 Mónica Fernández-Cortés†
Mónica Fernández-Cortés† Daniel Delgado-Bellido
Daniel Delgado-Bellido F. Javier Oliver
F. Javier Oliver- CSIC, CIBERONC, Instituto de Parasitología y Biomedicina López Neyra, Granada, Spain
Blood vessels supply all body tissues with nutrients and oxygen, take away waste products and allow the arrival of immune cells and other cells (pericytes, smooth muscle cells) that form part of these vessels around the principal endothelial cells. Vasculogenic mimicry (VM) is a tumor blood supply system that takes place independently of angiogenesis or endothelial cells, and is associated with poor survival in cancer patients. Aberrant expression of VE-cadherin has been strongly associated with VM. Even more, VE-cadherin has constitutively high phosphorylation levels on the residue of Y658 in human malignant melanoma cells. In this review we focus on non-endothelial VE-cadherin and its post-translational modifications as a crucial component in the development of tumor VM, highlighting the signaling pathways that lead to their pseudo-endothelial and stem-like phenotype and the role of tumor microenvironment. We discuss the importance of the tumor microenvironment in VM acquisition, and describe the most recent therapeutic targets that have been proposed for the repression of VM.
Background
The concept of neovascularization was described for the first time in 1787 in the context of developmental biology. The term angiogenesis was coined early in the 20th century but was not applied to tumor biology until decades later (1). Angiogenesis can arise in a variety of forms during cancer, namely sprouting angiogenesis, intussusceptive microvascular growth, and glomeruloid microvascular proliferation (2). Emerging studies have shown that a few tumors can grow without need of angiogenesis even in hypoxic conditions, while other tumors display both angiogenic and non-angiogenic regions (3). Vasculogenic mimicry refers to the ability of cancer cells to organize themselves into vascular-like structures for the obtention of nutrients and oxygen independently of normal blood vessels or angiogenesis.
Research in VM has often been surrounded by skepticism to a certain extent. The reason for this controversy is usually the difficulty to distinguish VM channels from endothelial blood vessels or blood lakes in vivo. Similarly, in vitro research presents inherent inconveniences, given the need to develop 3-D models where tumor cells can develop capillary-like structures. Most in vitro research is based on cell cultures using matrigel. However, the tubular structures observed in this model might not always represent in vivo VM. Table 1 shows a few articles where VM is thoroughly described in various cancer types in vivo and in vitro (4–10, 12, 13).

Table 1. Main tumor types where VM has been reported and main inhibitors described against VM.
The unique arrangement of VM grids simulates embryonic vasculogenesis, suggesting that malignant tumor cells acquire an embryonic-like phenotype. Gene expression analysis showed that aggressive tumor cells capable of VM display a diversified gene profile, expressing genes from multiple cell types such as epithelial cells, fibroblasts and endothelial cells (16, 17). Nevertheless, the molecular mechanisms that give rise to VM remain largely unknown.
Vascular endothelial cadherin (VE-cadherin), also known as cadherin 5 or CD144, is a cell-cell adhesion protein commonly expressed by endothelial cells. Phosphorylation of VE-cadherin at a number of residues can modulate endothelial junction stability and permeability in different contexts (18). VE-cadherin was believed to be specific for endothelial cells (ECs), though in the past few decades it has been found in a wide variety of tumors, where it crucially contributed to aggressiveness and VM. In fact, VE-cadherin was found in highly aggressive tumor cells but it was not expressed by their poorly aggressive counterparts. Moreover, VE-cadherin down-regulation leads to the inhibition of VM formation (19). In this mini-review, we focus on the new findings on the role of extravascular VE-cadherin and its different localizations in the acquisition of the VM phenotype. In addition, we discuss the influence of the tumor microenvironment, stromal cells and mural cells in the development of VM.
