 Irene Scarfò
Irene Scarfò Matthew J. Frigault
Matthew J. Frigault Marcela V. Maus
Marcela V. Maus
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 16 April 2019
Sec. Hematologic Malignancies
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.00259
This article is part of the Research Topic Cutaneous T-cell Lymphomas View all 8 articles
Cutaneous T cell lymphomas (CTCL) are a heterogeneous group of malignancies characterized by the expansion of a malignant T cell clone. Chimeric Antigen Receptor (CAR) T cell therapy has shown impressive results for the treatment of B-cell tumors, but several challenges have prevented this approach in the context of T cell lymphoma. These challenges include the possibilities of fratricide due to shared T-cell antigens, T cell immunodeficiency, and CAR transduction of malignant cells if CAR T are manufactured in the autologous setting. In this review, we discuss these and other challenges in detail and summarize the approaches currently in development to overcome these challenges and offer cellular targeting of T cell lymphomas.
Cutaneous T-cell lymphomas (CTCL) are a heterogeneous group of malignancies of T-cell origin that occur primarily in the skin. They are the second most common form of extranodal Non-Hodgkin lymphomas (NHL) and their incidence has been increasing over the time (1). Mycosis fungoides (MF) and Sézary syndrome (SS) are the most common subtypes, which represent about 70–75% of CTCL (2, 3); other frequent types include primary cutaneous CD30+ T-cell lymphoproliferative disorders, adult T-cell leukemia/lymphoma and a portion of peripheral T-cell lymphoma not otherwise specified (PTCL-NOS). Although both cutaneous and/or systemic therapies have been developed for these tumors, long-term outcomes are characterized by high relapse rates with advanced forms of CTCL considered incurable. Recent progress has been made using immunotherapy approaches. These include the development of monoclonal antibodies, such as brentuximab vedotin, which has shown clinical efficacy leading to FDA approval in certain disease sub-types (4–6). Here, we discuss recent advances in adoptive Chimeric Antigen Receptor (CAR) T cell therapy and summarize the challenges and opportunities for the treatment of CTCL.
Immunotherapy has emerged as groundbreaking approach for the treatment of cancer and includes monoclonal antibodies, immune checkpoint blockade, tumor vaccines, and most recently, CAR T cell therapy. CAR-T cell therapy redirects a poly-clonal T-cell population against a tumor specific antigen in an MHC independent fashion. Based on impressive phase I/II data, CAR-T cell therapies are now approved for CD19+ B-cell malignancies including pediatric acute lymphoblastic leukemia (B-ALL) and large cell lymphoma (7, 8). Although B-cell aplasia is a common and manageable side effect of CD19 directed CAR-T cell therapy, T-cell aplasia is less tolerable in the long term, and can be life-threatening if it is not reversed or ameliorated. Furthermore, in the case of CTCL, malignant and normal T cells express many shared surface markers, limiting our ability to differentially target these populations. For this reason, special considerations have to be made when designing CARs for the treatment of T-cell lineage malignancies. First, permanent T cell aplasia is not acceptable, but approaches using either transient CAR T cell expression or persistence or suicide genes could be used to eliminate the CAR cells and allow for T cell immune reconstitution (Figure 1A). Second, killing of CAR-expressing cells by each other, known as fratricide, can undermine the ex/in vivo expansion of modified T cells and the generation of CAR T cell products (Figure 1B). Third, circulating tumor cells can contaminate leukapheresis products and be transduced with CARs during manufacturing, which could be associated with a growth advantage for the transduced tumor cells or resistance to CAR-T cell mediated cytotoxicity (Figure 1C). This phenomenon has been recently documented in a B-ALL patient relapsed after CTL019 treatment (9), whereby transduction of the tumor cells with the CAR led to “masking” the expression of the CD19 target antigen and therefore resistance to the CAR T cell-mediated killing. All these aspects need to be considered for the development of CAR T cell therapy against CTCL. However, the unmet need in T cell lymphomas is great, and effective treatments would represent a significant therapeutic advance.
