 Imogen M. Roth1
Imogen M. Roth1 Agadha C. Wickremesekera1,2
Agadha C. Wickremesekera1,2 Susrutha K. Wickremesekera1,3
Susrutha K. Wickremesekera1,3 Paul F. Davis1
Paul F. Davis1 Swee T. Tan1,4*
Swee T. Tan1,4*- 1Gillies McIndoe Research Institute, Wellington, New Zealand
- 2Department of Neurosurgery, Wellington Regional Hospital, Wellington, New Zealand
- 3Upper Gastrointestinal, Hepatobiliary and Pancreatic Section, Department of General Surgery, Wellington Regional Hospital, Wellington, New Zealand
- 4Wellington Regional Plastic, Maxillofacial and Burns Unit, Hutt Hospital, Wellington, New Zealand
Cancer stem cells (CSCs) are proposed to be the cells that initiate tumorigenesis and maintain tumor development due to their self-renewal and multipotency properties. CSCs have been identified in many cancer types and are thought to be responsible for treatment resistance, metastasis, and recurrence. As such, targeting CSCs specifically should result in durable cancer treatment. One potential option for targeting CSCs is by manipulation of the renin-angiotensin system (RAS) and pathways that converge on the RAS with numerous inexpensive medications currently in common clinical use. In addition to its crucial role in cardiovascular and body fluid homeostasis, the RAS is vital for stem cell maintenance and differentiation and plays a role in tumorigenesis and cancer prevention, suggesting that these roles may converge and result in modulation of CSC function by the RAS. In support of this, components of the RAS have been shown to be expressed in many cancer types and have been more recently localized to the CSCs in some tumors. Given these roles of the RAS in tumor development, clinical trials using RAS inhibitors either singly or in combination with other therapies are underway in different cancer types. This review outlines the roles of the RAS, with respect to CSCs, and suggests that the presence of components of the RAS in CSCs could offer an avenue for therapeutic targeting using RAS modulators. Due to the nature of the RAS and its crosstalk with numerous other signaling pathways, a systems approach using traditional RAS inhibitors in combination with inhibitors of bypass loops of the RAS and other signaling pathways that converge on the RAS may offer a novel therapeutic approach to cancer treatment.
Cancer Stem Cells
As in normal tissue, tumors consist of diverse cell populations. The cellular heterogeneity observed in tumors has led to the suggestion that cancer may be sustained by cancer stem cells (CSCs), which, like normal embryonic stem cells (ESCs), are able to self-renew and undergo differentiation into multiple cell types. This is supported by several observations in cancer biology, including that only some tumor cells can recapitulate a tumor when xenografted into immunodeficient mice, and that tumors grown from tumorigenic cells consist of a mixed population of both tumorigenic and non-tumorigenic cancer cells (1). CSCs are thought to arise from either resident adult stem cells which have acquired oncogenic mutations or from progenitor cells which have an unlimited ability to replicate. CSCs share the properties of differentiation, self-renewal and homeostatic control with normal stem cells (1), express stem cell markers (2), and have subverted self-renewal pathways of normal stem cells (3).
The CSC concept proposes that cancer develops from a small subset of cells which can generate all the heterogeneous cell types seen within the tumor, including generating more CSCs as well as differentiated cancer cells. It has been shown in numerous tumor types that expression of certain markers can define populations of cancer cells which are able to generate a tumor, as well as their ability to respond to or resist cancer therapies, suggesting CSCs are present within these tumors (4). Studies transplanting mouse tumors into compatible wild-type mice have also shown that the cancer cells differ in their tumorigenic capacity, as only a small population of cancer cells are able to form tumors (4).
Given that adult human stem cells themselves are a diverse pool of cells expressing different markers, it is unsurprising that CSCs are also mixed populations of cells and are phenotypically and functionally diverse, and that the same tumor can contain multiple pools of CSCs (5). CSC diversity has also resulted in the emergence of a hierarchy, with a slow-cycling pool of cells giving rise to both a rapidly cycling population and non-proliferative cells, suggesting that targeting the cells with the potential to produce multiple types of tumor cells would be a beneficial approach to cancer treatment (6, 7). Heterogeneity within CSCs extends beyond tumorigenic potential and encompasses genetic and epigenetic changes as well as local environmental determinants and temporal and spatial differences (8). These differences have implications for effective therapies, as some cancer cells have been shown to resist chemotherapy and radiotherapy, and it has been suggested that they could be specifically targeted for differentiation as a therapeutic approach (8). Importantly, there is a level of plasticity within this system, as differentiated non-tumorigenic cancer cells can revert to CSCs (9, 10). This could be advanced by changes in the local environment driven by cues including hypoxia and inflammatory mediators to induce epithelial-mesenchymal transition and de-differentiation to increase the “stemness” of the tumor (5). This heterogeneity conferred by plasticity can result in treatment resistance (11).
CSCs have been shown to be capable of surviving radiotherapy and chemotherapy, which have no effect on the ability of the CSCs to regrow tumors (12). This resistance to radiotherapy is thought to occur by several mechanisms, including activation of DNA repair mechanisms, through activation of Wnt/β-catenin signaling, reactive oxygen species generation, and activation of other pro-survival signaling pathways (12). Resistance to chemotherapeutic agents is thought to occur via the use of drug efflux pumps and the expression of metabolic mediators (12). In addition, the quiescent, slow-cycling nature of CSCs is also likely to confer resistance to conventional treatments such as chemotherapy and radiotherapy which target rapidly dividing cells. The ability of CSCs to resist conventional cancer treatments has been well documented in breast cancer. Irradiation of mouse mammary primary epithelial cells enriches for progenitor cells (13), and breast cancer cells from patients following neoadjuvant chemotherapy are enriched for self-renewing cells (14). Furthermore, the number of CSCs and their ability to form mammospheres in culture is increased following chemotherapy of breast cancer patients (15) and Trastuzumab treatment of a breast cancer cell line (16).
Given their ability to generate a diverse cell population within a tumor and their ability to resist conventional cancer treatments, CSCs are proposed to be the cause of loco-regional recurrence and distant metastasis, and consequently treatment failure. This has implications for cancer therapy and suggests that the CSCs should be targeted for effective and durable cancer treatment. Consequently, several treatments targeting CSCs are currently in use in the clinic, with the main strategies being inhibiting key signaling pathways or directly targeting CSCs (17). These therapies include targeting CSC markers, such as CD44 and CD133, which have shown promise in a pre-clinical setting and therapies targeting these markers are in current clinical trials for acute myeloid leukemia and recurrent solid tumors, including liver, brain, pancreatic, breast, and colorectal cancers (18). In addition, a vaccination-based strategy against CSCs is in clinical trials for glioblastoma and other brain tumors (18), demonstrating the diverse approaches taken to target these cells. Given that CSCs express a unique set of markers, another approach toward identifying and eliminating these cells is to characterize other common features of CSCs and exploit these features for therapeutic targeting using drugs in common use, such as via modulation of signaling pathways such as the renin-angiotensin system (RAS).
The Renin-Angiotensin System
Physiological Control of Blood Pressure and Fluid Balance
The RAS is an endocrine system crucial for the maintenance of homeostasis, as it regulates blood pressure and fluid balance via a signaling network (Figure 1). Physiologically, the RAS is activated in response to either reduced blood volume or blood pressure, and acts to restore homeostasis through the release of renin from the kidneys. Pro-renin is converted to active renin by binding to the pro-renin receptor (PRR). Renin then cleaves angiotensinogen, which is normally synthesized and released by the liver, giving rise to angiotensin I (ATI). ATI is then converted to angiotensin II (ATII) by angiotensin converting enzyme (ACE). Aminopeptidase A converts ATII to angiotensin III, and together they act on ATII receptors 1 and 2 (ATIIR1 and ATIIR2). These receptors have divergent actions, with ATIIR1 driving vasoconstriction and inhibiting renin to restore blood pressure, and ATIIR2 acting to promote vasodilation. Angiotensin 1-7 (Ang1-7) is the cleavage product of ATII and affects cardiovascular functions by binding to the G-protein coupled receptor MAS. However, there is considerable redundancy in the pathway with bypass loops involving proteases such as cathepsins B, D, and G, and the convergence of other signaling pathways on the RAS itself, including inflammatory pathways and Wnt/β-catenin signaling (Figure 1). Given the importance of the RAS for maintaining blood pressure, numerous modulators that inhibit the RAS at different points in the pathway have been developed (Figure 2). These groups of RAS inhibitors are commonly used in the clinic for the treatment of hypertension and include β-blockers, ACE inhibitors (ACEI), and ATIIR1 blockers (ARBs) as well as newer agents targeting other points in the pathway (e.g., renin inhibitors, chymase inhibitors, ATIIR2 inhibitors), inhibitors targeting bypass loops in the RAS pathway (e.g., cathepsin inhibitors), and inhibitors used in other canonical signaling pathways that converge on the RAS (e.g., Wnt/β-catenin inhibitors, metformin, and non-steroidal anti-inflammatory drugs) (Figure 2).
