 Jing Jin1†
Jing Jin1† Xu Wu2,3†
Xu Wu2,3† Jianhua Yin2,3
Jianhua Yin2,3 Mingxing Li2,3
Mingxing Li2,3 Jing Shen2,3Jing Li4
Jing Shen2,3Jing Li4 Yueshui Zhao2,3
Yueshui Zhao2,3 Qijie Zhao2,3Jingbo Wu1Qinglian Wen1Chi Hin Cho2,3
Qijie Zhao2,3Jingbo Wu1Qinglian Wen1Chi Hin Cho2,3 Tao Yi5*Zhangang Xiao2*Liping Qu6
Tao Yi5*Zhangang Xiao2*Liping Qu6- 1Department of Oncology, The Affiliated Hospital of Southwest Medical University, Southwest Medical University, Luzhou, China
- 2Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, China
- 3South Sichuan Institute of Translational Medicine, Luzhou, China
- 4Department of Oncology and Hematology, Hospital (T.C.M) Affiliated to Southwest Medical University, Luzhou, China
- 5School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, China
- 6College of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu, China
The introduction of targeted therapy is the biggest success in the treatment of cancer in the past few decades. However, heterogeneous cancer is characterized by diverse molecular alterations as well as multiple clinical profiles. Specific genetic mutations in cancer therapy targets may increase drug sensitivity, or more frequently result in therapeutic resistance. In the past 3 years, several novel targeted therapies have been approved for cancer treatment, including drugs with new targets (i.e., anti-PD1/PDL1 therapies and CDK4/6 inhibitors), mutation targeting drugs (i.e., the EGFR T790M targeting osimertinib), drugs with multiple targets (i.e., the EGFR/HER2 dual inhibitor neratinib) and drug combinations (i.e., encorafenib/binimetinib and dabrafenib/trametinib). In this perspective, we focus on the most up-to-date knowledge of targeted therapy and describe how genetic mutations influence the sensitivity of targeted therapy, highlighting the challenges faced within this era of precision medicine. Moreover, the strategies that deal with mutation-driven resistance are further discussed. Advances in these areas would allow for more targeted and effective therapeutic options for cancer patients.
Introduction
Targeted therapies usually present with high selectivity, target precisely to specific gene or protein, and exert a biological function with minimal side effects (1), which has distinguished them from most conventional non-specific chemotherapeutic drugs. Targeted therapy has thus been regarded as the biggest success in the treatment of cancer in the past few decades. Many novel promising agents have been experimentally designed and developed and are increasingly entering clinical evaluation. However, the frequently observed alterations in the drug targets have posed a big challenge to successful cancer treatment. Genetic mutations in cancer are resulted from both inherited and environmental factors. In a recent report, it is demonstrated that a large proportion of cancer-related mutations are due to randomized DNA replication errors (2). Notably, the mutations in cancer therapy targets can greatly affect drug sensitivity. Mutation-driven drug resistance is very common in cancer. The efficacy of targeted therapy is thus largely dependent on the mutation profile of tumors in patients. Accurate molecular and genetic profiling of tumor cells is becoming a routine practice before the introduction of targeted therapy in patients.
In recent years, great progress has been made in targeted therapy discovery. Notably, many new drugs are designed primarily based on specific genetic background. For instance, nearly 40–50% of metastatic cutaneous melanoma possess v-raf murine sarcoma viral oncogene homolog B1 (BRAF) mutations (3), and ~90% of these BRAF mutations are caused by substitution of glutamic acid for valine at codon 600 (V600E) (4). Two selective BRAF inhibitors vemurafenib and dabrafenib were approved for the treatment of patients with BRAF-V600E mutation, showing improved progression-free survival (5). In November 2018, the U.S. Food and Drug Administration (FDA) approved an inhibitor of tropomyosin receptor kinases (TRKs), larotrectinib, for treatment of any type of solid tumors with TRK gene fusion (6). This is the second targeted therapy approved not for specific cancer types but for any cancers with specific mutations. Targeted therapies are becoming more precise.