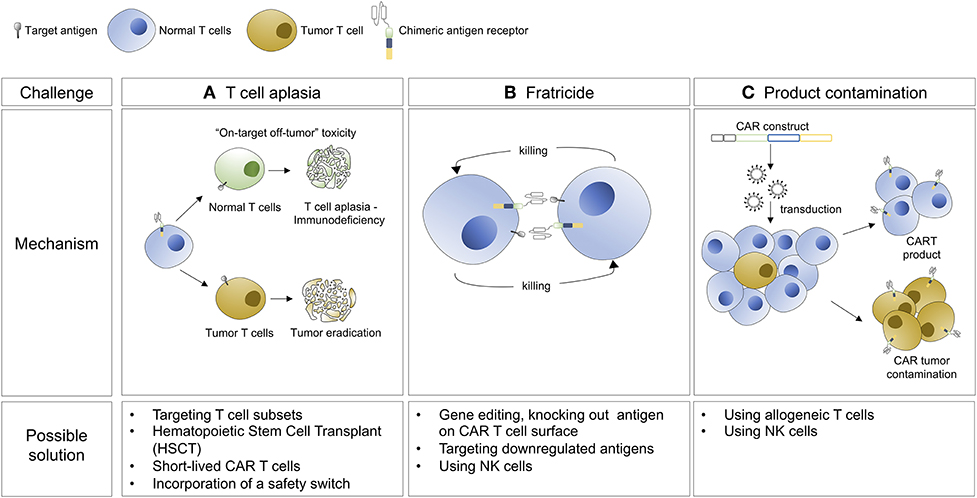
Figure 1. Hurdles associated with the development of CAR T cell therapy for the treatment of CTCL and possible solutions.
It has been difficult to identify targets uniquely expressed on malignant but not on normal T cells. One strategy has been to target molecules expressed by a subpopulation of T cells, or which are downregulated when T cells are activated. This approach has been adopted for the design of CAR against CD4, CD5, CD7, CD30, CD37, CCR4, and the 2 alleles of the T cell receptor beta chains (TRBC1/TRBC2) (Table 1).
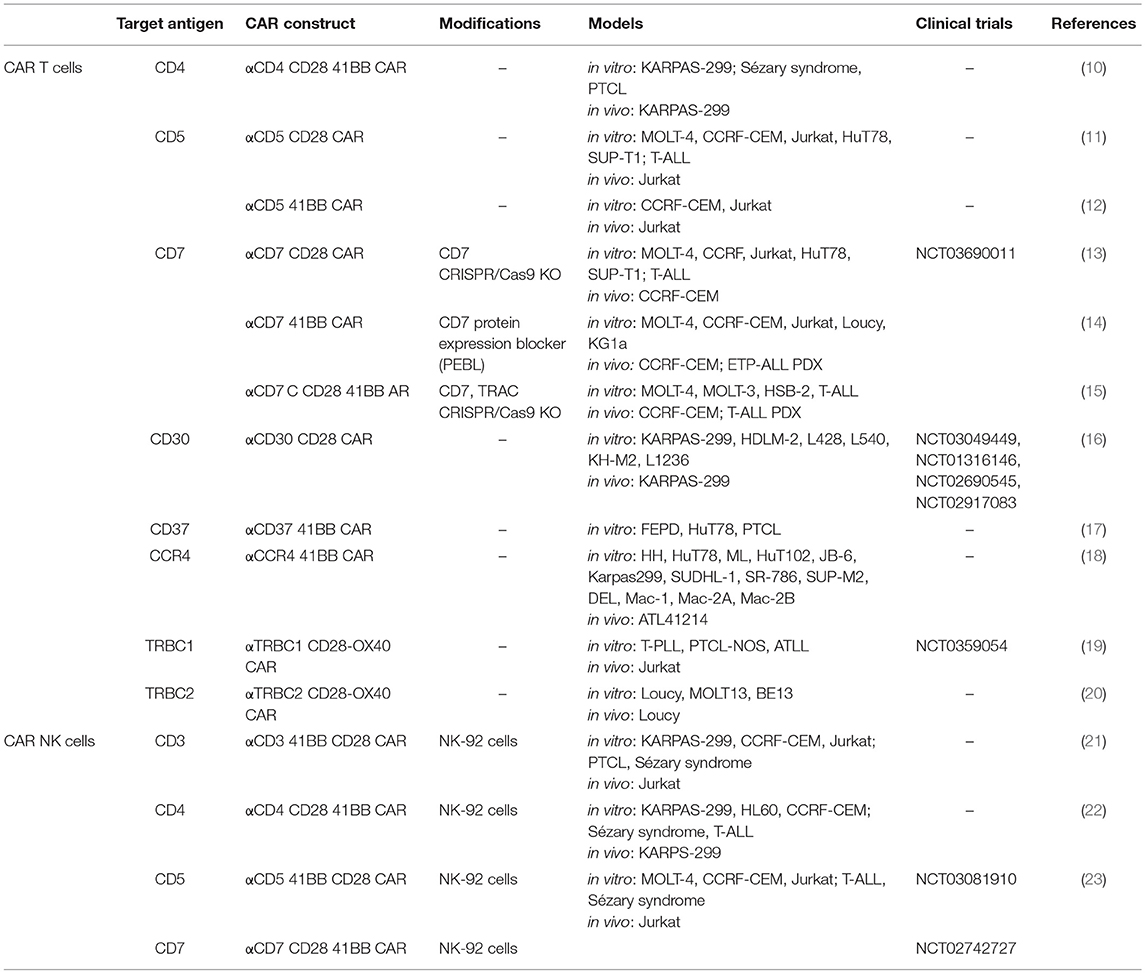
Table 1. CAR T/NK cells for the treatment of CTCL.
CD4 was one of the first molecules selected as target for CAR T cell therapy design. It is highly and uniformly expressed by T cell lymphomas, including CTCL which are predominantly comprised of peripheral CD4 positive T cells. In 2016, Pinz and colleagues showed preclinical efficacy of anti-CD4 CAR T cells in vitro and in a xenograft mouse model of ALCL (10). Although this approach demonstrated the potential for CAR-T cells in ALCL, ongoing CD4 depletion could lead to a T cell immunodeficiency similar to that observed in the acquired immunodeficiency syndrome (AIDS) induced by the human immunodeficiency virus (HIV).
CD5 is another highly expressed antigen on malignant T cells (24, 25). In normal mature T cells, it has a costimulatory role in synergy with CD28 and TCR/CD3 (26–28); previous studies have shown that its expression is post-transnationally regulated (29). Anti-CD5 CAR T cells have been tested in two configurations. The first, designed by Mamonkin et al. included CD28 as costimulatory domain and showed a transient fratricide and a limited bystander killing of normal T cells due indeed to surface downregulation of CD5 protein (11). These CAR T cells demonstrated preclinical activity in vitro against different TCL and T-ALL cell lines, including the HUT78 Sézary syndrome