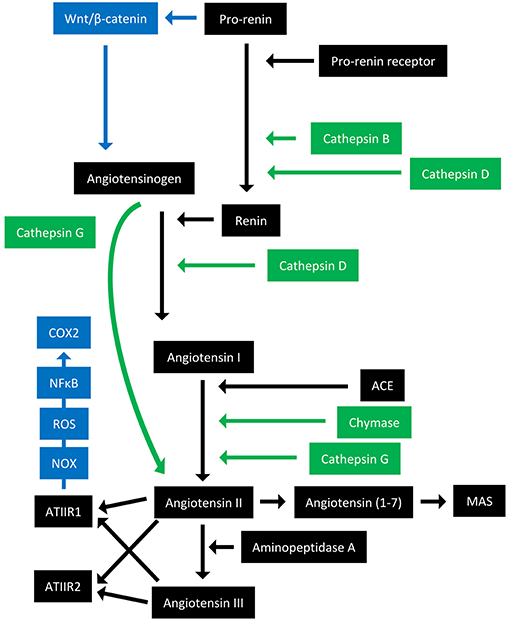
Figure 1. Overview of the renin-angiotensin system with its bypass loops and convergent signaling pathways. The renin-angiotensin system (black) regulates blood pressure, stem cell differentiation, and tumor development. Bypass loops of the RAS involving enzymes such as chymase and cathepsins B, D, and G (green) provide redundancy, while convergent inflammatory and developmental signaling pathways (blue) have multiple roles and effects. Angiotensinogen (AGN) is physiologically synthesized and released by the liver and is cleaved by renin to form angiotensin I (ATI). Renin is formed following binding of pro-renin to the pro-renin receptor. ATI is converted to angiotensin II (ATII) by angiotensin converting enzyme (ACE). ATII interacts with the G-protein coupled receptors ATII receptor 1 (ATIIR1) and ATII receptor 2 (ATIIR2) to restore homeostasis, via vasoconstriction and vasodilation, respectively. ATII can also give rise to angiotensin III via the action of aminopeptidase A, and Angiotensin 1–7 which binds and activates the G-protein coupled receptor MAS. Cathepsins B and D are also renin-activating enzymes that convert pro-renin to renin. Cathepsin D converts AGN to ATI, and cathepsin G converts ATI to ATII or AGN directly to ATII. Chymase converts ATI to ATII. Pro-renin also induces Wnt/β-catenin signaling in a feedback loop. ATIIR1 can also result in inflammatory signaling via the NOX-ROS-NFκB-COX2 signaling axis. ROS, reactive oxygen species.
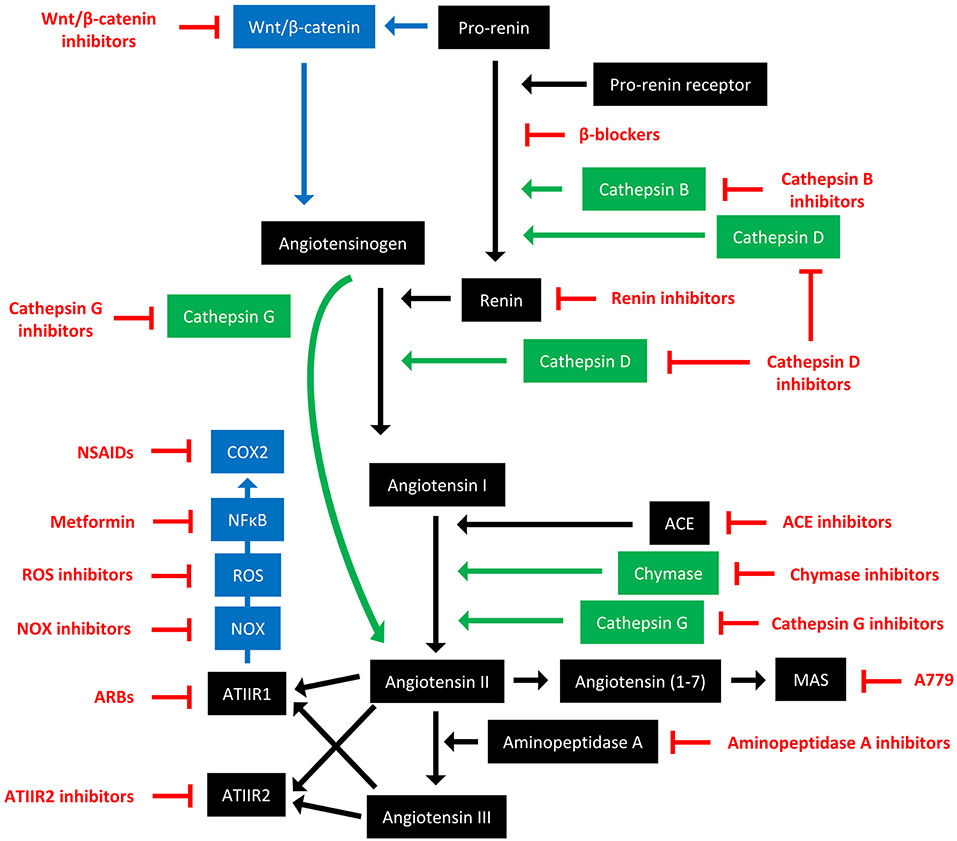
Figure 2. The renin-angiotensin system and its bypass loops and converging signaling pathways can be targeted at different points. The renin-angiotensin system (black) regulates blood pressure, stem cell differentiation, and tumor development. Bypass loops in the system involving cathepsins and chymase (green) provide redundancy, while convergent inflammatory and development signaling pathways (blue) have multiple roles and effects. Multiple points of the pathway can be targeted by specific inhibitors (red). ACE, angiotensin converting enzyme; ARBs, ATIIR1 blockers; ROS, reactive oxygen species; NSAIDS, non-steroidal anti-inflammatory drugs.
Stem Cell Differentiation
Alongside its crucial role in fluid volume regulation, the RAS is also important for stem cell maintenance and differentiation in several cell types. ATII expression drives the differentiation of mesenchymal stem cells into adipocytes (19), while other components of the RAS drive differentiation into insulin producing cells (20). ACE is required for hemangioblast expansion, and modulation of ATIIR1 or ATIIR2 signaling can direct the fate of the blasts toward either an endothelial or hematopoietic lineage (21). The RAS also plays a role in hematopoiesis (22, 23), vasculogenesis (24), erythropoiesis (25, 26), and myeloid differentiation (27).
Importantly, the RAS not only acts to promote stem cell differentiation in diverse cell populations, but also appears to act in a feedback loop with Wnt/β-catenin signaling, where pro-renin receptor (PRR) can induce Wnt/β-catenin (28), and components of the RAS themselves are targets of Wnt/β-catenin (29). Wnt/β-catenin signaling is crucial for embryonic development and induces differentiation of pluripotent stem cells into progenitor cells (30). Given that Wnt signaling is also involved in cancer development (31), and downstream Wnt targets include the CSC markers CD44 and c-Myc (32), it may be that CSCs require activation of Wnt signaling (33). This suggests that RAS modulators could be employed in these cells to indirectly inhibit Wnt signaling and its effects.
Having identified these roles in normal stem cell maintenance and differentiation and feedback loops with a canonical developmental signaling pathway, it may be that the expression of the RAS also plays a role in the regulation or function of CSCs.
Retrospective Studies and Clinical Trials Indicate Potential Benefit of RAS Modulators in Reducing Cancer Risk
The widespread use of RAS modulators as anti-hypertensives and their potential effect on cancer risk have been extensively documented. A seminal study has shown that the use of ACEI and ARBs is associated with a reduced risk of developing some cancer types, particularly cancers affecting women (34). Many other retrospective population studies have reported differing effects on cancer risk depending on the cancer type, cohort characteristics, and the RAS inhibitor used. As a result, several meta-analyses have been undertaken (35), again with differing results, which could be due to the nature of the original studies included and inherent publication bias. Aside from the reported effects on cancer risk, many retrospective population studies have also assessed the effect of RAS inhibitors on cancer death. Again, these results have been mixed, though meta-analyses have indicated that β-blocker use is not associated with survival in breast cancer patients (36), and a meta-analysis looking at ACEI use in all cancers showed no effect on cancer survival (37). A more recent meta-analysis looking at the use of different RAS inhibitors in all cancers showed that RAS inhibitor use extended overall, progression-free and disease-free survival (38). This is mainly due to ARBs and not ACEI use, with some site-specific effects. These studies need to be interpreted critically and with caution as they do not prove causality and the effect on cancer risk and mortality could be due to other factors. It may also be that a defined patient group will derive benefit from these treatments and that a more holistic approach of targeting the RAS in cancer is required to achieve a sustained treatment for patients. Due to the nature of the RAS, with its inherent bypass loops conferring redundancies, and the presence of many other pathways that converge on the RAS, it is likely that a multi-faceted approach to target the RAS will be required for effective cancer treatment.