In this perspective, we focus on the updated knowledge of targeted therapy in the last 3 years and describe how genetic mutations influence sensitivity of targeted therapy, highlighting the challenges faced within this era of precision medicine. Moreover, the strategies dealing with mutation-driven resistance are further discussed.
Influence of Genetic Mutation on Sensitivity of Targeted Therapy
It is well-acknowledged that mutations in therapeutic targets can increase or decrease drug sensitivity (Table 1). The main challenge of targeted therapy today is the identification of particular cancer mutations which affect efficacy of targeted therapies as well as the identification of a specific group of patients most likely or unlikely to respond to certain targeted therapies. Despite the great challenges, in the last 3 years, we have seen significant progress in targeted therapy (Table 2), largely owing to the rise of large-scale sequencing technology and big data analysis. Several novel targets, including the programmed death-1/programmed death-ligand 1 (PD1/PDL1) and cyclin-dependent kinases 4 and 6 (CDK4/6), have been validated, with several new targeted drugs being approved. Some newly approved drugs are directly designed to deal with some known activating mutations, such as the T790M mutation in epidermal growth factor receptor (EGFR). Moreover, many new findings have been added to our knowledge of how mutations influence targeted therapies [e.g., the inhibitors of human epidermal growth factor receptor 2 (HER2) and anaplastic lymphoma kinase (ALK)]. Here, based on the most updated research in the last 3 years, we summarize the recent advances of several targeted therapies.
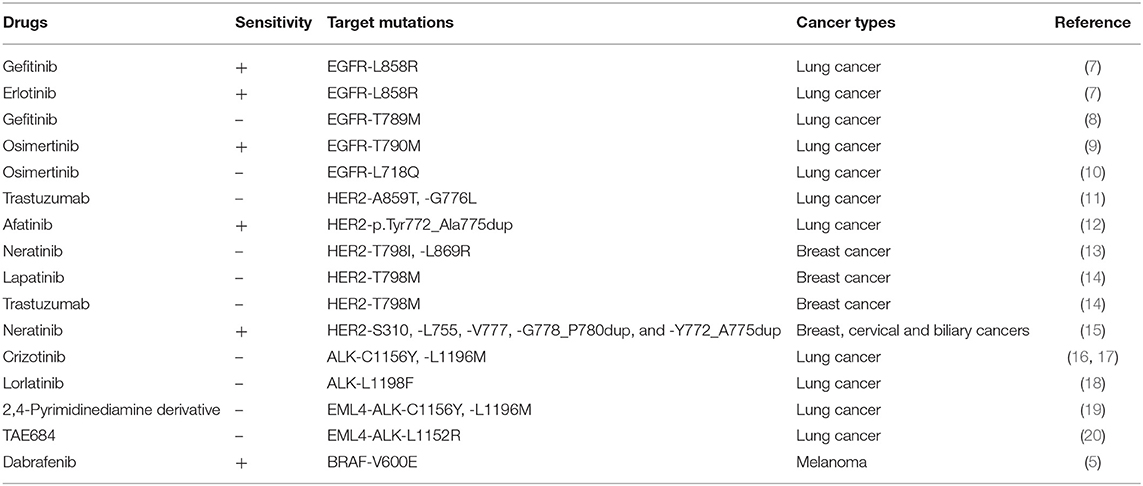
Table 1. Therapeutic response of targeted therapy in mutant cancers.
