 Meghan A. Rice
Meghan A. Rice Sanjay V. Malhotra
Sanjay V. Malhotra Tanya Stoyanova
Tanya Stoyanova- 1Department of Radiology, Canary Center at Stanford for Cancer Early Detection, Stanford University, Palo Alto, CA, United States
- 2Department of Radiation Oncology, Stanford University, Palo Alto, CA, United States
Prostate cancer is the most commonly diagnosed cancer affecting men in the United States. The prostate is a hormone-dependent gland in which androgen hormones testosterone and dihydrotestosterone bind to and activate the androgen receptor, initiating nuclear translocation of androgen receptor and a subsequent signaling cascade. Due to the androgen dependency of the prostate, androgen deprivation therapies have emerged as first line treatment for aggressive prostate cancer. Such therapies are effective until the point at which prostate cancer, through a variety of mechanisms including but not limited to generation of ligand-independent androgen receptor splice variants, or intratumoral androgen production, overcome hormone deprivation. These cancers are androgen ablation resistant, clinically termed castration resistant prostate cancer (CRPC) and remain incurable. First-generation antiandrogens established androgen receptor blockade as a therapeutic strategy, but these therapies do not completely block androgen receptor activity. Efficacy and potency have been improved by the development of second-generation antiandrogen therapies, which remain the standard of care for patients with CRPC. Four second-generation anti-androgens are currently approved by the Food and Drug Administration (FDA); abiraterone acetate, enzalutamide, and recently approved apalutamide and darolutamide. This review is intended to provide a thorough overview of FDA approved second-generation antiandrogen discovery, treatment application, strategies for combination therapy to overcome resistance, and an insight for the potential future approaches for therapeutic inhibition of androgen receptor.
Introduction
Prostate cancer (PC) has long been the most commonly diagnosed non-cutaneous cancer in men in the United States, and currently has the second highest cancer-associated deaths after lung cancer (1). Through the process of annual digital rectal exams (DREs), prostate specific antigen (PSA) screening and follow-up biopsies, PC is often diagnosed in an actionable time-frame. When diagnosed early, PC grows slowly, and therefore, men can be monitored via active surveillance (also termed “watchful waiting”) for some time prior to medical intervention to limit overtreatment and increase quality of life. Localized PC is primarily treated by either radical prostatectomy and/or radiation therapy and is often well-managed on these regiments. Metastatic or recurrent PC is usually treated with hormonal therapy, or androgen deprivation therapy (ADT), also termed therapeutic castration. These therapeutic interventions work by inhibiting the testosterone production of the testes and prostate tumors blocking AR, which is largely impactful as over 80% of PC is androgen dependent (2). ADT includes luteinizing hormone-releasing hormone (LHRH) agonists and antagonists, and AR blockers such as bicalutamide. Patients undergoing ADT have excellent initial responses, however most cases result in relapse within a few years due to alternative mechanisms of androgen receptor (AR) signaling, AR amplification or alternative splicing, intratumoral androgen production, or adrenal gland testosterone production at which time the disease is termed castration resistant PC (CRPC) and it is currently incurable (3).
CRPC is often metastatic and is responsible for the majority of PC-associated deaths. Treatment options for CRPC are antiandrogen therapies, taxane-based chemotherapies, sipuleucel-T (provenge) vaccine, or radium-223. Antiandrogens differ from LHRH antagonists by blocking specific aspects of androgen signaling. The first-generation antiandrogens bicalutamide, nilutamide, or flutamide exclusively target AR translocation to the nucleus and prevent downstream signaling, while second-generation antiandrogens enzalutamide, apalutamide and darolutamide improve upon this mechanism, and abiraterone acetate prevents androgen biosynthesis.
The original discovery of a link between androgen ablation and prostatic disease was made in 1786 by John Hunter who demonstrated effects of surgical castration on animals and secondary sex organ sizes (4, 5). However, it was not until 1941 that Charles Huggins and Clarence Hodges discovered androgen deprivation to be an effective treatment of PC (6). In contrast to prostate biology, studying the AR has only reached thorough understanding in the past 30 years, with the first sequence of AR being cloned and mapped to the X chromosome in 1988 by Lubahn et al. (7) Canonical signaling through the AR occurs by androgen ligand activation of the AR (Figure 1). The most common of which are testosterone and its derivative dihydrotestosterone (DHT) which is converted by 5-alpha-reductase, though DHT binds AR with 2–5 times higher affinity than testosterone (8, 9). AR is composed of four distinct domains: the N-terminal domain, DNA binding domain (DBD), a hinge region which allows for N- and C-terminal interaction, and a C-terminal ligand binding domain (LBD) (10). Prior to activation, AR is located in the cytoplasm bound to several chaperone proteins, members of the heat-shock protein family (Figure 1). Androgens bind to the AR ligand binding domain releasing AR chaperones and allowing AR to homodimerize and translocate to the nucleus where it acts as a transcription factor for androgen responsive genes such as PSA and others (Figure 1). Many aspects of AR signaling allow for therapeutic exploitation, such as sequestration of DHT ligands that activate AR, blockade of AR N-C terminal interaction, disruption of AR co-activator interaction, and prevention of AR nuclear translocation (11, 12).
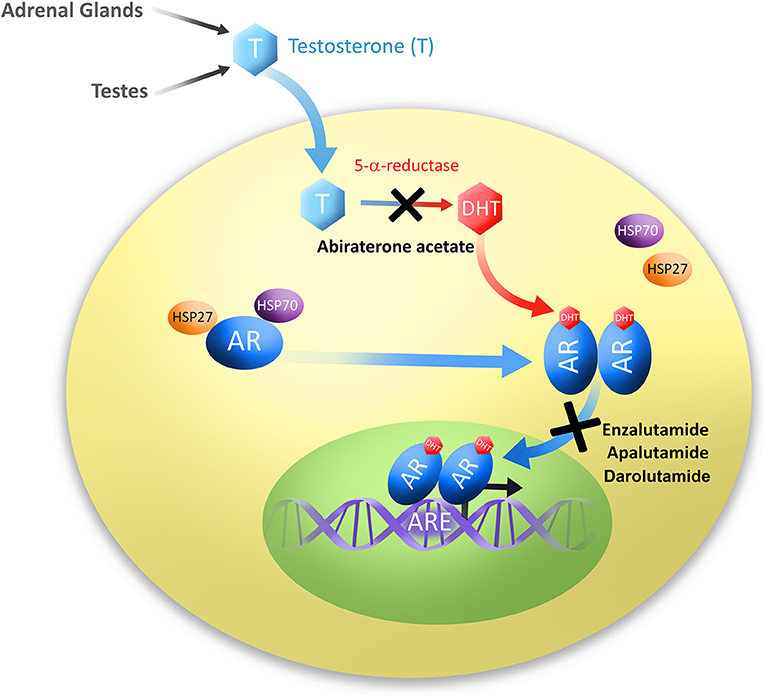
Figure 1. Diagram of androgen production and subsequent signaling through the androgen receptor. Testosterone (T) is produced in the testes and adrenal glands. Testosterone is then converted to its most common and active metabolite, dihydrotestosterone (DHT) by 5α-reductase. Androgens, usually DHT, bind to the androgen receptor (AR), dissociating chaperone proteins, members of the heat shock protein family HSP27 and HSP70. Ligand-bound AR molecules homodimerize and translocate to the nucleus where they bind to androgen response elements (ARE), and act as transcription factors to signal downstream targets. Second-generation antiandrogens are illustrated at their points of pathway disruption; Abiraterone acetate prevents androgen biosynthesis, and Enzalutamide, Apalutamide and Darolutamide prevent AR translocation to the nucleus.