Despite these disparate observations in retrospective population studies, the data around the involvement of the RAS in tumor models is clear, leading to many clinical trials using RAS inhibitors and the development of new targeted agents (39–41). Several of these studies have trialed ARBs in cancer patients, with Losartan being shown to enhance the efficacy of chemotherapy and improve overall survival in ovarian cancer patients (42). Another ARB, Candesartan, has been shown to decrease prostate specific antigen levels in hormone-refractory prostate cancer patients (43), and is tolerated in advanced pancreatic cancer (44, 45). The ACEI Captopril is tolerated in patients with advanced cancer (46), and has been shown to reduce biochemical recurrence in prostate cancer patients (47), while Perindopril reduced the risk of recurrence of hepatocellular carcinoma as a combination therapy with other non-traditional treatments (48, 49). Several trials have targeted the Ang1-7/MAS axis in breast cancer before or after chemotherapy (50), and in metastatic sarcoma, where it is well tolerated (51), and a number of advanced solid tumors where it provides benefit for some patients (52).
β-blockers work by blocking β-adrenergic receptors to prevent neurotransmitter binding. This prevents renin secretion and its actions and subsequently results in lowered blood pressure (Figure 2). The non-selective β-blocker Propranolol has been shown in several case reports to be efficacious in treating angiosarcoma (53), and in combination with chemotherapy treatment induced responses in seven patients with advanced angiosarcoma (54). Another study showed that addition of Propranolol or another non-selective β-blocker Carvedilol to treatment regimens for metastatic angiosarcoma improved progression-free and overall survival (55). Propranolol has also been used in a proof of concept study in multiple myeloma patients receiving hematopoietic cell transplantations (56), and in a prospective cohort study in melanoma patients where its use was associated with reduced recurrence (57).
While these trials have demonstrated promise for targeting the RAS in cancer treatment, the mechanisms by which this is achieved are yet to be elucidated. Current clinical trials and the development of new RAS targets should help to further define which patient groups may benefit from these treatments.
In vitro and in vivo Cancer Models Rationalize the RAS as a Therapeutic Target
Given the potential effects on reducing cancer risk observed in retrospective population studies, expression of components of the RAS have been assessed in many different tumor types to clarify the potential role of the RAS in tumorigenesis (Table 1).
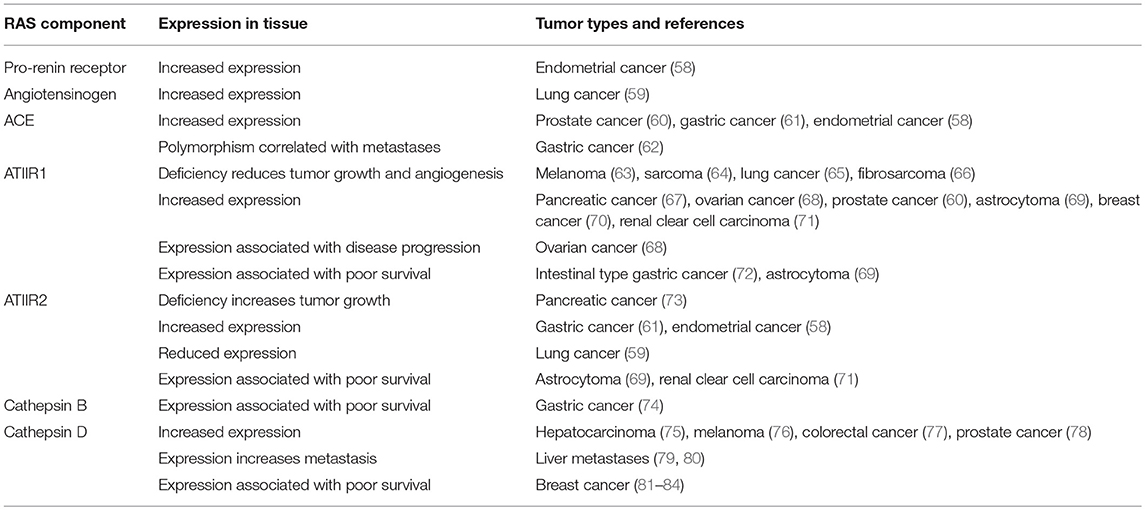
Table 1. Components of the RAS are expressed in tumors.
These studies have helped define the role of the RAS in tumorigenesis, and collectively show that components of the RAS are expressed in many different cancer types (39, 85). The effects on tumor growth, angiogenesis, metastasis and survival indicate that the RAS plays a role in the hallmarks of cancer (39, 86, 87). It is also thought to contribute to an immunosuppressive microenvironment in tumors and reduce infiltration of tumor-associated macrophages (88). The increased expression of components of the RAS in different cancer types may contribute to tumorigenesis and the poor clinical outcome seen in some cancer types. This suggests that regulation of the RAS may be a general mechanism for cancer prevention and warrants further investigation to understand the precise underlying mechanisms.
Given that the RAS is over-expressed in many cancer types and the use of RAS modulators may affect cancer risk and cancer survival, numerous studies have assessed the effect of RAS inhibitors in vitro and on tumor models in vivo. These have focused on β-blockers (Table 2), ACEI (Table 3), and ARBs (Table 4) to assess the role of the RAS in tumor development.
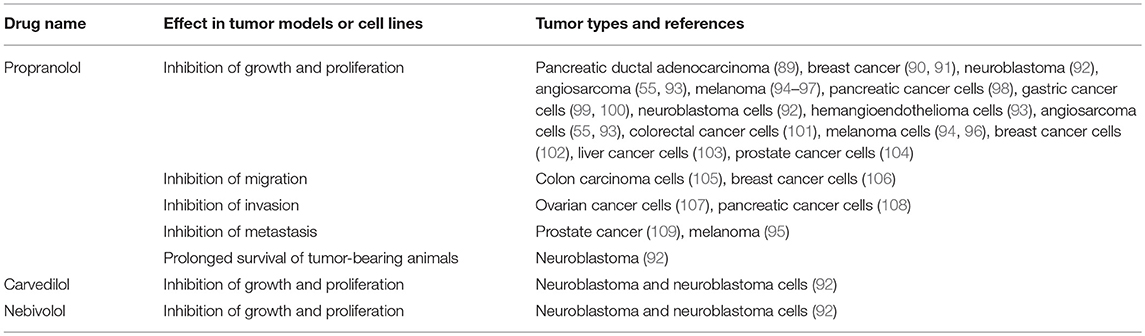
Table 2. β-blockers inhibit tumorigenesis in cell and animal models.
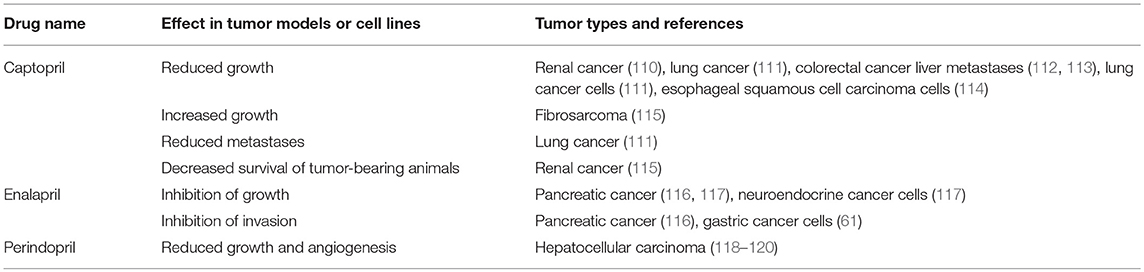
Table 3. ACE inhibitors inhibit tumorigenesis in cell and animal models.
