 Mohammad Reza Javan
Mohammad Reza Javan Arezou Khosrojerdi
Arezou Khosrojerdi Seyed Mohammad Moazzeni
Seyed Mohammad Moazzeni- Department of Immunology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
Tumor microenvironment interacts with tumor cells, establishing an atmosphere to contribute or suppress the tumor development. Among the cells which play a role in the tumor microenvironment, mesenchymal stem cells (MSCs) have been demonstrated to possess the ability to orchestrate the fate of tumor cells, drawing the attention to the field. MSCs have been considered as cells with double-bladed effects, implicating either tumorigenic or anti-tumor activity. On the other side, the promising potential of MSCs in treating human cancer cells has been observed from the clinical studies. Among the beneficial characteristics of MSCs is the natural tumor-trophic migration ability, providing facility for drug delivery and, therefore, targeted treatment to detach tumor and metastatic cells. Moreover, these cells have been the target of engineering approaches, due to their easily implemented traits, in order to obtain the desired expression of anti-angiogenic, anti-proliferative, and pro-apoptotic properties, according to the tumor type. Tumor angiogenesis is the key characteristic of tumor progression and metastasis. Manipulation of angiogenesis has become an attractive approach for cancer therapy since the introduction of the first angiogenesis inhibitor, namely bevacizumab, for metastatic colorectal cancer therapy. This review tries to conclude the approaches, with focus on anti-angiogenesis approach, in implementing the MSCs to combat against tumor cell progression.
Introduction
Cancer is considered as one of the top life-threatening complications, which is responsible for a quarter of human mortalities (1). The common cancer therapies, such as surgery, chemotherapy and radiotherapy, are usually symptomatic and passive in their procedures. In spite of improved treatment approaches, most of the cancer cells do not respond properly to traditional therapeutics. The major hindrance in limiting the efficacy of traditional cancer therapeutics is tumor specificity. As a consequence, it is important to look for efficient therapeutic strategies specifically targeting malignancies (2).
MSCs are the first choice of stem cells for application in clinical medicine. A number of these cells have been extracted from different tissues, such as the heart, brain, and kidney, and have demonstrated the potential as attractive candidates for treating various types of diseases (3, 4). The potential of MSC to differentiate into various cell types, and the simplicity of expansion in vitro, have raised the interests toward their therapeutic applications to treat human diseases. They can be extracted from adult human tissues and have the ability for self-renewal and development into mesenchymal lineages. They are re-grafted following proliferation and manipulation in vitro. After re-implantation, they gain the potential to suppress the immune system. Furthermore, they can be adapted into the re-grafted tissue architecture and promoted to progeny with regard to both stem cells and lineage of daughters in the microenvironment. MSCs show pathotropic migratory characteristics, providing them as potential tools for application in targeted delivery vectors for tumor therapy (3, 4). With the advent of specific anticancer drugs and considering the capacity of MSCs in tumor-targeted delivery and incorporation, a new research area has emerged with the aim of developing efficient cancer therapy using manipulated MSCs.
Tumor angiogenesis is an approach for tumor cells for their proliferation (5). The observations that several growth factors and mediators of the extracellular matrix account for tumor angiogenesis raised attention for the utilization of targeted anti-angiogenic therapy of cancers (6). On the other side, different types of stem cells have been modulated to express anti-angiogenic factors (3).
In this review, after general characterization of MSCs, we go through the approaches for application of MSCs in treatment of cancers. Alternately, with respect to the possible anti-angiogenic properties of MSCs in normal human physiology, we proposed a putative straightforward application of these cells in cancer therapy.
Characteristics of MSCs
MSCs are a group of adult stem cells, which are naturally developed in the human body. They were first identified in the stromal matrix of bone marrow by Friedenstein et al. (7, 8). The comprehensive nature and localization of MSCs in vivo remain poorly known. Other than bone marrow, MSCs have been found in a number of other adult and fetal tissues, such as heart, amniotic fluid, skeletal muscle, synovial tissue, adipose tissue, pancreas, placenta, cord blood and circulating blood. It has been suggested that basically all organs containing connective tissue possess MSCs (9). Among the stem cells, MSCs are the most investigated and the best-defined stem cells. MSCs are primitive cells, which originate from the mesodermal germ layer and were classically known as progenitors developing to connective tissues, skeletal muscle cells, and cells of the vascular system. MSCs can develop into cells of the mesodermal lineage, like bone, fat and cartilage cells, but they have the potential to differentiate into endodermic and neuroectodermic lineages. In fact, bone marrow-derived MSCs are a heterogeneous population (10). Because of their supposed capacity of self-renewal and differentiation, bone marrow-derived stromal cells were first regarded as stem cells and named MSCs (11), despite some controversy regarding their nomenclature (12). MSCs have emerged as considerable biomedical sources as a result of their multilineage potential (13). Due to their easy acquisition, fast ex vivo proliferation and the feasibility of autologous transplantation, MSCs became the first choice of stem cells to be applied in the clinical regenerative medicine. They may provide important potentials for cell survival in injured tissues, with or without direct participation in long-term tissue repairmen procedures (14). MSCs can modify the response of immune cells and therefore are linked with immune-related disorders, especially autoimmune settings (15, 16). MSCs have been shown to have specific tumor-oriented migration as well as incorporation capacity in several preclinical models, demonstrating the potential for MSCs to be used as favorable carriers for anticancer compounds (17). Bone marrow-derived MSCs obtained from other tissues, like adipose tissue, can also be potentially utilized as anticancer gene vehicles for cancer treatment (18, 19). MSCs show both pro- and anti-cancer features (20), providing “double-edged sword” characteristics in their interaction with tumor cells. However, if MSCs are suitably manipulated with anticancer genes they could be used as a favorable “single-edged sword” against cancer cells.
Origin of MSCs
MSCs can be extracted from adult human tissues and have the potential for self-renewal and differentiation into mesenchymal lineages, such as chondrocytic, osteocytic, and adipogenic. The harvesting of MSC generally does not comply with ethical issues and is less invasive than other sources, for example neural stem cells (3). MSCs have the potential to develop into tissue types of other lineages, both within or across germ lines (21). The highest degree of lineage plasticity has been implicated in bone marrow-derived MSCs, which are capable of giving rise to virtually all cell types upon implantation into early blastocysts and are relatively easy to manipulate in vitro (22, 23). To date, most of the preclinical studies have been done with bone marrow-derived MSCs, which might not be the best-suited source available for the clinical applications. The harvesting of bone marrow requires invasive steps which yields a small number of cells, and the number, differentiation potential, and life span of bone marrow-derived MSCs reduces alongside with the age of the patient (24, 25). Two other accessory sources for harvesting MSCs that have received significant attention are adipose tissue and umbilical cord blood. MSCs derived from adipose have become a highly attractive alternative in recent years, mainly due to the ease of tissue collection, high initial cell yields, and favorable proliferation ability in vitro (26). The expansion and differentiation capacity as well as the immunophenotype of MSCs obtained from adipose tissue are nearly the same as those extracted from bone marrow (27). Immunogenicity of allogeneic and xenogeneic MSCs isolated from adipose tissue has been shown not to be a problematic issue for their therapeutic applications, at least in recurrent spontaneous abortion (28). Moreover, MSC therapy could modulate the immune responses in a beneficial way (29). In fact, MSC therapy modulated the balance of helper T (Th) 1/Th2 cytokines production toward increased Th2 type cytokines (30).
Umbilical cord blood and Wharton's jelly have been shown to be a rich source of MSCs (31). As the cells are isolated after removal of the placenta, the extraction procedure is completely non-invasive and simple. The cells in the adherent layer have been demonstrated to have a fibroblastiod morphology, which express the same surface markers as bone marrow-derived MSCs, including CD13, CD29, CD49e, CD54, and CD90 (32). MSCs with umbilical cord blood origin have the potential to expand at a higher speed in relation to bone marrow and adipose tissue-derived MSCs (27, 33). This issue may be somewhat due to higher telomerase activity (34).
Adult dental pulp (DP) is another source of MSC, which has been explored and validated. DP is a vascular connective tissue like mesenchymal tissue. Stem cells isolated from DP have a phenotype similar to the adult bone marrow-derived MSCs that express mesenchymal progenitor-related antigens, including SH2, SH3, SH4, CD166, and CD29. Moreover, the DP and bone marrow-derived stem cell populations show similar profiles of gene expression (35, 36). DP-derived MSCs are obtained from a very accessible tissue source, which is further expandable using deciduous teeth, that show stem cell-like properties, such as efficient multilineage differentiation and self-renewal. Furthermore, their capacity to stimulate osteogenesis could be beneficial in clinical use for implantology (37). DP-derived MSCs can show potential clinical applications in autologous in vivo stem cell transplantation in order to rebuild the calcified tissue. Immunomodulatory function of DP-derived MSCs is suitable for suppression of T cell-mediated responses in case of allogeneic transplantation of bone marrow (36, 37).
Colonization and Migration of MSCs
The very first key step for MSC homing to tumors is their mobilization from the bone marrow and other organs. Endogenous MSCs have been demonstrated to mobilize from the bone marrow and other tissues to the peripheral blood during different injury conditions, such as normoxia, hypoxia, and inflammatory conditions (38, 39) (Figure 1). The mechanisms underlying MSC migration across the endothelium and homing to the target tissues are not fully described. Nonetheless, it is known that a normal role of MSC is the potential to migrate to and repair wounded tissue. This feature of wound healing originates with migration toward inflammatory signals released by the wounded tissue (40). Several inflammatory mediators that are produced by wounds are found in the tumor microenvironment and are believed to be involved in attracting MSCs to these sites (41). Furthermore, we indicated that pro-inflammatory cytokines were able to upregulate the efficacy of MSCs in a cell-based therapy of various complications (42). A bulk of studies has demonstrated that migration is mediated through cytokines secreted by MSCs that are involved with G-protein coupled receptor (GPCR), growth factor receptors, and various cytokine/receptor pairs SDF-1/CXCR4, SCF-c-Kit, HGF/c- Met, VEGF/VEGFR, PDGF/PDGFR, HMGB1/RAGE, and MCP-1/CCR2 [N (43)]. Stromal cell-derived factor SDF-1 and its receptor, namely CXC chemokine receptor-4 (CXCR4), are major mediators of stem cell infiltration to the tumor microenvironment. The significance of the interactions between secreted SDF-1 and cell surface CXCR4 for stem cell migration has been displayed by studies in which the function of either the receptor or the cytokine was downregulated (44–46). CXCR4 and SDF-1 simultaneous blockade in vivo in mice resulted in significant reduction in the migration of transplanted stem cells to tumor locations and regions of demyelination, implicating that the SDF-1/CXCR4 signaling pathway is important for effective pathotropism quality of the stem cell therapy (44). Other studies with blockades of both CXCR4 and transforming growth factor (TGF)-β receptor have shown that CXCR4 is required for endogenous MSC colonization into tumors, differentiation to myofibroblasts, and survival of MSCs (47). MSCs express a wide range of chemokine receptors, such as CXCR1, CXCR3, CXCR4, CXCR5, CXCR6, chemokine receptor (CCR)-1, CCR2, CCR3, CCR4, CCR5, and CCR9, among others. Studies have shown that chemokines such as CXCL12, CXCL13, CXCL16, and their related receptors can promote the bidirectional migration of MSCs to and create the bone marrow niche. Specific chemokines and their receptors play a role in the unidirectional migration of MSCs. In effective homing of MSC into the bone marrow, CXCL16 plays vital role, whereas CCL22 has the strongest chemotactic effect in mobilizing MSCs from the bone marrow into the circulation (48). Both CCL2 and CXCL16 are expressed in tumor tissues, such as lung carcinoma, hepatocellular carcinoma, colorectal cancer, and brain and ovarian cancers, hence it is possible that they play a role in the migration of MSC into tumors (49–51). Transactivation of cell receptor is a mechanism which is thought to be involved in migration and plays a role in the activation of one receptor through another. While it plays an important role in processes like cellular migration and apoptosis, its deregulation can cause pathological settings like cancer. On the other side, transactivation of various growth factor receptors, such as epidermal growth factor receptor (EGFR) by GPCRs, has been observed in multiple cellular model systems, therefore illustrating the plausible role of GPCR in tumor tropism by means of receptor transactivation (52–54). A suggested mechanism of receptor transactivation involves the activation of membrane-tethered growth factors, like EGFR, by direct interaction with GPCR, like CXCR4 (55). This procedure is accompanied by matrix metalloproteinases (MMPs), like MMP-2 and MMP-9 (56). MMPs are enzymes with proteinases activity that are required to proteolytically process precursor proteins like growth factors, adhesion molecules, cytokines, and their receptors. Migration of MSCs through bone marrow endothelium has been reported to be mediated by MMP-1 as well as the tissue inhibitor of metalloproteinase-3 (57). Another report demonstrated that elevated levels of MMP-2 were responsible for C1q complement protein-mediated migration of umbilical cord blood-derived MSC into harmed tissue (58). Studies on MSC behavior show that MSCs are attracted to the sites of irradiation, and that local irradiation might enhance the specificity of MSCs to be migrated and engrafted (59). Taken together, these observations suggest a strong validation for the development of therapeutic approaches that employ the tumoritropic characteristics of MSCs by engineering them into delivery vehicles for antitumor compounds.