The first therapeutic attempts at AR inhibition were the development of first-generation antiandrogens; that is, steroidal analogs meant to block AR ligand activation. However, developed resistance to these inhibitors became quickly apparent. It is believed that the primary drug resistance mechanisms arise from AR amplification, point mutations, expression of AR splice variants which are ligand independent, intratumoral androgen production or downstream signaling mechanisms (3). It was also determined that in the presence of excess AR, as is commonplace in the context of aggressive PC, first-generation AR antagonists undergo a switch acting as agonists promoting tumor progression in preclinical models of PC (13). This agonist switching results in increased AR mRNA and protein levels, leading to resistance to antiandrogen therapy (14). These changes in pharmacological properties led to the need for second-generation antiandrogens, specifically looking to compounds that would retain antagonistic properties when exposed to excess AR. Further, first-generation antiandrogens are almost completely penetrable by AR mutations, and in the event of bicalutamide discontinuation, a common observation is androgen withdrawal syndrome in which tumors would regress due to AR mutations and agonistim of bicalutamide (15).
Prior to development of second-generation antiandrogens, the most effective treatment strategy for CRPC included taxane chemotherapeutic agent docetaxel, and subsequently cabazitaxel (16, 17). Another therapy option included immunotherapy with the sipuleucel-T (Provenge) vaccine. Sipuleucel-T is the first dendritic cell-based cancer vaccine based on Dr. Edgar G. Engleman's approach in which patient dendritic cells are harvested and pulsed with recombinant prostatic acid phosphatase fusion protein (PAP) and administered back to the host, stimulating autoimmunity to the PC (18). Sipuleucel-T was ultimately approved by the FDA on April 29, 2010 after the success of clinical trials (19).
The development and FDA approval of second-generation, or next-generation antiandrogen treatments has since significantly altered the armamentarium of PC, specifically in treatment of CRPC. Second-generation antiandrogens have increased specificity to the androgen receptor over other steroidal receptors, act at a higher affinity than the previous generation, are exclusively antagonistic to the AR, and in turn elicit no androgen withdrawal syndrome. These compounds have greatly increased patient lifespan, extended metastasis free overall survival, decreased circulating and intratumoral androgens and serum PSA. Even with these advances CRPC has not been eradicated. It is important to fully understand the successes and downfalls in implementation of antiandrogen therapy to patients to look to the future of CRPC treatment.
Androgen Biosynthesis Inhibition
In contrast to androgen receptor blockade, another method of androgen signaling inhibition is the upstream blockade of androgen production. Androgens are produced in the testes, the adrenal glands as well as intratumorally. They are processed by the cytochrome p450 enzyme 17R-hydroxylase-17,20-lyase (CYP17A1). Androgens are then released and circulate through the body. Targeting the biosynthesis mechanism of testosterone through inhibition of CYP17A1, also produced in the testes and adrenal glands, would inhibit production of DHT and decrease endogenous androgen levels. The FDA approved abiraterone acetate will be described in this section, but it is not the only member of this class of inhibitors. Ketoconazole, an FDA approved antifungal compound is also an androgen biosynthesis inhibitor. However, ketoconazole has toxicity due to non-specificity to CYP17A1, though it has also been tested as treatment in PC (20).
Other compounds that have been tested as biosynthesis inhibitors for PC include orteronel (TAK-700), a non-steroidal inhibitor of 17,20-lyase that was tested in clinical trials for metastatic CRPC, as well as galeteronel/TOK-001/VN-124. Galeteronel is a unique dual androgen antagonist and biosynthesis inhibitor that was well-tolerated in Phase 1 trials (21, 22). However, neither compound was able to meet their clinical trial endpoints and have been since terminated, leaving abiraterone acetate as the only currently approved compound in this class for CRPC treatments (23, 24).
Abiraterone Acetate
Other names: Zytiga, Yonsa.
Discovery
The discovery of abiraterone, predecessor to abiraterone acetate, was fueled by the potential of CYP17 inhibition as a treatment for PC. Previously the antifungal ketoconazole which also inhibits CYP17 was determined to inhibit PC growth, with the limitations of low potency, causing adrenal insufficiency, and a general short half-life in the body (25, 26). Abiraterone/CB7598 was a success in that it decreased testosterone production throughout the body and did in fact target CYP17. Abiraterone underwent clinical trials for treatment of advanced PC. However, abiraterone had high toxicity due to the non-specific inhibition of other members of the CYP family. In an attempt to limit toxicity, 20 new compounds were synthesized with variations on the original abiraterone structure to ultimately inhibit the side effects of treatment associated with steroidal hormone interactions. The steroidal scaffold of abiraterone was replaced with non-steroidal cores. One of the compounds, abiraterone acetate, was among them, demonstrating comparable activity to abiraterone with even higher selectivity for CYP17 (27–29).
Abiraterone acetate was the first of the second-generation antiandrogens to be approved by the FDA in April 2011 (Table 1). In contrast to canonical antiandrogens which target androgen receptor, abiraterone acetate is an androgen biosynthesis inhibitor, and the only one approved by the FDA for use in PC at this time (Figure 1 and Table 1) As such, it targets cytochrome P450 enzyme 17R-hydroxylase-17,20-lyase (CYP17). CYP17 processes testosterone and is produced in the testes and adrenal glands. Therefore, inhibition of CYP17 prevents androgen production in both locations and was therefore predicted to be more effective in androgen dependent PC than Gonadotropin-releasing hormone (GnRH) analogs (ADT).
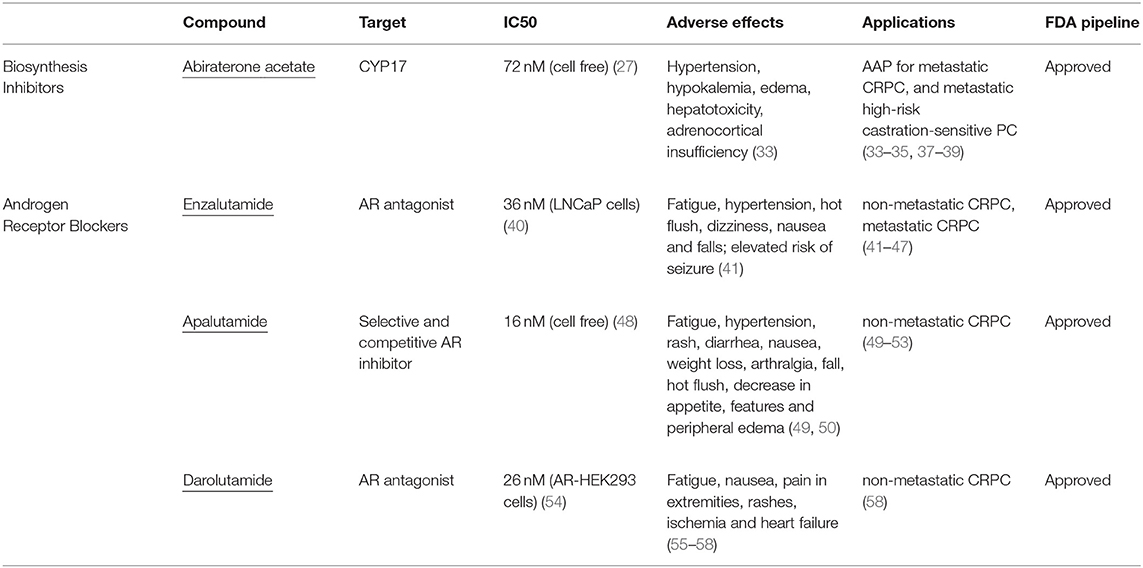
Table 1. Pharmacological and clinical properties of FDA approved second-generation anti-androgens.
Clinical Trials and Path to FDA Approval
Phase 1 and 2 studies were performed on a small cohort of 30 chemotherapy-naïve patients, some of which had previously undergone ketoconazole treatment (NCT00473746) (30–32). No dose-limiting toxicities were observed, and many patients benefited from decreased circulating androgens (33). This study found that the main side effects of abiraterone acetate were associated with low levels of mineralocorticoids, including hypokalemia, fluid retention, and hypertension. These events are ameliorated in combination with glucocorticoids such as prednisone, which has become a standard combination therapy.