VE-Cadherin in Non-Endothelial Context
VE-cadherin is a single pass transmembrane protein typically found in endothelium, where it takes part in adherents junctions (20). VE-cadherin has been extensively studied with reference to vascular adhesion, but its function during VM in aggressive tumor cells is not fully understood. The structure of VE-cadherin contains five extracellular calcium-dependent domains (aminoacid residues 46-599) which can establish cis homodimers with another VE-cadherin molecule. Similarly, VE-cadherin can form trans dimers binding through the ECDI-ECDIV domains. The intracellular domain of VE-cadherin can undergo several post-translational modifications and 13 different residues of VE-cadherin have been reported to undergo phosphorylation in humans. Of these, residues Y658, S665, Y685, and Y731 have drawn most of the attention. They have been implicated in the intracellular dynamics of the cytoskeleton that control endothelial permeability. In particular, phosphorylation of VE-cadherin can trigger junctional changes via VE-cadherin internalization. As a result, vascular permeability is increased, allowing intravasation and extravasation of different cell types, including tumor cells (21). Recent investigations demonstrated that focal adhesion kinase (FAK) can phosphorylate VE-cadherin at Y658 in tumor-associated ECs, pinpointing the importance of FAK in regulating EC barrier function and hence tumor metastasis (22). FAK is a cytoplasmic tyrosine kinase co-activated by vascular endothelial growth factor receptor (VEGFR) 2 and integrin in the control of vascular permeability (23). Recently, we reported that human aggressive melanoma cells have a constitutively high FAK-dependent phosphorylation of VE-cadherin at Y658 (pY658-VEC). pY658-VEC interacts with p120-catenin and the transcriptional repressor kaiso in the nucleus. The inhibition of FAK led to the release of kaiso, promoting its recruitment to kaiso binding sites and therefore repressing kaiso target genes. Moreover, the repression of kaiso target genes CCDN1 and WNT11 abrogated VM. In that line, uveal melanoma cells genetically deficient for VE-cadherin (either through CRISPR/Cas9 technology or after silencing of VE-cadherin) lost the ability to develop VM. Even more, the rescue of WT-VE-cadherin reverted the ability to form VM; in contrast, expression of the non-phosphorylated Y658F-VE-cadherin blunted in vitro VM (24) (see Figure 1).
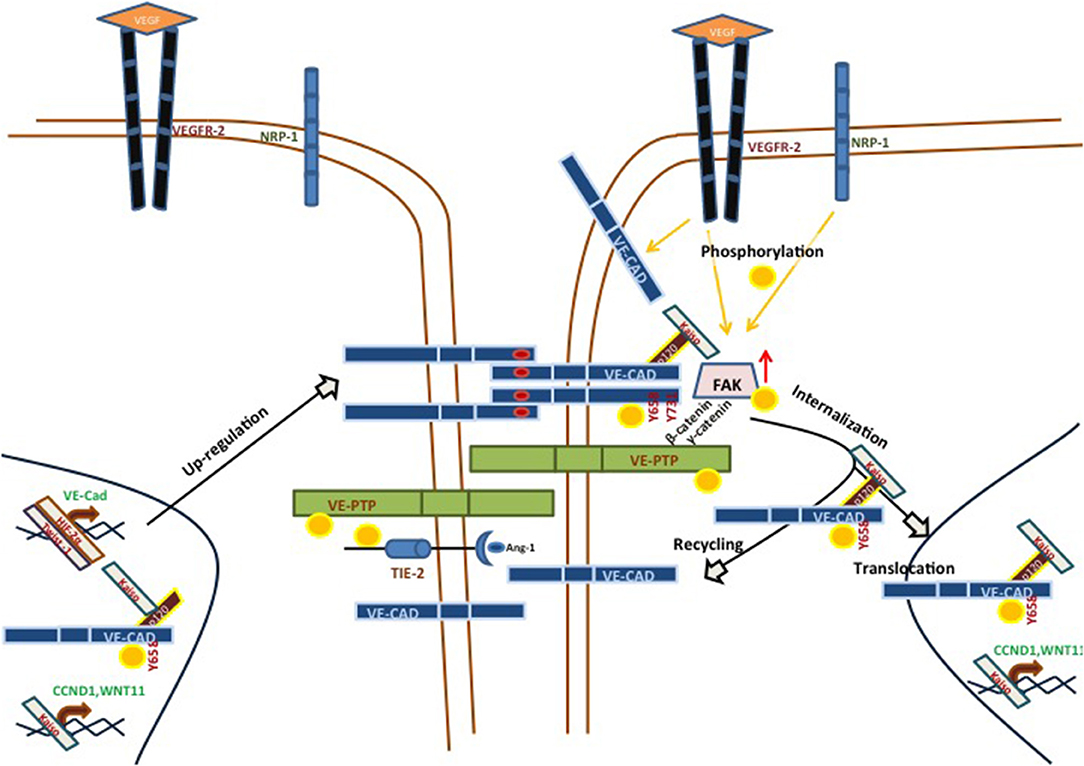