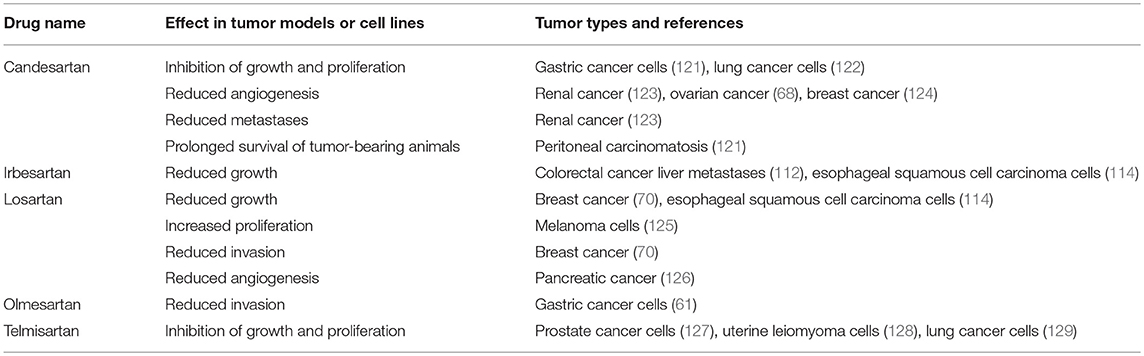
Table 4. ARBs inhibit tumorigenesis in cell and animal models.
Studies investigating β-blockers in cancer (Table 2) have largely used the β-blocker Propranolol and have shown that across a wide range of cancer types, Propranolol inhibits the growth of tumors and tumor cells. This suggests that Propranolol could be repurposed for cancer treatment (130, 131), as has been the case for the benign vascular tumor infantile hemangioma for which it is an effective treatment (132–134).
Given the effects of β-blockers on cancer and cancer cell growth, other studies have investigated the impact of other classes of drugs that modulate the RAS on neoplastic processes. One of these classes is ACEIs, which block the action of ACE and hence downstream production of ATII (Figure 2). Studies looking at ACEIs (Table 3) are extensive and demonstrate that this class of drugs (including Captopril, Enalapril, and Perindopril) appear to prevent tumor growth and invasion in many different tumor types and models.
Another broad class of drugs that modulate the RAS are ARBs, which block ATIIR1 (Figure 2). Studies using ARBs to assess cancer development in cell and animal models (Table 4) have also shown that different drugs within this class (Candesartan, Irbesartan, Losartan, Olmesartan, and Telmisartan) inhibit tumor development across several tumor types.
These studies underscore the complex nature of the RAS and suggests that different RAS modulators may have different effects in different tumor types. Taken together, they suggest that anti-hypertensive drugs which target the RAS have shown promise for repurposing in the cancer setting. Across several classes of drugs (β-blockers, ACEIs, and ARBs) in both in vitro and in vivo models, they have been shown to reduce tumor cell growth, migration, invasion, and metastasis in numerous cancer types. These processes comprise many of the characteristics of CSCs and of the hallmarks of cancer (135), and are consistent with the expression of some components of the RAS in high-grade disease and the associated poor survival (Table 1). This suggests that there is merit in repurposing RAS inhibitors for cancer treatment. Many clinical trials using this approach are currently underway, despite limited functional work and mechanistic understanding about how this approach might work in cancer patients. With the development of new agents targeting specific parts of the pathway, including the bypass loops, and the refinement of existing drugs, new opportunities are emerging for modulating the RAS pathway, either in combination with current therapies or by targeting the entire RAS and its bypass loops, and pathways converging on the RAS.
Cancer Stem Cells Express Components of the RAS
Given the well characterized role of the RAS in both stem cell maintenance and tumorigenesis, it is possible that these functions are directed by RAS signaling in CSCs. In order to demonstrate this, it is important to first establish that CSCs express both CSC markers and components of the RAS. This has been shown to be the case in numerous cancer types, including glioblastoma (136–138), metastases to the liver from colon adenocarcinoma (139, 140), head and neck cutaneous squamous cell carcinoma (141), and oral cavity squamous cell carcinoma affecting the buccal mucosa (142, 143), oral tongue (144–146), and lip (147, 148). In addition, components of the RAS have also been demonstrated on the tumor stem cells of benign tumors such as meningioma (149, 150), infantile hemangioma (151, 152), and pyogenic granuloma (153). Importantly, the expression of cathepsins B, D, and G in some of these cancer types (74, 138, 140, 146, 150) suggests the presence of bypass loops of the RAS which could circumvent the action of traditional RAS inhibitors and offer a potential explanation for the differing findings of cancer risk and cancer survival with long-term use of traditional RAS inhibitors. Given the presence of components of the RAS in CSCs in these cancers, it is possible that the expression of these components is controlling the differentiation and function of the CSCs within these tumors.
Despite the indirect evidence from retrospective population studies and more substantial direct evidence from in vitro studies and in vivo tumor models, very little is known about the mechanism by which RAS modulators influence tumor development. Although expression of components of the RAS has been demonstrated in CSCs, their function and how they might respond to RAS modulators has yet to be characterized. However, the fact that many clinical trials involving targeting of the RAS in cancer have taken place and are currently underway underscores the role of RAS in tumorigenesis and the need for further investigations into this system. Importantly, the findings in tumor model systems are seen consistently across a broad range of tumor types, suggesting its common role in cancer biology which may be affected through CSCs and their functions.
The expression of both components of the RAS and CSC markers in several cancer types may indicate that the CSCs may be a novel therapeutic target through modulation of the RAS. It is possible that a multi-faceted strategy simultaneously targeting multiple critical points of the RAS and related signaling pathways may result in durable cancer treatments by altering CSC function. Indeed, Phase II trials in metastatic renal cell carcinoma using either Perindopril or Candesartan in combination with other agents, including a cyclooxygenase-2 inhibitor have shown potential for stabilizing the disease and reducing recurrence (154). Propranolol treatment in combination with a cyclooxygenase-2 inhibitor is well tolerated in breast cancer patients and transcriptional profiling showed the combination reduced markers of invasion and inflammation (155). Furthermore, targeting other pathways which converge on the RAS may also prove worthwhile, as Metformin selectively kills CSCs in mouse breast cancer models (156), and targeting Wnt signaling is known strategy for CSC elimination (157, 158).
Conclusion
The involvement of the RAS in both tumor development and stem cell maintenance suggests that these roles may converge on CSC maintenance and function. Given the ability of CSCs to promote cell migration, invasion and metastasis (17), and the reduction of these processes by RAS inhibitors in vitro and in vivo, it may be that the success of RAS inhibitors in reducing cancer risk and improving cancer survival is due to their effects on CSCs. In support of this, components of the RAS and enzymes that constitute bypass loops of the RAS have been shown to be expressed in CSCs of several different cancer types. This offers an avenue for targeted therapies using RAS inhibitors, modulators of the bypass loops, and agents targeting other signaling pathways that converge on the RAS. Importantly, RAS inhibitors are commonly available, well tolerated and inexpensive and have been shown to be effective in controlling tumor growth in several settings. However, many of these studies have relied on immortalized cancer cell lines and xenograft tumor models, and in order to better understand the mechanisms of these drugs and the discrepancies observed in their effects clinically, models closer to the patient need to be employed. In addition, the nature of the RAS and its crosstalk with other pathways means a system-wide approach simultaneously targeting multiple key steps of the RAS is needed to achieve effective cancer control.
Author Contributions
IR drafted the manuscript. AW, SW, PD, and ST commented on the manuscript. All authors approved the manuscript.
Conflict of Interest Statement
PD and ST are inventors of the of the patent Cancer Stem Cells (US15/503025) and the PCT patent Cancer Therapeutic (PCT/NZ2018/050006), and the provisional patent application Novel Pharmaceutical Compositions for Cancer Therapy (US/62/711709).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. (2007) 58:267–84. doi: 10.1146/annurev.med.58.062105.204854
2. Kim WT, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Rep. (2017) 50:285–98. doi: 10.5483/BMBRep.2017.50.6.039
3. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414:105–11. doi: 10.1038/35102167
4. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. (2009) 138:822–9. doi: 10.1016/j.cell.2009.08.017
5. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. (2012) 22:457–72. doi: 10.1038/cr.2012.13
6. Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg. (2016) 3:21. doi: 10.3389/fsurg.2016.00021
7. Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature. (2017) 549:227–32. doi: 10.1038/nature23666
8. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. (2012) 21:283–96. doi: 10.1016/j.ccr.2012.03.003
9. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. (2011) 108:7950–5. doi: 10.1073/pnas.1102454108
10. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. (2011) 146:633–44. doi: 10.1016/j.cell.2011.07.026
11. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
12. Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. (2008) 26:2839–45. doi: 10.1200/JCO.2007.15.1829
13. Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. (2007) 104:618–23. doi: 10.1073/pnas.0606599104
14. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. (2007) 131:1109–23. doi: 10.1016/j.cell.2007.10.054
15. Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. (2008) 100:672–9. doi: 10.1093/jnci/djn123
16. Reim F, Dombrowski Y, Ritter C, Buttmann M, Hausler S, Ossadnik M, et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res. (2009) 69:8058–66. doi: 10.1158/0008-5472.CAN-09-0834
17. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
18. Annett S, Robson T. Targeting cancer stem cells in the clinic: current status and perspectives. Pharmacol Ther. (2018) 187:13–30. doi: 10.1016/j.pharmthera.2018.02.001
19. Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension. (2006) 48:1095–102. doi: 10.1161/01.HYP.0000248211.82232.a7
20. Sadik NA, Metwally NS, Shaker OG, Soliman MS, Mohamed AA, Abdelmoaty MM. Local renin-angiotensin system regulates the differentiation of mesenchymal stem cells into insulin-producing cells through angiotensin type 2 receptor. Biochimie. (2017) 137:132–8. doi: 10.1016/j.biochi.2017.03.002
21. Zambidis ET, Park TS, Yu W, Tam A, Levine M, Yuan X, et al. Expression of angiotensin-converting enzyme. (CD143) identifies and regulates primitive hemangioblasts derived from human pluripotent stem cells. Blood. (2008) 112:3601–14. doi: 10.1182/blood-2008-03-144766
22. Hubert C, Savary K, Gasc JM, Corvol P. The hematopoietic system: a new niche for the renin-angiotensin system. Nat Clin Pract Cardiovasc Med. (2006) 3:80–5. doi: 10.1038/ncpcardio0449
23. Park TS, Zambidis ET. A role for the renin-angiotensin system in hematopoiesis. Haematologica. (2009) 94:745–7. doi: 10.3324/haematol.2009.006965
24. Khakoo AY, Sidman RL, Pasqualini R, Arap W. Does the renin-angiotensin system participate in regulation of human vasculogenesis and angiogenesis? Cancer Res. (2008) 68:9112–5. doi: 10.1158/0008-5472.CAN-08-0851
25. Walter J. Does captopril decrease the effect of human recombinant erythropoietin in haemodialysis patients? Nephrol Dial Transplant. (1993) 8:1428.