cells, but only partial clearance of T-ALL xenograft tumor, suggesting a lack of CAR-T cell persistence. For this reason, Mamonkin and colleagues designed a second version of the CAR using 4-1BB as costimulatory domain. Interestingly, they reported a higher fratricide when expanding 4-1BB CAR T cells compared to CD28 CAR T cells. The authors demonstrated that 4-1BB upregulates ICAM-1 molecule increasing the stability of the immunological synapse and consequent killing (12). In order to regulate CD5 targeted killing, the authors put their 4-1BB CAR under an inducible promoter allowing for transient expression and therefore killing. This approach demonstrated complete elimination of T-ALL xenograft tumors, but raised concerns about the clinical safety and the immunogenicity of transactivator proteins. Moreover, CD5 is not expressed by many malignant T cell clones and can be easily down regulated, potentially leading to antigen escape.
CD7 is a transmembrane glycoprotein which is a primary marker for acute T-ALL and is highly expressed in a subset of T cell lymphomas (24, 30, 31). In normal tissues, CD7 expression is confined to T and natural killer (NK) cells. Recently, various groups have independently shown the potential of targeting CD7, however, all the studies reported a lack of CD7 downregulation on effector T-cells which resulted in extensive fratricide. Given the near universal expression of CD7 on normal T-cells, Gomes-Silva et al. used CRISPR/Cas9 system to disrupt the CD7 locus. Genetic knockout (KO) of CD7 led to normal expansion of CD7 specific CAR T cells without detectible fratricide of gene disrupted T cells. More importantly, they also demonstrated that anti-CD7 CAR T cells retained anti-viral activity in vitro which may provide protection in the context of T and NK ablation (13). These data led to the opening of a first in human phase I clinical trial (NCT03690011) of CD7 specific CAR-T cells in T cell leukemia and lymphoma.