Figure 1. Main signaling pathways implicated in VE-cadherin/VM-positive cells. VE-cadherin can be phosphorylated by VEGFR-2, alone or in a NRP-1-dependent manner, in response to different VEGF soluble factors. These stimuli lead to the phosphorylation of VE-cadherin at Y658 and its subsequent internalization. pY658-VEC can interact with p120 and Kaiso. This complex prevents Kaiso from binding its target genes (CCND1, WNT11), promoting VM formation in aggressive melanoma models. VE-PTP may be an important factor in the maintenance of VE-cadherin phosphorylation status, and catenins p120 or β-catenin may influence the VE-PTP/VE-cadherin axis. On the other hand, VM-positive aggressive melanoma cells showed an up-regulation in a number of proteins, as compared with poorly melanoma cells. One of these proteins was TIE-1, which may be a target of VE-PTP, suggesting an implication in the TIE/angiopoietin pathway.
The intracellular domain V of VE-cadherin is necessary to bind vascular endothelial protein tyrosine phosphatase (VE-PTP) (25), in a plakoglobin (γ-catenin)-dependent way (26, 27). VE-PTP is an endothelial receptor-type phosphatase which was first described in relation to its implications in embryonic vasculature. VE-PTP-deficient mice undergo vasculogenesis but still die at the embryonic stage due to angiogenesis malfunction (28). VE-PTP may have many other implications, such as ocular vascular pathology (29), blood vessels development (30), breast cancer vasculature and metastatic progression (31). VE-PTP is also involved with the TIE1/2-Angiopoietin pathway. Different laboratories have shown that targeting VE-PTP with a specific inhibitor (AKB-9978, Aerpio Pharmaceuticals) activates TIE2 and stabilizes the ocular vasculature in ischemic/inflammation models (29). AKB-9778 induced TIE2 phosphorylation, directly as well as via ANG1. Furthermore, other signaling components of the TIE2 pathway, such as ERK, AKT, and eNOS (see Figure 1), also display increased phosphorylation (29). In 2015, Gong et al. reported that hypoxia increased the expression of VE-PTP in acute lung injury in a HIF-2α-dependent manner (32). All of these functions of VE-PTP have always been studied in endothelial models, though the implications of the VE-PTP/VE-cadherin axis in a VM context remain unknown. In view of the anomalously high levels of phospho-VE-cadherin in cells undergoing VM and the role of VE-PTP in the exquisite maintenance of VE-cadherin phosphorylation, new studies should address the function of this phosphatase in regulating vasculogenic mimicry.
Tumor Microenvironment and VM
The complexity of tumors has been increasingly acknowledged in the past decades, to the point where numerous articles published in the cancer research field are no longer focused exclusively on cancer cells. On the contrary, the different components of the tumor microenvironment have received ever greater attention.