For metastatic PC, abiraterone acetate was tested in chemotherapy refractive PC (COU-AA-301; NCT00638690) and chemotherapy-naïve patients (COU-AA-302; NCT00887198). COU-AA-301 enrolled 1,195 patients over 147 sites in 13 countries. Patients with metastatic CRPC were included with progression following chemotherapy on docetaxel, and observed an overall increase in median overall survival from 10.4 months in placebo controlled group to 15.4 months (34, 35). Progression free survival increased as measured by PSA (increased from 6.6 to 8.5 months), and radiologic progression (increased from 3.6 to 5.6 months) (35). In COU-AA-302, abiraterone acetate and prednisone were provided for metastatic CRPC patients who had not previously failed chemotherapy and was also highly effective (36–38). In this trial of 1,088 enrolled patients, overall survival increased from 10.9 to 14.8 months, time to PSA progression increased 6.6 to 10.2 months, and progression-free survival increased 3.6 to 5.6 months compared against placebo-prednisone group (36).
While abiraterone remains an effective clinical therapy to date, new discoveries are still underway to further optimize effectiveness, harnessing more active metabolites found in CRPC patients as primary therapies such as in the case of Δ4-abiraterone (D4A) (39).
Androgen Receptor Blockers
The most utilized mechanism of antiandrogens is blockade of AR signaling by sequestration of AR itself, thus preventing nuclear translocation and subsequent signaling to AR target genes (Figure 1 and Table 1). While first-generation antiandrogens also fit this classification, as a group, second-generation androgen receptor blockers no longer exhibit agonist/antagonist switch, androgen withdrawal syndrome, and have decreased patient toxicities. The main new side effect associated with androgen receptor blockade is increased risk of seizure. This is due to penetrance of the compounds through the blood-brain barrier (BBB) and subsequent inhibition of the γ-aminobutyric acid receptor (GABAA). Enzalutamide discussed herein was the first of the class to be characterized and has the highest risk of seizures at low doses. The recently approved apalutamide and darolutamide provide reductions in brain penetrance and reduced association with clinical risk of seizure, demonstrating promising advances in the field of antiandrogen therapy.
Enzalutamide
Other names: MDV3100, XTANDI.
Discovery
Enzalutamide was the first characterized second-generation antiandrogen. Developed in the laboratories of Drs. Charles Sawyers and Michael Jung, enzalutamide was isolated from a mutagenic screen of non-steroidal agonist RU59063 (40). Compounds were tested for their potential as antagonists of AR and then optimized based on stability and bioavailability. These studies led to the discoveries of MDV3100 and RD162, which demonstrated 5-8-fold greater affinity to AR, and only slightly less affinity than AR ligand DHT. Enzalutamide also inhibits nuclear translocation of AR, as well as inhibits AR DNA binding and coactivator recruitment (Figure 1 and Table 1) (40). Importantly, these compounds did not exhibit agonistic behavior to AR when used in an AR saturated environment in contrast to their first-generation predecessors (40). Ultimately enzalutamide was prioritized over RD162 for clinical development based on favorable drug properties (59).
Clinical Trials and Path to FDA Approval
In 2012, enzalutamide was approved by the FDA for the treatment of men with metastatic CRPC (Table 1). Enzalutamide exhibited greater overall survival over placebo treated patients in clinical trials of patients who relapsed on chemotherapy (AFFIRM trial; NCT00974311), from 13.6 to 18.4 months, as well as radiographic progression-free survival in first line therapy for chemotherapy-naïve patients (PREVAIL trial; NCT01212991), increased from 14 to 65% (42, 43). In the PREVAIL study, enzalutamide was beneficial to patients irrespective of visceral disease, or metastatic sites (low or high volume bone disease as well as lymph node only metastasis) (44). In a head-to-head trial (STRIVE trial; NCT01664923), enzalutamide and first-generation antiandrogen bicalutamide were directly compared. In this trial, treatment with enzalutamide significantly reduced the risk of PC progression and PC-associated death compared to bicalutamide in both metastatic and non-metastatic CRPC (45). FDA approval of enzalutamide was later expanded to include non-metastatic CRPC following positive results of the Phase 3 PROSPER trial (NCT020032924), demonstrating improved metastasis-free survival, which has largely replaced overall survival as a more accurate representation of disease progression in aggressive PC (46, 47).
An advancement from first-generation antiandrogens to enzalutamide was the observation that there was no observed androgen withdrawal syndrome following treatment with enzalutamide (60). Clinically enzalutamide was generally well-tolerated, and the most common side effects included fatigue, arthralgia, and constipation (41). One new complication associated with high dosages of enzalutamide is a risk of increased seizures stemming from the binding to and inhibition of the γ-aminobutyric acid receptor (GABAA), which was an important object of investigation in development of later therapies such as apalutamide (41).
Apalutamide
Other names: ARN-509, JNJ-56021927, Erleada.
Discovery
Building upon the knowledge gained from development of enzalutamide, a more recent addition to the antiandrogen class was apalutamide, which is also a next-generation AR antagonist. Apalutamide was demonstrated to bind with high affinity to the ligand-biding domain of the AR leading to inhibition of its translocation to the nucleus and DNA binding. Apalutamide is a synthetic compound generated based on structure-activity relationship (SAR)-guided chemistry (Figure 1 and Table 1). This was important in identifying compounds which would remain completely antagonistic in the presence of AR expression. Apalutamide is a biaryl thiohydantoin and its selection as candidate was based upon assessment of antagonistic vs. agonistic activity of AR in LNCaP cells overexpressing AR (48). In vitro, apalutamide bound AR with 7–10 fold higher affinity than bicalutamide with an IC50 value of 150 nmol/L. Apalutamide binds AR in the same ligand-binding domain as bicalutamide but with greater affinity, and unlike bicalutamide, retains full antagonist activity in the setting of AR overexpression (48). Furthermore, apalutamide was shown to be selective for AR over other nuclear hormone receptors (48). Ultimately apalutamide was considered an ideal candidate for preclinical assessment having a long serum half-life, low systemic clearance and high oral bioavailability, while demonstrating dramatic effects in reduction of tumor volume in preclinical models of CRPC (48). Further, apalutamide was also effective on non-castrated animals in preclinical studies suggesting it may also be effective prior to castration resistance.
The advantages of apalutamide over enzalutamide included greater efficacy and higher tumor to plasma ratio and four-fold lower concentrations in the central nervous system, suggesting its decreased ability to permeate the blood brain barrier (BBB), potentially indicating a lower risk of seizure activity (48).
Clinical Trials and Path to FDA Approval
Phase I testing was performed on a small cohort of 30 patients with metastatic CRPC and was well-tolerated at all doses (51). Ultimately, the dose at 240 mg/d was selected for further testing in phase II studies. This study was successful based on responses in PSA levels and disease control of patients, delaying time to cancer progression. After selection of patient dose from the Phase I trial, it was proceeded by a multicenter phase 2 study enrolling non-metastatic CRPC at risk of progression as defined by a PSA ≥ equal to 8 ng/ml or PSA doubling time <10 months (NCT01171898) (52). The study was deemed highly successful noting responses in PSA in addition to prolonged metastasis free survival and used to support the design of the phase 3 apalutamide SPARTAN trial (NCT01946204) (52, 53). The SPARTAN trial was an enormous international undertaking spanning 332 centers across 26 countries with 1,207 patients (53). Apalutamide increased metastasis free survival from 16.2 to 40.5 months, with results favorable enough to unblind the trial and offer placebo patients the new therapy. Based on these data apalutamide was approved under priority FDA review for non-metastatic CRPC in 2018 (Table 1).
The safety of apalutamide for metastatic CRPC was evaluated in patients who had either undergone treatment with abiraterone acetate plus prednisone (AAP), or AAP-naïve patients (ARN-509-001; NCT01171898). Apalutamide was again safe, well-tolerated, and demonstrated therapeutic benefit in in patients with metastatic CRPC. Apalutamide treatment was incredibly effective in patients naïve for AAP treatment with an 80% reduction in PSA decline compared to a 22% decline in post-AAP therapy patients (49). The TITAN trial was designed to test apalutamide in metastatic CRPC, newly diagnosed without prior therapies (NCT02489318). Similar to the non-metastatic trial, the TITAN study has recently been unblinded to allow patients to switch to apalutamide treatment based on improvements to progression-free survival as well as overall survival (50).
Darolutamide
Other names: ODM-201, BAY1841788, NUBEQA.
