 Hongming Zhang
Hongming Zhang Runzhe Chen
Runzhe Chen Xiyong Wang3
Xiyong Wang3
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 24 July 2019
Sec. Pharmacology of Anti-Cancer Drugs
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00538
Platinum-based chemotherapy is recommended as the first-line treatment regimen for patients with advanced non-small-cell lung cancer (NSCLC). Lobaplatin (LBP), a third-generation platinum anti-neoplastic agent, has shown an improved efficacy. This study is aimed to investigate the mechanisms of LBP-induced apoptosis in the A549 p53 wild-type cell line. The Cell Counting Kit-8 assay (CCK-8), flow cytometry (FCM), Western blot, xenograft tumor models, terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL), and RNA interference were used in this study. Our results showed that the proliferation of A549 cells could be inhibited by LBP. At lower concentrations, LBP triggered cell cycle arrest at the G1 phase in A549 cells. LBP could also induce apoptosis of A549 cells. LBP also increased the expression of PARP and Bax and the cleavage of caspase-3, caspase-8, and caspase-9 and reduced Bcl-2 expression. In vivo experiment confirmed that LBP could inhibit tumor growth in the A549 xenograft models and induce apoptosis. Apoptosis of A549 cells was decreased after transfected with p53 shRNA or treated with reactive oxygen species inhibitor NAC and p38MAPK inhibitor SB203580, suggesting that the p53/ROS/p38MAPK pathway appeared to mediate the LBP-induced apoptosis of A549 cells. Our data demonstrate that LBP could be a promising candidate for the treatment of NSCLC with wild-type p53.
Lung cancer is the leading cause of cancer-related death worldwide (1), with non-small-cell lung cancer (NSCLC) accounting for 85% of lung cancer cases (2). Platinum-based doublet regimens have been used as a standard first-line therapy against NSCLC. There are two kinds of platinum-based agents widely used in NSCLC: cisplatin (DDP) and carboplatin (CBP). However, severe toxicity and drug resistance after utilization often occur. Therefore, exploration of new effective platinum-based agents with low toxicity for the treatment of NSCLC is urgently required.
Lobaplatin (LBP) is a third-generation platinum compound (Figure 1A) and has shown improved stability, a broader spectrum, and higher efficacy compared with DDP and CBP (3). As a result, it has been approved by the China Food and Drug Administration (CFDA) for the treatment of inoperable, metastatic breast cancer, small cell lung cancer (SCLC), and chronic myelocytic leukemia (CML) (4). Multiple studies have demonstrated its anticancer activity against other types of cancer (5–15).
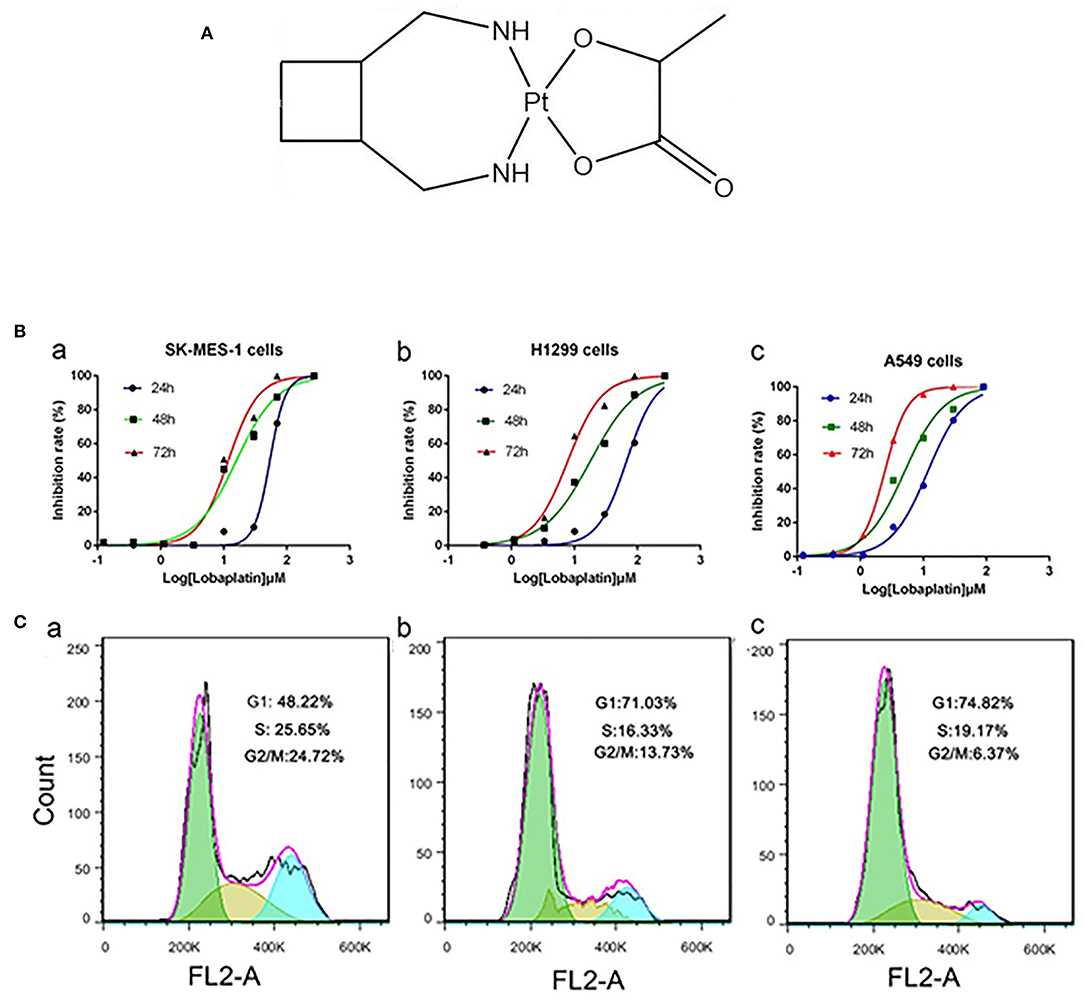
Figure 1. LBP inhibited the proliferation of human NSCLC cells and induced the cell cycle progression of A-549 cells. (A) Chemical structure of LBP. (B) Cells were exposed to various concentrations of LBP for 24, 48, or 72 h followed by analysis with a CCK-8 assay. (a) SK-MES-1, (b) NCI-H1299, (c) A-549. All of the assays were performed in triplicate. All of the assays were performed in triplicate. (C) A549 cells were exposed to various concentrations of LBP (a) 0 μM, (b) 6 μM, and (c) 12 μM for 24 h followed by analysis of the cell cycle by FCM.
At present, very few studies have been conducted on LBP in the treatment of NSCLC. Manegold et al. (16) studied the treatment efficacy of LBP in 39 advanced NSCLC patients, and only one of the 33 evaluable cases had a partial response. This report indicated that a single treatment with LBP is hardly effective against NSCLC in Caucasian patients. Moreover, the combined treatment of LBP with vinorelbine was reported to have a response rate (RR) of 35.1% and a disease control rate (DCR) of 78.4% in an open, single-experiment group, multiple-centered phase-II clinical research trial of patients with advanced NSCLC in China (17). While the efficacy of LBP therapy in Chinese patients has been clinically validated, less has been revealed about the underlying molecular mechanism responsible for the antitumor effects of LBP in NSCLC.
The anticancer mechanism of DDP includes the induction of oxidative stress, apoptosis, enhancement of reactive oxygen species (ROS) accumulation, activation of p38 mitogen-activated protein kinases (MAPKs), and regulation of p53 (18). As a similar mechanism to that of DDP, we hypothesized that LBP might function through a similar molecular mechanism to that of DDP. P53 is a tumor suppressor gene that encodes a transcription factor that functions in response to multiple stresses, regulating the cell cycle and apoptosis by adjusting the expression profiles of many genes (19). Activated p53 can prevent the propagation of cells carrying oncogenic lesions via a multitude of methods, i.e., induction of growth arrest, senescence or apoptosis, modulation of tumor stroma, angiogenesis, and modification of the metabolism (20). In lung cancer, it has been revealed that p53 mutations occur in up to 46% of adenocarcinoma cases and 81% of squamous cell cancer cases (21). Platinum-based agents' effects on tumor cells with either wild-type or mutated p53 remain controversial, as some studies have reported that wild-type tumor cells were more sensitive to chemotherapeutic drugs (22), while another study has indicated that [Pt(BDIQQ)]Cl, another kind of platinum-based agent, showed similar cytotoxicity in A549 cells with or without wild-type p53 gene (23). St. Germain et al. (24) showed that the MAPK pathway was involved in the apoptosis of tumor cells induced by DDP. Additionally, DDP was reported to trigger apoptosis in colon cancer cells via a p53/ROS/p38MAPK/p53 loop (22). Thus, we hypothesized that the p53/ROS/p38MAPK apoptotic pathway has been involved in LBP-induced apoptosis in A549 cells with wild-type p53.