26. Vlahakos DV, Balodimos C, Papachristopoulos V, Vassilakos P, Hinari E, Vlachojannis JG. Renin-angiotensin system stimulates erythropoietin secretion in chronic hemodialysis patients. Clin Nephrol. (1995) 43:53–9.
27. Shen XZ, Bernstein KE. The peptide network regulated by angiotensin converting enzyme. (ACE) in hematopoiesis. Cell Cycle. (2011) 10:1363–9. doi: 10.4161/cc.10.9.15444
28. Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. (2010) 327:459–63. doi: 10.1126/science.1179802
29. Zhou L, Li Y, Hao S, Zhou D, Tan RJ, Nie J, et al. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J Am Soc Nephrol. (2015) 26:107–20. doi: 10.1681/ASN.2014010085
30. Nusse R. Wnt signaling and stem cell control. Cell Res. (2008) 18:523–7. doi: 10.1038/cr.2008.47
32. Kim JH, Park SY, Jun Y, Kim JY, Nam JS. Roles of Wnt target genes in the journey of cancer stem cells. Int J Mol Sci. (2017) 18:E1604. doi: 10.3390/ijms18081604
33. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. (2010) 327:1650–3. doi: 10.1126/science.1186624
34. Lever AF, Hole DJ, Gillis CR, McCallum IR, McInnes GT, MacKinnon PL, et al. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet. (1998) 352:179–84. doi: 10.1016/S0140-6736(98)03228-0
35. Wuerzner G, Burnier M, Waeber B. Critical review of cancer risk associated with angiotensin receptor blocker therapy. Vasc Health Risk Manag. (2011) 7:741–7. doi: 10.2147/VHRM.S13552
36. Cardwell CR, Pottegard A, Vaes E, Garmo H, Murray LJ, Brown C, et al. Propranolol and survival from breast cancer: a pooled analysis of European breast cancer cohorts. Breast Cancer Res. (2016) 18:119. doi: 10.1186/s13058-016-0782-5
37. Sipahi I, Chou J, Mishra P, Debanne SM, Simon DI, Fang JC. Meta-analysis of randomized controlled trials on effect of angiotensin-converting enzyme inhibitors on cancer risk. Am J Cardiol. (2011) 108:294–301. doi: 10.1016/j.amjcard.2011.03.038
38. Sun H, Li T, Zhuang R, Cai W, Zheng Y. Do renin-angiotensin system inhibitors influence the recurrence, metastasis, and survival in cancer patients?: Evidence from a meta-analysis including 55 studies. Medicine. (2017) 96:e6394. doi: 10.1097/MD.0000000000006394
39. George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. (2010) 10:745–59. doi: 10.1038/nrc2945
40. Unger T, Dahlof B. Compound 21, the first orally active, selective agonist of the angiotensin type 2 receptor. (AT2): implications for AT2 receptor research and therapeutic potential. J Renin Angiotensin Aldosterone Syst. (2010) 11:75–7. doi: 10.1177/1470320309347792
41. Bader M, Santos RA, Unger T, Steckelings UM. New therapeutic pathways in the RAS. J Renin Angiotensin Aldosterone Syst. (2012) 13:505–8. doi: 10.1177/1470320312466519
42. Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci USA. (2019) 116:2210–9. doi: 10.1073/pnas.1818357116
43. Uemura H, Hasumi H, Kawahara T, Sugiura S, Miyoshi Y, Nakaigawa N, et al. Pilot study of angiotensin II receptor blocker in advanced hormone-refractory prostate cancer. Int J Clin Oncol. (2005) 10:405–10. doi: 10.1007/s10147-005-0520-y
44. Nakai Y, Isayama H, Ijichi H, Sasaki T, Kogure H, Yagioka H, et al. Phase I trial of gemcitabine and candesartan combination therapy in normotensive patients with advanced pancreatic cancer: GECA1. Cancer Sci. (2012) 103:1489–92. doi: 10.1111/j.1349-7006.2012.02311.x
45. Nakai Y, Isayama H, Ijichi H, Sasaki T, Takahara N, Ito Y, et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Invest New Drugs. (2013) 31:1294–9. doi: 10.1007/s10637-013-9972-5
46. Jones PH, Christodoulos K, Dobbs N, Thavasu P, Balkwill F, Blann AD, et al. Combination antiangiogenesis therapy with marimastat, captopril and fragmin in patients with advanced cancer. Br J Cancer. (2004) 91:30–6. doi: 10.1038/sj.bjc.6601897
47. Ronquist G, Frithz G, Wang YH, Lindeborg T. Captopril may reduce biochemical. (prostate-specific antigen) failure following radical prostatectomy for clinically localized prostate cancer. Scand J Urol Nephrol. (2009) 43:32–6. doi: 10.1080/00365590802468875
48. Yoshiji H, Noguchi R, Toyohara M, Ikenaka Y, Kitade M, Kaji K, et al. Combination of vitamin K2 and angiotensin-converting enzyme inhibitor ameliorates cumulative recurrence of hepatocellular carcinoma. J Hepatol. (2009) 51:315–21. doi: 10.1016/j.jhep.2009.04.011
49. Yoshiji H, Noguchi R, Ikenaka Y, Kaji K, Aihara Y, Yamazaki M, et al. Combination of branched-chain amino acids and angiotensin-converting enzyme inhibitor suppresses the cumulative recurrence of hepatocellular carcinoma: a randomized control trial. Oncol Rep. (2011) 26:1547–53. doi: 10.3892/or.2011.1433
50. Rodgers KE, Oliver J, diZerega GS. Phase I/II dose escalation study of angiotensin 1-7 [A(1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother Pharmacol. (2006) 57:559–68. doi: 10.1007/s00280-005-0078-4
51. Savage PD, Lovato J, Brosnihan KB, Miller AA, Petty WJ. Phase II trial of angiotensin-(1-7) for the treatment of patients with metastatic Sarcoma. Sarcoma. (2016) 2016:4592768. doi: 10.1155/2016/4592768
52. Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1-7), an endogenous antiangiogenic hormone. Clin Cancer Res. (2009) 15:7398–404. doi: 10.1158/1078-0432.CCR-09-1957
53. Wagner MJ, Cranmer LD, Loggers ET, Pollack SM. Propranolol for the treatment of vascular sarcomas. J Exp Pharmacol. (2018) 10:51–8. doi: 10.2147/JEP.S146211
54. Pasquier E, Andre N, Street J, Chougule A, Rekhi B, Ghosh J, et al. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. (2016) 6:87–95. doi: 10.1016/j.ebiom.2016.02.026
55. Amaya CN, Perkins M, Belmont A, Herrera C, Nasrazadani A, Vargas A, et al. Non-selective beta blockers inhibit angiosarcoma cell viability and increase progression free- and overall-survival in patients diagnosed with metastatic angiosarcoma. Oncoscience. (2018) 5:109–19. doi: 10.18632/oncoscience.413
