–21. doi: 10.5301/ijao.5000465
59. Sawada K, Ohdo M, Ino T, Nakamura T, Numata T, Shibata H, et al. Safety and tolerability of nafamostat mesilate and heparin as anticoagulants in leukocytapheresis for ulcerative colitis: post hoc analysis of a large-scale, prospective, observational study. Ther Apher Dial. (2016) 20:197–204. doi: 10.1111/1744-9987.12357
60. Kim HS, Lee KE, Oh JH, Jung CS, Choi D, Kim Y, et al. Cardiac arrest caused by nafamostat mesilate. Kidney Res Clin Pract. (2016) 35:187–9. doi: 10.1016/j.krcp.2015.10.003
61. Li Q, Nacion K, Bu H, Lin F. The complement inhibitor FUT-175 suppresses T cell autoreactivity in experimental autoimmune encephalomyelitis. Am J Pathol. (2009) 175:661–7. doi: 10.2353/ajpath.2009.081093
62. Poe M, Wu JK, Blake JT, Zweerink HJ, Sigal NH. The enzymatic activity of human cytotoxic T-lymphocyte granzyme A and cytolysis mediated by cytotoxic T-lymphocytes are potently inhibited by a synthetic antiprotease, FUT-175. Arch Biochem Biophys. (1991) 284:215–8. doi: 10.1016/0003-9861(91)90286-R
63. Li C, Wang J, Fang Y, Liu Y, Chen T, Sun H, et al. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav Immun. (2016) 56:230–45. doi: 10.1016/j.bbi.2016.03.019
64. Inagaki H, Nonami T, Kurokawa T, Takeuchi Y, Okuda N, Nakao A, et al. Effects of nafamostat mesilate, a synthetic protease inhibitor, on immunity and coagulation after hepatic resection. Hepatogastroenterology. (1999) 46:3223–8.
65. Waki K, Yamada A. Blockade of high mobility group box 1 augments antitumor T-cell response induced by peptide vaccination as a co-adjuvant. Cancer Sci. (2016) 107:1721–9. doi: 10.1111/cas.13084
66. Lv M, Zeng H, He Y, Zhang J, Tan G. Dexmedetomidine promotes liver regeneration in mice after 70% partial hepatectomy by suppressing NLRP3 inflammasome not TLR4/NFkappaB. Int Immunopharmacol. (2018) 54:46–51. doi: 10.1016/j.intimp.2017.10.030
67. Kanangat S, Seder CW, Pergande MR, Lobato GC, Fhied CL, Raouf MF, et al. Circulating histocompatibility antigen (HLA) gene products may help differentiate benign from malignant indeterminate pulmonary lesions. Hum Immunol. (2018) 79:558–63. doi: 10.1016/j.humimm.2018.04.003
68. Liu Z, Zhao Y, Fang J, Cui R, Xiao Y, Xu Q. SHP2 negatively regulates HLA-ABC and PD-L1 expression via STAT1 phosphorylation in prostate cancer cells. Oncotarget. (2017) 8:53518–30. doi: 10.18632/oncotarget.18591
69. Lin PL, Wu TC, Wu DW, Wang L, Chen CY, Lee H. An increase in BAG-1 by PD-L1 confers resistance to tyrosine kinase inhibitor in non-small cell lung cancer via persistent activation of ERK signalling. Eur J Cancer. (2017) 85:95–105. doi: 10.1016/j.ejca.2017.07.025
70. Sasidharan Nair V, Toor SM, Ali BR, Elkord E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin Ther Targets. (2018) 22:547–57. doi: 10.1080/14728222.2018.1471137
71. Smithy JW, Moore LM, Pelekanou V, Rehman J, Gaule P, Wong PF, et al. Nuclear IRF-1 expression as a mechanism to assess “Capability” to express PD-L1 and response to PD-1 therapy in metastatic melanoma. J Immunother Cancer. (2017) 5:25. doi: 10.1186/s40425-017-0229-2
72. Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A, et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. (2017) 141:1402–12. doi: 10.1002/ijc.30834
73. Tornatore L, Sandomenico A, Raimondo D, Low C, Rocci A, Tralau-Stewart C, et al. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell. (2014) 26:495–508. doi: 10.1016/j.ccr.2014.07.027
74. Boutaffala L, Bertrand MJ, Remouchamps C, Seleznik G, Reisinger F, Janas M, et al. NIK promotes tissue destruction independently of the alternative NF-kappaB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ. (2015) 22:2020–33. doi: 10.1038/cdd.2015.69