Low oxygen concentration in tumors, commonly known as tumor hypoxia, has been repeatedly associated with malignancy, metastasis and therapy resistance in cancer (33). Hypoxia has been linked to VM by many research groups as well (15). In hepatocellular carcinoma, hypoxia promoted VM through transcriptional co-activation of Bcl-2 and Twist1 (34). Nuclear co-expression of Bcl2 and Twist1 correlated with VE-cadherin expression in tumor cells. In fact, VE-cadherin gene expression can be induced by hypoxia, specifically by hypoxia inducible factor (HIF) 2α (35). Though it was first described in ECs, hypoxia-driven expression of VE-cadherin has been reported in a large number of tumor types too (36–38), where it is always involved in a promotion of the VM phenotype.
Hypoxia has been shown to promote VM through other signaling pathways apart from VE-cadherin. For instance, reactive oxygen species (ROS)-mediated stabilization of HIF1α activated the met proto-oncogene, which induced in vitro tube formation on matrigel in melanoma cells (39). Moreover, HIF1- and HIF2α promoted in vitro tube formation on matrigel through the upregulation of vascular endothelial growth factors (VEGF) C and D, as well as VEGF receptor (VEGFR)3 (40). In triple negative breast cancer, hypoxia increased the subpopulation of CD133+ cells (commonly regarded as cancer stem cells) through a Twist1-mediated mechanism. This population shift seemed to enhance tube formation, since CD133+ cells were found to line the VM-like tubes (41). In addition, HIF1α could promote tube formation in hepatocellular carcinoma by up-regulating lysyl oxidase like 2 (LOXL2) (42, 43), which is involved in collagen cross-linkage during extracellular matrix (ECM) remodeling.
ECM per se can play a fundamental role in regulating VM. The NC11 domain in collagen XVI could trigger tube formation in oral squamous cell carcinoma, since it could induce VEGFR1/2 expression (43). On the contrary, the presence of collagen I altered the vascular potential of pancreatic ductal adenocarcinoma (PDAC) CSCs, decreasing the secretion of pro-angiogenic factors and the expression of VEGFR2, altogether hindering VM formation in PDAC (44). Furthermore, Velez et al. showed that ECM architecture can influence VM; in particular, collagen matrices with small pores and short fibers induced β-integrin expression and hence VM (45).
The importance of non-cancer cells within the tumor stromal is slowly gaining attention in the study of VM as well. Tumor-associated macrophages (TAMs) seemed to promote VM formation in glioblastoma multiforme, namely increasing the expression of cyclooxygenase 2 in the tumor cells (46). Cancer-associated fibroblasts (CAFs) can be determinant in VM formation too. In a recent study, vasculogenic murine melanoma cells were injected in mice carrying a CAF-specific deletion for the matricellular protein CCN2. As a result, the absence of fibroblast-derived CCN2 reduced tumor vasculature, including VM (47). Finally, a recent publication by Thijssen et al. (48) showed that PAS+ tissues in human cutaneous melanoma stained positive for pericyte marker α-smooth muscle actin (αSMA) within the ECM networks lined by tumor cells. Furthermore, when VM+ tumor cells were co-cultured with pericytes, there was a stabilization of the VM networks for up to 96 h. Pericyte recruitment to VM networks was shown to be dependent on PDBF-B signaling, whereas the addition of STI-571 (imatinib mesylate) to inhibit PDGF receptor hindered VM as well as tumor growth.
Targeting VM and Perspectives
A meta-analysis of 22 clinical studies derived from data concerning VM and 5-year survival of 3,062 patients across 15 cancer types showed that tumor VM is correlated with poor prognosis (49). Anti-angiogenic therapies (preferably, antibodies against VEGF receptor bevacizumab and related) against tumor development have had limited results so far. Therefore, the development of novel anti-tumor neovascularization strategies is of vital importance, expanding the targets from conventional angiogenesis to all the alternative mechanisms recently discovered, such as VM (50).
A unique small molecular compound with particular interest in anti-VM cancer treatment is CVM-1118, which is currently undergoing clinical trials (NCT03582618). CVM-1118 is classified as a phenyl-quinoline derivative, whose core structure displays potent anti-neoplastic and anti-mutagenic properties (51).