LBP (Hainan Changan International Pharmaceutical Co., Ltd.) was dissolved in dimethyl sulfoxide (DMSO) to obtain 2.5 mM stock solutions (in vitro) or in physiological saline (in vivo). The stock solutions were kept frozen in aliquots at −20°C. DMSO, N-acetyl-L-cysteine (NAC), and SB203580 were obtained from Sigma-Aldrich (St. Louis, MO, USA). The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). The Annexin V-FITC Apoptosis Detection Kit was purchased from BD (Franklin Lakes, NJ, USA). The TUNEL staining kit was purchased from Roche (Basel, Switzerland). The ROS detection kit was purchased from Beyotime Institute of Biotechnology (Jiangsu, People's Republic of China). Antibodies against caspase-3, caspase-8, caspase-9, Bcl-2, p53, p-p38MAPK, poly-adenosine diphosphate (ADP) ribose polymerase (PARP), and GAPDH were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against Bax was purchased from Proteintech Group Inc. (Chicago, IL, USA). GIPZ Lentiviral shRNA was purchased from Horizon Discovery (Cambridge, UK).
A549, SK-MES-1, NCI-H1299, and 293T cells were acquired from Cobioer (Jiangsu, People's Republic of China). SK-MES-1 cells were cultured in MEM medium supplemented with 10% (v/v) heat-inactivated newborn calf serum (Thermo Fisher Scientific) with 1 ×105 U/L penicillin G and 1 ×105 U/L streptomycin in a humidified incubator at 37°C and 5% CO2. NCI-H1299, A549, and A549/control-shRNA cells were cultured in RPMI l640 medium supplemented with 10% (v/v) heat-inactivated newborn calf serum (Thermo Fisher Scientific) with 1 ×105 U/L penicillin G and 1 ×105 U/L streptomycin in a humidified incubator at 37°C and 5% CO2. 293T cells were cultured in DMEM medium supplemented with 10% (v/v) heat-inactivated newborn calf serum (Thermo Fisher Scientific) with 1 ×105 U/L penicillin G and 1 ×105 U/L streptomycin in a humidified incubator at 37°C and 5% CO2.
The cytotoxic effect of LBP on NSCLC was determined by the CCK-8 assay. Cells were seeded in a 96-well culture plate at a density of 4 ×104 cells/well and were cultured prior to exposure to a series of concentrations of LBP for 24, 48, and 72 h. A total of 10 μl of the CCK-8 solution was added to each well and incubated for an additional 4 h. The absorbance was measured at a 450-nm wavelength using the ELX 800 Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA). The measurement was performed three times for each concentration. The 50% inhibitory concentration (IC50) was determined by non-linear regression fit of the mean values of the data obtained.
The effect of LBP on human A549 cell cycle distribution was determined by FCM analysis. Approximately 3 ×105 cells were harvested at room temperature after pretreatment with various LBP concentrations for 24 h. Cells were then collected, washed twice with phosphate-buffered saline (PBS), and fixed with ice-cold 70% ethanol at 4°C overnight. After centrifugation, cells were resuspended in PBS containing 50 μg/ml RNase and incubated at 37°C for 30 min. Then, the cells were stained with 100 μl of 100 μg/ml propidium iodide (PI) and analyzed by a flow cytometer (C6; BD). The cell cycle distribution was analyzed according to the standard procedures. The distribution of cell cycle phases (G0/G1, S, or G2/M phase) was determined using Flow Jo.
Approximately 3 ×105 A549 cells/L were harvested after pretreatment with various reagents for 24, 48, and 72 h. Then, cells were trypsinized with EDTA-free trypsinogen, washed twice in PBS, and resuspended in 400 μl of 1× binding buffer. Next, 5 μl of annexin V-FITC and 5 μl of PI were added. After incubation at room temperature for 15 min in the dark, cells were then analyzed by FCM (C6; BD).
A549 cells were seeded in a 96-well culture plate at a density of 3 ×105 cells/well, with a coverslip (containing poly-lysine) in each well. Wells were incubated overnight and assigned to groups treated with 0, 12, or 24 μM LBP. After 24 h of treatment, cells were washed with PBS and fixed with formaldehyde for 30 min. Then, cells were again washed with PBS three times for 5 min each time. To perform the staining, 2.5 μg/ml DAPI was added in the dark for 10 min. After staining, cells were again washed three times with PBS for 5 min each time. Then, 10 μl of antifade was applied on the slide before a cell-containing slip was placed on the slide. Samples were then observed with a fluorescence microscope (400×) and photographed.
Western blot was performed as previously described (25). The code of caspase3 is #9662 (CST). The code of caspase9 is #9502 (CST). The code of caspase8 is #9494 (CST). The code of p-p38MAPK is #4511 (CST). The code of PARP is #9532 (CST). The code of p53 is #2527 (CST). The code of Bcl-2 is #2870 (CST). The code of GAPDH is #5174 (CST). The code of Bax is #50599-2-Ig (Proteintech). All primary antibodies and secondary were diluted to 1000 ×.
Five-weeks-old male BALB/Ca nude mice were purchased from Shanghai Laboratory Animal Center (Shanghai, China). A xenograft of A549 cells was established by inoculating viable A549 cells (107 cells/100 μl PBS per mouse) into the right flanks of the nude mice. When the average tumor volume reached ~100 mm3, the nude mice were randomly divided into two groups (5 mice per group). The experimental group was treated with LBP (d1, d8, 12 mg/kg) via the tail vein, and the control group was treated with saline (d1, d8, and saline only) via the tail vein. The body weight of each mouse was recorded twice a week. Tumor size was measured every other day. Tumor volume was calculated with a caliper (calculated volume = shortest diameter2 × longest diameter/2). The mice were sacrificed after 21 days, and the tumors were excised and stored at −80°C until further analysis. The relative tumor volume (RTV) of each mouse was determined by the formula RTV = Vt/V0, where Vt is the volume at each measurement and V0 is the volume at the initial treatment. The therapeutic effect of a given compound was expressed in terms of T/C (%), which was calculated by the formula T/C (%) = mean RTV of the treated group/mean RTV of the control group ×100%. All of the animal experiments were conducted following protocols approved by the animal ethics committee of the Medical School, Southeast University, and animal care was provided in accordance with institutional guidelines.
Formalin-fixed tumor tissues were embedded in paraffin before being sectioned. A TUNEL system was used to evaluate apoptosis in the tumor sections that were placed on slides according to the manufacturer's protocol. Tissue sections were analyzed to detect the localized green fluorescence of the apoptotic cells and the blue fluorescence of the cell nuclei. Images were acquired and photographed using an Olympus IX51 fluorescence microscope (400×).
Intracellular hydrogen peroxide levels were monitored by FCM after staining with 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). Briefly, cells in a logarithmic growth phase (3 ×105 cells in each well) were treated with various reagents for various periods of time and were then labeled with 2.5 μM DCFH-DA for 20 min. Next, the cells were trypsinized, washed with PBS, and then analyzed by FCM (Becton Dickinson). The percentage of cells displaying increased dye uptake was used to reflect an increase in ROS level.
Specific shRNA and control shRNA were designed and synthesized by Nanjing Kebai Biotech. Co. Ltd. (Nanjing, China). A BLAST search was performed with the National Center for Biotechnology Information (NCBI) database to ensure that the shRNA constructs were targeting only human p53. The sequence of the p53 shRNA was 5′-TACACATGTAGTTGTAGTG-3′, while the sequence of the nonsense-p53 shRNA (NS-p53-shRNA) was 5′-TTCTCCGAACGTGTCACGT-3′. The oligonucleotides were annealed and cloned into the pGIPZ-CMV-GFP-Puro, as described by the manufacturer. A total of 2.5 μg of pGIPZ constructs containing shRNAs and 7.5 μg of packaging plasmid were used to transfect 293T cells following the calcium phosphate precipitation method. After 48 h, lentiviruses containing targeted gene shRNA were collected and used to transfect A549 cells according to the manufacturer's instructions.