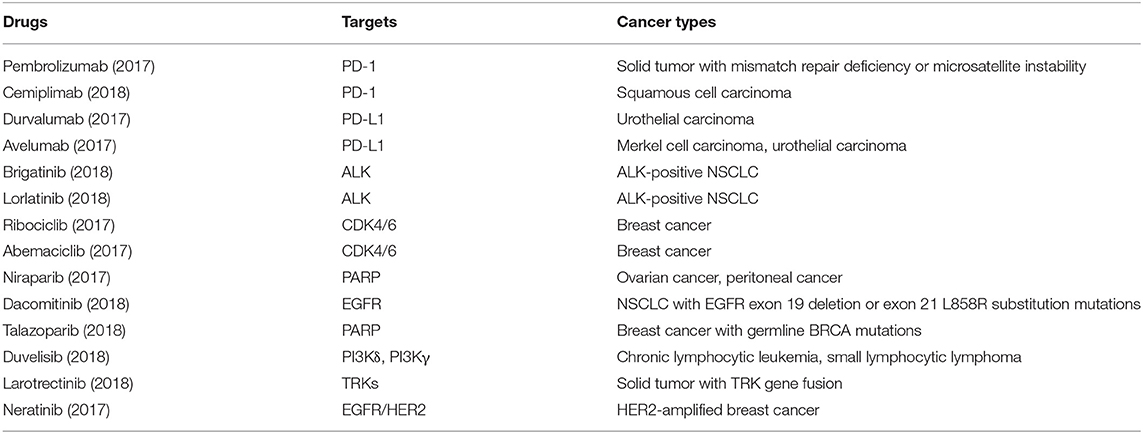
Table 2. Cancer targeted therapy approved by FDA in 2017 and 2018.
Anti-PD1/PDL1 Therapies
So far, there are a total of 6 anti-PD1/PDL1 therapies that have been approved by the FDA. Notably, in 2017, a PD1 antibody pembrolizumab was approved for the treatment of any solid tumor with a mismatch repair deficiency or a microsatellite instability. Monotherapy of PD1/PDL1 blockade has received great success in many types of cancers (21, 22). However, there are certain patients that are gradually developing resistance after an initial response (23). Mutation-driven resistance of anti-PD1/PDL1 therapies has recently been studied in a small number of cancer patients. Zaretsky et al. reported that mutations of JAK1/JAK2 led to the desensitization of cancer cells to IFN-γ and contributed to an acquired resistance of pembrolizumab in patients with melanoma (23). Moreover, in one resistant patient, a frame-shift deletion in exon 1 of the β-2-microglobulin was detected, which may result in the loss of expression of surface the MHC class I (23). More studies are advocated to explore the acquired resistance of immune checkpoint inhibitors.
Resistance of CDK4/6 Inhibitors
Currently, three CDK 4/6 selective targeting inhibitors, palbociclib, ribociclib, and abemaciclib have been approved to treat breast cancer. CDK4/6 inhibitors are increasingly used in clinical settings, but patients eventually show disease progression and the major reasons remain unclear (24). Dysregulation of cyclin D1-CDK4/6-retinoblastoma (Rb) pathway has been implicated in hormone receptor positive (HR+) breast cancer and in chemotherapeutic drug-resistance. Rb is usually intact in HR+ breast cancer and is important for the efficacy of CDK4/6-inhibitors in the treatment of breast cancer (25). It is indicated that T47D cells that become resistant to CDK4/6 inhibitors, develop CCNE1 amplification or Rb1 loss (26). Moreover, the acquisition of multiple de novo somatic Rb1 mutations in metastatic breast cancer patients may result in the emergence of a resistance to CDK 4/6 inhibitors (24). Until now, there has been no report on CDK4/6 mutations in cancer patients and their effect on efficacy of CDK4/6 inhibitors.