As mentioned above, pharmacological inhibition of activity of FAK/Y658 VE-cadherin with PF-271 may represent a new therapeutic opportunity in the repression of genes involved with VM promotion in cancer cells. Similarly, inhibition of the VE-PTP/TIE-2 pathway with AKB-9778 could open new ways to control the capacity to form pseudo-vessels by vascular mimicry cells. Finally, new treatments targeting mural cells, such as pericytes, could also have therapeutic value. It is the case of targeting PDGF-B axis with STI-571, which proved useful in VM mice models. Targeting VM with specific molecular compounds combined with front-line therapies may represent the best approach to obtain a good prognosis in patients in the future.
Author Contributions
MF-C and DD-B designed and wrote the review. FO designed, wrote and coordinated the review.
Funding
This work was supported by the grants from the Spanish Ministry of Economy and Competitiveness SAF2015-70520-R, Fundación Domingo Martínez and the Spanish Ministry of Science and Technology RTI2018-098968-B-I00 and CIBERONC ISCIII CB16/12/00421 to FO.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Natale G, Bocci G, Lenzi P. Looking for the word “Angiogenesis” in the history of health sciences: from ancient times to the first decades of the twentieth century. World J Surg. (2017) 41:1625–34. doi: 10.1007/s00268-016-3680-1
2. Dome B, Hendrix MJ, Paku S, Tovari J, Timar J. Alternative vascularization mechanisms in cancer: pathology and therapeutic implications. Am J Pathol. (2007) 170:1–15. doi: 10.2353/ajpath.2007.060302
3. Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol. (2017) 241:362–74. doi: 10.1002/path.4845
4. Liu S, Ni C, Zhang D, Sun H, Dong X, Che N, et al. S1PR1 regulates the switch of two angiogenic modes by VE-cadherin phosphorylation in breast cancer. Cell Death Dis. (2019) 10:200. doi: 10.1038/s41419-019-1411-x
5. Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene. (2013) 32:544–53. doi: 10.1038/onc.2012.85
6. Qi L, Song W, Liu Z, Zhao X, Cao W, Sun B. Wnt3a promotes the vasculogenic mimicry formation of colon cancer via Wnt/beta-catenin signaling. Int J Mol Sci. (2015) 16:18564–79. doi: 10.3390/ijms160818564
7. El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain. (2010) 133:973–82. doi: 10.1093/brain/awq044
8. Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N, et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology. (2010) 51:545–56. doi: 10.1002/hep.23311
9. Xia Y, Cai XY, Fan JQ, Zhang LL, Ren JH, Li ZY, et al. The role of sema4D in vasculogenic mimicry formation in non-small cell lung cancer and the underlying mechanisms. Int J Cancer. (2019) 144:2227–38. doi: 10.1002/ijc.31958
10. Dunleavey JM, Xiao L, Thompson J, Kim MM, Shields JM, Shelton SE, et al. Vascular channels formed by subpopulations of PECAM1+ melanoma cells. Nat Commun. (2014) 5:5200. doi: 10.1038/ncomms6200
11. Yang J, Zhu D-M, Zhou X-G, Yin N, Zhang Y, Zhang Z-X, et al. HIF-2α promotes the formation of vasculogenic mimicry in pancreatic cancer by regulating the binding of Twist1 to the VE-cadherin promoter. Oncotarget. (2017) 8:47801–15. doi: 10.18632/oncotarget.17999
12. Racordon D, Valdivia A, Mingo G, Erices R, Aravena R, Santoro F, et al. Structural and functional identification of vasculogenic mimicry in vitro. Sci Rep. (2017) 7:6985. doi: 10.1038/s41598-017-07622-w
13. Tang J, Wang J, Fan L, Li X, Liu N, Luo W, et al. cRGD inhibits vasculogenic mimicry formation by down-regulating uPA expression and reducing EMT in ovarian cancer, Oncotarget. (2016) 7:24050–62. doi: 10.18632/oncotarget.8079