Statistical analysis was performed using the SPSS 22.0 software (IBM Corporation, Armonk, NY) package for Windows. Statistical significance was calculated using unpaired Student's t-test, with P < 0.05 indicating statistical significance.
The CCK-8 assay was performed to evaluate the effects of LBP on the proliferation of A549, NCI-H1299, and SK-MES-1 cells. The cells were treated with LBP at concentrations of 270, 90, 30, 10, 3.3, 1.1, 0.37, or 0.123 μM for 24, 48, or 72 h. As shown in Figure 1B, LBP reduced the proliferation of cells in a dose- and time-dependent manner. The IC50 values of LBP for SK-MES-1 cells were 63.47 ± 1.03, 15.77 ± 1.09, and 12.0 ± 1.08 μM when the cells were treated for 24, 48, and 72 h, respectively. The IC50 values for NCI-H1299 cells were 67.26 ± 1.06, 17.98 ± 1.05, and 7.85 ± 1.01 μM for the 24-, 48-, and 72-h treatment times, respectively, while the IC50 values for A549 cells were 11.69 ± 1.05, 5.02 ± 1.11, and 2.43 ± 1.01 μM for the 24-, 48-, and 72-h treatment times, respectively. It seems that LBP had a notably low IC50 in A549 cells with wild-type p53. Therefore, the A549 cell line was chosen for the next experiments to investigate the anticancer mechanism of LBP. Based on the IC50 of LBP, 6 and 12 μM were selected to be the experimental concentrations for the cell cycle arrest experiments, while 12 and 24 μM concentrations were selected for apoptosis detection, DAPI stain, and Western blot of A549 cells.
FCM analysis was performed to assess the cell cycle of cells treated with low concentrations of LBP (6 and 12 μM). After 24 h of treatment, 72.03 ± 1.53% of the cells remained in G1 phase under 6 μM LBP treatment (Figure 1C-b), while 73.78 ± 1.26% of the cells remained in G1 phase under 12 μM LBP treatment (Figure 1C-c). In the control group (Figure 1C-a), only 47.47 ± 1.59% of cells were detected in this phase. Additionally, the percentage of cells in the G2 or S phases was strikingly decreased in the experimental groups (P < 0.05). Our data showed that LBP could induce G1 phase cell cycle arrest at both 6 and 12 μM concentrations.
The Annexin V-FITC/PI Apoptosis Detection Kit was used to quantitatively determine whether LBP could induce A-549 cell apoptosis. The result showed that after treatment with 12 μM LBP for 24 h, 20.2 ± 1.3% of the experimental A549 cells developed apoptosis, while 44.5 ± 1.6% of the cells were affected at a concentration of 24 μM LBP. After we doubled the treatment time, apoptotic proportion of A549 cells treated with LBP at concentrations of 12 and 24 μM increased to 55.3 ± 1.2% and 70.0 ± 0.9%, respectively, suggesting that LBP could induce A549 cell apoptosis in a time- and dose-dependent manner (P < 0.05, Figure 2A). Next, we performed DAPI staining to observe the morphological changes of the cell nuclei during the process of LBP-induced apoptosis. After being treated with 12 μM LBP for 24 h, nuclei were observed to be condensed and deformed. When cells were treated with 24 μM LBP for 24 h, fewer nuclei were found intact in the same field of vision than were found in the cells treated with 12 μM LBP, and apoptosis bodies were found (Figure 2B). We then performed Western blot to study the expression levels of apoptosis-related proteins. After being treated with LBP at 12 or 24 μM LBP for 24 h, expression levels of clv-PARP, Bax, and clv-caspase-3,8,9 were upregulated and Bcl-2 was downregulated all in a dose-dependent manner (P < 0.05, Figure 2C).
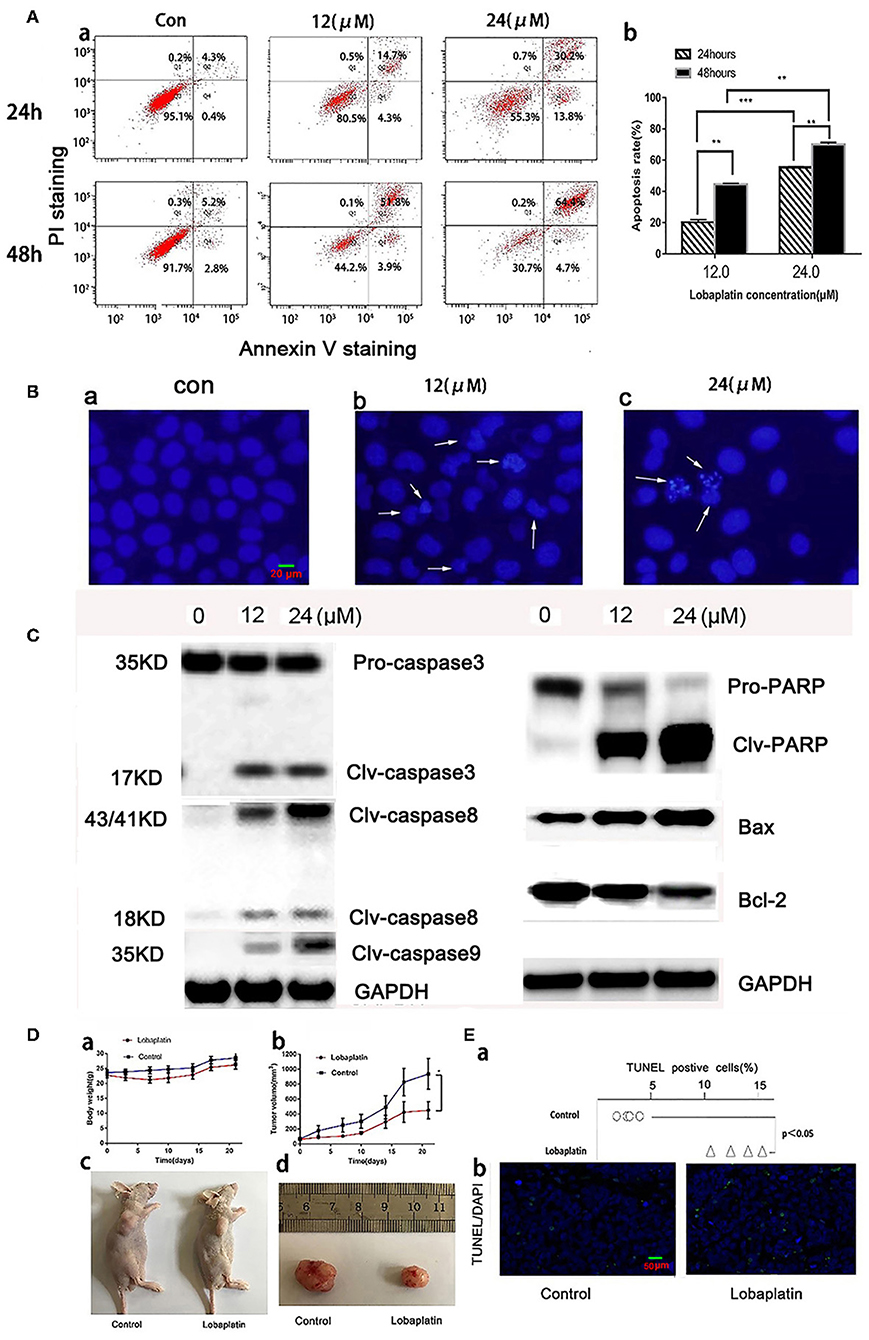
Figure 2. Induction of apoptosis in A549 cells by LBP in vitro and in vivo. (A,a) A549 cells were treated with LBP at the indicated concentrations for 24 and 48 h. The apoptosis rates were calculated by adding the percentages of Q2 and Q4, which appeared to be annexin V-positive. (b) The apoptosis rate was analyzed statistically. The data are expressed as means ± SD. **P < 0.01 and ***P < 0.001. (B) After being treated with LBP at concentrations of 0 μM (a), 12 μM (b), and 24 μM (c), A549 cells were stained with DAPI at a concentration of 2.5 μg/ml for 10 min and were then observed through a fluorescence microscope (400×) for morphological changes in their nuclei. (C) Expression levels of PARP, Bax, Bcl-2, and clv-caspase-3/8/9 affected by LBP at concentrations of 0, 12, and 24 μM for 24 h. GAPDH was used as an internal reference. (D) The antitumor effect of LBP on A-549 human xenograft models. (a) The average body weight of each group was expressed as the mean ± SD. (b) Tumor size comparison between the treatment group (n = 5), which was given LBP at 12 mg/kg via caudal vein injection, and the control group, which was treated with normal saline (P < 0.05). (c) Image of mice with A-549 xenograft tumors after 21 days of treatment. (d) At the experimental end point, tumors were dissected and photographed. For (c,d), only one mouse is shown as an example for each group. (E,a) The TUNEL-positive rate of each tumor was analyzed. (b) Representative images of TUNEL/DAPI staining of tumor tissues of the different groups (magnification 200×). *P < 0.05.