EGFR and Different Generation of Tyrosine Kinase Inhibitors (TKIs)
EGFR is a prevalent target in several human cancers, such as lung, breast, colorectal, thyroid, and melanoma cancer. In lung cancer, several generations of small-molecular inhibitors have been developed to target the EGFR tyrosine kinases (27), such as inhibitors gefitinib, erlotinib, osimertinib, and necitumumab. The EGFR mutation in non-small cell lung cancer (NSCLC) was first identified in 2004, and the major missense and deletion mutation of EGFR in NSCLC occurs in the tyrosine kinase-coding domain in exons 18–21 (28). The L858R mutation in the exon 21 and exon-19 frame deletion are the most commonly detected mutation types of EGFR, representing 50 and 40% of tumor patients, respectively (7). These two types of mutations are sensitive to EGFR tyrosine kinase inhibitors (TKIs) in NSCLC. The first-generation TKIs, gefitinib and erlotinib, have a high selective inhibitory activity against both wild-types and these sensitive mutant EGFR (29). Previous studies show that gefitinib and erlotinib are important for the first-line treatment of NSCLC patients with the sensitive EGFR mutations (30, 31). On the other hand, another mutation T790M, a secondary EGFR mutation emerging in NSCLC, can lead to the resistance of more than half of patients' TKIs treatment (32). Very recently, the third-generation TKI inhibitor osimertinib has been approved to effectively target to EGFR T790M mutation with a response rate of 61% in NSCLC, significantly extending the overall survival in patients with the T790M mutation (9). However, the further mutation of a residue in the P-loop (L718Q) has been found to cause resistance to osimertinib (10). Nevertheless, though diverse EGFR mutations are present, the overall survival of lung cancer patients is markedly improved with TKI therapy.
HER2 and Its Inhibitors
In breast cancer, the overall HER2 mutation rate is ~1.6% (25 out of 1,499 patients). In a study by Bose et al., seven HER2 somatic mutations including G309A, D769H, D769Y, V777L, P780ins, V842I, and R896C, have been identified as activating mutations (33). Several patients with HER2 activating mutations are resistant to the reversible HER2 inhibitor lapatinib, but sensitive to the irreversible HER2 inhibitor neratinib. Neratinib as a dual inhibitor of HER2 and EGFR was approved by FDA in 2017. It has been shown that the HER2 L755S mutation results in an acquired resistance to lapatinib in breast cancer, which could be overcome by the neratinib (34). In another study, the HER2-T798I gatekeeper mutation in breast cancer patients with a AHER2-L869R mutation was identified as a mechanism of acquired resistance to neratinib (13). The trial of neratinib has also been conducted in colorectal cancer (CRC) patients. The HER2 gene amplification and mutation in CRC can lead to the resistance of EGFR-targeted therapies cetuximab and panitumumab (35, 36). A negative effect of neratinib monotherapy has recently been confirmed in 12 CRC patients with different tumors harboring HER2 mutations (15). There were no positive therapeutic response and the median PFS was only 1.8 months, indicating that monotherapy with neratinib is ineffective. The underlying mechanisms still require further investigations.
ALK and Different Generation of ALK Inhibitors
ALK has long been identified as a therapeutic target in cancer. The first ALK inhibitor crizotinib was approved by the FDA in 2011 (37). Although most NSCLC patients respond to this drug, tumors become resistant after 1–2 years of treatment. Around 1/3 of crizotinib-resistant tumors harbor mutations within the ALK kinase domain. The most commonly observed mutations of L1196M and G1269A lead to a decreased affinity for crizotinib (38). Other ALK point mutations, such as L1152R, C1156Y, I1171T, F1174L, G1202R, and S1206Y, are also associated with crizotinib resistance (39). Another oncoprotein of fusion-type tyrosine kinase, the EML4-ALK, results from the inversion within the short arm of the human chromosome 2 in 4–5% of cases of NSCLC (40). Two mutations of EML4-ALK, C1156Y, and L1196M, confer a significant resistance to ALK inhibitors, such as crizotinib and PDD (2,4-pyrimidinediamine derivative) (19). The EML4-ALK C1156Y mutation can contribute to a higher resistance to PDD than those in the L1196M mutant form. It is reported that a candidate ALK inhibitor TAE684 can bind to these mutant kinases, which may have potency in overcoming the mutation-driven resistance (41). The new generation ALK inhibitors lorlatinib and brigatinib were approved in 2018 for the treatment of patients with ALK-rearranged NSCLC. Lorlatinib has been demonstrated to inhibit resistant ALK mutations, including ALK G1202R (16). However, Shaw et al. showed that an ALK L1198F mutation together with the C1156Y mutation results in the resistance of lorlatinib in a patient with metastatic ALK-rearranged NSCLC (18). However, the L1198F mutation re-sensitized crizotinib treatment of a resistant tumor. It was demonstrated that both lorlatinib and brigatinib can overcome crizotinib resistance in NSCLC patients (42, 43). Moreover, when brigatinib was combined with anti-EGFR antibody, it was effective against EGFR triple-mutant cells in vitro and in vivo (44).