Discovery
Darolutamide is a unique AR antagonist generated as part of a synthetic molecule library, specifically interested in inhibiting AR nuclear translocation (Figure 1 and Table 1). In competitive AR binding assays darolutamide or the active metabolite (ORM-15341) markedly increased potency over enzalutamide and apalutamide in Ki and IC50 values (54). Uniquely, darolutamide and ORM-15341 each individually inhibited wild-type AR as well as clinically relevant AR mutations AR(F876L), which trigger enzalutamide and apalutamide antagonist to agonist switch, as well as AR(W742L) and AR(T877A) which cause bicalutamide agonist switch (54). This inhibition of all known AR mutations is a large advancement for the field of PC treatment that may lead to decreased instance of therapeutic resistance (61). Further, darolutamide inhibited in vivo growth of VCaP CRPC xenografts, and exhibited low penetrance of the BBB in mice and rats (54, 62).
Clinical Trials and Path to FDA Approval
Phase 1 and 2 studies tested safety in patients with CRPC, performed in the ARADES (55, 56) (NCT01429064) and ARAFOR (NCT01784757) (57) trials [and reviewed in (63)]. Darolutamide proved safe and well-tolerated, in addition to reducing tumor volume and PSA levels in both trials (55–57). Darolutamide was approved by the FDA on July 30, 2019 with fast track designation for use in non-metastatic CRPC, making it the most recently FDA approved PC therapy. Approval was granted based on performance in the ARAMIS trial (NCT02200614) (58). The study encompassed 1,509 patients as a multicenter, double-blind tral. Metastasis free survival was 40.4 months in darolutamide treated patients compared to 18.5 months in placebo treated patients. Current clinical recommendation is that darolutamide treatment be combined with ADT or bilateral orchiectomy for non-metastatic CRPC. Darolutamide provides promising reductions in brain penetrance, as well as effectively inhibiting all known AR mutations (61). Ongoing trials include the phase 3 ARASENS trial in which darolutamide plus ADT are combined for treatment of metastatic hormone-sensitive PC (NCT02799602), with an anticipated endpoint in 2022.
Resistance and Combination Therapy Strategies
Resistance
Therapeutic resistance is not a new problem specific to antiandrogen therapy. Resistance mechanisms that evade therapy are a consistent medical challenge in ailments from oncology, in solid cancers and hematological malignancies, to viral infections such as human immunodeficiency virus (HIV). The first instance of clinical combination therapy was in HIV as an answer to the rapid rate of mutation of the disease-causing retrovirus. Termed highly active antiretroviral therapy (HAART), it combined multiple antiretroviral drugs preventing the growth of the virus, and is still used to date (64). Early targeted cancer therapies had great success such as in treatment with Gleevec (imatinib) in leukemia, which was thought to be a “magic-bullet” for cancer therapy (65). While still widely utilized, Imatinib commonly results in resistant disease (66). Oncogenic treatments have begun following a similar trend to HAART therapy to prevent cancer resistance or sensitize to already refractory therapies by targeting multiple pathways essential to oncogenesis such as growth and angiogenesis (67).
Since the treatment with first-generation antiandrogens, resistance associated with androgen withdrawal has been commonly characterized. Greater than 95% of testosterone production in the male body is generated in the testes, yet adrenal, prostatic, and intratumoral androgens play a great role in resistance as minute overexpression of AR is enough to counteract androgen withdrawal, sensitizing tumors to small amounts of androgen to activate AR signaling [reviewed (68–70)]. Further, genetically one-third of CRPC patients have AR amplification (71).
Mutation is one common mechanism by which androgen withdrawal experiences resistance. It was first reported in 2003 by Hara et al. who discovered a mutation of AR which could cause resistance to bicalutamide by allowing an AR antagonist to agonist switch (15). Several AR point mutations associated with first-generation antiandrogens include T877A association with flutamide resistance (15), as well as W741C associated with bicalutamide agonistic switch (72). Subsequently, other clinically relevant mutations of AR (F876L and F877L) were also found to cause an AR agonistic response in enzalutamide and apalutamide, respectively (73, 74). Darolutamide has clinically shown to act as a full antagonist toward AR F876L among other mutations, making a strong platform for darolutamide to quickly gain traction as standard of care in non-metastatic CRPC now that it received FDA approval (54). One method of abiraterone resistance comes from intratumoral generation of androgens and CYP17A1 production, selecting for increased intratumoral production, in conjunction with a progesterone responsive mutant AR (T877A). These tumors are still responsive to steroids and may benefit from switching therapies to androgen receptor blockade (75). Abiraterone treatment increases progesterone, activating AR mutations previously associated with flutamide resistance (T878A) (76).
While second-generation antiandrogens have taken a huge stride in treatment of advanced PC, 20–40% of patients still do not respond to these therapies, as measured by PSA (34, 36, 42, 77), and of the patients that respond, resistance will undoubtedly occur given enough time. Androgen receptor splice variants, such as AR-V7 commonly occur in clinical specimens, and approximately 75% of CRPC cases (78, 79). AR splice variants missing the ligand binding domain signal independently of AR ligand stimulation and are therefore among the most common resistance mechanisms of antiandrogen therapy (80). Ligand binding mutations have been characterized in as many as 20% of patients who have progressed following first-generation antiandrogen treatment (81, 82). Current diagnostic testing can determine AR-V7 expression among other biomarkers from patient needle biopsies (oncotype Dx) to predict a Genomic Prostate Score (GPS) associated with aggressive PC for clinical decision-making (83–85). Further, other studies have followed AR-V7 levels in circulating tumor cells of metastatic CRPC patients and correlated with risk of recurrence, with follow-up PSA serum levels (80).
Mechanisms which have been proposed to prevent therapeutic resistance include drug cycling, intermittent hormonal therapy as opposed to continuous, and combination therapy.
Combination Therapy Strategies
Combination therapies have become more common in recent years in many medical fields, especially oncology. Due to resistance mechanisms, including those just described, antiandrogens are often not prescribed as a single therapy. Antiandrogens were originally given in combination with ADT in a combined androgen blockade (CAB). Prior to the development of second-generation antiandrogens, first-generation antiandrogens were already being combined with medical androgen suppression. A large meta-analysis performed on data from PC patients who received combined androgen receptor blockade consisting of androgen suppression with a first-generation antiandrogen noted a mild improvement in 5-year survival (86). Another strategy in CAB entails combination of androgen receptor blockers with biosynthesis inhibitors. The reasoning behind which being abiraterone acetate working through a different mechanism may sensitize enzalutamide resistant cancers. Enzalutamide plus abiraterone was tested in patients undergoing resistance to enzalutamide in the PLATO trial (NCT01995513) under the assumption that inhibiting androgen synthesis would sensitize patients to enzalutamide (87). Abiraterone acetate with prednisone has also been safely tested with apalutamide successfully in clinical trial with observed antitumor effects in ongoing clinical trials of metastatic CRPC (NCT02257736) (88, 89). Apalutamide with standard ADT is also ongoing in the TITAN trial for metastatic CRPC and to date has improved progression-free survival as well as overall survival vs. ADT alone (90). Finally, in LATITUDE trial, Abiraterone acetate plus prednisone combined with ADT lengthened time of progression free survival 14.8–33 months (NCT01715285) (91).
In contrast to other more rapidly dividing cancers, chemotherapy for PC treatments is usually performed later in the disease progression. However, in late stage PC, taxane chemotherapy is a common standard of care. Chemotherapy has long since been given in conjunction with ADT such as in the CHAARTED trial specifically treating docetaxel with ADT for patients with high volume disease (NCT00309985) (92). Looking forward, chemotherapy treatment is now being tested with antiandrogens. Such as in the STAMPEDE trial; a unique multi-arm multi-stage clinical trial attempting to assay multiple treatment strategies. Within, STAMPEDE includes groups of patients receiving combination of ADT with radiotherapy, docetaxel and abiraterone and ADT with enzalutamide, abiraterone and prednisolone (NCT00268476). Another example is the treatment of apalutamide and everolimus in patients with metastatic CRPC (49).