To study LBP's antitumor efficacy against A549 cells in vivo, we further generated a model of nude mice bearing human A549 tumors. The data showed that the experimental group weighed slightly less than the control group did, but there was no significant difference (P > 0.05). The mice lost some weight right after each time they were treated with LBP, which indicated that LBP might cause a mild gastrointestinal reaction (Figure 2D-a). By the end of the experiment, the average of the LBP group tumor was 451 ± 113 mm3 while it was 936 ± 205 mm3 in the control group (Figure 2D-b). The T/C value was 55.2%, and the tumor inhibition rate was 47.9% and Supplementary Figures 1, 2 (Figures 2D-c,d), suggesting that LBP could inhibit the growth of the A549 tumor. TUNEL/DAPI staining indicated the apoptosis rate of 15.5 ± 2.1% in the treatment group, whereas the percentage was only 4.8 ± 0.5% in the control group (P < 0.05) (Figures 2E-a,b). Taken together, our results indicated that LBP inhibited the growth of A549 tumors in mice by inducing the apoptosis.
To test whether ROS are involved in the pathway through which LBP induces A549 cell apoptosis, we examined ROS levels in A549 cells after treating the cells with 24 μM LBP. We performed the ROS analysis with both DCFH-DA staining analysis with a fluorescence microscope and FCM. From the resulting images, it can be inferred that ROS release increased as the treatment time increased (Figures 3A–G), with an apparent burst at 6 h (P < 0.05) (Figure 3H). FCM analysis also confirmed this finding (P < 0.05) (Figure 3I).
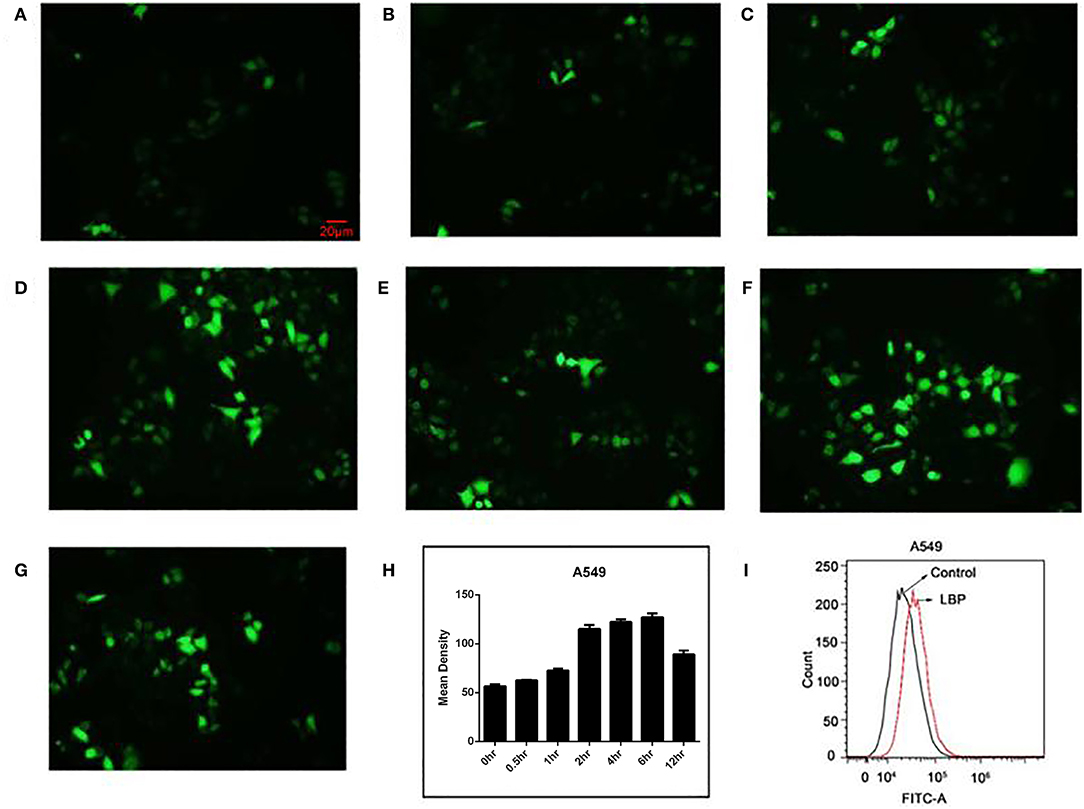
Figure 3. ROS release in A549 cells after LBP treatment. With DCFH-DA staining, the levels of ROS are shown at (A) 0 h, (B) 0.5 h, (C) 1 h, (D) 2 h, (E) 4 h, (F) 6 h, and (G) 12 h. (H) The levels of ROS were analyzed statistically. (I) A549 cells were treated with 24 μM LBP for 6 h. FCM analysis of intracellular ROS using a DFCH-DA probe.
To determine whether LBP-induced apoptosis requires p53, stable cell lines transfected with the plasmid-based shRNA expression system targeting p53 mRNA were established, and the sensitivity of these cell lines to LBP was then surveyed. As shown in Figure 4A, the p53 protein level of A549/p53-shRNA was efficiently knocked down compared to the expression in the A549 parental control or A549/NS-shRNA (vector control) cells. Subsequently, A549/NS-shRNA and A549/p53-shRNA cells were exposed to LBP at concentrations ranging from 0.0123 to 90 μM for 24, 48, or 72 h (Figure 4B). The IC50 of LBP for A549/p53-shRNA cells was 39.54 ± 1.02 μM for the 24-h treatment, 27.89 ± 1.13 μM for the 48-h treatment, and 10.93 ± 1.08 μM for the 72-h treatment. However, the IC50 of LBP for A549/NS-shRNA cells was 11.48 ± 1.12 μM for the 24-h treatment, 5.14 ± 0.94 μM for the 48-h treatment, and 2.49 ± 0.90 μM for the 72-h treatment. Compared to the A549 cells, the A549/NS-shRNA cells showed similar sensitivity to LBP (P > 0.05). Hence, A549/p53-shRNA cells with p53 knockdown became less sensitive to LBP, as shown by their higher IC50 values (P < 0.05). Sequentially, FCM analysis in association with annexin V-PI double staining was performed to investigate the apoptosis of A549/NS-shRNA and A549/p53-shRNA cells treated with LBP. The results showed that after 24 h, 38.6 ± 1.1% of the 24 μM-LBP-treated A549/NS-shRNA24h cells underwent apoptosis, but no significant apoptosis observed in A549/p53-shRNA cells (Figure 4C). Our findings suggested that A549/p53-shRNA cells were more resistant to LBP-induced apoptosis than A549/NS-shRNA cells (P < 0.05). To further evaluate whether LBP could also modify the expression level of p38MAPK, expression levels of p53 and p-p38MAPK were measured in A549/NS-shRNA and A549/p53-shRNA cells that were treated with 24 μM LBP for 24 h. As shown in Figure 4D, LBP could upregulate the expression levels of p53 and p-p38MAPK in A549/NS-shRNA cells (P < 0.05) but not in A549/p53-shRNA cells (P > 0.05). Interestingly, p-p38MAPK was found to have a higher expression level in A549/p53-shRNA cells than in A549/NS-shRNA cells with no LBP treatment.