56. Knight JM, Kerswill SA, Hari P, Cole SW, Logan BR, D'Souza A, et al. Repurposing existing medications as cancer therapy: design and feasibility of a randomized pilot investigating propranolol administration in patients receiving hematopoietic cell transplantation. BMC Cancer. (2018) 18:593. doi: 10.1186/s12885-018-4509-0
57. De Giorgi V, Grazzini M, Benemei S, Marchionni N, Botteri E, Pennacchioli E, et al. Propranolol for off-label treatment of patients with melanoma: results from a Cohort study. JAMA Oncol. (2018) 4:e172908. doi: 10.1001/jamaoncol.2017.2908
58. Delforce SJ, Lumbers ER, Corbisier de Meaultsart C, Wang Y, Proietto A, Otton G, et al. Expression of renin-angiotensin system. (RAS) components in endometrial cancer. Endocr Connect. (2017) 6:9–19. doi: 10.1530/EC-16-0082
59. Goldstein B, Trivedi M, Speth RC. Alterations in gene expression of components of the renin-angiotensin system and its related enzymes in lung cancer. Lung Cancer Int. (2017) 2017:6914976. doi: 10.1155/2017/6914976
60. Uemura H, Hasumi H, Ishiguro H, Teranishi J, Miyoshi Y, Kubota Y. Renin-angiotensin system is an important factor in hormone refractory prostate cancer. Prostate. (2006) 66:822–30. doi: 10.1002/pros.20407
61. Carl-McGrath S, Ebert MP, Lendeckel U, Rocken C. Expression of the local angiotensin II system in gastric cancer may facilitate lymphatic invasion and nodal spread. Cancer Biol Ther. (2007) 6:1218–26. doi: 10.4161/cbt.6.8.4412
62. Rocken C, Lendeckel U, Dierkes J, Westphal S, Carl-McGrath S, Peters B, et al. The number of lymph node metastases in gastric cancer correlates with the angiotensin I-converting enzyme gene insertion/deletion polymorphism. Clin Cancer Res. (2005) 11:2526–30. doi: 10.1158/1078-0432.CCR-04-1922
63. Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest. (2003) 112:67–75. doi: 10.1172/JCI200316645
64. Fujita M, Hayashi I, Yamashina S, Fukamizu A, Itoman M, Majima M. Angiotensin type 1a receptor signaling-dependent induction of vascular endothelial growth factor in stroma is relevant to tumor-associated angiogenesis and tumor growth. Carcinogenesis. (2005) 26:271–9. doi: 10.1093/carcin/bgh324
65. Imai N, Hashimoto T, Kihara M, Yoshida S, Kawana I, Yazawa T, et al. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab Invest. (2007) 87:189–98. doi: 10.1038/labinvest.3700504
66. Clere N, Corre I, Faure S, Guihot AL, Vessieres E, Chalopin M, et al. Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int J Cancer. (2010) 127:2279–91. doi: 10.1002/ijc.25234
67. Fujimoto Y, Sasaki T, Tsuchida A, Chayama K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett. (2001) 495:197–200. doi: 10.1016/S0014-5793(01)02377-8
68. Suganuma T, Ino K, Shibata K, Kajiyama H, Nagasaka T, Mizutani S, et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res. (2005) 11:2686–94. doi: 10.1158/1078-0432.CCR-04-1946
69. Arrieta O, Pineda-Olvera B, Guevara-Salazar P, Hernandez-Pedro N, Morales-Espinosa D, Ceron-Lizarraga TL, et al. Expression of AT1 and AT2 angiotensin receptors in astrocytomas is associated with poor prognosis. Br J Cancer. (2008) 99:160–6. doi: 10.1038/sj.bjc.6604431
70. Rhodes DR, Ateeq B, Cao Q, Tomlins SA, Mehra R, Laxman B, et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc Natl Acad Sci USA. (2009) 106:10284–9. doi: 10.1073/pnas.0900351106
71. Dolley-Hitze T, Jouan F, Martin B, Mottier S, Edeline J, Moranne O, et al. Angiotensin-2 receptors. (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br J Cancer. (2010) 103:1698–705. doi: 10.1038/sj.bjc.6605866
72. Rocken C, Rohl FW, Diebler E, Lendeckel U, Pross M, Carl-McGrath S, et al. The angiotensin II/angiotensin II receptor system correlates with nodal spread in intestinal type gastric cancer. Cancer Epidemiol Biomarkers Prev. (2007) 16:1206–12. doi: 10.1158/1055-9965.EPI-05-0934
73. Doi C, Egashira N, Kawabata A, Maurya DK, Ohta N, Uppalapati D, et al. Angiotensin II type 2 receptor signaling significantly attenuates growth of murine pancreatic carcinoma grafts in syngeneic mice. BMC Cancer. (2010) 10:67. doi: 10.1186/1471-2407-10-67
74. Ebert MP, Kruger S, Fogeron ML, Lamer S, Chen J, Pross M, et al. Overexpression of cathepsin B in gastric cancer identified by proteome analysis. Proteomics. (2005) 5:1693–704. doi: 10.1002/pmic.200401030
75. Brouillet JP, Hanslick B, Maudelonde T, Pivat MT, Grenier J, Blanc F, et al. Increased plasma cathepsin D concentration in hepatic carcinoma and cirrhosis but not in breast cancer. Clin Biochem. (1991) 24:491–6. doi: 10.1016/S0009-9120(05)80008-6
76. Podhajcer OL, Bover L, Bravo AI, Ledda MF, Kairiyama C, Calb I, et al. Expression of cathepsin D in primary and metastatic human melanoma and dysplastic nevi. J Invest Dermatol. (1995) 104:340–4. doi: 10.1111/1523-1747.ep12665371
77. Tumminello FM, Gebbia N, Pizzolanti G, Russo A, Bazan V, Leto G. Cathepsin D content in colorectal cancer. Correlation with cathepsin D activity and other biological parameters: a preliminary report. Oncology. (1995) 52:237–42. doi: 10.1159/000227464
78. Ross JS, Nazeer T, Figge HL, Fisher HA, Rifkin MD. Quantitative immunohistochemical determination of cathepsin D levels in prostatic carcinoma biopsies. Correlation with tumor grade, stage, PSA level, and DNA ploidy status. Am J Clin Pathol. (1995) 104:36–41. doi: 10.1093/ajcp/104.1.36
79. Garcia M, Derocq D, Pujol P, Rochefort H. Overexpression of transfected cathepsin D in transformed cells increases their malignant phenotype and metastatic potency. Oncogene. (1990) 5:1809–14.
80. Rochefort H, Capony F, Garcia M. Cathepsin D: a protease involved in breast cancer metastasis. Cancer Metastasis Rev. (1990) 9:321–31. doi: 10.1007/BF00049522
81. Spyratos F, Maudelonde T, Brouillet JP, Brunet M, Defrenne A, Andrieu C, et al. Cathepsin D: an independent prognostic factor for metastasis of breast cancer. Lancet. (1989) 2:1115–8. doi: 10.1016/S0140-6736(89)91487-6
82. Thorpe SM, Rochefort H, Garcia M, Freiss G, Christensen IJ, Khalaf S, et al. Association between high concentrations of Mr 52,000 cathepsin D and poor prognosis in primary human breast cancer. Cancer Res. (1989) 49:6008–14.