Strategies for Overcoming Mutation-Driven Resistance
Mutations in cancer therapy targets usually result in the loss of functions and the accumulation of dysfunctional proteins in tumors (45). Moreover, many mutants have oncogenic gain-of-function (GOF) activities including increased tumor proliferation, metastasis and drug resistance (46). Notably, tumor cells that receive targeted therapy may lead to an overactivation of the by-pass signaling pathways to develop resistance. In most cases, multiple alterations are observed in a resistant tumor.
Recently, many strategies dealing with mutation-driven drug resistance have been proposed and evaluated both experimentally and clinically. The traditional chemotherapy concept of “one ligand to one receptor” for a biological response is inadequate. The treatment of a particular type of cancer with the prescriptive drugs involves many special genes, interacting with their respective targets and triggering a series of biological responses. The concept of using multi-drug therapy and seeking multifunctional compounds that can efficiently interact with various targets might be feasible (47). Currently, to overcome mutation-driven drug resistance, the main strategies include: (1) the design of new mutation-targeted compounds to restore wide-type protein activities, to delete mutants or to influence downstream targets; (2) the application of combinational therapy or new compounds for multiple targeting. Here, we give some examples of how to overcome mutation-driven resistance of targeted therapy.
Dacomitinib, an Irreversible Pan-ERBB Inhibitor, Targeting EGFR Activating Mutants
Recently, dacomitinib was approved to use for metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations. In a randomized, multicenter, open-label, phase III trial (ARCHER 1050), the patients with newly diagnosed advanced NSCLC and one EGFR mutation (exon 19 deletion or L858R) received a 45 mg/day dose of oral dacomitinib or a 250 mg/day gefitinib for 28 days. In the dacomitinib group, the progression-free survival (14.7 months, 95% CI 11.1–16.6) was significantly longer than that in the gefitinib group (9.2 months, 95% CI 9.1–11.0) (48). This investigation supports the dacomitinib as the first line therapy for EGFR-mutation NSCLC patients.
Dacomitinib is initially designed for irreversible pan-ERBB inhibition. As a small-molecule covalent binding inhibitor of enzymatically active HER family tyrosine kinases EGFR and HER2, it may act as a potent inhibitor of EGFR T790M mutation (49). Additionally, dacomitinib significantly inhibits both wild-type and the gefitinib-resistant ERBB2 mutation in lung cancer. Based on an in-depth investigation, dacomitinib is an effective drug that may treat NSCLC patients with a T790M-related acquired resistance to gefitinib or erlotinib (8). It has been indicated that dacomitinib significantly improves progression-free survival in EGFR-mutation NSCLC patients and is considered as a new treatment option for this population.
Encorafenib/Binimetinib and Dabrafenib/Trametinib for Dual Inhibition of BRAF and MEK
The FDA approved dabrafenib plus trametinib for the anaplastic thyroid cancer (ATC) with BRAF-V600E mutation in May 2018, as well as for the adjuvant treatment of BRAF V600E/K-mutated melanoma in April 2018. Previous studies revealed that dabrafenib plus trametinib have shown substantial antitumor activity in patients with previously treated BRAF-V600E mutated metastatic NSCLC and untreated BRAFV-600E mutated NSCLC (50, 51). Trametinib is an orally administered MEK1/MEK2 inhibitor that suppresses RAF-dependent MEK phosphorylation and persistently inhibits phosphorylated ERK (a substrate of MEK) (52). Dabrafenib is a reversible and high-efficiency ATP-competitive inhibitor of RAF kinases, especially the mutant BRAF (53). Subbiah et al. reported that the overall response rate of dabrafenib plus trametinib applied in BRAF V600E-mutated ATC (complete reaction plus partial reaction to the best overall response) is 69% (54). In contrast to BRAF inhibitor monotherapy, it has longer progression-free survival and overall survival. Overall, the most common adverse events include fatigue, pyrexia and nausea (54), consistent with previous reports in advanced or metastatic melanoma and NSCLC (50). Dabrafenib plus trametinib is the first regimen approved to have significant clinical efficacy in BRAF V600E-mutated ATC.