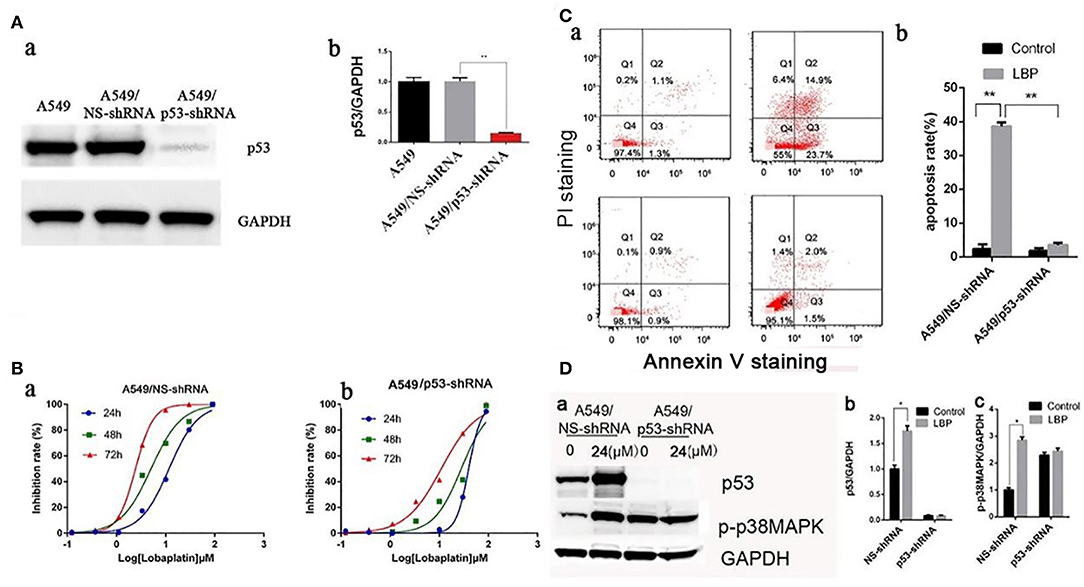
Figure 4. LBP induces apoptosis by upregulating p53 and p38MAPK. (A) Establishment of stable p53-knockdown clones. The plasmid containing the p53-shRNA sequence was constructed and transfected into A549 cells, and the stable lines harboring vector control (A549/NS-shRNA) or p53-shRNA vector were isolated as described in the Materials and Methods section. The p53 protein expression levels were determined by Western blot. (B) Cells were exposed to various concentrations (0.00123–90 μM) of LBP for 24, 48, or 72 h followed by analysis with a CCK-8 assay. (a) A549/NS-shRNA, (b) A549/p53-shRNA. (C,a) A549/NS-shRNA and A549/p53-shRNA cells were treated with LBP at the indicated concentrations for 24 h. The apoptosis rates were calculated by adding the percentages of Q2 and Q4, which appeared to be annexin V-positive. (b) The apoptosis rate was analyzed statistically. The data are expressed as the means ± SD. (D,a) Western blot results detecting expression levels of p53 and p-p38MAPK in A549/NS-shRNA and A549/p53-shRNA cells treated with 24 μM LBP for 24 h. GAPDH was used as an internal reference. (b) Protein expression levels (relative to GAPDH) were determined. The data are expressed as the means ± SD. *P < 0.05 and **P < 0.01.
Based on previous results, we applied an ROS inhibiting agent, NAC, to determine whether ROS are crucial for LBP-induced apoptosis. After pretreatment with NAC, cells were incubated with 24 μM LBP for 24 h, followed by FCM. As shown in Figure 5A, the apoptosis rate was reduced from 40.7 ± 2.4% to 9.6 ± 2.1% (P < 0.05). Our findings suggested that ROS participated in the process of LBP-induced apoptosis.
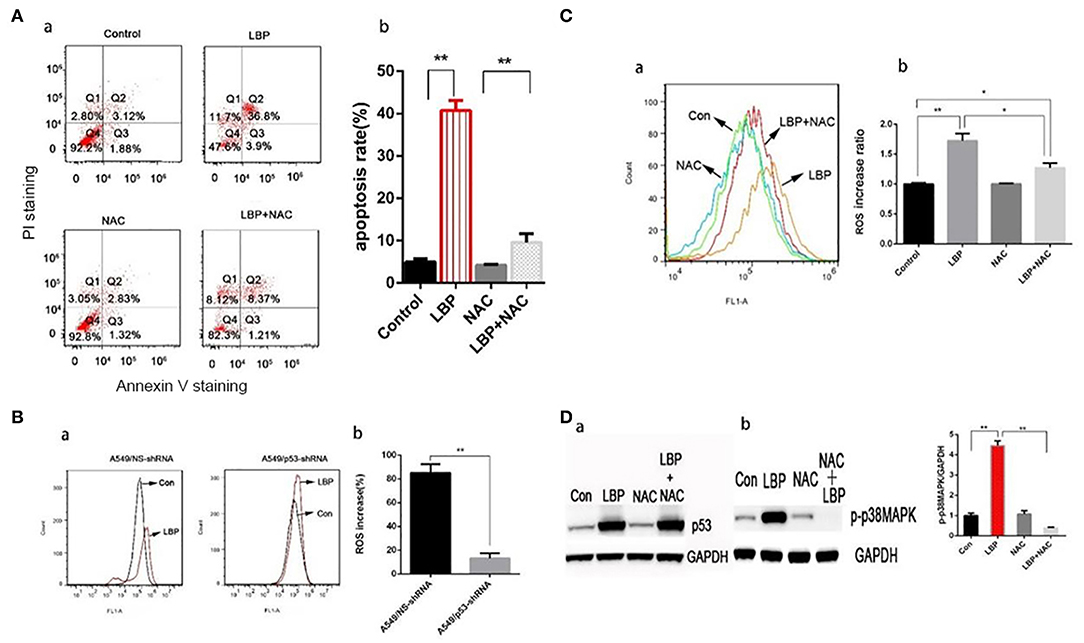
Figure 5. LBP-induced p53 and p-p38MAPK expression, ROS release, and apoptosis are affected by NAC. (A,a) A549 cells were treated with 5 mM NAC for 4 h and 5 mM NAC for 4 h before treatment with 24 μM LBP for 24 h, followed by FCM analysis. The apoptosis rates were calculated by adding the percentages of Q2 and Q4. (b) The apoptosis rate was analyzed statistically. The data are expressed as the means ± SD. *P < 0.05 and **P < 0.01. (B) FCM analysis of intracellular ROS using the DFCH-DA probe. A549/NS-shRNA and A549/p53-shRNA cells were treated with LBP at a concentration of 24 μM for 6 h. The value shown in (b) is the percentage of increase. (C) FCM analysis of intracellular ROS using the DFCH-DA probe. A549 cells were treated with 5 mM NAC for 4 h and 5 mM NAC for 4 h before treatment with 24 μM LBP for 6 h. Con indicates the control group. *P < 0.05, **P < 0.01. (D) Western blot detecting (a) p53 and (b) p-p38MAPK expression levels with and without NAC. A549 cells were pre-treated with 5 mM NAC for 4 h before treatment for 24 h with 24 μM LBP.
With DCFH-DA staining followed by FCM, we measured the ROS levels in A549/NS-shRNA and A549/p53-shRNA cells treated with LBP at a concentration of 24 μM for 6 h. While only a slight upregulation of ROS release occurred in the A549/p53-shRNA cells, A549/NS-shRNA cells generated a much higher level of ROS under LBP treatment. The percentage of ROS released was significantly higher in the A549/NS-shRNA cells than in the A549/p53-shRNA cells (P < 0.05, Figure 5B). NAC can partially suppress LBP-induced ROS release (P < 0.05, Figure 5C). To investigate the precise connection between p53, ROS, and p38MAPK in the pathway of LBP-induced apoptosis, we applied NAC and SB203580, a p38MAPK inhibiting agent, to the cells before LBP treatment to monitor the expression levels of p53 and p38MAPK. The results were shown in Figure 5D. It is implied that LBP at a concentration of 24 μM could upregulate the expression of p53 and p-p38MAPK (P < 0.05). Treatment with NAC prevented p-p38MAPK activation (P < 0.05), while p53 levels remained unchanged (P > 0.05). This result suggested that the p53/ROS/p38MAPK pathway was involved in the LBP-induced apoptosis.
P38MAPK plays vital roles in many stress reaction pathways in human cells. P38MAPK has been reported to be involved in DDP's antitumor mechanism (22). In our experiment, LBP at a concentration of 24 μM can induce a significant upregulation of p-p38MAPK in A549 cells (P < 0.05), which was inhibited by the application of SB203580 (P > 0.05, Figure 6D). To further examine the role of p38MAPK in LBP-induced A549 cell apoptosis, we applied SB203580 to the cells before they were treated with 24 μM LBP for 24 h followed by FCM for apoptosis analysis. The results showed that pretreatment with SB203580 reduced the LBP-induced apoptosis rate in A549 cells from 40.7 ± 2.4% to 18.1 ± 1.6% (P < 0.05, Figure 6A). Our findings indicated that p38MAPK participates in the process of LBP-induced apoptosis. Next, we performed DCFH-DA staining followed by FCM to detect ROS accumulation in A549 cells treated with or without SB203580 before LBP treatment. The results showed that SB203580 failed to alleviate the accumulation of ROS induced by LBP (P > 0.05, Figure 6B). Moreover, our findings indicated that SB203580 could not inhibit the upregulation of p53 triggered by 24 μM LBP (P > 0.05, Figure 6C).