83. Tandon AK, Clark GM, Chamness GC, Chirgwin JM, McGuire WL. Cathepsin D and prognosis in breast cancer. N Engl J Med. (1990) 322:297–302. doi: 10.1056/NEJM199002013220504
84. Foekens JA, Look MP, Bolt-de Vries J, Meijer-van Gelder ME, van Putten WL, Klijn JG. Cathepsin-D in primary breast cancer: prognostic evaluation involving 2810 patients. Br J Cancer. (1999) 79:300–7. doi: 10.1038/sj.bjc.6690048
85. Deshayes F, Nahmias C. Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab. (2005) 16:293–9. doi: 10.1016/j.tem.2005.07.009
86. Wegman-Ostrosky T, Soto-Reyes E, Vidal-Millan S, Sanchez-Corona J. The renin-angiotensin system meets the hallmarks of cancer. J Renin Angiotensin Aldosterone Syst. (2015) 16:227–33. doi: 10.1177/1470320313496858
87. Ishikane S, Takahashi-Yanaga F. The role of angiotensin II in cancer metastasis: potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem Pharmacol. (2018) 151:96–103. doi: 10.1016/j.bcp.2018.03.008
88. Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: implications for immunotherapy. Sci Transl Med. (2017) 9:eaan5616. doi: 10.1126/scitranslmed.aan5616
89. Al-Wadei HA, Al-Wadei MH, Schuller HM. Prevention of pancreatic cancer by the beta-blocker propranolol. Anticancer Drugs. (2009) 20:477–82. doi: 10.1097/CAD.0b013e32832bd1e3
90. Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, et al. Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. (2011) 2:797–809. doi: 10.18632/oncotarget.343
91. Lamkin DM, Sung HY, Yang GS, David JM, Ma JC, Cole SW, et al. alpha2-Adrenergic blockade mimics the enhancing effect of chronic stress on breast cancer progression. Psychoneuroendocrinology. (2015) 51:262–70. doi: 10.1016/j.psyneuen.2014.10.004
92. Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, et al. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. (2013) 108:2485–94. doi: 10.1038/bjc.2013.205
93. Stiles JM, Amaya C, Rains S, Diaz D, Pham R, Battiste J, et al. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS ONE. (2013) 8:e60021. doi: 10.1371/journal.pone.0060021
94. Wrobel LJ, Le Gal FA. Inhibition of human melanoma growth by a non-cardioselective β-blocker. J Invest Dermatol. (2015) 135:525–31. doi: 10.1038/jid.2014.373
95. Jean Wrobel L, Bod L, Lengagne R, Kato M, Prevost-Blondel A, Le Gal FA. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget. (2016) 7:77825–37. doi: 10.18632/oncotarget.12833
96. Zhou C, Chen X, Zeng W, Peng C, Huang G, Li X, et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget. (2016) 7:68314–27. doi: 10.18632/oncotarget.11599
97. Maccari S, Buoncervello M, Rampin A, Spada M, Macchia D, Giordani L, et al. Biphasic effects of propranolol on tumour growth in B16F10 melanoma-bearing mice. Br J Pharmacol. (2017) 174:139–49. doi: 10.1111/bph.13662
98. Zhang D, Ma Q, Shen S, Hu H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: the study of beta-adrenoceptor antagonist's anticancer effect in pancreatic cancer cell. Pancreas. (2009) 38:94–100. doi: 10.1097/MPA.0b013e318184f50c
99. Liao X, Che X, Zhao W, Zhang D, Bi T, Wang G. The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor kappaB signaling. Oncol Rep. (2010) 24:1669–76. doi: 10.3892/or_00001032
100. Liao X, Che X, Zhao W, Zhang D, Long H, Chaudhary P, et al. Effects of propranolol in combination with radiation on apoptosis and survival of gastric cancer cells in vitro. Radiat Oncol. (2010) 5:98. doi: 10.1186/1748-717X-5-98
101. Coelho M, Moz M, Correia G, Teixeira A, Medeiros R, Ribeiro L. Antiproliferative effects of β-blockers on human colorectal cancer cells. Oncol Rep. (2015) 33:2513–20. doi: 10.3892/or.2015.3874
102. Montoya A, Amaya CN, Belmont A, Diab N, Trevino R, Villanueva G, et al. Use of non-selective β-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget. (2017) 8:6446–60. doi: 10.18632/oncotarget.14119
103. Wang F, Liu H, Wang F, Xu R, Wang P, Tang F, et al. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol Med Rep. (2018) 17:5213–21. doi: 10.3892/mmr.2018.8476
104. Brohee L, Peulen O, Nusgens B, Castronovo V, Thiry M, Colige AC, et al. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci Rep. (2018) 8:7050. doi: 10.1038/s41598-018-25340-9
105. Masur K, Niggemann B, Zanker KS, Entschladen F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. (2001) 61:2866–9.
106. Wilson JM, Lorimer E, Tyburski MD, Williams CL. β-Adrenergic receptors suppress Rap1B prenylation and promote the metastatic phenotype in breast cancer cells. Cancer Biol Ther. (2015) 16:1364–74. doi: 10.1080/15384047.2015.1070988
107. Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, et al. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. (2006) 12:369–75. doi: 10.1158/1078-0432.CCR-05-1698
108. Guo K, Ma Q, Wang L, Hu H, Li J, Zhang D, et al. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol Rep. (2009) 22:825–30. doi: 10.3892/or_00000505
109. Palm D, Lang K, Niggemann B, Drell TL, Masur K, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. (2006) 118:2744–9. doi: 10.1002/ijc.21723
110. Hii SI, Nicol DL, Gotley DC, Thompson LC, Green MK, Jonsson JR. Captopril inhibits tumour growth in a xenograft model of human renal cell carcinoma. Br J Cancer. (1998) 77:880–3. doi: 10.1038/bjc.1998.145
111. Attoub S, Gaben AM, Al-Salam S, Al Sultan MA, John A, Nicholls MG, et al. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann N Y Acad Sci. (2008) 1138:65–72. doi: 10.1196/annals.1414.011
112. Neo JH, Malcontenti-Wilson C, Muralidharan V, Christophi C. Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model of colorectal cancer liver metastases. J Gastroenterol Hepatol. (2007) 22:577–84. doi: 10.1111/j.1440-1746.2006.04797.x
113. Neo JH, Ager EI, Angus PW, Zhu J, Herath CB, Christophi C. Changes in the renin angiotensin system during the development of colorectal cancer liver metastases. BMC Cancer. (2010) 10:134. doi: 10.1186/1471-2407-10-134
114. Chen YH, Huang CH, Lu HI, Chen CH, Huang WT, Hsieh MJ, et al. Prognostic impact of renin-angiotensin system blockade in esophageal squamous cell carcinoma. J Renin Angiotensin Aldosterone Syst. (2015) 16:1185–92. doi: 10.1177/1470320314535275
115. Wysocki PJ, Kwiatkowska EP, Kazimierczak U, Suchorska W, Kowalczyk DW, Mackiewicz A. Captopril, an angiotensin-converting enzyme inhibitor, promotes growth of immunogenic tumors in mice. Clin Cancer Res. (2006) 12:4095–102. doi: 10.1158/1078-0432.CCR-05-2489
116. Fendrich V, Chen NM, Neef M, Waldmann J, Buchholz M, Feldmann G, et al. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut. (2010) 59:630–7. doi: 10.1136/gut.2009.188961
117. Fendrich V, Lopez CL, Manoharan J, Maschuw K, Wichmann S, Baier A, et al. Enalapril and ASS inhibit tumor growth in a transgenic mouse model of islet cell tumors. Endocr Relat Cancer. (2014) 21:813–24. doi: 10.1530/ERC-14-0175
118. Yoshiji H, Kuriyama S, Kawata M, Yoshii J, Ikenaka Y, Noguchi R, et al. The angiotensin-I-converting enzyme inhibitor perindopril suppresses tumor growth and angiogenesis: possible role of the vascular endothelial growth factor. Clin Cancer Res. (2001) 7:1073–8.
119. Yoshiji H, Yoshii J, Ikenaka Y, Noguchi R, Yanase K, Tsujinoue H, et al. Suppression of the renin-angiotensin system attenuates vascular endothelial growth factor-mediated tumor development and angiogenesis in murine hepatocellular carcinoma cells. Int J Oncol. (2002) 20:1227–31. doi: 10.3892/ijo.20.6.1227
120. Noguchi R, Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Yanase K, et al. Combination of interferon-beta and the angiotensin-converting enzyme inhibitor, perindopril, attenuates murine hepatocellular carcinoma development and angiogenesis. Clin Cancer Res. (2003) 9:6038–45.
121. Kinoshita J, Fushida S, Harada S, Yagi Y, Fujita H, Kinami S, et al. Local angiotensin II-generation in human gastric cancer: correlation with tumor progression through the activation of ERK1/2, NF-kappaB and survivin. Int J Oncol. (2009) 34:1573–82. doi: 10.3892/ijo_00000287
122. Rasheduzzaman M, Park SY. Antihypertensive drug-candesartan attenuates TRAIL resistance in human lung cancer via AMPK-mediated inhibition of autophagy flux. Exp Cell Res. (2018) 368:126–35. doi: 10.1016/j.yexcr.2018.04.022
123. Miyajima A, Kosaka T, Asano T, Asano T, Seta K, Kawai T, et al. Angiotensin II type I antagonist prevents pulmonary metastasis of murine renal cancer by inhibiting tumor angiogenesis. Cancer Res. (2002) 62:4176–9.