75. Yoon WH, Jung YJ, Kim TD, Li G, Park BJ, Kim JY, et al. Gabexate mesilate inhibits colon cancer growth, invasion, and metastasis by reducing matrix metalloproteinases and angiogenesis. Clin Cancer Res. (2004) 10:4517–26. doi: 10.1158/1078-0432.CCR-04-0084
76. Brandi G, Tavolari S, De Rosa F, Di Girolamo S, Agostini V, Barbera MA, et al. Antitumoral efficacy of the protease inhibitor gabexate mesilate in colon cancer cells harbouring KRAS, BRAF and PIK3CA mutations. PLoS ONE. (2012) 7:e41347. doi: 10.1371/journal.pone.0041347
77. Yuhara H, Ogawa M, Kawaguchi Y, Igarashi M, Shimosegawa T, Mine T. Pharmacologic prophylaxis of post-endoscopic retrograde cholangiopancreatography pancreatitis: protease inhibitors and NSAIDs in a meta-analysis. J Gastroenterol. (2014) 49:388–99. doi: 10.1007/s00535-013-0834-x
78. Okumura K, Kiyohara Y, Komada F, Iwakawa S, Hirai M, Fuwa T. Improvement in wound healing by epidermal growth factor (EGF) ointment. I. Effect of nafamostat, gabexate, or gelatin on stabilization and efficacy of EGF. Pharm Res. (1990) 7:1289–93.
79. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. (2011) 2:135–64. doi: 10.18632/oncotarget.240
80. Chiang CY, Ulrich RL, Ulrich MP, Eaton B, Ojeda JF, Lane DJ, et al. Characterization of the murine macrophage response to infection with virulent and avirulent Burkholderia species. BMC Microbiol. (2015) 15:259. doi: 10.1186/s12866-015-0593-3
81. Wang J, Li C, Chen T, Fang Y, Shi X, Pang T, et al. Nafamostat mesilate protects against acute cerebral ischemia via blood-brain barrier protection. Neuropharmacology. (2016) 105:398–410. doi: 10.1016/j.neuropharm.2016.02.002
82. Chen T, Wang J, Li C, Zhang W, Zhang L, An L, et al. Nafamostat mesilate attenuates neuronal damage in a rat model of transient focal cerebral ischemia through thrombin inhibition. Sci Rep. (2014) 4:5531. doi: 10.1038/srep05531
83. Choi S, Kwon HJ, Song HJ, Choi SW, Nagar H, Piao S, et al. Nafamostat mesilate promotes endothelium-dependent vasorelaxation via the Akt-eNOS dependent pathway. Korean J Physiol Pharmacol. (2016) 20:539–45. doi: 10.4196/kjpp.2016.20.5.539
84. Lin CC, Lin LJ, Wang SD, Chiang CJ, Chao YP, Lin J, et al. The effect of serine protease inhibitors on airway inflammation in a chronic allergen-induced asthma mouse model. Mediators Inflamm. (2014) 2014:879326. doi: 10.1155/2014/879326
85. Inman RD, Chiu B. Nafamostat mesylate, a serine protease inhibitor, demonstrates novel antimicrobial properties and effectiveness in Chlamydia-induced arthritis. Arthritis Res Ther. (2012) 14:R150. doi: 10.1186/ar3886
86. Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI, et al. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother. (2016) 60:6532–9. doi: 10.1128/AAC.01043-16
Keywords: nafamostat mesylate, antitumor activities, signaling pathways, efficacy, toxicity
Citation: Chen X, Xu Z, Zeng S, Wang X, Liu W, Qian L, Wei J, Yang X, Shen Q, Gong Z and Yan Y (2019) The Molecular Aspect of Antitumor Effects of Protease Inhibitor Nafamostat Mesylate and Its Role in Potential Clinical Applications. Front. Oncol. 9:852. doi: 10.3389/fonc.2019.00852
Received: 08 May 2019; Accepted: 19 August 2019;
Published: 03 September 2019.
Edited by:
Christian Celia, Università degli Studi G. d'Annunzio Chieti e Pescara, ItalyReviewed by:
Claudia Maletzki, University Hospital Rostock, GermanyLoredana Bergandi, University of Turin, Italy
Copyright © 2019 Chen, Xu, Zeng, Wang, Liu, Qian, Wei, Yang, Shen, Gong and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanliang Yan, eWFueXVhbmxpYW5nJiN4MDAwNDA7Y3N1LmVkdS5jbg==
†These authors have contributed equally to this work