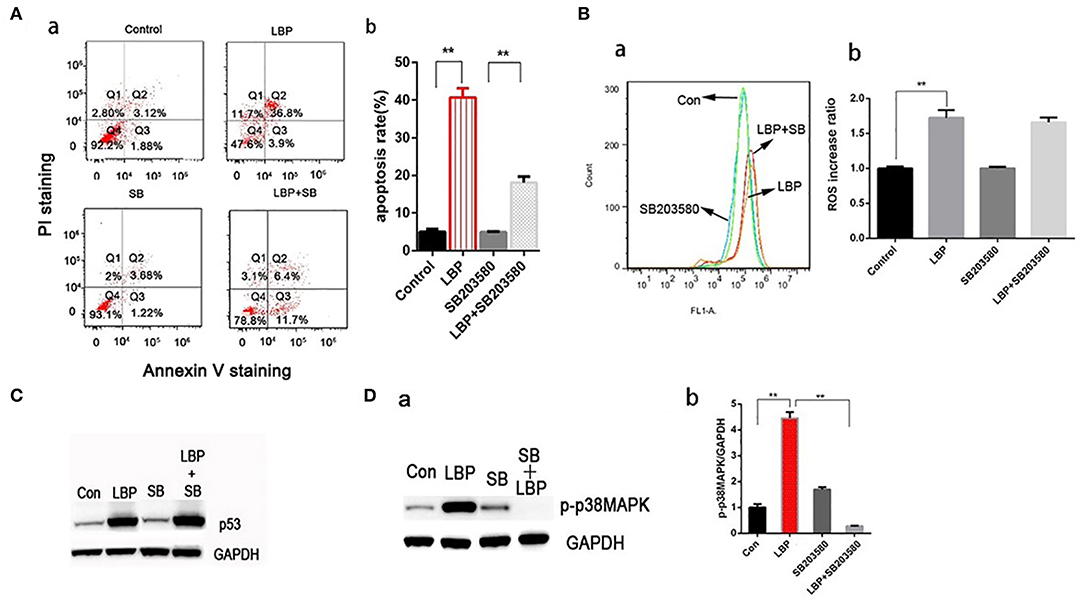
Figure 6. P-p38MAPK is involved in the mechanism of LBP-induced apoptosis. (A,a) A549 cells were treated with 12.5 μM SB203580 for 1 h and 12.5 μM SB203580 for 1 h before treatment with 24 μM LBP for 24 h followed by FCM. (b) The apoptosis rate was analyzed statistically. The data are expressed as the means ± SD. *P < 0.05 and **P < 0.01. (B) FCM analysis of intracellular ROS using the DFCH-DA probe. A549 cells were treated with 12.5 μM SB203580 for 1 h and 12.5 μM SB203580 for 1 h before treatment with 24 μM LBP for 6 h. Con indicates the control group. *P < 0.05, **P < 0.01. (C) Western blot detecting p53 expression levels with and without SB203580 treatment. A549 cells were pre-treated with 12.5 μM SB203580 for 1 h before treatment for 24 h with 24 μM LBP. (D) Western blot detecting p-p38MAPK expression levels with and without SB203580 treatment. A549 cells were pretreated with 12.5 μM SB203580 for 1 h before treatment for 24 h with 24 μM LBP.
In this study, we observed that LBP can induce the apoptosis of NSCLC cells with different p53 genotypes, and A549 cells with wild-type p53 were the most sensitive to LBP-induced apoptosis. LBP was also found to reduce the tumor burden in the A549 xenograft model. Importantly, the underlying mechanisms of LBP-induced apoptosis were revealed, involving the p53/ROS/p38MAPK signaling pathway.
Lung cancer cells with different p53 genotypes were treated with LBP. A549 cells with wild-type p53 were more sensitive to LBP. Additionally, we measured the 48-h IC50 values of LBP for NCI-H1299 and A549 cells, which were 18.00 ± 1.05 and 5.02 ± 1.11 μM, respectively. In comparison, DDP was reported to have IC50 values of 25.21 ± 4.38 and 21.88 ± 3.21 μM for NCI-H1299 and A549 cells, respectively (26). Thus, LBP showed more powerful effects than DDP on A549 and NCI-H1299 cells.
Cell cycle arrest is one of the mechanisms for cell growth inhibition induced by many anticancer drugs. Many platinum-based drugs, including DDP, CBP, oxaliplatin, and nedaplatin (27–29), have been found to induce cell cycle arrest. In this study, LBP was also found to trigger G1 phase cell cycle arrest. Wang et al. (14) found that LBP could induce G1 phase cell cycle arrest in cholangiocarcinoma cells. In SMMC-7721 cells, LBP was found to cause arrest in the G1 phase within 24–48 h of treatment and G2/M arrest within 36–48 h of treatment (30). It was also reported that 3 μM LBP led to S phase arrest in A549 cells (31). In general, LBP can induce cell cycle arrest, which may vary by phases according to different cell types, dosages, and/or treatment times.
Apoptosis is one of the most important anticancer mechanisms (32). Many chemotherapeutic compounds, like DDP (26) and CBP (33), have been found to inhibit tumor growth by inducing apoptosis. With DAPI staining, we observed condensation and margination of chromatin in the A549 cells that were treated with LBP for 24 h. In the FCM analysis, the LBP-induced apoptosis rates were found to be dose- and time-dependent. TUNEL staining confirmed that LBP could induce A549 cell apoptosis in vivo. LBP can also induce a dose-dependent upregulation of PARP, which was also proven to be regulated by DDP (26). The expression levels of clv-caspase-3/8/9 were also indicated by the Western blot results to be elevated by LBP in A549 cells in a dose-dependent manner. Thus, both internal and external apoptotic pathways are employed by LBP to induce A549 cell apoptosis.
In the in vivo study, LBP could effectively inhibit tumor growth in mice bearing A549 tumors through the induction of apoptosis, with a relative T/C of 55.2% and an inhibitory rate of 47.9%. Harstrick et al. (34) applied LBP to mice bearing H12.1 tumors. LBP was applied at a dosage of 12 mg/kg when the tumor size reached 1–1.5 cm3 on days 1, 3, and 5, yielding a T/C value of 39.1%. It seems that the effectiveness of LBP depends on the timing, frequency, and dosage of treatment; these factors will require further investigation.
In normal physiological conditions, ROS are natural by-products of aerobic respiration and metabolism. Some anticancer agents, such as gemcitabine (35), pemetrexed (36), gefitinib (37), paclitaxel (38, 39), vinorelbine (40), docetaxel (41), CBP (42), and oxaliplatin (43), work by generating ROS. The overaccumulation of ROS induces cell apoptosis through internal and external apoptotic pathways (44). LBP can upregulate the ROS level in human BGC-823 stomach cancer cells (6). Our study showed that LBP at a concentration of 24 μM was able to trigger ROS accumulation in A549 cells, with a burst at 6 h. The LBP-induced ROS release and apoptosis in A549 cells could be partially attenuated by NAC. This finding indicates that ROS play a role in the A549 cell apoptosis induced by LBP.