In June 2018, the FDA approved the combination of BRAF inhibitor encorafenib and the MEK inhibitor binimetinib for treatment of patients with unresectable or metastatic melanoma with a BRAF-V600E or -V600K mutation. It is the third BRAF/MEK inhibitor combination approved following the dabrafenib/trametinib and vemurafenib/cobimetinib combinations (55). The main adverse events for encorafenib plus binimetinib when applied to BRAF-V600 mutant melanoma are gastrointestinal reactions, including nausea, diarrhea and vomiting. Additionally, this combination has a lower calorific value and photosensitivity than other available BRAF-MEK inhibitor combinations do (56). Considerable evidence supports that the median progression-free survival was 14.9 months with encorafenib plus binimetinib, compared with 7.3 months with vemurafenib (57). Therefore, it is an effective therapeutic option in patients with unresectable or metastatic melanoma, with a BRAF V600E or V600K mutation.
Conclusions
In the new era of targeted therapy, treatment options are increasingly based on the precise molecular and genetic profiling of tumor cells (58). Currently, the main challenge for further novel drug development in targeted therapy is the clarification of specific molecular mechanisms underlying the varied forms of tumors in clinic. It has been acknowledged that cancer is caused by a set of driver mutations. In this regard, it is of great significance to: (1) identify and validate key mutant genes and proteins in cancers as new targets; (2) identify patients most likely and unlikely to benefit from certain targeted therapies; (3) evaluate the mechanism of mutation-driven drug resistance. In past decades, several key mutations which influence drug sensitivity have been identified in various cancers. In order to deal with mutation-driven drug resistance, new methods and drugs have been discovered and approved for clinical use (47). Even so, detailed individualized treatment strategies targeting specific tumorigenesis and drug resistant mechanisms are still required. Advances in these areas would allow for more targeted and effective therapeutic options for more cancer patients.
Author Contributions
ML, JS, JL, YZ, JW, LQ, and QW were responsible for the review of the literature. JJ, JY, XW, QZ, CC, and ML wrote the manuscript. XW and LQ drew the Tables. XW, TY, and ZX designed the study and contributed with the valuable discussion and revision of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant nos. 81503093, 81602166, and 81672444) and the Joint Funds of the Southwest Medical University and Luzhou (2016LZXNYD-T01, 2017LZXNYD-Z05, and 2017LZXNYD-J09). The funding from National Natural Science Foundation of China (Grant nos. 81503093) will cover open access fee.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Scotti L, Mendonca Junior FJ, Ishiki HM, Ribeiro FF, Singla RK, Barbosa Filho JM, et al. Docking studies for multi-target drugs. Curr Drug Targ. (2017) 18:592–604. doi: 10.2174/1389450116666150825111818
2. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. (2017) 355:1330–4. doi: 10.1126/science.aaf9011
3. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. (2005) 353:2135–47. doi: 10.1056/NEJMoa050092
4. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. (2002) 417:949–54. doi: 10.1038/nature00766
5. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2012) 380:358–65. doi: 10.1016/s0140-6736(12)60868-x
6. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
7. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. (2005) 97:339–46. doi: 10.1093/jnci/dji055
8. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. (2005) 352:786–92. doi: 10.1056/NEJMoa044238
9. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. (2017) 376:629–40. doi: 10.1056/NEJMoa1612674
10. Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, et al. L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chem Sci. (2018) 9:2740–9. doi: 10.1039/c7sc04761d
11. Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med. (2006) 354:2619–21. doi: 10.1056/NEJMc060020
12. De Greve J, Teugels E, Geers C, Decoster L, Galdermans D, De Mey J, et al. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer. (2012) 76:123–7. doi: 10.1016/j.lungcan.2012.01.008
13. Hanker AB, Brewer MR, Sheehan JH, Koch JP, Sliwoski GR, Nagy R, et al. An acquired HER2(T798I) gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discov. (2017) 7:575–85. doi: 10.1158/2159-8290.cd-16-1431
14. Rexer BN, Ghosh R, Narasanna A, Estrada MV, Chakrabarty A, Song Y, et al. Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin Cancer Res. (2013) 19:5390–401. doi: 10.1158/1078-0432.ccr-13-1038
15. Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. (2018) 554:189–94. doi: 10.1038/nature25475
16. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. (2015) 21:2227–35. doi: 10.1158/1078-0432.ccr-14-2791
17. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. (2014) 370:1189–97. doi: 10.1056/NEJMoa1311107
18. Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. (2016) 374:54–61. doi: 10.1056/NEJMoa1508887
19. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. (2010) 363:1734–9. doi: 10.1056/NEJMoa1007478
20. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. (2011) 71:6051–60. doi: 10.1158/0008-5472.can-11-1340
21. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. (2013) 369:134–44. doi: 10.1056/NEJMoa1305133
22. Peters S, Kerr KM, Stahel R. PD-1 blockade in advanced NSCLC: a focus on pembrolizumab. Cancer Treat Rev. (2018) 62:39–49. doi: 10.1016/j.ctrv.2017.10.002
23. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
24. Condorelli R, Spring L, O'Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. (2018) 29:640–5. doi: 10.1093/annonc/mdx784
25. Cancer Genome Atlas Network. (2012). Comprehensive molecular portraits of human breast tumours. Nature. 490, 61–70. doi: 10.1038/nature11412
26. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. (2016) 76:2301–13. doi: 10.1158/0008-5472.can-15-0728
27. Renhowe PA. Inhibitors of growth factor receptor kinase-dependent signaling pathways in anticancer chemotherapy–clinical progress. Curr Opin Drug Discov Devel. (2002) 5:214. doi: 10.2174/0929867024606957
28. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. (2004) 101:13306–11. doi: 10.1073/pnas.0405220101
29. Morin MJ. From oncogene to drug: development of small molecule tyrosine kinase inhibitors as anti-tumor and anti-angiogenic agents. Oncogene. (2000) 19:6574–83. doi: 10.1038/sj.onc.1204102
30. Inoue A, Kobayashi K, Usui K, Maemondo M, Okinaga S, Mikami I, et al. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J Clin Oncol. (2009) 27:1394–400. doi: 10.1200/JCO.2008.18.7658
31. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. (2010) 362:2380–8. doi: 10.1056/NEJMoa0909530
32. Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. (2005) 37:1315–6. doi: 10.1038/ng1671
33. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. (2013) 3:224–37. doi: 10.1158/2159-8290.cd-12-0349
34. Xu X, De Angelis C, Burke KA, Nardone A, Hu H, Qin L, et al. HER2 Reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2(+) breast cancer. Clin Cancer Res. (2017) 23:5123–34. doi: 10.1158/1078-0432.ccr-16-2191
35. Dienstmann R, Tabernero J. Spectrum of gene mutations in colorectal cancer: implications for treatment. Cancer J. (2016) 22:149–55. doi: 10.1097/ppo.0000000000000191
36. Ross JS, Fakih M, Ali SM, Elvin JA, Schrock AB, Suh J, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. (2018) 124:1358–73. doi: 10.1002/cncr.31125
37. Ou SH. Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des Devel Ther. (2011) 5:471–85. doi: 10.2147/dddt.s19045
38. Kimura AGM. Molecularly targeted therapy: past, present and future. Chemotherapy 1:105. doi: 10.4172/2167-7700.1000105
39. Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. (2011) 78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x
40. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/nature05945
41. Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. (2011) 17:7394–74. doi: 10.1158/1078-0432.ccr-11-1648
42. Zhang S, Anjum R, Squillace R, Nadworny S, Zhou T, Keats J, et al. The potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. (2016) 22:5527–38. doi: 10.1158/1078-0432.ccr-16-0569
43. Shaw AT, Felip E, Bauer TM, Besse B, Navarro A, Postel-Vinay S, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. (2017) 18:1590–9. doi: 10.1016/s1470-2045(17)30680-0
44. Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun. (2017) 8:14768. doi: 10.1038/ncomms14768
45. Iranzo J, Martincorena I, Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proc Natl Acad Sci USA. (2018) 115:E6010–9. doi: 10.1073/pnas.1803155115
46. Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J Mol Biol. (2017) 429:1595–606. doi: 10.1016/j.jmb.2017.03.030
47. Wang L, Wang H, Song D, Xu M, Liebmen M. New strategies for targeting drug combinations to overcome mutation-driven drug resistance. Semin Cancer Biol. (2017) 42:44–51. doi: 10.1016/j.semcancer.2016.11.002
48. Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. (2017) 18:1454–66. doi: 10.1016/s1470-2045(17)30608-3
49. Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. (2007) 67:11924–32. doi: 10.1158/0008-5472.can-07-1885
50. Chen G, McQuade JL, Panka DJ, Hudgens CW, Amin-Mansour A, Mu XJ, et al. Clinical, molecular, and immune analysis of dabrafenib-trametinib combination treatment for BRAF inhibitor-refractory metastatic melanoma: a phase 2 clinical trial. JAMA Oncol. (2016) 2:1056–64. doi: 10.1001/jamaoncol.2016.0509
51. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. (2017) 18:1307–16. doi: 10.1016/s1470-2045(17)30679-4
52. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. (2011) 17:989–1000. doi: 10.1158/1078-0432.ccr-10-2200
53. Puszkiel A, Noe G, Bellesoeur A, Kramkimel N, Paludetto MN, Thomas-Schoemann A, et al. Clinical pharmacokinetics and pharmacodynamics of dabrafenib. Clin Pharmacokinet. (2019) 58:451–67. doi: 10.1007/s40262-018-0703-0
54. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. (2018) 36:7–13. doi: 10.1200/jco.2017.73.6785
55. Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib orencorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. (2018b) 19:1315–27. doi: 10.1016/s1470-2045(18)30497-2
56. Sullivan RJ, Weber JS, Patel SP, Dummer R, Miller WH, Cosgrove D, et al. A phase Ib/II study of BRAF inhibitor (BRAFi) encorafenib (ENCO) plus MEK inhibitor (MEKi) binimetinib (BINI) in cutaneous melanoma patients naive to BRAFi treatment. J Clin Oncol. (2015) 33:9007–9007. doi: 10.1200/jco.2015.33.15_suppl.9007
57. Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. (2018) 19:603–15. doi: 10.1016/s1470-2045(18)30142-6
Keywords: targeted therapy, cyclin-dependent kinases 4/6, somatic mutation, resistance, EGFR, PD-1/PD-L1
Citation: Jin J, Wu X, Yin J, Li M, Shen J, Li J, Zhao Y, Zhao Q, Wu J, Wen Q, Cho CH, Yi T, Xiao Z and Qu L (2019) Identification of Genetic Mutations in Cancer: Challenge and Opportunity in the New Era of Targeted Therapy. Front. Oncol. 9:263. doi: 10.3389/fonc.2019.00263
Received: 25 January 2019; Accepted: 22 March 2019;
Published: 16 April 2019.
Edited by:
Yan-yan Yan, Shanxi Datong University, ChinaReviewed by:
Shengpeng Wang, University of Macau, ChinaMaria Munoz Caffarel, Biodonostia Health Research Institute, Spain
Tinghong Ye, Sichuan University, China
Copyright © 2019 Jin, Wu, Yin, Li, Shen, Li, Zhao, Zhao, Wu, Wen, Cho, Yi, Xiao and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Yi, eWl0YW9AaGtidS5lZHUuaGs=
Zhangang Xiao, eHpnNTU1ODk4QGhvdG1haWwuY29t
†These authors have contributed equally to the work