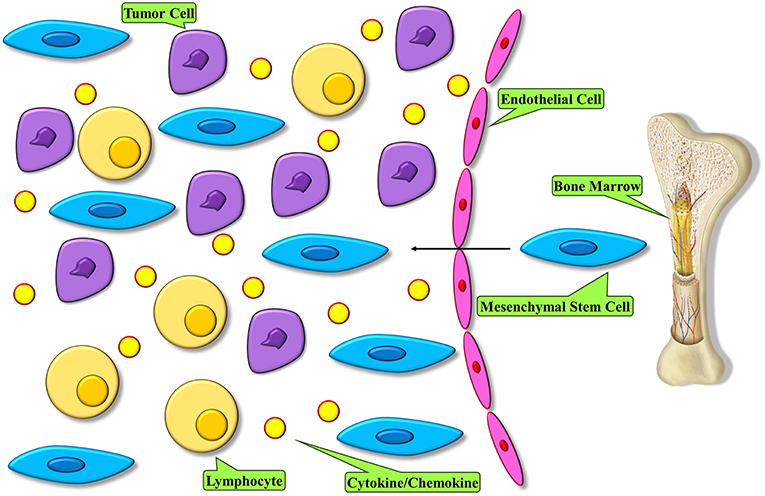
Figure 1. MSC homing within tumors. Tumor microenvironment modulates an inflammatory condition and releases mediators to recruit MSCs into tumor. These cells become the major constituent of the tumor microenvironment.
MSC Behavior in Tumor Microenvironment
MSCs represent various functions in distinct microenvironments, through secreting several mediators (Figure 2), due to diversity of the signaling pathways that stimulate these cells. The MSCs applied for clinical treatment are predominantly naïve MSCs driven from normal tissue sources and are harvested in vitro. Naïve MSCs have the potential to interrupt tumor cell growth when co-cultured with tumor cells in vitro. A study demonstrated that naïve MSCs inhibited the proliferation of solid tumor and leukemia cell lines. The inhibitory impression of naïve MSCs was observed to be dose-dependent, as proportions of naïve MSCs were accompanied with higher levels of inhibition (60). The possible mechanism of tumor inhibition via naïve MSCs was attributed to the soluble factors secreted by these cells, including Dickkopf-related protein 1 (DKK1), which plays a role in the inhibition of Wnt signaling pathways in tumor cells. Interruption in the Wnt pathway downregulated Cyclin D2 and c-Myc and upmodulated expression of P27KIP1 and P21CIP1, leading to tumor cell cycle suppression (61–64). Moreover, naïve MSCs can also activate apoptosis in tumor cells (65) through upregulation of caspase 3 (61). Naïve MSCs, by inhibiting angiogenesis, can indirectly interrupt tumor expansion. The mechanisms underlying angiogenesis interruption by naïve MSCs are apoptosis in vascular endothelial cells (66, 67) and impairing the vascular network construction (68). Nonetheless, adverse roles of naïve MSCs have also been reported, as these cells were related to enhanced tumor angiogenesis in colon cancer cell lines (69, 70), MSC differentiation to vascular endothelial cells in melanoma (71), and promotion of cancer stem cells (CSCs) that enhance tumorigenesis, metastasis and recurrence of tumors (72) like breast cancer (73), increasing the expansion of gastric cancer cell lines (72, 74). Among the other unwanted influences of naïve MSCs in tumors are enhancement of tumor cell migration mediated by release of chemokines, such as CXCR4 (75), CCL5 (76, 77), vascular cell adhesion molecules (VCAMs), and intercellular adhesion molecules (ICAMs) (78). Additionally, MSCs were observed to suppress immune responses, leading to tumor development (79). Naïve MSCs have been associated with bidirectional effects on tumor growth and development, and different investigations have been accompanied by discrepancies on whether MSCs are involved in tumor promotion or suppression. The divergence in observed outcomes may stem from differences in experimental settings, in which different observations may be achieved by exerting in vivo or in vitro experiments.

Figure 2. Different cytokines and mediators secreted from MSCs during cross-talk with tumor cells that are involved in three major interfaces of homing, growth, and stemness of tumor cells.
The other type of MSCs, the tumor-derived MSCs (T-MSCs), demonstrate phenotype similarity with naïve MSCs but exhibit stronger immunosuppressive activity and proliferative capacity than naïve MSCs (80). MSCs isolated from mouse lymphomas (L-MSCs) compared with bone marrow-derived MSCs (BM-MSCs) promoted tumor cell proliferation vigorously, which was related to release of CCL2 by L-MSCs, resulting in recruitment of immunosuppressive cells, such as F4/80+ macrophages, CD11b+Ly6G+ neutrophils, and CD11b+ Ly6C+ monocytes to lymphoid tissues (81). Moreover, MSCs derived from breast cancer tissues relative to MSCs from normal breast tissues present higher amounts of immunosuppressive mediators such as TGF-β1, IL-4, and IL-10. Additionally, MSCs isolated from breast cancer tissues co-cultured with peripheral blood mononuclear cells (PBMCs) led to the development of CD4+CD25hiFoxp3+ regulatory T cells (82). Furthermore, MSCs derived from pediatric sarcomas demonstrated inhibitory effects of the cytotoxic functions of natural killer (NK) cells through downregulating the activation receptors of NK cells (83). On the other side, T-MSCs have also disadvantageous ramifications. T-MSCs exert a function in epithelial-to-mesenchymal transition (EMT), as pancreatic cancer revealed upregulation of vimentin and downregulation of E-cadherin (84). Moreover, MSCs from prostate cancer and breast cancer cell lines overexpress CCL5, which in turn up-modulated the expression of genes involved in EMT, such as zinc finger E-box-binding homeobox 1, CXCR4, and Snail (76, 85). As well, T-MSCs obtained from ovarian cancer increased the proliferation of tumor cells (86). Additionally, T-MSCs promoted the proportion of CSCs, as observed in ovarian cancer cell, probably because of upregulation of bone morphogenetic proteins (BMPs), such as BMP2, BMP4, and BMP6 in ovarian cancer-derived MSCs (86). Taken together, in contrast to naïve MSCs, T-MSCs demonstrate further capacities in promotion of tumor progression.
The Potential of MSCs For Tumor Therapy
MSCs with no modification have been demonstrated to have antitumor effects both in vitro and in different mouse models of tumors. This is in part due to the factors released by MSC that have antitumor functions that can reduce the proliferation of melanoma, glioma, lung cancer, hepatoma, and breast adenocarcinoma (63, 87–89). BM-MSCs injected intravenously in a mouse model of Kaposi's sarcoma were shown to be colonized in sites of tumorigenesis and potently inhibit tumor development (90). MSCs have also been observed to have anti-angiogenic properties both in vitro and in mouse model of melanoma (66). Moreover, direct MSC injection into subcutaneous melanoma in mice triggered apoptosis and diminished tumor growth (66). Furthermore, umbilical cord blood-derived MSCs have been used as naïve cells for the therapy of glioblastoma multiforme (GBM). Umbilical cord blood-derived stem cells expressing high levels of CD44 and CD133 cells underwent apoptosis following co-culturing with GBM cells (91). Additionally, treatment of glioma cells with umbilical cord blood-derived stem cells caused inhibition of focal adhesion kinase (FAK)-mediated angiogenesis (92), upregulation of phosphatase and tensin homolog (PTEN) expression in glioma cells, and down-modulation of Akt and PI3K signaling pathway molecules in the nude mice tumors, resulting in the inhibition of migration as well as wound healing characteristics of the glioma cells (93). Alternately, this eventuated in downregulation of XIAP activating caspase-3 and caspase-9 to stimulate apoptosis in glioma cells (94). Another investigation surveyed a comparative study of umbilical cord blood-derived MSCs and MSCs from other sources and showed diminished GBM growth via tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) (95). In consideration of these observations, MSCs have been modified mainly to overexpress target genes in order to express or secrete a selective favorable therapeutic molecule for targeted treatment of various cancer types. These compounds mainly include interleukins, interferons, pro-apoptotic proteins, and anti-angiogenesis agents.
Interleukins
Interleukins are cytokines that modulate immune responses and have been suggested to show antitumor properties through either direct tumoricidal effects or positive regulation of the endogenous immune system against tumor cells (96). Nonetheless, lack of tumor-targeted delivery has diminished their application for cancer treatment (97). With respect of results, the delivery of interleukins by MSCs has been investigated. Engineered MSCs for expressing interleukins have been employed to enhance the anticancer immune surveillance by activating cytotoxic lymphocytes and NK cells (96). Moreover, MSCs expressing interleukin (IL)-12 have been indicated to hinder the metastasis into the lymph nodes and other internal organs as well as promoted tumor cell apoptosis in animal models with pre-established metastases of melanoma, breast, and hepatoma tumor cells (98). IL-12 expressing MSCs were observed to have antitumor functions in mice with cervical tumors (99) and renal cell carcinomas (100). These studies disclosed sustained expression of interferon (IFN)-γ and IL-12 in serum and tumor microenvironment. Additionally, umbilical cord blood-derived MSCs were successfully used as systems to deliver IL-12 to contribute in the malignant glioma therapy (101). As such, the transplantation of MSCs secreting IL-18 was previously indicated to increase T cell infiltration and long-term antitumor responses in mice with non-invasive and invasive gliomas (102). On the other hand, umbilical cord blood-derived MSCs were manipulated to express IL-21 that demonstrated therapeutic efficacy in mice with ovarian cancer xenografts (103). Moreover, the efficacy of IL-24 expressing MSCs as a therapeutic cytokine delivery tool for lung cancer was assessed. Human umbilical cord-derived MSCs modulated to deliver IL-24 inhibited the exacerbation of lung cancer cells by stimulation of apoptosis and arrest of cell cycle (104). Furthermore, this survey demonstrated that injection of MSCs secreting IL-24 repressed xenograft tumor growth and showed anti-angiogenic effects both in vitro and in vivo evaluations (104). These observations suggest that employing MSCs as vehicles to deliver interleukin have promising effects in treatment of various cancers.