P53 is thought to be involved in the maintenance of genome integrity. DNA damage can trigger a rapid upregulation of p53 expression (45). Once activated, p53 is able to affect a number of transcription factors, such as ROS (19) and MAPKs (18). MAPKs, including ERK, p38, and JNK, are components of signaling cascades that respond to extracellular stimuli by targeting transcription factors, resulting in the regulation of gene expression. DDP has also been reported to employ both p38MAPK and JNK pathways to kill human cervical cancer cells (46). CBP can induce the apoptosis of human cervical cancer cells through the p53/ERK pathway (47). LBP has an anti-tumor mechanism where it binds to guanine and adenine in double-strand DNA, creating a Pt–DNA conjugate compromising genome integrity, which is similar to that of DDP (48). Thus, we continued to examine whether p53 and p38MAPK are involved in the induction of apoptosis by LBP. First, our experiments showed that treatment with 24 μM LBP for 24 h elevated p53 expression in A549 cells. LBP had a much higher IC50 for A549/p53-shRNA cells than for A549/NS-shRNA cells. Moreover, LBP at a concentration of 24 μM resulted in an apoptosis rate of 45% in p53/NS-shRNA cells, while no apparent apoptosis was observed in A549/p53-shRNA cells. Our findings demonstrated that A549 cells were less sensitive to LBP without the presence of the p53 gene. In comparison, Suntharalingam et al. (23) reported that the IC50 of a novel platinum-based agent, [Pt(BDIQQ)]Cl, was the same in A549 and A549/p53−/− cells, which indicates that the inhibition of tumors is p53-independent. Gefitinib, reported by Chang et al. (49), was more active in A549 cells with wild-type p53 than on A549/p53-shRNA cells, while NCI-H1299 with p53+/+ cells was sensitive to gefitinib. In another study (30), Huh-7 cells with a mutated p53 gene were less sensitive to LBP than were SMMC-7721 and Bel-7402 cells but showed similar sensitivity to HePG2 cells; all three of these cell lines are liver cancer cells with wild-type p53. Based on these studies, it can be inferred that whether p53 is key to cancer cells' sensitivity to platinum-based drugs relies on the types of cells and/or drugs. Overall, p53 is required for LBP-induced A549 apoptosis. Second, we found that LBP could not lead to an upregulation of p-p38MAPK in A549/NS-shRNA cells. However, in A549/p53-shRNA cells, the p-p38MAPK expression level was noticeably higher than in A549/NS-shRNA cells, an observation that requires further investigation. We found that NAC and SB203580 were able to downregulate the p-p38MAPK expression level in A549/p53-shRNA cells. SB203580 showed no interference with the upregulation of p53 and ROS induced by LBP. However, pretreatment with SB203580 reduced the LBP-induced apoptosis rate in A549 cells. Overall, it is indicated that p38MAPK is also required for the LBP-induced apoptosis of A549 cells. At last, we found that a slight upregulation of ROS release occurred in the A549/p53-shRNA cells after LBP treatment. The expression of p53 is not affected by the application of NAC. LBP can also trigger the upregulation of p38MAPK, which is inhibited by applying NAC to the cells. By far, we draw the conclusion that LBP induced A549 apoptosis through the p53/ROS/p38MAPK pathway. This pathway has been proved to be involved in the induction of HCT-116 cell apoptosis by DDP (22) and in the inhibition of breast cancer metastasis by theaflavin (50). Both studies reported a downregulation of p53 after the treatment with SB203580. But in this study, no such effect of SB203580 on p53 expression was found, which is consistent with results by Sanchez-Prieto et al. (51). The difference still needs to be well-investigated.
In summary, our findings revealed that LBP could arrest the cell cycle in the G1 phase in A549 cells, significantly inhibit the growth of A549 cells in vitro and in vivo, and induce cell apoptosis by upregulating the expression levels of Bax, PARP, clv-caspase-3,−8, and−9 and downregulating the expression of Bcl-2. We verified for the first time that the p53/ROS/p38MAPK signaling pathway was involved in LBP-induced apoptosis. Taken together, our findings clearly concluded that LBP is a promising candidate for the treatment of NSCLC.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
All of the animal experiments were conducted following protocols approved by the Animal Ethics Committee of the Medical School, Southeast University, and animal care was provided in accordance with Institutional Guidelines.
HoZ, RC, and XZ designed the study. HoZ and XW performed the experiments. HoZ, HaZ, and JC analyzed the data. HoZ and RC wrote the manuscript.
This work was supported by the Wu Jieping Medical Foundation Clinical Scientific Research Fund (No. 320.6750.15069).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00538/full#supplementary-material
Supplementary Figure 1. Image of mice of LBP treatment group with A549 xenograft.
Supplementary Figure 2. Image of mice of control group with A549 xenograft.
1. DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin. (2019) 69:211–33. doi: 10.3322/caac.21555
2. Houston KA, Henley SJ, Li J, White MC, Richards TB. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004–2009. Lung Cancer. (2014) 86: 22–8. doi: 10.1016/j.lungcan.2014.08.001
3. Voegeli R, Schumacher W, Engel J, Respondek J. Hilgard P. D-19466, a new cyclobutane–platinum complex with antitumor activity. J Cancer Res Clin Oncol. (1990) 116:439–42. doi: 10.1007/BF01612990
4. Monneret C. Platinum anticancer drugs. From serendipity to rational design. Ann Pharm Fr. (2011) 69:286–95. doi: 10.1016/j.pharma.2011.10.001
5. Du L, Fei Z, Song S, Wei N. Antitumor activity of Lobaplatin against esophageal squamous cell carcinoma through caspase-dependent apoptosis and increasing the Bax/Bcl-2 ratio. Biomed Pharmacother. (2017) 95:447–52. doi: 10.1016/j.biopha.2017.08.119
6. Li Y, Liu B, Yang F, Yu Y, Zeng A, Ye T, et al. Lobaplatin induces BGC-823 human gastric carcinoma cell apoptosis via ROS-mitochondrial apoptotic pathway and impairs cell migration and invasion. Biomed Pharmacother. (2016) 83:1239–46. doi: 10.1016/j.biopha.2016.08.053
7. Zhou NN, Zhao YY, Zhai LZ, Ruan CM, Yang YP, Huang Y, et al. The efficacy and toxicity of Lobaplatin-contained chemotherapy in extensive-stage small-cell lung cancer. J Cancer. (2018) 9:2232–6. doi: 10.7150/jca.24557
8. Peng S, Yang QX, Zhang T, Lu MJ, Yang G, Liu ZY, et al. Lobaplatin-TACE combined with radioactive 125I seed implantation for treatment of primary hepatocellular carcinoma. Asian Pac J Cancer Prev. (2014) 15:5155–60. doi: 10.7314/APJCP.2014.15.13.5155
9. He MK, Le Y, Li QJ, Yu ZS, Li SH, Wei W, et al. Hepatic artery infusion chemotherapy using mFOLFOX versus transarterial chemoembolization for massive unresectable hepatocellular carcinoma: a prospective non-randomized study. Chin J Cancer. (2017) 36:83. doi: 10.1186/s40880-017-0251-2
10. He MK, Zou RH, Wei W, Shen JX, Zhao M, Zhang YF, et al. Comparison of stable and unstable ethiodized oil emulsions for transarterial chemoembolization of hepatocellular carcinoma: results of a single-center double-blind prospective randomized controlled trial. J Vasc Interv Radiol. (2018) 29:1068–77.e2. doi: 10.1016/j.jvir.2018.03.027
11. Sternberg CN, de Mulder P, Fossa S, Kaye S, Roberts T, Pawinsky A, et al. Lobaplatin in advanced urothelial tract tumors. The Genitourinary Group of the European Organization for Research and Treatment of Cancer (EORTC). Ann Oncol. (1997) 8:695–6. doi: 10.1023/A:1008269432176
12. Kirpensteijn J, Teske E, Kik M, Klenner TRutteman GR. Lobaplatin as an adjuvant chemotherapy to surgery in canine appendicular osteosarcoma: a phase II evaluation. Anticancer Res. (2002) 22:2765–70.
13. Yang F, Yu Y, Lei Q, Zeng A, Li Y, Xie Y, et al. Lobaplatin arrests cell cycle progression, induces apoptosis and impairs migration and invasion in B16-F10 melanoma cell line in vitro. Biomed Pharmacother. (2015) 69:402–8. doi: 10.1016/j.biopha.2014.12.011
14. Wang Z, Tang X, Zhang Y, Qi R, Li Z, Zhang K, et al. Lobaplatin induces apoptosis and arrests cell cycle progression in human cholangiocarcinoma cell line RBE. Biomed Pharmacother. (2012) 66:161–6. doi: 10.1016/j.biopha.2011.09.008
15. Li X, Ran L, Fang W, Wang D. Lobaplatin arrests cell cycle progression, induces apoptosis and alters the proteome in human cervical cancer cell Line CaSki. Biomed Pharmacother. (2014) 68:291–7. doi: 10.1016/j.biopha.2013.10.004
16. Manegold C, Drings P, Gatzemeier U, Pawel JV, Fiebig HH, Queißer W, et al. Lobaplatin (D-19466) in patients with advanced non-small-cell lung cancer: a trial of the association for medical oncology (AIO) Phase II study group. Onkologie. (1996) 19:248–51. doi: 10.1159/000218806
17. Yang L-Q, Shi Y, Qin S-K, Qian J, Han B-H, Pan L-X, et al. Clinical study of lobaplatin combined with navelbine for advanced non-small cell lung cancer. Chin Clin Oncol. (2006) 11:890–4.