124. Chen X, Meng Q, Zhao Y, Liu M, Li D, Yang Y, et al. Angiotensin II type 1 receptor antagonists inhibit cell proliferation and angiogenesis in breast cancer. Cancer Lett. (2013) 328:318–24. doi: 10.1016/j.canlet.2012.10.006
125. Renziehausen A, Wang H, Rao B, Weir L, Nigro CL, Lattanzio L, et al. The renin angiotensin system. (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene. (2019) 38:2320–36. doi: 10.1038/s41388-018-0563-y
126. Noguchi R, Yoshiji H, Ikenaka Y, Namisaki T, Kitade M, Kaji K, et al. Synergistic inhibitory effect of gemcitabine and angiotensin type-1 receptor blocker, losartan, on murine pancreatic tumor growth via anti-angiogenic activities. Oncol Rep. (2009) 22:355–60. doi: 10.3892/or_00000445
127. Funao K, Matsuyama M, Kawahito Y, Sano H, Chargui J, Touraine JL, et al. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol Rep. (2008) 20:295–300. doi: 10.3892/or_00000006
128. Isobe A, Takeda T, Sakata M, Miyake A, Yamamoto T, Minekawa R, et al. Dual repressive effect of angiotensin II-type 1 receptor blocker telmisartan on angiotensin II-induced and estradiol-induced uterine leiomyoma cell proliferation. Hum Reprod. (2008) 23:440–6. doi: 10.1093/humrep/dem247
129. Zhang S, Wang Y. Telmisartan inhibits NSCLC A549 cell proliferation and migration by regulating the PI3K/AKT signaling pathway. Oncol Lett. (2018) 15:5859–64. doi: 10.3892/ol.2018.8002
130. Pantziarka P, Bouche G, Sukhatme V, Meheus L, Rooman I, Sukhatme VP. Repurposing Drugs in Oncology. (ReDO)-Propranolol as an anti-cancer agent. Ecancermedicalscience. (2016) 10:680. doi: 10.3332/ecancer.2016.680
131. Pantziarka P, Bryan BA, Crispino S, Dickerson EB. Propranolol and breast cancer-a work in progress. Ecancermedicalscience. (2018) 12:ed82. doi: 10.3332/ecancer.2018.ed82
132. Leaute-Labreze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taieb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. (2008) 358:2649–51. doi: 10.1056/NEJMc0708819
133. Tan ST, Itinteang T, Leadbitter P. Low-dose propranolol for infantile haemangioma. J Plast Reconstr Aesthet Surg. (2011) 64:292–9. doi: 10.1016/j.bjps.2010.06.010
134. Leaute-Labreze C, Hoeger P, Mazereeuw-Hautier J, Guibaud L, Baselga E, Posiunas G, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. (2015) 372:735–46. doi: 10.1056/NEJMoa1404710
135. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
136. Bradshaw A, Wickremesekera A, Brasch HD, Chibnall AM, Davis PF, Tan ST, et al. Cancer stem cells in glioblastoma multiforme. Front Surg. (2016) 3:48. doi: 10.3389/fsurg.2016.00048
137. Bradshaw AR, Wickremesekera AC, Brasch HD, Chibnall AM, Davis PF, Tan ST, et al. Glioblastoma multiforme cancer stem cells express components of the renin-angiotensin system. Front Surg. (2016) 3:51. doi: 10.3389/fsurg.2016.00051
138. Koh SP, Wickremesekera AC, Brasch HD, Marsh R, Tan ST, Itinteang T. Expression of cathepsins B, D, and G in isocitrate dehydrogenase-wildtype glioblastoma. Front Surg. (2017) 4:28. doi: 10.3389/fsurg.2017.00028
139. Humphries HN, Wickremesekera SK, Marsh RW, Brasch HD, Mehrotra S, Tan ST, et al. Characterization of cancer stem cells in colon adenocarcinoma metastasis to the liver. Front Surg. (2017) 4:76. doi: 10.3389/fsurg.2017.00076
140. Mehrotra S, Wickremesekera SK, Brasch HD, Van Schaijik B, Marsh RW, Tan ST, et al. Expression and localization of cathepsins B, D and G in cancer stem cells in liver metastasis from colon adenocarcinoma. Front Surg. (2018) 5:40. doi: 10.3389/fsurg.2018.00040
141. Nallaiah S, Lee VMY, Brasch HD, de Jongh J, Schaijik BV, Marsh R, et al. Cancer stem cells within moderately differentiated head and neck cutaneous squamous cell carcinoma express components of the renin-angiotensin system. J Plast Reconstr Aesthet Surg. (2018). doi: 10.1016/j.bjps.2018.11.013. [Epub ahead of print].
142. Featherston T, Yu HH, Dunne JC, Chibnall AM, Brasch HD, Davis PF, et al. Cancer stem cells in moderately differentiated buccal mucosal squamous cell carcinoma express components of the renin-angiotensin system. Front Surg. (2016) 3:52. doi: 10.3389/fsurg.2016.00052
143. Yu HH, Featherston T, Tan ST, Chibnall AM, Brasch HD, Davis PF, et al. Characterization of cancer stem cells in moderately differentiated buccal mucosal squamous cell carcinoma. Front Surg. (2016) 3:46. doi: 10.3389/fsurg.2016.00046
144. Baillie R, Itinteang T, Yu HH, Brasch HD, Davis PF, Tan ST. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma. J Clin Pathol. (2016) 69:742–4. doi: 10.1136/jclinpath-2015-203599
145. Itinteang T, Dunne JC, Chibnall AM, Brasch HD, Davis PF, Tan ST. Cancer stem cells in moderately differentiated oral tongue squamous cell carcinoma express components of the renin-angiotensin system. J Clin Pathol. (2016) 69:942–5. doi: 10.1136/jclinpath-2016-203736
146. Featherston T, Marsh RW, van Schaijik B, Brasch HD, Tan ST, Itinteang T. Expression and localization of cathepsins B, D, and G in two cancer stem cell subpopulations in moderately differentiated oral tongue squamous cell carcinoma. Front Med. (2017) 4:100. doi: 10.3389/fmed.2017.00100
147. Ram R, Brasch HD, Dunne JC, Davis PF, Tan ST, Itinteang T. The identification of three cancer stem cell subpopulations within moderately differentiated lip squamous cell carcinoma. Front Surg. (2017) 4:12. doi: 10.3389/fsurg.2017.00012
148. Ram RS, Brasch HD, Dunne JC, Davis PF, Tan ST, Itinteang T. Cancer stem cells in moderately differentiated lip squamous cell carcinoma express components of the renin-angiotensin system. Front Surg. (2017) 4:30. doi: 10.3389/fsurg.2017.00030
149. Shivapathasundram G, Wickremesekera AC, Brasch HD, Marsh R, Tan ST, Itinteang T. Expression of embryonic stem cell markers on the microvessels of WHO grade I meningioma. Front Surg. (2018) 5:65. doi: 10.3389/fsurg.2018.00065
150. Rahman RMA, van Schaijik B, Brasch HD, Marsh RW, Wickremesekera AC, Johnson R, et al. Expression of cathepsins B, D, and G in WHO grade I meningioma. Front Surg. (2019) 6:6. doi: 10.3389/fsurg.2019.00006
151. Itinteang T, Brasch HD, Tan ST, Day DJ. Expression of components of the renin-angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J Plast Reconstr Aesthet Surg. (2011) 64:759–65. doi: 10.1016/j.bjps.2010.08.039
152. Itinteang T, Withers AH, Leadbitter P, Day DJ, Tan ST. Pharmacologic therapies for infantile hemangioma: is there a rational basis? Plast Reconstr Surg. (2011) 128:499–507. doi: 10.1097/PRS.0b013e31821b63a0
153. Papali'i-Curtin JC, Brasch HD, van Schaijik B, de Jongh J, Marsh RW, Tan ST, et al. Expression of components of the renin-angiotensin system in pyogenic granuloma. Front Surg. (2019) 6:13. doi: 10.3389/fsurg.2019.00013
154. Tatokoro M, Fujii Y, Kawakami S, Saito K, Koga F, Matsuoka Y, et al. Phase-II trial of combination treatment of interferon-alpha, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor. (I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci. (2011) 102:137–43. doi: 10.1111/j.1349-7006.2010.01756.x
155. Shaashua L, Shabat-Simon M, Haldar R, Matzner P, Zmora O, Shabtai M, et al. Perioperative COX-2 and β-adrenergic blockade improves metastatic biomarkers in breast cancer patients in a phase-II randomized trial. Clin Cancer Res. (2017) 23:4651–61. doi: 10.1158/1078-0432.CCR-17-0152
156. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
157. Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. (2010) 16:3153–62. doi: 10.1158/1078-0432.CCR-09-2943
Keywords: cancer stem cells, renin-angiotensin system, stem cell differentiation, tumorigenesis, bypass loops
Citation: Roth IM, Wickremesekera AC, Wickremesekera SK, Davis PF and Tan ST (2019) Therapeutic Targeting of Cancer Stem Cells via Modulation of the Renin-Angiotensin System. Front. Oncol. 9:745. doi: 10.3389/fonc.2019.00745
Received: 08 May 2019; Accepted: 24 July 2019;
Published: 08 August 2019.
Edited by:
Cyril Corbet, Catholic University of Louvain, BelgiumReviewed by:
Aamir Ahmad, Mitchell Cancer Institute, United StatesMasahiro Hitomi, Cleveland Clinic, Lerner Research Institute, United States
Copyright © 2019 Roth, Wickremesekera, Wickremesekera, Davis and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swee T. Tan, swee.tan@gmri.org.nz