Radiotherapy is a common treatment for localized PC as well as palliative treatment for bone metastases. Enzalutamide plus radiation in vitro sensitizes cells to radiation (93). In trial COU-AA-31(NCT00638690), abiraterone acetate with radiation were safely co-administered to patients with a perceived advantage in palliative bone metastasis response. Abiraterone acetate with radiotherapy has been further tested in two separate phase 3 clinical trials including ERA-223 (NCT02043678), and in the ongoing PEACE 1 trial as well, though this trial uniquely combines ADT, abiraterone, docetaxel and radiation therapy (GETUG-AFU-21; NCT01957436) (94).
Aside from the discovery of sipuleucel-T, immunotherapy for PC treatment is not a heavily researched avenue. This is largely because PC often does not have high levels of acquired genetic mutations. Even still, several cancer vaccines and immunomedics have been utilized in combination with antiandrogen treatment to date. Specifically, in second-generation antiandrogens, sipuleucel-T has undergone clinical trial in combination with abiraterone acetate plus prednisone (AAP) (P11-3; NCT01487863). In this trial sipuleucel-T was administered safely either concurrently or sequentially with AAP, and patients had similar immunologic prime-boost effects, commonly associated with effectiveness of the vaccine (95). While impact on overall survival is still to be determined, the ability to overlap two successful non-interfering therapies often has additive effects. Enzalutamide is being tested in preclinical combination with a poxviral-based metastasis vaccine which largely targets the transcription factor Twist, commonly associated with mesenchymal phenotyping and increased epithelial to mesenchymal transition (EMT) in cancer (96, 97). Finally, pembrolizumab (Keytruda), an FDA approved anti-PD-1 antibody is a well-known T-cell checkpoint inhibitor antibody commonly used in treatment of melanoma among other cancers. Pembrolizumab has undergone independent testing in two small clinical trials for metastatic CRPC with initial outcome of disease stabilization at 50% when cycles were completed (KEYNOTE-199; NCT02787005) (98). However when administered with enzalutamide, pembrolizumab demonstrated a beneficial response in PSA in 3/10 patients (NCT02312557) (99) [Many additional clinical trials involving first-generation antiandrogens or LHRH antagonists are reviewed in (100)].
Second-generation antiandrogens have been explored as cotreatments with therapeutics targeting other pathways important in PC growth, etc., some with very promising results. Several tested combinations include treatment with PARP inhibition exploiting mutations in DNA repair pathways. Olaparib plus abiraterone acetate increased clinical benefit in metastatic CRPC (NCT01972217) (101, 102). Abiraterone in conjunction with anti-apoptotic XIAP inhibitors exhibited success in preclinical studies (103). Cabozantinib, a kinase inhibitor with activity against MET and VEGFR2 among others, has been tested as a single agent therapy in refractory metastatic CRPC in the COMET-1 trial (NCT01605227) (104). Cabozantinib did not demonstrate a decrease in PSA levels but may be beneficial in treating and preventing skeletal burden. However, cabozantinib treatment with abiraterone acetate in metastatic CRPC exhibited strong preclinical synergy (105, 106). Abiraterone acetate with added inhibition of autophagy has synergized in vitro (107). Niclosamide (anti-helminthic) has strong preclinical efficacy as a PC treatment via potent inhibition of AR-V7, and sensitizes PC to abiraterone acetate, but combined with enzalutamide as a phase 1 trial had observed toxicity (108–110). Chromosomal relocations of the ETS-related gene (ERG) resulting in excess ERG signaling predominantly through joining with transmembrane serine protease 2 (TMPRSS2) (111). TMPRSS2-ERG fusions are very common in PC (111). An inhibitor of ERG, ERGi-USU inhibits ERG-positive PC cell growth, and combined with enzalutamide exhibits additive effects (112). Monoamine oxidase A inhibitors (MAOAIs), typically used to treat depression have shown to moderate PC growth and inhibit metastasis due to high concentration of MAOA in the prostate (113, 114). MAOAI combined with enzalutamide synergizes and sensitizes cells to enzalutamide treatment (115). MAOAI is also in clinical trial for non-metastatic PC clinical trials (NCT02217709). Finally, inhibitors of gamma secretase which regulates cleavage of cell surface receptors including Notch receptors among others, in combination with enzalutamide or abiraterone in vitro inhibits PC cell growth, migration and invasion, as well as sensitizes enzalutamide resistant cells and xenografts to enzalutamide treatment (116–119).
Conclusions
The future of healthcare is moving toward personalized precision medicine rather than blanketed therapy to determine best practice treatment strategies for individual patients. Genomic sequencing of PC patient samples led to the discovery of mutations involving genes such as SPOP, BRCA1, or BRCA2, FOXA1, chromosomal translocations of TMPRSS2:ERG/ETS, and point mutations of AR itself (111, 120, 121). Current analyses depict the majority of PC tumors as having actionable mutations, either as aberrations, point mutations, chromosomal translocations or single nucleotide polymorphisms (SNPs) (122). Aside from AR, actionable targets represent the pathways of DNA repair, Wnt signaling, PI3K pathway, RAF kinases and CDK inhibitors.
Another shift in treatment paradigm for aggressive PC is the increased incidence of neuroendocrine PC (NEPC). It is believed the increasing frequency of NEPC in patients (currently up to 25% of CRPC patients) (123), commonly characterized by a loss of AR and increased neuroendocrine markers such as synaptophysin and chromogranin A, results from excess modulation of androgen signaling. Being AR negative, this aggressive disease is resistant to antiandrogen therapy and currently incurable. Further, a new double negative phenotype (DNPC) has been described which is negative for AR as well as the neuroendocrine phenotype (124). Rather, DNPC requires signaling through fibroblast growth factor receptor (FGFR) and the mitogen-activated protein kinase (MAPK) pathway (124). These cancers currently have no effective therapies.
New treatments targeting different aspects of the androgen receptor signaling cascade have more exploratory potential. The ideal antiandrogen compound would be a directed inhibitor toward the N-terminal AR domain inhibiting all AR isoforms described to date including full length AR as well as ligand-independent splice variants. 5α-reductase inhibitors finasteride and dutasteride are in development, and part of standard of care treatment for benign prostatic hyperplasia (BPH). However, in PC, the use of these compounds which prevent the conversion of testosterone to DHT, have been minimally implemented and could be explored further [reviewed in (125)]. Similarly, new therapies such as orteronel and galeterone are also under development, and darolutamide performed well-enough in end point clinical trials to recently receive expedited approval by FDA for non-metastatic CRPC, but may extend its treatment to more aggressive patient populations in the future. While treatment regiments for some of these compounds have not reached initial guidelines from clinical trials, these and other compounds will still continue to make their way through the clinical pipeline and hopefully alter the course of PC treatment.
Potential for antiandrogens as standalone therapeutic agents seems to have plateaued for use in advanced PC. Until such time as antiandrogens are able to inhibit all splice variants of the androgen receptor, it is far more likely that the next wave of therapeutic investigation will be focused on the combination of antiandrogen therapy with other treatments such as chemotherapy.
Author Contributions
MR and TS conceptualized the manuscript. MR researched the literature. MR, SM, and TS wrote and reviewed the manuscript.
Funding
TS was supported by the Canary Foundation, the National Institute of Health/National Cancer Institute (NCI) R03CA230819, the U.S. Army Medical Research Acquisition Activity through the Congressionally Directed Medical Research Program (CDMRP) under Award No. W81XWH1810323, the McCormick and Gabilan Faculty Award. MR was supported by the U.S. Army Medical Research Acquisition Activity, through the Congressionally Directed Medical Research Program (CDMRP) under Award No. W81XWH1810141. SM was supported by the National Institute of Health R01 DK114174 and the Stanford Cancer Institute. Opinions, interpretations, conclusions and recommendations are those of the authors and not necessarily endorsed by the funding agencies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Would like to thank the Canary Foundation for their continued support and Alexander Ksoll for the adaptation of the artwork in Figure 1.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
2. Denis LJ, Griffiths K. Endocrine treatment in prostate cancer. Semin Surg Oncol. (2000) 18:52–74. doi: 10.1002/(SICI)1098-2388(200001/02)18:1<52::AID-SSU8>3.0.CO;2-6
3. Knudsen KE, Kelly WK. Outsmarting androgen receptor: creative approaches for targeting aberrant androgen signaling in advanced prostate cancer. Expert Rev Endocrinol Metab. (2011) 6:483–93. doi: 10.1586/eem.11.33
4. Hunter J. The Works of John Hunter, F.R.S.: With Notes. Vol. 2, Cambridge Library Collection - History of Medicine. Cambridge: Cambridge University Press (2015). Available online at: https://www.cambridge.org/core/books/works-of-john-hunter-frs/8152753ABB19452360E8C810EA34DC52
5. Denmeade SR, Isaacs JT, Comprehensive K. A history of prostate cancer treatment. Nat Rev Cancer. (2014) 2:389–96. doi: 10.1038/nrc801
6. Huggins C, Hodges CV. Studies on prostatic cancer i. the effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. (1941) 22:232–40.
7. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. (1988) 240:327–30. doi: 10.1126/science.3353727
8. Zhou Z, Lane MV, Kemppainen JA, French FS, Wilson EM. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. (1995) 9:208–18. doi: 10.1210/mend.9.2.7776971
9. Wright AS, Douglas RC, Thomas LN, Lazier CB, Rittmaster RS. Androgen-induced regrowth in the castrated rat ventral prostate: role of 5α-reductase. Endocrinology. (1999) 140:4509–15. doi: 10.1210/endo.140.10.7039
10. MacLean HE, Warne GL, Zajac JD. Localization of functional domains in the androgen receptor. J Steroid Biochem Mol Biol. (1997) 62:233–42. doi: 10.1016/S0960-0760(97)00049-6
11. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. (2005) 23:8253–61. doi: 10.1200/JCO.2005.03.4777
12. Andriole GL, Crawford ED, Grubb RL, Buys SS, Chia D, Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. (2009) 360:1310–9. doi: 10.1056/NEJMoa0810696
13. Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, et al. Switch from antagonist to agonist of the androgen receptor blocker bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. (1999) 81:242–51. doi: 10.1038/sj.bjc.6690684
14. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. (2004) 10:33–9. doi: 10.1038/nm972
15. Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. (2003) 63:149–53.
16. Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. (2008) 26:242–5. doi: 10.1200/JCO.2007.12.4008
17. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels J-P, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. (2010) 376:1147–54. doi: 10.1016/S0140-6736(10)61389-X
18. Fong L, Brockstedt D, Benike C, Breen JK, Strang G, Ruegg CL, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol. (2001) 167:7150–6. doi: 10.4049/jimmunol.167.12.7150
19. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
20. Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. (1983) 71:1495–9. doi: 10.1172/JCI110903
21. Brawer MK. New treatments for castration-resistant prostate cancer: highlights from the 44th annual meeting of the American society of clinical oncology, may 30-june 3, 2008, chicago, IL. Rev Urol. (2008) 10:294–6.
22. Montgomery RB, Eisenberger MA, Rettig M, Chu F, Pili R, Stephenson J, et al. Phase I clinical trial of galeterone (TOK-001), a multifunctional antiandrogen and CYP17 inhibitor in castration resistant prostate cancer (CRPC). J Clin Oncol. (2012) 30(Suppl. 15):4665. doi: 10.1200/jco.2012.30.15_suppl.4665
23. Fizazi K, Jones R, Oudard S, Efstathiou E, Saad F, de Wit R, et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy. J Clin Oncol. (2015) 33:723–31. doi: 10.1200/JCO.2014.56.5119
24. Njar VCO, Brodie AMH. Discovery and Development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem. (2015) 58:2077–87. doi: 10.1021/jm501239f
25. Harris KA, Weinberg V, Bok RA, Kakefuda M, Small EJ. Low dose ketoconazole with replacement doses of hydrocortisone in patients with progressive androgen independent prostate cancer. J Urol. (2002) 168:542–5. doi: 10.1016/S0022-5347(05)64675-7
26. Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017.alpha.-Hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. (1995) 38:2463–71. doi: 10.1021/jm00013a022
27. Pinto-Bazurco Mendieta MAE, Negri M, Jagusch C, Müller-Vieira U, Lauterbach T, Hartmann RW. Synthesis, biological evaluation, and molecular modeling of abiraterone analogues: novel CYP17 inhibitors for the treatment of prostate cancer. J Med Chem. (2008) 51:5009–18. doi: 10.1021/jm800355c
28. Bryce A, Ryan CJ. Development and clinical utility of abiraterone acetate as an androgen synthesis inhibitor. Clin Pharmacol Ther. (2012) 91:101–8. doi: 10.1038/clpt.2011.275
29. Schweizer MT, Antonarakis ES. Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: a new era of hormonal therapies is born. Ther Adv Urol. (2012) 4:167–78. doi: 10.1177/1756287212452196
30. Attard G, Reid AHM, A'Hern R, Parker C, Oommen NB, Folkerd E, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. (2009) 27:3742–8. doi: 10.1200/JCO.2008.20.0642
31. Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. (2008) 26:4563–71. doi: 10.1200/JCO.2007.15.9749
32. Attard G, Reid AHM, de Bono JS. Abiraterone acetate is well tolerated without concomitant use of corticosteroids. J Clin Oncol. (2010) 28:e560–1. doi: 10.1200/JCO.2010.29.5170
33. Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. (2010) 28:1481–8. doi: 10.1200/JCO.2009.24.1281
34. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. (2011) 364:1995–2005. doi: 10.1056/NEJMoa1014618
35. Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2012) 13:983–92. doi: 10.1016/S1470-2045(12)70379-0
36. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. (2012) 368:138–48. doi: 10.1056/NEJMoa1209096
37. Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PFA, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. (2015) 16:152–60. doi: 10.1016/S1470-2045(14)71205-7
38. Miller K, Carles J, Gschwend JE, Van Poppel H, Diels J, Brookman-May SD. The Phase 3 COU-AA-302 study of abiraterone acetate plus prednisone in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: stratified analysis based on pain, prostate-specific antigen, and gleason score. Eur Urol. (2018) 74:17–23. doi: 10.1016/j.eururo.2017.08.035
39. Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. (2015) 523:347–51. doi: 10.1038/nature14406
40. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. (2009) 324:787–90. doi: 10.1126/science.1168175
41. Graff JN, Gordon MJ, Beer TM. Safety and effectiveness of enzalutamide in men with metastatic, castration-resistant prostate cancer. Exp Opin Pharmacother. (2015) 16:749–54. doi: 10.1517/14656566.2015.1016911
42. Scher HI, Fizazi K, Saad F, Taplin M-E, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
43. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. (2014) 371:424–33. doi: 10.1056/NEJMoa1405095
44. Evans CP, Higano CS, Keane T, Andriole G, Saad F, Iversen P, et al. The PREVAIL study: primary outcomes by site and extent of baseline disease for enzalutamide-treated men with chemotherapy-naïve metastatic castration-resistant prostate cancer. Eur Urol. (2016) 70:675–83. doi: 10.1016/j.eururo.2016.03.017
45. Penson DF, Armstrong AJ, Concepcion R, Agarwal N, Olsson C, Karsh L, et al. Enzalutamide versus bicalutamide in castration-resistant prostate cancer: the STRIVE trial. J Clin Oncol. (2016) 34:2098–106. doi: 10.1200/JCO.2015.64.9285
46. Tombal B, Saad F, Penson D, Hussain M, Sternberg CN, Morlock R, et al. Patient-reported outcomes following enzalutamide or placebo in men with non-metastatic, castration-resistant prostate cancer (PROSPER): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. (2019) 20:556–69. doi: 10.1016/S1470-2045(18)30898-2
47. Xie W, Regan MM, Buyse M, Halabi S, Kantoff P, Sartor O, et al. Metastasis-free survival is a strong Surrogate of overall survival in localized prostate cancer. J Clin Oncol. (2017) 35:3097–104. doi: 10.1200/JCO.2017.73.9987
48. Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. (2012) 72:1494–503. doi: 10.1158/0008-5472.CAN-11-3948
49. Rathkopf DE, Antonarakis ES, Shore ND, Tutrone RF, Alumkal JJ, Ryan CJ, et al. Safety and Antitumor Activity of Apalutamide (ARN-509) in metastatic castration-resistant prostate cancer with and without prior abiraterone acetate and prednisone. Clin Cancer Res. (2017) 23:3544–51. doi: 10.1158/1078-0432.CCR-16-2509