Interferons
Interferon (IFN)-β has shown anti-proliferative and pro-apoptotic effects on tumor cells (104–106). Therapeutic efficacy of IFN-β has been limited because of toxicity related to systemic administration. To resolve this issue, human MSCs have been engineered to express IFN-β, hence these cells carried out targeted delivery to treat metastatic melanoma and breast models (107, 108), lung metastasis (109, 110), and gliomas (45). Intraperitoneal injection of IFN-β expressing MSCs in mice bearing ovarian xenografts led to complete destruction of tumors in 70% of treated mice (111). These observations validated the potential of MSCs to be employed as targeted delivery vehicle for the production of IFN-β inside the tumor tissue. Furthermore, the intravenous injection of MSCs expressing IFN-β significantly decreased prostate tumor weight and promoted animal survival in comparison to controls (112). Amniotic fluid (AF)-derived MSCs were studied to deliver the IFN-β to the involved site of neoplasia in a bladder tumor model. This investigation indicated that tumor growth was limited and survival of mice was prolonged following administration of AF-derived MSCs secreting IFN-β (113). In previous studies, human adult MSCs engineered for expressing INF-β demonstrated in vivo efficacy of these cells to treat the solid melanomas in nude mice (107). The antitumor effects of engineered MSCs expressing INF-β in therapy of central nervous system (CNS) tumors were displayed by evaluating if human MSC could be efficient in tracking the murine brain tumors when administered intravenously (45). Through engineering the MSCs to secrete INF-β, this strategy could be carried out to elicit antitumor effects, as administration of human MSCs secreting INF-β led to enhanced murine survival. Moreover, the efficacy of MSCs secreting IFN-β in a model of prostate cancer and lung metastasis was investigated (110). Targeted colonization of MSCs secreting IFN-β was observed at sites of lung tumors with pulmonary metastases, and this led to inhibition of tumor growth. The antitumor effects of IFN-α, which shows multifunctional regulatory effects, have also been studied. Oftentimes, IFN-α is utilized as a supplementary therapy to destroy the micrometastatic deposits in cases with high risk of systemic recurrence after therapy with other strategies (114, 115). IFN-α expressing MSCs can be efficiently targeted inside the tumor tissue in mice model of plasmacytoma. On the other side, administration of MSCs engineered to express IFN-α through subcutaneous way significantly limited tumor growth in vivo and increased the survival of the mice by triggering apoptosis in tumor cells and by diminishing the growth of tumor vessels (115, 116). Moreover, the systemic administration of MSCs expressing IFN-α decreased tumor cell growth and considerably increased the survival rate because of intensified tumor cell apoptosis and decreased development of tumor blood vessels in a mouse model of metastatic melanoma (108, 115).
Pro-apoptotic Proteins
TRAIL is known as an endogenous member of the TNF ligand family that ligates to its death domain, which bears receptors for Dr4 and Dr5 and triggers apoptosis by activation of caspases, particularly in cancer cells without effect on other cell types (117). Studies have indicated the therapeutic effects of engineered MSCs to express TRAIL in both cell lines or animal models of tumors, such as gliomas (118–120), colorectal carcinoma (121), and lung, breast, squamous, and cervical cancers (122). These investigations disclosed the favorable TRAIL effects in stimulation of apoptosis and a further reduction of tumor cell survival. Studies also indicated that adipose tissue-derived MSCs engineered to express TRAIL targeted several tumor cell lines of human cervical carcinoma, colon cancer, and pancreatic cancer. Following administration into mice, TRAIL expressing adipose tissue-derived MSCs migrated to tumors and triggered apoptosis in tumor cells. Moreover, this targeted delivery resulted in no significant toxicities to normal cells (123). Similarly, adipose tissue-derived MSCs engineered to express TRAIL demonstrated anti-myeloma activities and significantly triggered in vitro killing of myeloma cell (124). TRAIL is a type II membrane protein, which needs enzymatic cleavage from its cell membrane anchoring site upon releasing into the microenvironment. In order to eliminate this cleavage step, attempts are on the way to re-design the TRAIL protein in order to achieve TRAIL-secreting cells with direct paracrine function. To engineer the secretable compound of TRAIL, researchers fused the extracellular domain of TRAIL and the extracellular domain of the hFlt3 ligand (125). This re-designed compound, namely S-TRAIL, is efficiently secreted into the tumor microenvironment and indicated excessive cytotoxic effects on tumor cells in comparison to the traditional TRAIL form (126, 127). Investigations disclosed that human MSCs are resistant toward the apoptosis mediated by TRAIL. The cell killing mechanism of MSCs engineered to express S-TRAIL is to trigger caspase-mediated apoptosis in glioma cell lines and glioblastoma stem cells (GBSC) in vitro (128).
Antagonists of Growth Factors
Antagonists of growth factors are beneficial choices for suppressing tumor development. Among the agents that inhibit the function of growth factors, there are a limited number of compounds that can be expressed in stem cells and released in the extracellular microenvironment. As an antagonist of hepatocyte growth factor (HGF) (129), NK4 is a strong suppressor of tumor growth as well as angiogenesis (130, 131). NK4 expressing MSCs has been evaluated in mice models with lung metastases (132). When NK4 expressing MSCs was systemically administered to mice, it localized to the sites of lung metastatic tumor and efficiently suppressed tumor progression/metastases in the lung. Inhibition of lymphangiogenesis and angiogenesis in the tumor tissues has been considered as the mechanism of anti-metastatic action of NK4-expressing MSCs.
Angiogenesis Perspectives For Cancer Therapy
Process of Angiogenesis
During embryonic procedures, formation of new blood vessels from pre-existing vasculature is known as a critical event resulting in formation of constant vasculature consisting of endothelial cells (ECs), mural cells, and basement membrane in adults (133). ECs are the most inner layer of vessel walls and are in direct interaction with mural cells along the length of the vessels. The basement membrane is a thin layer that coats the entire length of ECs (134). Vasculogenesis in normal physiological conditions is very stable and is rarely seen in adult individuals. However, adults may develop angiogenesis in cyclical growth of vessels in the ovarian corpus luteum as well as during pregnancy (135). Angiogenesis is defined to be an important event in pathological settings like tissue repair during wound healing as well as during tumor growth (136). Nonetheless, in comparison to normal vessels, the structure and components of tumor vessels are distinct (137). Tumor vessels are disorganized and mazy and do not possess the hierarchy of arterioles, capillaries, and venules. Because the ECs are not in immediate contact with mural cells and are connected to basement membrane loosely in tumor constructions, they are leakier. Overexpression of PlGF, CD109, CD137, and CD276 have been identified in ECs of tumor tissues in comparison to ECs of non-tumor tissues. Although numerous studies have investigated the cellular and molecular features of ECs in tumors, the source of these cells remains controversial in tumors (138). A worthwhile amount of data in certain models recognized endothelial progenitor cells (EPCs) to be directly contributing to tumor angiogenesis (138). Regardless of cellular origin of vasculature in tumors, stimulation and initiation of angiogenesis need a shift to activation and upmodulation of angiogenesis stimulators alongside with suppression of angiogenesis inhibitors. Among the most important stimulatory factors of angiogenesis are MMPs, vascular endothelial growth factor-A (VEGF-A), placenta growth factor (PlGF), fibroblast growth factor (FGF), and hepatocyte growth factor (HGF) (139, 140). On the other side, IL-12, thrombospondins (THSBs), endostatin, and angiostatin are important endogenous inhibitors of angiogenesis (141). In consideration of all, it seems that angiogenesis plays an important role in physiological homeostasis as well as in tumor growth and development.
Inhibition of the VEGF Pathway as a Major Approach of Anti-angiogenesis Tumor Therapy
With respect of the involvement of angiogenesis in tumor progression and expansion, targeting tumor angiogenesis procedures as a therapeutic tool has long been a putative approach. In fact, suppression of growth factors/signaling pathways required for growth and progression of ECs is considered as a practical strategy to inhibit tumor vasculogenesis (136, 142). Vascular endothelial growth factor (VEGF) has been established as a key modulator of angiogenesis in both physiological and pathological conditions (143). Given the genetic background, a VEGF allele loss, leading to angiogenesis defects, has been observed to be the cause of death during embryonic life (143). Furthermore, VEGF-null embryonic stem cells were shown to be unable to construct teratoma in the recipient upon administration in the testis capsule, implicating the important role of VEGF and thereby angiogenesis in tumor growth and expansion. Among many factors (144), hypoxia-inducible factor (HIF-1a), which binds to VEGF promoter, is one of the major regulators of VEGF expression (145). A hypoxia-conducive environment in different areas of tumors leads to overexpression of VEGF in tumor microenvironment, implicating on accelerated proliferation of tumor cells and faulty blood flow (146). On the other hand, VEGF is highly present in numerous human tumor microenvironments (147), whereas VEGFR1, VEGFR2, and VEGFR3 are overexpressed in tumor associated ECs (148).
To further shed light on the role of VEGF in tumor angiogenesis and devise a therapeutic approach, human tumor-bearing mice were treated with an anti-VEGF neutralizing monoclonal antibody, which significantly suppressed the tumor growth in the mice (149). Bevacizumab, a humanized variant of a VEGF neutralizing monoclonal antibody (150), was approved by the FDA (Food & Drug Administration) in 2003 as a first anti-angiogenic factor as a supplement to therapy with standard of care (SOC) in patients with metastatic colorectal cancer (151). Moreover, bevacizumab was then confirmed to have beneficial effects in treatment of patients with non-small-cell lung cancer (152) and metastatic breast cancer (153). After these prosperous observations, a number of VEGF pathway inhibitory agents have been under different stages of clinical trials, which subject either VEGF or its receptor. VEGF-TrapR1R2, which is a chimeric soluble receptor of VEGF, harbors functional compartments of VEGFR1 and VEGFR2 (154), which have the potential to bind to and thereby neutralize circulating VEGF. Furthermore, VEGF-TrapR1R2 has indicated to be surpassed in anti-tumor properties and function in comparison to other VEGF receptor blockers such as DC101, as observed through preclinical models. On the other hand, inhibition of the VEGF pathway by means of VEGF receptor blockades also suppresses the tumor development (155, 156). As an instance, receptor tyrosine kinase inhibitors (RTKIs) have been evaluated, which is receptor tyrosine kinases inhibitor and targets VEGF and other molecules involved in VEGF signaling pathways. Among the most clinically promising RTKIs are linifanib (157, 158), cabozantinib (159), axitinib (160), tivozatinib (161), vendatanib (162), sunitinib (163), pazopanib (164), and sorafenib (165). While clinical measurements imply that there are beneficial effects of these compounds on tumor treatment, the precise mechanism of action of these anti-angiogenic agents in cancer therapy is still obscure. Angiogenesis inhibitors targeting the VEGF pathway have been indicated in preclinical models to repress tumor development through inhibition of tumor angiogenesis. Nonetheless, other studies show that anti-angiogenesis agents may function through vascular normalization, in which these agents target the non-functional vessels in tumors, leading to diminished blood flow, enhanced tumor oxygenation, and better delivery of cytotoxic drugs to the tumor tissue (166). It should be noted that the results of preclinical models have been in contrast with those of clinical trials with respect to VEGF inhibitors. Clinical trials have indicated different results between the efficacy of monoclonal antibodies in comparison to small anti-angiogenesis inhibitors. Bevacizumab in combination with SOC indicated favorable results in patients with metastatic breast cancer (153), colorectal cancer (151), and non-small cell lung cancer (152). It can be concluded that development of VEGF-related and anti-angiogenic compounds provided a promising therapeutic tool for cancer disorders and opened new horizons within this context. In addition to VEGF inhibitors, other classes of anti-angiogenesis agents, namely vascular disrupting agents (VDAs), have been evaluated. VDAs function in suppressing the tumor progression through triggering vascular collapse, resulting in hypoxia and thereby necrosis of tumor cells (167). As a VDA, ASA404, which is a flavonoid compound, induced apoptosis in tumor ECs, leading to a blockade of blood flow in tumor tissues. Currently, the ASA404 is under clinical evaluations in patients with non-small cell lung cancer (168).