18. Dasari ST, Chounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Clin Pharmacol. (2014) 740:364–78. doi: 10.1016/j.ejphar.2014.07.025
19. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. (2000) 408:307–10. doi: 10.1038/35042675
20. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
21. Gibbons DL, Byers LA, Kurie JM. Smoking, p53 mutation, and lung cancer. Mol Cancer Res. (2014) 12:3–13. doi: 10.1158/1541-7786.MCR-13-0539
22. Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis. (2007) 12:1733–42. doi: 10.1007/s10495-007-0082-8
23. Suntharalingam K, Wilson JJ, Lin W, Lippard SJ. A dual-targeting, p53-independent, apoptosis-inducing platinum(II) anticancer complex, [Pt(BDI(QQ))]Cl. Metallomics. (2014) 6:437–43. doi: 10.1039/C3MT00364G
24. St.Germain C, Niknejad N, Ma L, Garbuio K, Hai T, Dimitroulakos J. Cisplatin induces cytotoxicity through the mitogen-activated protein kinase pathways and activating transcription factor 3. Neoplasia. (2010) 12:527–38. doi: 10.1593/neo.92048
25. Zhang H, Chen R, Yang S, Liu W, Li K, Zhang H, et al. Lobaplatin for the treatment of SK-MES-1 lung squamous cell line in vitro and in vivo. Onco Targets Ther. (2016) 9:4215–24. doi: 10.2147/OTT.S108032
26. Wang LH, Li Y, Yang SN, Wang FY, Hou Y, Cui W, et al. Gambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-kappaB and MAPK/HO-1 signalling. Biomed Pharmacother. (2014) 110:341–52. doi: 10.1038/bjc.2013.752
27. Pasetto LM, D'Andrea MR, Brandes AA, Rossi E, Monfardini S. The development of platinum compounds and their possible combination. Crit Rev Oncol Hematol. (2006) 60:59–75. doi: 10.1016/j.critrevonc.2006.02.003
28. Voland C, Bord A, Peleraux A, Penarier G, Carriere D, Galiegue S, et al. Repression of cell cycle-related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol Cancer Ther. (2006) 5:2149–57. doi: 10.1158/1535-7163.MCT-05-0212
29. Chatterjee S, Rhee YH, Ahn JC. Sulforaphene-carboplatin combination synergistically enhances apoptosis by disruption of mitochondrial membrane potential and cell cycle arrest in human non-small cell lung carcinoma. J Med Food. (2016) 19:860–9. doi: 10.1089/jmf.2016.3675
30. Wu Q, Qin SK, Teng FM, Chen CJ, Wang R. Lobaplatin arrests cell cycle progression in human hepatocellular carcinoma cells. J Hematol Oncol. (2010) 3:43. doi: 10.1186/1756-8722-3-43
31. Xie CY, Xu YP, Jin W, Lou LG. Antitumor activity of lobaplatin alone or in combination with antitubulin agents in non-small-cell lung cancer. Anticancer Drugs. (2012) 23:698–705. doi: 10.1097/CAD.0b013e328352cc10
32. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. (2002) 108:153–64. doi: 10.1016/S0092-8674(02)00625-6
33. Hu XL, Feng JH, Pham TA, Ma HY, Ma MX, Song R, et al. Identification of amentoflavone as a potent highly selective PARP-1 inhibitor and its potentiation on carboplatin in human non-small cell lung cancer. Phytomedicine. (2018) 50:88–98. doi: 10.1016/j.phymed.2018.09.012
34. Harstrick A, Bokemeyer C, Scharnofkse M, Hapke G, Reile D, Schmoll HJ. Preclinical activity of a new platinum analogue, lobaplatin, in cisplatin-sensitive and -resistant human testicular, ovarian, and gastric carcinoma cell lines. Cancer Chemother Pharmacol. (1993) 33:43–7. doi: 10.1007/BF00686021
35. Fu Z, Cheng X, Kuang J, Feng H, Chen L, Liang J, et al. CQ sensitizes human pancreatic cancer cells to gemcitabine through the lysosomal apoptotic pathway via reactive oxygen species. Mol Oncol. (2018) 12:529–44. doi: 10.1002/1878-0261.12179
36. Oien DB, Garay T, Eckstein S, Chien J. Cisplatin and pemetrexed activate AXL and AXL inhibitor BGB324 enhances mesothelioma cell death from chemotherapy. Front Pharmacol. (2017) 8:970. doi: 10.3389/fphar.2017.00970
37. Okon IS, Coughlan KA, Zhang M, Wang Q, Zou MH. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. (2015) 290:9101–10. doi: 10.1074/jbc.M114.631580
38. Sanchez-Carranza JN, Diaz JF, Redondo-Horcajo M, Barasoain I, Alvarez L, Lastres P, et al. Gallic acid sensitizes paclitaxel-resistant human ovarian carcinoma cells through an increase in reactive oxygen species and subsequent downregulation of ERK activation. Oncol Rep. (2018) 39:3007–14. doi: 10.3892/or.2018.6382
39. Gong X, Smith JR, Swanson HM, Rubin LP. Carotenoid lutein selectively inhibits breast cancer cell growth and potentiates the effect of chemotherapeutic agents through ROS-mediated mechanisms. Molecules. (2018) 23:E905. doi: 10.3390/molecules23040905
40. Chiu WH, Luo SJ, Chen CL, Cheng JH, Hsieh CY, Wang CY, et al. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem Pharmacol. (2012) 83:1159–71. doi: 10.1016/j.bcp.2012.01.016
41. Spencer BH, McDermott CM, Chess-Williams R, Christie D, Anoopkumar-Dukie S. Prazosin but not tamsulosin sensitises PC-3 and LNCaP prostate cancer cells to docetaxel. Pharmacology. (2018) 102:10–8. doi: 10.1159/000488713
42. Pandey V, Chaube B, Bhat MK. Hyperglycemia regulates MDR-1, drug accumulation and ROS levels causing increased toxicity of carboplatin and 5-fluorouracil in MCF-7 cells. J Cell Biochem. (2011) 112:2942–52. doi: 10.1002/jcb.23210
43. Santoro V, Jia R, Thompson H, Nijhuis A, Jeffery R, Kiakos K, et al. Role of reactive oxygen species in the abrogation of oxaliplatin activity by cetuximab in colorectal cancer. J Natl Cancer Inst Monographs. (2016) 108:djv394. doi: 10.1093/jnci/djv394
44. Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci. (2007) 96:2181–96. doi: 10.1002/jps.20874
45. Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. (2006) 13:951–61. doi: 10.1038/sj.cdd.4401916
46. Brozovic A, Fritz G, Christmann M, Zisowsky J, Jaehde U, Osmak M, et al. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int J Cancer. (2004) 112:974–85. doi: 10.1002/ijc.20522
47. Singh S, Upadhyay AK, Ajay AK, Bhat MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett. (2007) 581:289–95. doi: 10.1016/j.febslet.2006.12.035
48. Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin–DNA adducts. Chem Rev. (1999) 99:2467–98. doi: 10.1021/cr980421n
49. Chang GC, Yu CT, Tsai CH, Tsai JR, Chen JC, Wu CC, et al. An epidermal growth factor inhibitor, Gefitinib, induces apoptosis through a p53-dependent upregulation of pro-apoptotic molecules and downregulation of anti-apoptotic molecules in human lung adenocarcinoma A549 cells. Eur J Clin Pharmacol. (2008) 600:37–44. doi: 10.1016/j.ejphar.2008.10.024
50. Adhikary A, Mohanty S, Lahiry L, Hossain DM, Chakraborty S, Das T. Theaflavins retard human breast cancer cell migration by inhibiting NF-kappaB via p53-ROS cross-talk. FEBS Lett. (2010) 584:7–14. doi: 10.1016/j.febslet.2009.10.081
Keywords: lobaplatin, non-small-cell lung cancer, p53, reactive oxygen species, p38MAPK, apoptosis
Citation: Zhang H, Chen R, Wang X, Zhang H, Zhu X and Chen J (2019) Lobaplatin-Induced Apoptosis Requires p53-Mediated p38MAPK Activation Through ROS Generation in Non-Small-Cell Lung Cancer. Front. Oncol. 9:538. doi: 10.3389/fonc.2019.00538
Received: 09 April 2019; Accepted: 03 June 2019;
Published: 24 July 2019.
Edited by:
Chiara Riganti, University of Turin, ItalyCopyright © 2019 Zhang, Chen, Wang, Zhang, Zhu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Zhu, emh1eGlhb2xpNjJAMTYzLmNvbQ==; Jibei Chen, MTYxMjhAc29odS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.