50. Janssen. Janssen Announces ERLEADA®(apalutamide) Phase 3 TITAN Study Unblinded as Dual Primary Endpoints Achieved in Clinical Program Evaluating Treatment of Patients with Metastatic Castration-Sensitive Prostate Cancer (2019). Available online at: https://bit.ly/2UuyCL1
51. Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. (2013) 31:3525–30. doi: 10.1200/JCO.2013.50.1684
52. Smith MR, Antonarakis ES, Ryan CJ, Berry WR, Shore ND, Liu G, et al. Phase 2 Study of the Safety and Antitumor Activity of Apalutamide (ARN-509), a potent androgen receptor antagonist, in the high-risk nonmetastatic castration-resistant prostate cancer cohort. Eur Urol. (2016) 70:963–70. doi: 10.1016/j.eururo.2016.04.023
53. Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. (2018) 378:1408–18. doi: 10.1056/NEJMoa1715546
54. Moilanen A-M, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. (2015) 5:12007. doi: 10.1038/srep12007
55. Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): An open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. (2014) 15:975–85. doi: 10.1016/S1470-2045(14)70240-2
56. Fizazi K, Massard C, Bono P, Kataja V, James N, Tammela TL, et al. Safety and antitumour activity of ODM-201 (BAY-1841788) in Castration-resistant, CYP17 inhibitor-naïve prostate cancer: results from extended follow-up of the ARADES Trial. Eur Urol Focus. (2017) 3:606–14. doi: 10.1016/j.euf.2017.01.010
57. Massard C, Penttinen HM, Vjaters E, Bono P, Lietuvietis V, Tammela TL, et al. Pharmacokinetics, antitumor activity, and safety of ODM-201 in patients with chemotherapy-naive metastatic castration-resistant prostate cancer: an open-label phase 1 study. Eur Urol. (2016) 69:834–40. doi: 10.1016/j.eururo.2015.09.046
58. Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. (2019) 380:1235–46. doi: 10.1056/NEJMoa1815671
59. Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, et al. Structure–activity relationship for thiohydantoin androgen receptor antagonists for Castration-Resistant Prostate Cancer (CRPC). J Med Chem. (2010) 53:2779–96. doi: 10.1021/jm901488g
60. Von Klot CA, Kuczyk MA, Merseburger AS. No androgen withdrawal syndrome for enzalutamide: a report of disease dynamics in the postchemotherapy setting. Eur Urol. (2014) 65:258–9. doi: 10.1016/j.eururo.2013.09.036
61. Fizazi K, Albiges L, Loriot Y, Massard C. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticancer Ther. (2015) 15:1007–17. doi: 10.1586/14737140.2015.1081566
62. Zurth C, Sandmann S, Trummel D, Seidel D, Gieschen H. Blood-brain barrier penetration of [14C]darolutamide compared with [14C]enzalutamide in rats using whole body autoradiography. J Clin Oncol. (2018) 36(6_suppl):345. doi: 10.1200/JCO.2018.36.6_suppl.345
63. Fizazi K, Smith MR, Tombal B. Clinical development of darolutamide: a novel androgen receptor antagonist for the treatment of prostate cancer. Clin Genitourin Cancer. (2018) 16:332–40. doi: 10.1016/j.clgc.2018.07.017
64. Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. (2004) 350:1023–35. doi: 10.1056/NEJMra025195
65. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. (2001) 344:1031–7. doi: 10.1056/NEJM200104053441401
66. Milojkovic D, Apperley JF. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. (2009) 15:7519–27. doi: 10.1158/1078-0432.CCR-09-1068
67. Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. (2013) 31:1592–605. doi: 10.1200/JCO.2011.37.6418
68. Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol. (2009) 27:36–41. doi: 10.1016/j.urolonc.2008.03.021
69. Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. (2010) 21:315–24. doi: 10.1016/j.tem.2010.01.002
70. Mostaghel EA, Montgomery B, Nelson PS. Castration-resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urol Oncol. (2009) 27:251–7. doi: 10.1016/j.urolonc.2009.03.016
71. Linja MJ, Visakorpi T. Alterations of androgen receptor in prostate cancer. J Steroid Biochem Mol Biol. (2004) 92:255–64. doi: 10.1016/j.jsbmb.2004.10.012
72. Veldscholte J, Ris-Stalpers C, Kuiper GGJM, Jenster G, Berrevoets C, Claassen E, et al. A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. (1990) 173:534–40. doi: 10.1016/S0006-291X(05)80067-1
73. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. (2013) 3:1020–9. doi: 10.1158/2159-8290.CD-13-0226
74. Rathkopf DE, Smith MR, Ryan CJ, Berry WR, Shore ND, Liu G, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol Off J Eur Soc Med Oncol. (2017) 28:2264–71. doi: 10.1093/annonc/mdx283
75. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, et al. Intratumoral De Novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. (2011) 71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532
76. Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. (2015) 21:1273–80. doi: 10.1158/1078-0432.CCR-14-1220
77. Scher HI, Beer TM, Higano CS, Anand A, Taplin M-E, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. (2010) 375:1437–46. doi: 10.1016/S0140-6736(10)60172-9
78. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. (2008) 68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594
79. Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. (2019) 129:192–208. doi: 10.1172/JCI122819
80. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. (2014) 371:1028–38. doi: 10.1056/NEJMoa1315815
81. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. (2012) 487:239–43. doi: 10.1038/nature11125
82. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. (2013) 63:920–6. doi: 10.1016/j.eururo.2012.08.053
83. Knezevic D, Goddard AD, Natraj N, Cherbavaz DB, Clark-Langone KM, Snable J, et al. Analytical validation of the oncotype DX prostate cancer assay - a clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genomics. (2013) 14:690. doi: 10.1186/1471-2164-14-690
84. Klein EA, Cooperberg MR, Magi-Galluzzi C, Simko JP, Falzarano SM, Maddala T, et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur Urol. (2014) 66:550–60. doi: 10.1016/j.eururo.2014.05.004
85. Cullen J, Rosner IL, Brand TC, Zhang N, Tsiatis AC, Moncur J, et al. A biopsy-based 17-gene genomic prostate score predicts recurrence after radical prostatectomy and adverse surgical pathology in a racially diverse population of men with clinically low- and intermediate-risk prostate cancer. Eur Urol. (2015) 68:123–31. doi: 10.1016/j.eururo.2014.11.030
86. Prostate Cancer Trialists' Collaborative Group. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Lancet. (2000) 355:1491–8. doi: 10.1016/S0140-6736(00)02163-2
87. Attard G, Borre M, Gurney H, Loriot Y, Andresen-Daniil C, Kalleda R, et al. Abiraterone alone or in combination with enzalutamide in metastatic castration-resistant prostate cancer with rising prostate-specific antigen during enzalutamide treatment. J Clin Oncol. (2018) 36:2639–46. doi: 10.1200/JCO.2018.77.9827
88. Posadas EM, Chi KN, De Wit R, De Jonge MJ, Attard G, Friedlander TW, et al. Phase Ib study of apalutamide (APA) with abiraterone acetate (AA) and prednisone (P) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Update on safety and efficacy. J Clin Oncol. (2017) 35(6_suppl):173. doi: 10.1200/JCO.2017.35.6_suppl.173
89. Rathkopf DE, Attard G, Efstathiou E, Yu MK, Griffin TW, Todd MB, et al. A phase 3 randomized, placebo-controlled double-blind study of ARN-509 plus abiraterone acetate (AA) in chemotherapy-naïve metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. (2015) 33:TPS5071. doi: 10.1200/jco.2015.33.15_suppl.tps5071