Inhibition of Signal Transduction by Targeting Receptor Tyrosine Kinases
Angiogenesis is mediated by ligation of growth factors to their receptors, which results in signal transduction through corresponding receptor tyrosine kinases (RTKs) (169). Three subfamilies of 14 RTK, namely Ang/Tie-2, VEGF/VEGFR-2, and Ephrin B2/EphB4 are particularly overexpressed in the endothelial cells. These three RTKs are essentially involved in the development of both vasculogenesis and angiogenesis (170). VEGF binding to VEGFR-2 induces signal transduction that eventuates in survival and proliferation of endothelial cells. Vessel stabilization and maturation as well as remodeling of vessel structure have been associated with angiogenic growth factors (Ang) and their tyrosine kinase receptor (Tie-2) (171). Moreover, Ephrin receptor tyrosine kinase (EphB4) and its transmembrane-type Ephrin B2 ligand play a role in vasculogenesis (172). The small molecules inhibiting the angiogenesis were (2-(3,4-Dihydroxyphenyl)-6,7-dimethylquinoxaline-HCl and (E)-3-(3,5-Diisopropyl-4-hydroxyphenyl)-2-[(3-phenyl-n-propyl) aminocarbonyl] acryl-onitrile), which were first identified in 1996. These molecules inhibited VEGF-dependent VEGF receptor-2 phosphorylation, and therefore hindered endothelial cell mitogenesis and blood vessel development (173). Targeting these RTKs is considered as a potentially therapeutic approach via designing novel anti-angiogenesis agents. For example, sorafenib, which is a multikinase inhibitor, interrupts tumor growth via antiproliferative and antiangiogenic effects. Sorafenib, which inhibits PTKs, has demonstrated antitumor activity in phase 3 trials in patients with advanced renal cell carcinoma and hepatocellular carcinoma (174). Moreover, a phase 3 trial of sunitinib, which is a RTK inhibitor, in patients with gastrointestinal stromal tumors has demonstrated promising outcomes (175).
MSCs and Anti-angiogenic Cancer Therapy
Through angiogenesis, tumors provide a way to continue to growth and progression (5). One of the approaches to employ anti-angiogenetic agents to limit the tumor growth and metastasis is to use MSCs as vehicles to deliver such agents. These manipulated MSCs demonstrated a tropism to cancer tissue and may deliver antiangiogenic compounds without adverse side effects (176). Moreover, BM-MSCs were indicated to be able to modulate the expression profile of chemokine genes (including CXCL10, CXCL3, CXCL6, and CCL-2) that play a role in angiogenesis (177). Nonetheless, prolonged systemic delivery of anti-angiogenic agents is related with toxicity, as well as poor blood supply, which decreases the delivery of chemotherapeutic drug agents to tumor cells (178). It has been identified that tumor-mediated angiogenesis stems from an aberrant regulation of both pro-angiogenic and antiangiogenic agents as well as via growth factors of the tumor microenvironment (6, 179). Endostatin, which is an important endogenous inhibitor of tumor angiogenesis, has been widely utilized in antiangiogenic therapy to treat various cancers (180). Human placenta-derived MSCs were engineered to deliver endostatin through adenoviral transduction and were then administered into nude mice. These engineered MSCs expressing the human endostatin exhibited homing to the tumor tissue and considerably diminished the tumor size, with no remarkable systemic toxic side effects. These beneficial effects in tumor therapy were due to enhanced tumor cell apoptosis, inhibited neovascularization, and hence diminished tumor cell proliferation and expansion (180). In addition, a phase II clinical study demonstrated that the delivery of anti-angiogenic agents normalized the abnormal structure and defective function of the blood vessels, which led to significantly reduced tumor-associated vasogenic brain edema (181). The vessel normalization was attributed to reduced vessel diameter and permeability (182, 183) as well as the increased mural cell coverage of the small vasculature (184). Studies show that MSCs possess the ability to home to tumor vasculature following intratumoral administration, suggesting favorable characteristics for targeted delivery of anti-angiogenic agents, especially in vascularized cancers (185).
Angiogenesis Inhibition; MSC or Other Antiangiogenic Factors
As discussed above, several antiangiogenic compounds, such as monoclonal antibodies against VEGF or VEGFR, have been addressed in cancer anti-angiogenic therapy. On the other side, MSCs have also been reported to modulate the tumor microenvironment to suppress angiogenesis or neovascularization. Each of these approaches has its pros and cons. The targeted delivery of MSCs provides a favorable characteristic in the treatment of solid tumors. Furthermore, MSCs can be engendered to deliver anti-angiogenetic agents, supporting an efficient MSC to treat angiogenesis. Nonetheless, MSCs may manifest with some unwanted properties. Among the critical concerns that requires precise evaluation before MSCs' clinical application in humans, is the safety issue. Although there are several clinical reports and current clinical trials, little has been revealed with respect to the long-term safety of MSCs in humans. Furthermore, the potential profibrogenic capacity of MSCs, as indicated in liver fibrosis triggered through human BM-MSCs (186), confers another limitation in their therapeutic application. Another issue is the heterogeneity of MSCs as a source for their application in clinical settings. MSCs are a heterogeneous pool of various cell populations which are characterized via cell surface molecules as well as by their ability to differentiate into several lineages, such as chondrocyte, osteoclast, or adipocyte. Heterogeneity of MSCs (obtained from donors as a cell source) may stem from aging, age, sex, genetics, and epigenetic factors (187). Another problem with application of MSCs to treat cancers with suppressing angiogenesis is that MSCs may confer a potential to develop angiogenesis, a characteristic in reverse to desirable purpose (188). Despite such obstacles, researchers have been surveying to increase the efficacy of MSC-based cancer therapy with respect to suppression of angiogenesis.
Putative Concept in Application of Anti-angiogenic Inhibitors and MSC Delivery Machine
Estrogen is the primary female sex hormone which regulates the growth, differentiation, and physiology of the reproductive process through binding to the estrogen receptor (ER). Estrogen plays a role in several tissues, including liver, bone, brain, and the cardiovascular system. Since estrogen's ligation to ER indicated diverse outcomes, disclosing the basis of ER functions at the molecular level has mainly been of interest in the aim of therapeutic interventions (189). Current evidence shows that estrogen induces angiogenesis by acting either directly on endothelial cells and/or indirectly on other endometrial cell types through several potential promoters. Estrogen stimulates VEGF expression in human uterine stromal cells (190, 191). Studies show that ER is a potentially favorable target to devise drugs for the treatment and prevention of the progression of breast cancer (192). Estrogen binding to ER can lead to enhanced proliferation of target cells; hence endocrine cancer therapy aims to block the interaction of estrogen with the ER. This aim is achieved through either blocking the production of estrogen or suppressing the conversion of steroidal precursors to estrogen. Selective estrogen receptor modulators (SERMs), such as tamoxifen and raloxifene, which are competitive inhibitors of estrogen, directly target ER. Manipulating the estrogen pathway is considered as the most effective therapy, with less toxic side effects for the treatment of hormone responsive breast cancers (193).
SERMs' function in select target tissues is like estrogen's but is like an anti-estrogen in other target tissues (194). The ideal SERM should act like an estrogen agonist in bone, liver, the cardiovascular system, and brain and an estrogen antagonist in the uterus and breast. On the other hand, the activity of tamoxifen relies on circulating levels of E2, which are in low levels in postmenopausal women and in high levels in premenopausal women. Tamoxifen treatment reduces bone density of premenopausal women but increases that of postmenopausal women (195). Furthermore, estrogenic activity of tamoxifen has been observed in the uterus, leading to high incidence of endometrial cancer in postmenopausal women (196). In prostate cancer, involvement of ER has been implicated. Moreover, overexpression of ERβ has been reported in the prostate (197). Within the prostate, ERβ is primarily found in the epithelium, while ERα is localized in the stroma (198). The prostate gland highly expresses ERβ, suggesting its susceptibility to the effects of estrogens (199). Application of estrogen therapy has been promising in ovarian cancers (200). The Women's Health Initiative (WHI) trial has provided evidence on the role of estrogen in colon carcinogenesis (201).
Together, given the role of estrogen and ERs in angiogenesis as well as promising therapeutic effects of inhibition of this pathway in several cancers, MSC application as a targeted delivery vehicle is suggested. Moreover, monoclonal antibodies against ERs can be employed to be directly present in the tumor microenvironment to inhibit angiogenesis. Additionally, administration of SERMs has indicated side effects in untargeted tissues. This way, estrogen/ER pathways are optimally manipulated to elicit better outcomes. This hypothesis is premature, which needs to be addressed in animal models and clinical trials. On the other side, progesterone also inhibits endothelial cell proliferation in vitro and arrests the cell cycle in the G0/G1 phase in human dermal endothelial cells (202). Progesterone receptors have been found in human endometrial endothelial cells and, when interacted with progesterone, suppressed VEGF-induced endothelial cell proliferation (203). Targeting the progesterone signaling pathway through engineered MSCs seems to be a plausible therapeutic tool for efficient cancer treatment, requiring extensive investigations.
The corpus luteum is characterized as a temporary endocrine structure in ovaries, which plays an important role in successful pregnancy in mammals. Upon rupture of follicle following ovulation, the corpus luteum is made from the debris and then is modulated through differentiation and growth. Studies indicate that the fast growth and development of the corpus luteum is contingent upon the angiogenesis process. A number of endogenous stimulatory and inhibitory factors are expressed in the corpus luteum to balance the luteal angiogenesis process (204). Pigment epithelium–derived factor (PEDF) is a physiological inhibitor of angiogenesis (205), which is mainly secreted by granulosa cells. Thrombospondins 1 and 2 are also antiangiogenic factors. Thrombospondin 1 binds to fibroblast growth factor 2 (FGF2) and then prevents its proangiogenic functions (206). Angiogenesis regulation mediated by thrombospondins is important for the diminishing of corpus luteum. Studies show that thrombospondins 1 expression was upregulated during luteolysis in an animal model (207). Additionally, prostaglandin PGF2a stimulates the overexpression of thrombospondins 1 and 2 as well as their receptor CD36 in the bovine corpus luteum. Inhibition of angiogenesis in the corpus luteum is mediated in response to the mentioned molecules in the presence of FGF2. However, inhibition was also seen in lack of FGF2 (208). These molecules play roles in inhibition of physiological angiogenesis in the corpus luteum, which might open new horizons in anti-angiogenesis therapy of cancer in the future, possibly by application of MSCs as vehicles to deliver these anti-angiogenetic mediators into the tumor tissue.
Recently, it has been established that exosomes derived from MSCs can deliver microRNAs (miRs) to target cells and, therefore, impress the angiogenesis. It was observed that incubation of MSCs with the exosome secretion blocker GW4869 decreased the levels of pro-angiomiRs in the MSC-derived medium (209). This observation led to the implication of the MSC-derived exosomes in inhibition of angiogenesis. In addition, an anti-tumor role of intra-tumoral injection of menstrual stem cell-derived exosomes was associated with an inhibitory impression on angiogenesis in oral squamous cell carcinoma (210). Moreover, MSCs were observed to suppress the angiogenesis potential of HUVEC through overexpressing miR-211, which downregulated the expression of Prox1 (211). Additionally, it was reported that MSC-derived exosomes, which were enriched with miR-16 (a miRNA targeting VEGF), could downregulate the expression of VEGF in tumor cells, resulting in suppression of angiogenesis both in vitro and in vivo (212). As a consequence, delivering exosomes expressing miRNAs (targeting angiogenetic molecules) by MSCs may also confer a potential tool in the suppression of angiogenesis.
Clinical Trials and MSCs-Based Therapy of Cancer
There is a paucity of registered clinical trials with the aim of cancer therapy via exertion of MSCs. The first clinical trial (a phase I/II clinical trial), namely Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1), applied MSC delivery of herpes simplex virus thymidine kinase (HSV-TK), which had prosperous results (213). There are two clinical trials focusing on ovarian cancer via application of MSCs. A phase I/II trial by Mayo Clinic purposed to recognize the adverse effects and well-suited dose of MSCs transfected with oncolytic measles virus encoding NIS (MV-NIS) and to evaluate its efficacy on patients with ovarian cancer. The University of Texas M.D. Anderson Cancer Center supported a phase I clinical trial that used human MSCs transfected with IFN-β to assess its safety and to identify the well-tolerable dose of such MSCs in treating ovarian cancer patients. In another phase I clinical test, aiming to investigate the safety and cancer-homing potential of MSCs, allogeneic BM-MSCs were administered in patients with prostate cancer. Nonetheless, the study revealed that MSCs could not efficiently home in on tumor sites to implement therapeutic functions (214). These studies have attempted to evaluate the potential of MSCs in treating various cancers; however, the results seem to be partially ineffective. That notwithstanding, further studies are required focusing on the efficacy and safety of such a therapeutic system through transforming the observations from bench (preclinical studies) toward the bed side (clinical uses).