14. Williamson SC, Metcalf RL, Trapani F, Mohan S, Antonello J, Abbott B, et al. Vasculogenic mimicry in small cell lung cancer. Nat Commun. (2016) 7:13322. doi: 10.1038/ncomms13322
15. van der Schaft DW, Hillen F, Pauwels P, Kirschmann DA, Castermans K, Egbrink MG, et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. (2005) 65:11520–8. doi: 10.1158/0008-5472.CAN-05-2468
16. Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. (2000) 406:536–40. doi: 10.1038/35020115
17. Seftor EA, Meltzer PS, Kirschmann DA, Pe'er J, Maniotis AJ, Trent JM, et al. Molecular determinants of human uveal melanoma invasion and metastasis. Clin Exp Metastasis. (2002) 19:233–46. doi: 10.1023/A:1015591624171
18. Breier G, Grosser M, Rezaei M. Endothelial cadherins in cancer. Cell Tissue Res. (2014) 355:523–7. doi: 10.1007/s00441-014-1851-7
19. Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci USA. (2001) 98:8018–23. doi: 10.1073/pnas.131209798
20. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. (2013) 26:441–54. doi: 10.1016/j.devcel.2013.08.020
21. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. (2008) 121:2115–22. doi: 10.1242/jcs.017897
22. Jean C, Chen XL, Nam JO, Tancioni I, Uryu S, Lawson C, et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol. (2014) 204:247–63. doi: 10.1083/jcb.201307067
23. Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. (2012) 22:146–57. doi: 10.1016/j.devcel.2011.11.002
24. Delgado-Bellido D, Fernandez-Cortes M, Rodriguez MI, Serrano-Saenz S, Carracedo A, Garcia-Diaz A, et al. VE-cadherin promotes vasculogenic mimicry by modulating kaiso-dependent gene expression. Cell Death Differ. (2018) 26:348–61. doi: 10.1038/s41418-018-0125-4
25. Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. (2002) 21:4885–95. doi: 10.1093/emboj/cdf497
26. Muramatsu F, Kidoya H, Naito H, Hayashi Y, Iba T, Takakura N. Plakoglobin maintains the integrity of vascular endothelial cell junctions and regulates VEGF-induced phosphorylation of VE-cadherin. J Biochem. (2017) 162:55–62. doi: 10.1093/jb/mvx001
27. Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. (2008) 205:2929–45. doi: 10.1084/jem.20080406
28. Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci USA. (2007) 104:3243–8. doi: 10.1073/pnas.0611510104
29. Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest. (2014) 124:4564–76. doi: 10.1172/JCI74527
30. Baumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, et al. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. (2006) 107:4754–62. doi: 10.1182/blood-2006-01-0141
31. Goel S, Gupta N, Walcott BP, Snuderl M, Kesler CT, Kirkpatrick ND, et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J Natl Cancer Inst. (2013) 105:1188–201. doi: 10.1093/jnci/djt164
32. Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, et al. HIF2alpha signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest. (2015) 125:652–64. doi: 10.1172/JCI77701
33. Nobre AR, Entenberg D, Wang Y, Condeelis J, Aguirre-Ghiso JA. The different routes to metastasis via hypoxia-regulated programs. Trends Cell Biol. (2018) 28:941–56. doi: 10.1016/j.tcb.2018.06.008
34. Sun B, Zhang D, Zhang S, Zhang W, Guo H, Zhao X. Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett. (2007) 249:188–97. doi: 10.1016/j.canlet.2006.08.016
35. Le Bras A, Lionneton F, Mattot V, Lelievre E, Caetano B, Spruyt N, et al. HIF-2alpha specifically activates the VE-cadherin promoter independently of hypoxia and in synergy with Ets-1 through two essential ETS-binding sites. Oncogene. (2007) 26:7480–9. doi: 10.1038/sj.onc.1210566