90. Chi KN, Chowdhury S, Radziszewski P, Lebret T, Ozguroglu M, Sternberg C, et al. TITAN: a randomized, double-blind, placebo-controlled, phase 3 trial of apalutamide (ARN-509) plus androgen deprivation therapy (ADT) in metastatic hormone-sensitive prostate cancer (mHSPC). Ann Oncol. (2016) 27(suppl_6):243–65. doi: 10.1093/annonc/mdw372.54
91. Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. (2017) 377:352–60. doi: 10.1056/NEJMoa1704174
92. Kyriakopoulos CE, Chen YH, Carducci MA, Liu G, Jarrard DF, Hahn NM, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: long-term survival analysis of the randomized phase III E3805 chaarted trial. J Clin Oncol. (2018) 36:1080–7. doi: 10.1200/JCO.2017.75.3657
93. Triggaiani L, Colosini A, Buglione M, Pasinetti N, Orizio F, Bardoscia L, et al. Exploring the role of enzalutamide in combination with radiation therapy: an in vitro study. Anticancer Res. (2018) 38:3487–92. doi: 10.21873/anticanres.12619
94. Smith M, Parker C, Saad F, Miller K, Tombal B, Ng QS, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2019) 20:408–19. doi: 10.1016/S1470-2045(18)30860-X
95. Small EJ, Lance RS, Gardner TA, Karsh LI, Fong L, McCoy C, et al. A Randomized Phase II Trial of Sipuleucel-T with concurrent versus sequential abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer. Clin Cancer Res. (2015) 21:3862–9. doi: 10.1158/1078-0432.CCR-15-0079
96. Kwilas AR, Ardiani A, Dirmeier U, Wottawah C, Schlom J, Hodge JW. A poxviral-based cancer vaccine the transcription factor twist inhibits primary tumor growth and metastases in a model of metastatic breast cancer and improves survival in a spontaneous prostate cancer model. Oncotarget. (2015) 6:28194–210. doi: 10.18632/oncotarget.4442
97. Ardiani A, Farsaci B, Rogers CJ, Protter A, Guo Z, King TH, et al. Combination therapy with a second-generation androgen receptor antagonist and a metastasis vaccine improves survival in a spontaneous prostate cancer model. Clin Cancer Res. (2013) 19:6205–18. doi: 10.1158/1078-0432.CCR-13-1026
98. Higa J, Wilenius K, Weidhaas JB, Larsen C, Lam RY, Turner J, et al. Pembrolizumab for recurrent or advanced prostate cancer. J Clin Oncol. (2018) 36:250. doi: 10.1200/JCO.2018.36.6_suppl.250
99. Graff JN, Alumkal JJ, Drake CG, Thomas GV, Redmond WL, Farhad M, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget. (2016) 7:52810–7. doi: 10.18632/oncotarget.10547
100. Gamat M, McNeel DG. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr Relat Cancer. (2017) 24:T297–310. doi: 10.1530/ERC-17-0145
101. Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2018) 19:975–86. doi: 10.1016/S1470-2045(18)30365-6
102. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
103. Danquah M, Duke CB, Patil R, Miller DD, Mahato RI. Combination therapy of antiandrogen and XIAP inhibitor for treating advanced prostate cancer. Pharm Res. (2012) 29:2079–91. doi: 10.1007/s11095-012-0737-1
104. Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. (2016) 34:3005–13. doi: 10.1200/JCO.2015.65.5597
105. Choudhury AD, Gray KP, Supko JG, Harshman LC, Taplin M-E, Pace AF, et al. A dose finding clinical trial of cabozantinib (XL184) administered in combination with abiraterone acetate in metastatic castration-resistant prostate cancer. Prostate. (2018) 78:1053–62. doi: 10.1002/pros.23662
106. Wang X, Huang Y, Christie A, Bowden M, Lee G-SM, Kantoff PW, et al. Cabozantinib inhibits abiraterone upregulation of IGFIR phosphorylation and enhances its anti–prostate cancer activity. Clin Cancer Res. (2015) 21:5578:87. doi: 10.1158/1078-0432.CCR-15-0824
107. Mortezavi A, Salemi S, Kranzbühler B, Gross O, Sulser T, Simon H-U, et al. Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J Urol. (2019) 37:351–8. doi: 10.1007/s00345-018-2385-5
108. Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. (2016) 7:32210–20. doi: 10.18632/oncotarget.8493
109. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. (2014) 20:3198–210. doi: 10.1158/1078-0432.CCR-13-3296
110. Schweizer MT, Haugk K, McKiernan JS, Gulati R, Cheng HH, Maes JL, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE. (2018) 13:e0198389. doi: 10.1371/journal.pone.0198389
111. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. (2005) 310:644–8. doi: 10.1126/science.1117679
112. Mohamed AA, Xavier CP, Sukumar G, Tan S-H, Ravindranath L, Seraj N, et al. Identification of a small molecule that selectively inhibits ERG-positive cancer cell growth. Cancer Res. (2018) 78:3659–71. doi: 10.1158/0008-5472.CAN-17-2949
113. Wu JB, Yin L, Shi C, Li Q, Duan P, Huang JM, et al. MAOA-Dependent activation of Shh-IL6-RANKL signaling network promotes prostate cancer metastasis by engaging tumor-stromal cell interactions. Cancer Cell. (2017) 31:368–82. doi: 10.1016/j.ccell.2017.02.003
114. Liao CP, Lin TP, Li PC, Geary LA, Chen K, Vaikari VP, et al. Loss of MAOA in epithelia inhibits adenocarcinoma development, cell proliferation and cancer stem cells in prostate. Oncogene. (2018) 37:517–9. doi: 10.1038/s41388-018-0325-x
115. Gaur S, Gross ME, Liao C-P, Qian B, Shih JC. Effect of Monoamine oxidase A (MAOA) inhibitors on androgen-sensitive and castration-resistant prostate cancer cells. Prostate. (2019) 79:667–77. doi: 10.1002/pros.23774
116. Rice MA, Hsu E-C, Aslan M, Ghoochani A, Su A, Stoyanova T. Loss of Notch1 activity inhibits prostate cancer growth and metastasis and sensitizes prostate cancer cells to anti-androgen therapies. Mol Cancer Ther. (2019) 18:1230–42. doi: 10.1158/1535-7163.MCT-18-0804
117. Farah E, Li C, Cheng L, Kong Y, Lanman NA, Pascuzzi PE, et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J Biol Chem. (2019). 294:8543–54. doi: 10.1074/jbc.RA118.006983
118. Cui J, Wang Y, Dong B, Qin L, Wang C, Zhou P, et al. Pharmacological inhibition of the Notch pathway enhances the efficacy of androgen deprivation therapy for prostate cancer. Int J Cancer. (2018) 143:645–56. doi: 10.1002/ijc.31346
119. Mohamed AA, Tan S-H, Xavier CP, Katta S, Huang W, Ravindranath L, et al. Synergistic activity with NOTCH inhibition and androgen ablation in ERG-positive prostate cancer cells. Mol Cancer Res. (2017) 15:1308–18. doi: 10.1158/1541-7786.MCR-17-0058
120. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat J-P, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. (2012) 44:685–9. doi: 10.1038/ng.2279
121. Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, et al. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet. (2003) 72:1–12. doi: 10.1086/345310
122. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. (2015) 161:1215–28. doi: 10.1016/j.cell.2015.06.053
123. Vlachostergios PJ, Puca L, Beltran H. Emerging variants of castration-resistant prostate cancer. Curr Oncol Rep. (2017) 19:32. doi: 10.1007/s11912-017-0593-6
124. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell. (2017) 32:474–89.e6. doi: 10.1016/j.ccell.2017.09.003
Keywords: prostate cancer, antiandrogens, CRPC, second-generation antiandrogens, enzalutamide, apalutamide, abiraterone acetate, darolutamide
Citation: Rice MA, Malhotra SV and Stoyanova T (2019) Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 9:801. doi: 10.3389/fonc.2019.00801
Received: 06 June 2019; Accepted: 07 August 2019;
Published: 28 August 2019.
Edited by:
George Kulik, Wake Forest University, United StatesReviewed by:
Hung-Ming Lam, University of Washington, United StatesKouji Izumi, Kanazawa University, Japan
Copyright © 2019 Rice, Malhotra and Stoyanova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tanya Stoyanova, stanya@stanford.edu