Conclusion and Future Challenges and Perspectives
MSCs are able to preferentially infiltrate into the solid and disseminated tumor tissues and interact with different cells in the tumor microenvironment. Additionally, they are easily available, non-immunogenic, and simply manipulated in vitro without needing to be immortalized; hence they are the most appropriate candidates for cell-based therapies in human cancers (Figure 3). Despite bone marrow, adipose, umbilical cord blood, and local tissues are suitable sources of autologous and allogeneic MSCs; however, their biological regulation and kinetics need to be further elucidated. A number of preclinical investigations in which MSCs have been utilized as vehicles to express a single therapeutic molecule for localized delivery have been explored in different cancer types. Although anti-angiogenic therapy has increased survival of cancer patients, the pooled results indicated that the overall survival improvement was limited (215, 216). The limitations of utilization of anti-angiogenesis therapy are due to several reasons. First, because the angiogenesis is a complicated procedure with involvement of a network of mechanisms, the tumor microenvironment plays a pivotal role in mediating resistance in treatment with angiogenesis inhibitors (217). Second, studies implicate that angiogenesis inhibitors culminating in vascular regression could elevate intratumoral hypoxia. This increased hypoxia results in radio-resistance, chemo-resistance and anti-angiogenesis resistance, since most of the pro-angiogenic factors with tumor aggression effect are mainly produced and released by tumor cells within a hypoxic environment (218, 219). Third, the systematic decrease of tumor vasculature and blood flow leads to reduced delivery of chemotherapeutic agents. These complications may result in tumor metastasis and negatively affect the clinical employment of angiogenesis inhibitors. Although our understandings of the mechanobiology of MSCs have improved greatly over the course of the past few years, we are still on the initiatory steps toward being armed with satisfactory knowledge of these cells and exertion of these cells in clinic. The gap here could be filled by novel hypotheses in the application of anti-angiogenic factors with less adverse effects and their related pre- and post-modulators. Achieving an optimal anti-angiogenic therapy needs complex solutions. Engineering MSCs to selectively deliver the anti-angiogenic molecules could still be promising. Additionally, exploring novel molecular targets contributes to the angiogenesis inhibitors as favorable cancer therapy agents in the future. Other than that, more clinical trials are mandatory to test for the efficacy and safety of such new strategies.
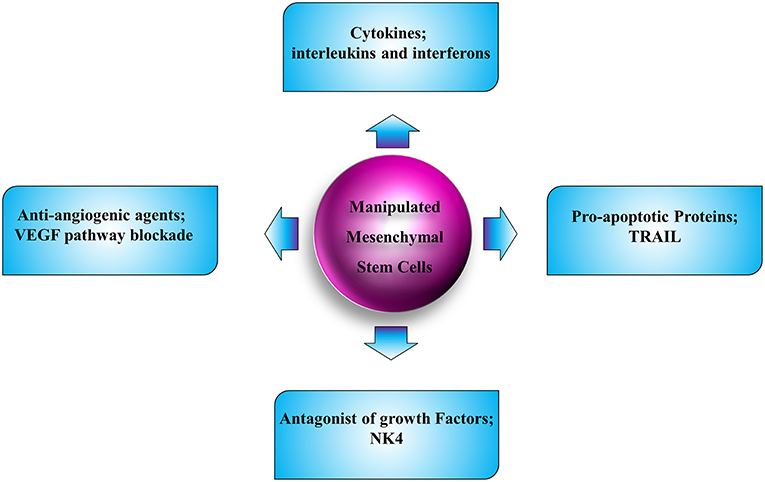
Figure 3. Schematic illustration of strategies for tumor therapy with employment of MSC.
Author Contributions
MJ performed the literature search and prepared the draft of the paper. AK drew the figures and read the manuscript critically. SM developed the main idea, designed the work, and read the manuscript critically.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. (2016) 66:7–30. doi: 10.3322/caac.21332
3. Corsten MF, Shah K. Therapeutic stem-cells for cancer treatment: hopes and hurdles in tactical warfare. Lancet Oncol. (2008) 9:376–84. doi: 10.1016/S1470-2045(08)70099-8
4. Teo AK, Vallier L. Emerging use of stem cells in regenerative medicine. Biochem J. (2010) 428:11–23. doi: 10.1042/BJ20100102
5. Jain RK, Di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. (2007) 8:610–22. doi: 10.1038/nrn2175
6. Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. (2011) 2:122–34. doi: 10.18632/oncotarget.234
7. Friedenstein A, Piatetzky-Shapiro I, Petrakova K. Osteogenesis in transplants of bone marrow cells. Development. (1966) 16:381–90.
8. Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. (1968) 6:230–247. doi: 10.1097/00007890-196803000-00009
9. Kalervo Väänänen H. Mesenchymal stem cells. Ann Med. (2005) 37:469–79. doi: 10.1080/07853890500371957
10. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. (2008) 8:726–36. doi: 10.1038/nri2395
12. Bexell D, Scheding S, Bengzon J. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol Ther. (2010) 18:1067–75. doi: 10.1038/mt.2010.58
13. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. (1999) 284:143–7. doi: 10.1126/science.284.5411.143
14. Le Blanc K, Pittenger M. Mesenchymal stem cells: progress toward promise. Cytotherapy. (2005) 7:36–45. doi: 10.1016/S1465-3249(05)70787-8
15. Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. (2007) 110:3499–506. doi: 10.1182/blood-2007-02-069716
16. Fiorina P, Jurewicz M, Augello A, Vergani A, Dada S, La Rosa S, et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol. (2009) 183:993–1004. doi: 10.4049/jimmunol.0900803
17. Loebinger MR, Janes SM. Stem cells as vectors for antitumour therapy. Thorax. (2010) 65:362–9. doi: 10.1136/thx.2009.128025
18. Morizono K, De Ugarte DA, Zhu M, Zuk P, Elbarbary A, Ashjian P, et al. Multilineage cells from adipose tissue as gene delivery vehicles. Hum Gene Ther. (2003) 14:59–66. doi: 10.1089/10430340360464714
19. Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue–derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. (2007) 67:6304–13. doi: 10.1158/0008-5472.CAN-06-4024
20. Mishra PJ, Mishra PJ, Glod JW, Banerjee D. Mesenchymal stem cells: flip side of the coin. Cancer Res. (2009) 69:1255–8. doi: 10.1158/0008-5472.CAN-08-3562
21. Anderson DJ, Gage FH, Weissman IL. Can stem cells cross lineage boundaries? Nat Med. (2001) 7:393–6. doi: 10.1038/86439
22. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature. (2001) 410:701–5. doi: 10.1038/35070587
23. Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. (2002) 418:41–9. doi: 10.1038/nature00870
24. Mueller SM, Glowacki J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional collagen sponges. J Cell Biochem. (2001) 82:583–90. doi: 10.1002/jcb.1174
25. Bentzon JF, Stenderup K, Hansen F, Schroder H, Abdallah B, Jensen T, et al. Tissue distribution and engraftment of human mesenchymal stem cells immortalized by human telomerase reverse transcriptase gene. Biochem Biophy Res Commun. (2005) 330:633–40. doi: 10.1016/j.bbrc.2005.03.072
26. Stewart MC, Stewart AA. Mesenchymal stem cells: characteristics, sources, and mechanisms of action. Vet Clin North Am. Equine Pract. (2011) 27:243–61. doi: 10.1016/j.cveq.2011.06.004
27. Kern S, Eichler H, Stoeve J, Klüter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. (2006) 24:1294–301. doi: 10.1634/stemcells.2005-0342
28. Rezaei F, Moazzeni SM. Comparison of the therapeutic effect of syngeneic, allogeneic, and xenogeneic adipose tissue-derived mesenchymal stem cells on abortion rates in A mouse model. Cell J. (2019) 21:92–8. doi: 10.22074/cellj.2019.5954
29. Sadighi-Moghaddam B, Salek Farrokhi A, Namdar Ahmadabad H, Barati M, Moazzeni S MJRS. Mesenchymal stem cell therapy prevents abortion in CBA/J × DBA/2 mating. Reprod Sci. (2018) 25:1261–69. doi: 10.1177/1933719117737848
30. Farrokhi AS, Zarnani AH, Moazzeni SMJTi. Mesenchymal stem cells therapy protects fetuses from resorption and induces Th2 type cytokines profile in abortion prone mouse model. Transpl Immunol. (2018) 47:26–31. doi: 10.1016/j.trim.2018.01.002
31. Prindull G, Ben-Ishay Z, Ebell W, Bergholz M, Dirk T, Prindull B. CFU-F circulating in cord blood. Ann Hematol. (1987) 54:351–9. doi: 10.1007/BF00626017
32. Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. (2000) 109:235–42. doi: 10.1046/j.1365-2141.2000.01986.x
33. Goodwin H, Bicknese AR, Chien S, Bogucki B, Oliver D, Quinn C, et al. Multilineage differentiation activity by cells isolated from umbilical cord blood: expression of bone, fat, and neural markers. Biol Blood Marrow Transplant. (2001) 7:581–8. doi: 10.1053/bbmt.2001.v7.pm11760145
34. Chang YJ, Shih DT, Tseng CP, Hsieh TB, Lee DC, et al. Disparate mesenchyme-lineage tendencies in mesenchymal stem cells from human bone marrow and umbilical cord blood. Stem cells. (2006) 24, 679–685. doi: 10.1634/stemcells.2004-0308
35. Shi S, Robey P, Gronthos S. Comparison of human dental pulp and bone marrow stromal stem cells by cDNA microarray analysis. Bone. (2001) 29:532–9. doi: 10.1016/S8756-3282(01)00612-3
36. Pierdomenico L, Bonsi L, Calvitti M, Rondelli D, Arpinati M, Chirumbolo G, et al. Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation. (2005) 80:836–42. doi: 10.1097/01.tp.0000173794.72151.88
37. Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: stem cells from human exfoliated deciduous teeth. Proc Nat Acad Sci USA. (2003) 100:5807–12. doi: 10.1073/pnas.0937635100
38. He Q, Wan C, Li G. Concise review: multipotent mesenchymal stromal cells in blood. Stem Cells. (2007) 25:69–77. doi: 10.1634/stemcells.2006-0335
39. Hong HS, Lee J, Lee E, Kwon YS, Lee E, Ahn W, et al. A new role of substance P as an injury-inducible messenger for mobilization of CD29+ stromal-like cells. Nat Med. (2009) 15:425–35. doi: 10.1038/nm.1909
40. Spaeth EL, Kidd S, Marini FC. Tracking inflammation-induced mobilization of mesenchymal stem cells. Stem Cell Mob Methods Protocol. (2012) 173–90. doi: 10.1007/978-1-61779-943-3_15
41. Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. (2008) 15:730–8. doi: 10.1038/gt.2008.39
42. Pourgholaminejad A, Aghdami N, Baharvand H, Moazzeni SMJC. The effect of pro-inflammatory cytokines on immunophenotype, differentiation capacity and immunomodulatory functions of human mesenchymal stem cells. Cytokine. (2016) 85:51–60. doi: 10.1016/j.cyto.2016.06.003
43. Momin NE, Vela GA, Zaidi H, Quiñones-Hinojosa A. The oncogenic potential of mesenchymal stem cells in the treatment of cancer: directions for future research. Curr Immunol Rev. (2010) 6:137–48. doi: 10.2174/157339510791111718
44. Imitola J, Raddassi K, Park KI, Mueller F-J, Nieto M, Teng YD, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1α/CXC chemokine receptor 4 pathway. Proc Nat Acad Sci USA. (2004) 101:18117–22. doi: 10.1073/pnas.0408258102
45. Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, et al. Human bone marrow–derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. (2005) 65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874
46. Son BR, Marquez-Curtis LA, Kucia M, Wysoczynski M, Turner AR, Ratajczak J, et al. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells. (2006) 24, 1254–1264. doi: 10.1634/stemcells.2005-0271
47. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
48. Smith H, Whittall C, Weksler B, Middleton J. Chemokines stimulate bidirectional migration of human mesenchymal stem cells across bone marrow endothelial cells. Stem Cells Dev. (2011) 21:476–86. doi: 10.1089/scd.2011.0025
49. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
50. Darash-Yahana M, Gillespie JW, Hewitt SM, Chen Y-YK, Maeda S, Stein I, et al. The chemokine CXCL16 and its receptor, CXCR6, as markers and promoters of inflammation-associated cancers. PLoS ONE. (2009) 4:e6695. doi: 10.1371/journal.pone.0006695
51. Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJM, Wesseling P, et al. Prognostic significance and mechanism of treg infiltration in human brain tumors. J Neuroimmunol. (2010) 225:195–9. doi: 10.1016/j.jneuroim.2010.05.020
52. Yahata Y, Shirakata Y, Tokumaru S, Yang L, Dai X, Tohyama M, et al. A novel function of angiotensin II in skin wound healing induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J Biol Chem. (2006) 281:13209–16. doi: 10.1074/jbc.M509771200
53. Maretzky T, Evers A, Zhou W, Swendeman SL, Wong P-M, Rafii S, et al. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. (2010) 2:229–229. doi: 10.1038/ncomms1232
54. Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC, Miller FJ. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovas Res. (2012) 93:406–13. doi: 10.1093/cvr/cvr308
55. Porcile C, Bajetto A, Barbieri F, Barbero S, Bonavia R, Biglieri M, et al. Stromal cell-derived factor-1α (SDF-1α/CXCL12) stimulates ovarian cancer cell growth through the EGF receptor transactivation. Exp Cell Res. (2005) 308:241–53. doi: 10.1016/j.yexcr.2005.04.024
56. Roelle S, Grosse R, Aigner A, Krell H-W, Czubayko F, Gudermann T. Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J Biol Chem. (2003) 278:47307–18. doi: 10.1074/jbc.M304377200
57. De Becker A, Van Hummelen P, Bakkus M, Broek IV, De Wever J, De Waele M, et al. Migration of culture-expanded human mesenchymal stem cells through bone marrow endothelium is regulated by matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-3. Haematologica. (2007) 92:440–9. doi: 10.3324/haematol.10475
58. Qiu Y, Marquez-Curtis LA, Janowska-Wieczorek A. Mesenchymal stromal cells derived from umbilical cord blood migrate in response to complement C1q. Cytotherapy. (2012) 14:285–95. doi: 10.3109/14653249.2011.651532
59. François S, Bensidhoum M, Mouiseddine M, Mazurier C, Allenet B, Semont A, et al. Local irradiation not only induces homing of human mesenchymal stem cells at exposed sites but promotes their widespread engraftment to multiple organs: a study of their quantitative distribution after irradiation damage. Stem Cells. (2006) 24:1020–9. doi: 10.1634/stemcells.2005-0260
60. Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. (2007) 21:304–10. doi: 10.1038/sj.leu.2404489
61. Lu Yr, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C, et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther. (2008) 7:245–51. doi: 10.4161/cbt.7.2.5296
62. Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. (2008) 269:67–77. doi: 10.1016/j.canlet.2008.04.032
63. Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, et al. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. (2008) 18:500–7. doi: 10.1038/cr.2008.40
64. Zhu Y, Sun Z, Han Q, Liao L, Wang J, Bian C, et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. (2009) 23:925–33. doi: 10.1038/leu.2008.384
65. Sun B, Roh K-H, Park J-R, Lee S-R, Park S-B, Jung J-W, et al. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy. (2009) 11:289–98. doi: 10.1080/14653240902807026
66. Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. (2009) 113:4197–205. doi: 10.1182/blood-2008-09-176198
67. Secchiero P, Zorzet S, Tripodo C, Corallini F, Melloni E, Caruso L, et al. Human bone marrow mesenchymal stem cells display anti-cancer activity in SCID mice bearing disseminated non-Hodgkin's lymphoma xenografts. PLoS ONE. (2010) 5:e11140. doi: 10.1371/journal.pone.0011140
68. Ho IA, Toh HC, Ng WH, Teo YL, Guo CM, Hui KM, et al. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells. (2013) 31:146–55. doi: 10.1002/stem.1247
69. Lin G, Yang R, Banie L, Wang G, Ning H, Li LC, et al. Effects of transplantation of adipose tissue-derived stem cells on prostate tumor. Prostate. (2010) 70:1066–73. doi: 10.1002/pros.21140
70. Huang W, Chang M, Tsai K, Hung M, Chen H, Hung S. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. (2013) 32:4343–54. doi: 10.1038/onc.2012.458
71. Suzuki K, Sun R, Origuchi M, Kanehira M, Takahata T, Itoh J, et al. Mesenchymal stromal cells promote tumor growth through the enhancement of neovascularization. Mol Med. (2011) 17:579. doi: 10.2119/molmed.2010.00157
72. Li Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol. (2013) 2:17. doi: 10.1186/2162-3619-2-17
73. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
74. Nishimura K, Semba S, Aoyagi K, Sasaki H, Yokozaki H. Mesenchymal stem cells provide an advantageous tumor microenvironment for the restoration of cancer stem cells. Pathobiology. (2012) 79:290–306. doi: 10.1159/000337296
75. Corcoran KE, Trzaska KA, Fernandes H, Bryan M, Taborga M, Srinivas V, et al. Mesenchymal stem cells in early entry of breast cancer into bone marrow. PLoS ONE. (2008) 3:e2563. doi: 10.1371/journal.pone.0002563
76. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. (2007) 449:557–63. doi: 10.1038/nature06188
77. Xu WT, Bian ZY, Fan QM, Li G, Tang TT. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. (2009) 281:32–41. doi: 10.1016/j.canlet.2009.02.022
78. Tsukamoto S, Honoki K, Fujii H, Tohma Y, Kido A, Mori T, et al. Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int J Oncol. (2012) 40:163–9. doi: 10.3892/ijo.2011.1220
79. Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. (2003) 102:3837–44. doi: 10.1182/blood-2003-04-1193
80. Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol. (2014) 7:14. doi: 10.1186/1756-8722-7-14
81. Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. (2012) 11:812–24. doi: 10.1016/j.stem.2012.08.013
82. Razmkhah M, Jaberipour M, Erfani N, Habibagahi M, Talei AR, Ghaderi A. Adipose derived stem cells (ASCs) isolated from breast cancer tissue express IL-4, IL-10 and TGF-β1 and upregulate expression of regulatory molecules on T cells: do they protect breast cancer cells from the immune response? Cell Immunol. (2011) 266:116–22. doi: 10.1016/j.cellimm.2010.09.005
83. Johann P-D, Vaegler M, Gieseke F, Mang P, Armeanu-Ebinger S, Kluba T, et al. Tumour stromal cells derived from paediatric malignancies display MSC-like properties and impair NK cell cytotoxicity. BMC Cancer. (2010) 10:501. doi: 10.1186/1471-2407-10-501
84. Liu Y, Han ZP, Zhang SS, Jing YY, Bu XX, Wang CY, et al. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J Biol Chem. (2011) 286:25007–15. doi: 10.1074/jbc.M110.213108
85. Luo J, Lee SO, Liang L, Huang C, Li L, Wen S, et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene. (2014) 33:2768–78. doi: 10.1038/onc.2013.233
86. McLean K, Gong Y, Choi Y, Deng N, Yang K, Bai S, et al. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J Clin Invest. (2011) 121:3206. doi: 10.1172/JCI45273
87. Maestroni G, Hertens E, Galli P. Factor (s) from nonmacrophage bone marrow stromal cells inhibit Lewis lung carcinoma and B16 melanoma growth in mice. Cell Mol Life Sci. (1999) 55:663. doi: 10.1007/s000180050322
88. Nakamura K, Ito Y, Kawano Y, Kurozumi K, Kobune M, Tsuda H, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. (2004) 11:1155–64. doi: 10.1038/sj.gt.3302276
89. Qiao C, Xu W, Zhu W, Hu J, Qian H, Yin Q, et al. Human mesenchymal stem cells isolated from the umbilical cord. Cell Biol Int. (2008) 32:8–15. doi: 10.1016/j.cellbi.2007.08.002
90. Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi's sarcoma. J Exp Med. (2006) 203:1235–47. doi: 10.1084/jem.20051921
91. Gondi CS, Veeravalli KK, Gorantla B, Dinh DH, Fassett D, Klopfenstein JD, et al. Human umbilical cord blood stem cells show PDGF-D–dependent glioma cell tropism in vitro and in vivo. Neuro-oncology. (2010) 12:453–65. doi: 10.1093/neuonc/nop049
92. Dasari VR, Kaur K, Velpula KK, Dinh DH, Tsung AJ, Mohanam S, et al. Downregulation of Focal Adhesion Kinase (FAK) by cord blood stem cells inhibits angiogenesis in glioblastoma. Aging. (2010) 2:791–803. doi: 10.18632/aging.100217
93. Dasari VR, Kaur K, Velpula KK, Gujrati M, Fassett D, Klopfenstein JD, et al. Upregulation of PTEN in glioma cells by cord blood mesenchymal stem cells inhibits migration via downregulation of the PI3K/Akt pathway. PLoS ONE. (2010) 5:e10350. doi: 10.1371/journal.pone.0010350
94. Dasari VR, Velpula KK, Kaur K, Fassett D, Klopfenstein JD, Dinh DH, et al. Cord blood stem cell-mediated induction of apoptosis in glioma downregulates X-linked inhibitor of apoptosis protein (XIAP). PLoS ONE. (2010) 5:e11813. doi: 10.1371/journal.pone.0011813
95. Akimoto K, Kimura K, Nagano M, Takano S, To'a Salazar G, Yamashita T, et al. Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells Dev. (2012) 22:1370–86. doi: 10.1089/scd.2012.0486
96. Okada H, Pollack IF. Cytokine gene therapy for malignant glioma. Expert Opin Biol Ther. (2004) 4:1609–20. doi: 10.1517/14712598.4.10.1609
97. Zhang X, Zhang L, Xu W, Qian H, Ye S, Zhu W, et al. Experimental therapy for lung cancer: umbilical cord-derived mesenchymal stem cell-mediated interleukin-24 delivery. Curr Cancer Drug Target. (2013) 13:92–102. doi: 10.2174/156800913804486665
98. Chen X, Lin X, Zhao J, Shi W, Zhang H, Wang Y, et al. A tumor-selective biotherapy with prolonged impact on established metastases based on cytokine gene-engineered MSCs. Mol Ther. (2008) 16:749–56. doi: 10.1038/mt.2008.3
99. Seo S, Kim K, Park S, Suh Y, Kim S, Jeun S, et al. The effects of mesenchymal stem cells injected via different routes on modified IL-12-mediated antitumor activity. Gene Ther. (2011) 18:488–95. doi: 10.1038/gt.2010.170
100. Gao P, Ding Q, Wu Z, Jiang H, Fang Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. (2010) 290:157–66. doi: 10.1016/j.canlet.2009.08.031
101. Ryu CH, Park S-H, Park SA, Kim SM, Lim JY, Jeong CH, et al. Gene therapy of intracranial glioma using interleukin 12–secreting human umbilical cord blood–derived mesenchymal stem cells. Hum Gene Ther. (2011) 22:733–43. doi: 10.1089/hum.2010.187
102. Xu X, Yang G, Zhang H, Prestwich GD. Evaluating dual activity LPA receptor pan-antagonist/autotaxin inhibitors as anti-cancer agents in vivo using engineered human tumors. Prostaglandins Other Lipid Med. (2009) 89:140–6. doi: 10.1016/j.prostaglandins.2009.07.006
103. Hu W, Wang J, Dou J, He X, Zhao F, Jiang C, et al. Augmenting therapy of ovarian cancer efficacy by secreting IL-21 human umbilical cord blood stem cells in nude mice. Cell Transpl. (2011) 20:669–80. doi: 10.3727/096368910X536509
104. Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-β compared with IFN-α2. Clinical Cancer Res. (2001) 7:1821–31.