36. Mao XG, Xue XY, Wang L, Zhang X, Yan M, Tu YY, et al. CDH5 is specifically activated in glioblastoma stemlike cells and contributes to vasculogenic mimicry induced by hypoxia. Neuro Oncol. (2013) 15:865–79. doi: 10.1093/neuonc/not029
37. Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, et al. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol. (2014) 133:575–83. doi: 10.1016/j.ygyno.2014.02.034
38. Tang N-N, Zhu H, Zhang H-J, Zhang W-F, Jin H-L, Wang L, et al. HIF-1α induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J Gastroenterol. (2014) 20:17894. doi: 10.3748/wjg.v20.i47.17894
39. Comito G, Calvani M, Giannoni E, Bianchini F, Calorini L, Torre E, et al. HIF-1alpha stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic Biol Med. (2011) 51:893–904. doi: 10.1016/j.freeradbiomed.2011.05.042
40. Spinella F, Caprara V, Di Castro V, Rosano L, Cianfrocca R, Natali PG, et al. Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J Mol Med. (2013) 91:395–405. doi: 10.1007/s00109-012-0956-2
41. Zhang Y, Sun B, Zhao X, Liu Z, Wang X, Yao X, et al. Clinical significances and prognostic value of cancer stem-like cells markers and vasculogenic mimicry in renal cell carcinoma. J Surg Oncol. (2013) 108:414–9. doi: 10.1002/jso.23402
42. Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F, et al. HIF-1alpha promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res. (2017) 36:60. doi: 10.1186/s13046-017-0533-1
43. Bedal KB, Grassel S, Spanier G, Reichert TE, Bauer RJ. The NC11 domain of human collagen XVI induces vasculogenic mimicry in oral squamous cell carcinoma cells. Carcinogenesis. (2015) 36:1429–39. doi: 10.1093/carcin/bgv141
44. Biondani G, Zeeberg K, Greco MR, Cannone S, Dando I, Dalla Pozza E, et al. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J. (2018) 285:2104–24. doi: 10.1111/febs.14471
45. Velez DO, Tsui B, Goshia T, Chute CL, Han A, Carter H, et al. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat Commun. (2017) 8:1651. doi: 10.1038/s41467-017-01556-7
46. Rong X, Huang B, Qiu S, Li X, He L, Peng Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget. (2016) 7:83976–86. doi: 10.18632/oncotarget.6930
47. Hutchenreuther J, Vincent K, Norley C, Racanelli M, Gruber SB, Johnson TM, et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. (2018) 74:52–61. doi: 10.1016/j.matbio.2018.06.003
48. Thijssen VL, Paulis YW, Nowak-Sliwinska P, Deumelandt KL, Hosaka K, Soetekouw PM, et al. Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J Pathol. (2018) 246:447–58. doi: 10.1002/path.5152
49. Cao Z, Bao M, Miele L, Sarkar FH, Wang Z, Zhou Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: a systemic review and meta-analysis. Eur J Cancer. (2013) 49:3914–23. doi: 10.1016/j.ejca.2013.07.148
50. Schnegg CI, Yang MH, Ghosh SK, Hsu M-Y. Induction of vasculogenic mimicry overrides VEGF-A silencing and enriches stem-like cancer cells in melanoma. Cancer Res. (2015) 75:1682–90. doi: 10.1158/0008-5472.CAN-14-1855
Keywords: vasculogenic mimicry, tumor microenvironment, metastasis, VE-cadherin, anti-angiogenesis therapeutic failure, cell plasticity
Citation: Fernández-Cortés M, Delgado-Bellido D and Oliver FJ (2019) Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 9:803. doi: 10.3389/fonc.2019.00803
Received: 21 June 2019; Accepted: 07 August 2019;
Published: 22 August 2019.
Edited by:
Laurence A. Marchat, National Polytechnic Institute, MexicoReviewed by:
Gabriele Multhoff, Technical University of Munich, GermanyFahd Al-Mulla, Genatak, Kuwait
Copyright © 2019 Fernández-Cortés, Delgado-Bellido and Oliver. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: F. Javier Oliver, am9saXZlckBpcGIuY3NpYy5lcw==
†These authors have contributed equally to this work