105. Wong VL, Rieman DJ, Aronson L, Dalton BJ, Greig R, Anzano MA. Growth-inhibitory activity of interferon-beta against human colorectal carcinoma cell lines. Int J Cancer. (1989) 43:526–30. doi: 10.1002/ijc.2910430331
106. Johns TG, Mackay IR, Callister KA, Hertzog PJ, Devenish RJ, Linnance AW. Antiproliferative potencies of interferons on melanoma cell lines and xenografts: higher efficacy of interferon β. J Nat Cancer Inst. (1992) 84:1185–90. doi: 10.1093/jnci/84.15.1185
107. Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-β delivery into tumors. Cancer Res. (2002) 62:3603–8.
108. Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Nat Cancer Inst. (2004) 96:1593–603. doi: 10.1093/jnci/djh299
109. Ren C, Kumar S, Chanda D, Chen J, Mountz JD, Ponnazhagan S. Therapeutic potential of mesenchymal stem cells producing interferon-α in a mouse melanoma lung metastasis model. Stem Cells. (2008) 26:2332–8. doi: 10.1634/stemcells.2008-0084
110. Ren C, Kumar S, Chanda D, Kallman L, Chen J, Mountz JD, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-β in a mouse prostate cancer lung metastasis model. Gene Ther. (2008) 15:1446–53. doi: 10.1038/gt.2008.101
111. Dembinski JL, Wilson SM, Spaeth EL, Studeny M, Zompetta C, Samudio I, et al. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy. (2013) 15:20–32. e22. doi: 10.1016/j.jcyt.2012.10.003
112. Wang G, Zhan Y, Hu H, Wang Y, Fu B. Mesenchymal stem cells modified to express interferon-β inhibit the growth of prostate cancer in a mouse model. J Int Med Res. (2012) 40:317–27. doi: 10.1177/147323001204000132
113. Bitsika V, Roubelakis MG, Zagoura D, Trohatou O, Makridakis M, Pappa KI, et al. Human amniotic fluid-derived mesenchymal stem cells as therapeutic vehicles: a novel approach for the treatment of bladder cancer. Stem Cells Dev. (2011) 21:1097–111. doi: 10.1089/scd.2011.0151
114. Grandér D, Einhorn S. Interferon and malignant disease-How does it work and why doesn't it always? Acta Oncol. (1998) 37:331–8. doi: 10.1080/028418698430548
115. Lens M. Cutaneous melanoma: interferon alpha adjuvant therapy for patients at high risk for recurrent disease. Dermatol Ther. (2006) 19:9–18. doi: 10.1111/j.1529-8019.2005.00051.x
116. Sartoris S, Mazzocco M, Tinelli M, Martini M, Mosna F, Lisi V, et al. Efficacy assessment of interferon-alpha–engineered mesenchymal stromal cells in a mouse plasmacytoma model. Stem Cells Dev. (2010) 20:709–19. doi: 10.1089/scd.2010.0095
117. Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. (2000) 256:58–66. doi: 10.1006/excr.2000.4840
118. Ehtesham M, Kabos P, Gutierrez MA, Chung NH, Griffith TS, Black KL, et al. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. (2002) 62:7170–4.
119. Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, John SY. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. (2002) 62:5657–63.
120. Kim S-K, Cargioli TG, Machluf M, Yang W, Sun Y, Al-Hashem R, et al. PEX-producing human neural stem cells inhibit tumor growth in a mouse glioma model. Clin Cancer Res. (2005) 11:5965–70. doi: 10.1158/1078-0432.CCR-05-0371
121. Mueller L, Luetzkendorf J, Widder M, Nerger K, Caysa H, Mueller T. TRAIL-transduced multipotent mesenchymal stromal cells (TRAIL-MSC) overcome TRAIL resistance in selected CRC cell lines in vitro and in vivo. Cancer Gene Ther. (2011) 18:229–39. doi: 10.1038/cgt.2010.68
122. Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. (2009) 69:4134–42. doi: 10.1158/0008-5472.CAN-08-4698
123. Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor–related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. (2010) 70:3718–29. doi: 10.1158/0008-5472.CAN-09-1865
124. Ciavarella S, Grisendi G, Dominici M, Tucci M, Brunetti O, Dammacco F, et al. In vitro anti-myeloma activity of TRAIL-expressing adipose-derived mesenchymal stem cells. Br J Haematol. (2012) 157:586–98. doi: 10.1111/j.1365-2141.2012.09082.x
125. Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. (2012) 64:739–48. doi: 10.1016/j.addr.2011.06.010
126. Shah K, Tung C-H, Yang K, Weissleder R, Breakefield XO. Inducible release of TRAIL fusion proteins from a proapoptotic form for tumor therapy. Cancer Res. (2004) 64:3236–42. doi: 10.1158/0008-5472.CAN-03-3516
127. Shah K, Bureau E, Kim DE, Yang K, Tang Y, Weissleder R, et al. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. (2005) 57:34–41. doi: 10.1002/ana.20306
128. Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, Van De Water JA, Mohapatra G, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Nat Acad Sci USA. (2009) 106:4822–7. doi: 10.1073/pnas.0806647106
129. Matsumoto K, Nakamura T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci. (2003) 94:321–7. doi: 10.1111/j.1349-7006.2003.tb01440.x
130. Cao Y. Molecular mechanisms and therapeutic development of angiogenesis inhibitors. Adv cancer Res. (2008) 100:113–31. doi: 10.1016/S0065-230X(08)00004-3
131. Cao Y, Cao R, Hedlund E-M. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med. (2008) 86:785–9. doi: 10.1007/s00109-008-0337-z
132. Kanehira M, Xin H, Hoshino K, Maemondo M, Mizuguchi H, Hayakawa T, et al. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. (2007) 14:894–903. doi: 10.1038/sj.cgt.7701079
133. Wilting J, Christ B. Embryonic angiogenesis: a review. Naturwissenschaften. (1996) 83:153–64. doi: 10.1007/BF01143056
134. Rhodin J, Fujita H. Capillary growth in the mesentery of normal young rats. Intravital video and electron microscope analyses. J Submicrosc cytol Pathol. (1989) 21:1–34.
135. Risau W, Flamme I. Vasculogenesis. Ann Rev Cell Dev Biol. (1995) 11:73–91. doi: 10.1146/annurev.cb.11.110195.000445
136. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
137. Hiratsuka S. Vasculogenensis, angiogenesis and special features of tumor blood vessels. Front Biosci. (2010) 16:1413–27. doi: 10.2741/3796
138. Ribatti D. The involvement of endothelial progenitor cells in tumor angiogenesis. J Cell Mol Med. (2004) 8:294–300. doi: 10.1111/j.1582-4934.2004.tb00319.x
139. Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophy Res Commun. (1989) 161:851–8. doi: 10.1016/0006-291X(89)92678-8
140. Khoury CC, Ziyadeh FN. Angiogenic factors. In: Lai KN, Tang SCW, editors. Diabetes and the Kidney. Basel: Karger Publishers (2011). p. 83–92. doi: 10.1159/000324950
141. Taraboletti G, Rusnati M, Rusnati L, Ragona G. Targeting tumor angiogenesis with TSP-1-based compounds: rational design of antiangiogenic mimetics of endogenous inhibitors. Oncotarget. (2010) 1:662–73. doi: 10.18632/oncotarget.200
142. Algire GH, Chalkley HW, Legallais FY, Park HD. Vasculae reactions of normal and malignant tissues in vivo. I. vascular reactions of mice to wounds and to normal and neoplastic transplants. J Nat Cancer Inst. (1945) 6:73–85 doi: 10.1093/jnci/6.1.73
143. Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. (2003) 9:669–76. doi: 10.1038/nm0603-669
144. Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. In: Housset C, editor. Seminars in liver disease. Paris: Thieme Medical Publishers (2010). p. 258–70. doi: 10.1055/s-0030-1255355
145. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. (1995) 270:1230–7. doi: 10.1074/jbc.270.3.1230
146. Brahimi-Horn C, Pouysségur J. The role of the hypoxia-inducible factor in tumor metabolism growth and invasion. Bulletin du Cancer. (2006) 93:10073–80.
147. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. (2008) 8:579–91. doi: 10.1038/nrc2403
148. Petrova TV, Bono P, Holnthoner W, Chesnes J, Pytowski B, Sihto H, et al. VEGFR-3 expression is restricted to blood and lymphatic vessels in solid tumors. Cancer Cell. (2008) 13:554–6. doi: 10.1016/j.ccr.2008.04.022
149. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. (1993) 362:841. doi: 10.1038/362841a0
150. Presta LG, Chen H, O'connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. (1997) 57:4593–99.
151. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. (2004) 350:2335–42. doi: 10.1056/NEJMoa032691
152. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel–carboplatin alone or with bevacizumab for non–small-cell lung cancer. N Engl J Med. (2006) 355:2542–50. doi: 10.1056/NEJMoa061884
153. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. (2007) 357:2666–76. doi: 10.1056/NEJMoa072113
154. Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Nat Acad Sci USA. (2002) 99:11393–8. doi: 10.1073/pnas.172398299
155. Ellis LM, Hicklin DJ. Pathways mediating resistance to vascular endothelial growth factor–targeted therapy. Clin Cancer Res. (2008) 14:6371–5. doi: 10.1158/1078-0432.CCR-07-5287
157. Jiang F, Albert DH, Luo Y, Tapang P, Zhang K, Davidsen SK, et al. ABT-869, a multitargeted receptor tyrosine kinase inhibitor, reduces tumor microvascularity and improves vascular wall integrity in preclinical tumor models. J Pharmacol Exp Therap. (2011) 338:134–42. doi: 10.1124/jpet.110.178061
158. Tan E-H, Goss GD, Salgia R, Besse B, Gandara DR, Hanna NH, et al. Phase 2 trial of Linifanib (ABT-869) in patients with advanced non-small cell lung cancer. J Thorac Oncol. (2011) 6:1418–25. doi: 10.1097/JTO.0b013e318220c93e
159. You W-K, Sennino B, Williamson CW, Falcón B, Hashizume H, Yao L-C, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. (2011) 71:4758–68. doi: 10.1158/0008-5472.CAN-10-2527
160. Ho TH, Jonasch E. Axitinib in the treatment of metastatic renal cell carcinoma. Future Oncol. (2011) 7:1247–53. doi: 10.2217/fon.11.107
161. Eskens FA, de Jonge MJ, Bhargava P, Isoe T, Cotreau MM, Esteves B, et al. Biologic and clinical activity of tivozanib (AV-951, KRN-951), a selective inhibitor of VEGF receptor-1,-2, and-3 tyrosine kinases, in a 4-week-on, 2-week-off schedule in patients with advanced solid tumors. Clin Cancer Res. (2011) 17:7156–63. doi: 10.1158/1078-0432.CCR-11-0411
162. Langmuir P, Yver A. Vandetanib for the treatment of thyroid cancer. Clin Pharmacol Therapeut. (2012) 91:71–80. doi: 10.1038/clpt.2011.272
163. O'Farrell A-M, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. (2003) 101:3597–605. doi: 10.1182/blood-2002-07-2307
164. Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schöffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European Organisation for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group (EORTC study 62043). J Clin Oncol. (2009) 27:3126–32. doi: 10.1200/JCO.2008.21.3223
165. Richly H, Henning B, Kupsch P, Passarge K, Grubert M, Hilger R, et al. Results of a phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann Oncol. (2006) 17:866–73. doi: 10.1093/annonc/mdl017
166. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. (2005) 307:58–62. doi: 10.1126/science.1104819
167. Cooney MM, van Heeckeren W, Bhakta S, Ortiz J, Remick SC. Drug insight: vascular disrupting agents and angiogenesis—novel approaches for drug delivery. Nat Clin Pract Oncol. (2006) 3:682–92. doi: 10.1038/ncponc0663
168. McKeage M, Von Pawel J, Reck M, Jameson M, Rosenthal M, Sullivan R, et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br J Cancer. (2008) 99:2006–12. doi: 10.1038/sj.bjc.6604808
169. Zheng X, Koh GY, Jackson T. A continuous model of angiogenesis: initiation, extension, and maturation of new blood vessels modulated by vascular endothelial growth factor, angiopoietins, platelet-derived growth factor-B, and pericytes. Discrete Contin. Dynam. Syst. Series B. (2013) 18:1109–54. doi: 10.3934/dcdsb.2013.18.1109
170. Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. (2011) 12:551–64. doi: 10.1038/nrm3176
171. Hudkins RL, Becknell NC, Zulli AL, Underiner TL, Angeles TS, Aimone LD, et al. Synthesis and biological profile of the pan-vascular endothelial growth factor receptor/tyrosine kinase with immunoglobulin and epidermal growth factor-like homology domains 2 (VEGF-R/TIE-2) inhibitor 11-(2-Methylpropyl)-12, 13-dihydro-2-methyl-8-(pyrimidin-2-ylamino)-4 H-indazolo [5, 4-a] pyrrolo [3, 4-c] carbazol-4-one (CEP-11981): a novel oncology therapeutic agent. J Med Chem. (2012) 55:903–13. doi: 10.1021/jm201449n
172. Oike Y, Ito Y, Hamada K, Zhang X-Q, Miyata K, Arai F, et al. Regulation of vasculogenesis and angiogenesis by EphB/ephrin-B2 signaling between endothelial cells and surrounding mesenchymal cells. Blood. (2002) 100:1326–33.
173. Strawn LM, McMahon G, App H, Schreck R, Kuchler WR, Longhi MP, et al. Flk-1 as a target for tumor growth inhibition. Cancer Res. (1996) 56:3540–5.
174. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Therapeut. (2008) 7:3129–40. doi: 10.1158/1535-7163.MCT-08-0013
175. Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST) Therapeutics and clinical risk management. Ther Clin Risk Manag. (2007) 3:341. doi: 10.2147/tcrm.2007.3.2.341
176. Ghaedi M, Soleimani M, Taghvaie NM, Sheikhfatollahi M, Azadmanesh K, Lotfi AS, et al. Mesenchymal stem cells as vehicles for targeted delivery of anti-angiogenic protein to solid tumors. J Gene Med. (2011) 13:171–80. doi: 10.1002/jgm.1552
177. Najafabadi MM, Shamsasenjan K, Akbarzadehlaleh P. The angiogenic chemokines expression profile of myeloid cell lines co-cultured with bone marrow-derived mesenchymal stem cells. Cell J. (2018) 20:19. doi: 10.22074/cellj.2018.4924
178. Toi M, Hoshina S, Takayanagi T, Tominaga T. Association of vascular endothelial growth factor expression with tumor angiogenesis and with early relapse in primary breast cancer. Cancer Sci. (1994) 85:1045–9. doi: 10.1111/j.1349-7006.1994.tb02904.x
179. Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. (1989) 339:58–61. doi: 10.1038/339058a0
180. Zheng L, Zhang D, Chen X, Yang L, Wei Y, Zhao X. Antitumor activities of human placenta-derived mesenchymal stem cells expressing endostatin on ovarian cancer. PLoS ONE. (2012) 7:e39119. doi: 10.1371/journal.pone.0039119
181. Batchelor TT, Sorensen AG, di Tomaso E, Zhang W-T, Duda DG, Cohen KS, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. (2007) 11:83–95. doi: 10.1016/j.ccr.2006.11.021
182. Kadambi A, Carreira CM, Yun CO, Padera TP, Dolmans DE, Carmeliet P, et al. Vascular endothelial growth factor (VEGF)-C differentially affects tumor vascular function and leukocyte recruitment. Cancer Res. (2001) 61:2404–8.
183. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. (2004) 64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074
184. Hormigo A, Gutin PH, Rafii S. Tracking normalization of brain tumor vasculature by magnetic imaging and proangiogenic biomarkers. Cancer Cell. (2007) 11:6–8. doi: 10.1016/j.ccr.2006.12.008
185. Bexell D, Gunnarsson S, Tormin A, Darabi A, Gisselsson D, Roybon L, et al. Bone marrow multipotent mesenchymal stroma cells act as pericyte-like migratory vehicles in experimental gliomas. Mol Ther. (2009) 17:183–90. doi: 10.1038/mt.2008.229
186. Russo FP, Alison MR, Bigger BW, Amofah E, Florou A, Amin F, et al. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. (2006) 130:1807–21. doi: 10.1053/j.gastro.2006.01.036
187. McLeod C, Mauck R. On the origin and impact of mesenchymal stem cell heterogeneity: new insights and emerging tools for single cell analysis. Eur Cell Mater. (2017) 34:217–31. doi: 10.22203/eCM.v034a14
188. Krueger TE, Thorek DL, Meeker AK, Isaacs JT, Brennen WN. Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate. (2019) 79:320–30. doi: 10.1002/pros.23738
189. Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J Med Chem. (2003) 46:883–908. doi: 10.1021/jm020449y
190. Hyder SM, Stancel GM. Regulation of angiogenic growth factors in the female reproductive tract by estrogens and progestins. Mol Endocrinol. (1999) 13:806–11. doi: 10.1210/mend.13.6.0308
191. Zygmunt M, Herr F, Münstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstetr Gynecol Reproduct Biol. (2003) 110:S10–S18. doi: 10.1016/S0301-2115(03)00168-4
192. Jensen EV, Jordan VC. The estrogen receptor. Clin Cancer Res. (2003) 9:1980–9. doi: 10.1172/JCI27987
193. Horwitz KB. The central role of progesterone receptors and progestational agents in the management and treatment of breast cancer. In: Seminars in Oncology. (1998), 14–19.
194. Jordan VC. Selective estrogen receptor modulation: a personal perspective. Cancer Res. (2001) 61:5683–7.
195. Powles TJ, Hickish T, Kanis JA, Tidy A, Ashley S. Effect of tamoxifen on bone mineral density measured by dual-energy x-ray absorptiometry in healthy premenopausal and postmenopausal women. J Clin Oncol. (1996) 14:78–84. doi: 10.1200/JCO.1996.14.1.78
196. Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Nat Cancer Inst. (2005) 97:1652–62. doi: 10.1093/jnci/dji372
197. Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. (1997) 138:863–70. doi: 10.1210/endo.138.3.4979
198. Weihua Z, Warner M, Gustafsson J-Å. Estrogen receptor beta in the prostate. Mol Cell Endocrinol. (2002) 193:1–5. doi: 10.1016/S0303-7207(02)00089-8
199. Chang WY, Prins GS. Estrogen receptor-ß: implications for the prostate gland. Prostate. (1999) 40:3181.
200. Makar A. Hormone therapy in epithelial ovarian cancer. Endocr Related Cancer. (2000) 7:85–93. doi: 10.1677/erc.0.0070085
201. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. (2002) 288:321–33. doi: 10.1001/jama.288.3.321
202. Vázquez F, Rodriguez-Manzaneque JC, Lydon JP, Edwards DP, O'Malley BW, Iruela-Arispe ML. Progesterone regulates proliferation of endothelial cells. J Biol Chem. (1999) 274:2185–92. doi: 10.1074/jbc.274.4.2185
203. Iruela-Arispe ML, Rodriguez-Manzaneque JC, Abu-Jawdeh G. Endometrial endothelial cells express estrogen and progesterone receptors and exhibit a tissue specific response to angiogenic growth factors. Microcirculation. (1999) 6:127–40. doi: 10.1080/713773947
204. Reynolds L, Redmer D. Growth and development of the corpus luteum. J Reproduct Fertil Suppl. (1998) 54:181–91.
205. Chuderland D, Ben-Ami I, Kaplan-Kraicer R, Grossman H, Komsky A, Satchi-Fainaro R, et al. Hormonal regulation of pigment epithelium-derived factor (PEDF) in granulosa cells. Mol Hum Reproduct. (2013) 19:72–81. doi: 10.1093/molehr/gas046
206. Colombo G, Margosio B, Ragona L, Neves M, Bonifacio S, Annis DS, et al. Non-peptidic Thrombospondin-1 Mimics as Fibroblast Growth Factor-2 Inhibitors an integrated strategy for the development of new antiangiogenic compounds. J Biol Chem. (2010) 285:8733–42. doi: 10.1074/jbc.M109.085605
207. Romero JJ, Antoniazzi AQ, Smirnova NP, Webb BT, Yu F, Davis JS, et al. Pregnancy-associated genes contribute to antiluteolytic mechanisms in ovine corpus luteum. Physiol Genom. (2013) 45:1095–108. doi: 10.1152/physiolgenomics.00082.2013
208. Zalman Y, Klipper E, Farberov S, Mondal M, Wee G, Folger JK, et al. Regulation of angiogenesis-related prostaglandin f2alpha-induced genes in the bovine corpus luteum. Biol Reproduct. (2012) 86:92. doi: 10.1095/biolreprod.111.095067
209. Gong M, Yu B, Wang J, Wang Y, Liu M, Paul C, et al. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget. (2017) 8:45200. doi: 10.18632/oncotarget.16778
210. Rosenberger L, Ezquer M, Lillo-Vera F, Pedraza PL, Ortúzar MI, González PL, et al. Stem cell exosomes inhibit angiogenesis and tumor growth of oral squamous cell carcinoma. Sci Rep. (2019) 9:663. doi: 10.1038/s41598-018-36855-6
211. Pan J, Wang X, Li D, Li J, Jiang Z. MSCs inhibits the angiogenesis of HUVECs through the miR-211/Prox1 pathway. J Biochem. (2019) 166, 17–113. doi: 10.1093/jb/mvz038
212. Lee J-K, Park S-R, Jung B-K, Jeon Y-K, Lee Y-S, Kim M-K, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE. (2013) 8:e84256. doi: 10.1371/journal.pone.0084256
213. Niess H, von Einem JC, Thomas MN, Michl M, Angele MK, Huss R, et al. Treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells (TREAT-ME1): study protocol of a phase I/II clinical trial. BMC Cancer. (2015) 15:237. doi: 10.1186/s12885-015-1241-x
214. Schweizer MT, Wang H, Bivalacqua TJ, Partin AW, Lim SJ, Chapman C, et al. A phase I study to assess the safety and cancer-homing ability of allogeneic bone marrow-derived mesenchymal stem cells in men with localized prostate cancer. Stem Cells Transl Med. (2019) 8:441–9. doi: 10.1002/sctm.18-0230
215. Hong S, Tan M, Wang S, Luo S, Chen Y, Zhang L. Efficacy and safety of angiogenesis inhibitors in advanced non-small cell lung cancer: a systematic review and meta-analysis. J Cancer Res Clin Oncol. (2015) 141:909–21. doi: 10.1007/s00432-014-1862-5
216. Li X, Zhu S, Hong C, Cai H. Angiogenesis inhibitors for patients with ovarian cancer: a meta-analysis of 12 randomized controlled trials. Curr Med Res Opin. (2016) 32:555–62. doi: 10.1185/03007995.2015.1131152
217. Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor–resistant human lung adenocarcinoma. J Clin Invest. (2011) 121:1313–28. doi: 10.1172/JCI42405
218. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. (2009) 15:220–31. doi: 10.1016/j.ccr.2009.01.027
Keywords: angiogenesis, mesenchymal stem cells, tumor microenvironment, cancer therapy, drug delivery
Citation: Javan MR, Khosrojerdi A and Moazzeni SM (2019) New Insights Into Implementation of Mesenchymal Stem Cells in Cancer Therapy: Prospects for Anti-angiogenesis Treatment. Front. Oncol. 9:840. doi: 10.3389/fonc.2019.00840
Received: 20 May 2019; Accepted: 15 August 2019;
Published: 28 August 2019.
Edited by:
Christina Annunziata, National Cancer Institute (NCI), United StatesReviewed by:
Dhiraj Kumar, University of Texas MD Anderson Cancer Center, United StatesKeqiang Zhang, City of Hope National Medical Center, United States
Copyright © 2019 Javan, Khosrojerdi and Moazzeni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seyed Mohammad Moazzeni, moazzeni@modares.ac.ir