A second group designed an elegant method to prevent membrane expression of CD7 protein called protein expression blocker (PEBL) by coupling an intracellular retention domain KDEL to an anti-CD7 single chain variable fragment. Transduction of anti-CD7 PEBL lead to abrogation of CD7 expression and inhibition of fratricide of PEBL CAR T cells. These modified T cells showed anti leukemic activity in cell-lines and patient derived xenograft (PDX) models of T-ALL (14). An additional advantage of this approach would be the possible direct translation to the clinic using existing current good manufacturing practice (cGMP) manufacturing processes.
Finally, Cooper and colleagues designed the first allogeneic CAR T cell product for the treatment of CD7 positive T cell tumors (15). They developed a CRISPR/Cas9 multiplex system targeting the T cell receptor alpha chain (TRAC) and CD7 genes. These UCART7 T cells were fratricide-resistant and killed PDX model of T-ALL in vivo without inducing xenogeneic graft-vs.-host disease (GvHD).
CD30 is a more selectively expressed T lineage marker, present in all anaplastic large cell lymphomas (ALCLs) and some T-ALL (32, 33). Antibody drug conjugates such as brentuximab vedotin have been used to treat cutaneous lymphomas and advanced stage mycosis fungoides with clinical success (34, 35). Despite this success, significant drug conjugate mediated toxicities, including neuropathy (62%) and cytopenias (upwards of 78%), as well as limited durability of response, have led to the development of anti-CD30 CAR T cells. Although the majority of CD30. CAR-T cell trials have focused on Hodgkin lymphomas, results of 2 ALCL patients treated with CD30.CAR-Ts are promising and include one complete remission for 9 months without impaired virus-specific immunity (16).
CCR4 is a chemokine receptor abundantly expressed on the surface of T cell tumors including T-ALL, PTCL, and CTCL and is responsible for the homing of malignant cells to the skin. Various anti-CCR4 monoclonal antibodies are in development including mogamulizumab which already has approval in Japan for the treatment of relapsed/refractory adult T-cell leukemia/lymphoma. CCR4 has been validated as a CAR-T target as Perera and colleagues have demonstrated the ability of an anti-CCR4-CAR to lyse multiple T-cell lines in vitro and clear T-ALL tumor in a xenograft mouse model (18). Furthermore, since CCR4 is also expressed by T regulatory (Tregs) cells, the authors speculate that anti-CCR4 CAR T cells would be able to eliminate Tregs cells which have typically an immunosuppressive role in the tumor microenvironment. Nevertheless, given the wide expression of CCR4, safety questions about the toxicity profile of these cells still need to be addressed.
CD37 is a four-passage transmembrane protein belonging to the tetraspanin superfamily. Our group and others have shown that it is expressed not only on B-cell tumors but on T cell malignancies as well (17). We generated CAR37 T cells and demonstrated specific in vitro killing of T cell lines, including HUT78 cells. We did not detect significant fratricide of CAR+ and/or untransduced T-cells in vitro lessening the concern for CAR-T mediated T cell aplasia. In vivo experiments and an upcoming phase I trial of CAR-37 will further evaluate CD37 as a therapeutic target.
Another promising approach is the targeting of the T-cell receptor beta-chain constant domain, type 1(TRBC1) or 2 (TRBC2). Maciocia and colleagues showed that T-cell malignancies are clonally restricted to either TRBC1 or TRBC2, akin to kappa/lambda clonality in multiple myeloma. They subsequently developed anti-TRBC1 CAR T cells and demonstrated specific killing of TRBC1 expressing normal and malignant T cells while sparing TRBC2 restricted cells (19). Given the near 50:50 distribution of TRBC1:TRBC2 expression, this approach offers the potential to spare an adequate number of normal T cells to maintain polyclonal T-cell immunity. An ongoing phase I/II clinical trial is evaluating the safety and efficacy of TRBC1 specific CAR-T cells in TRBC1 positive malignancies (NCT03590574). This group is also developing a novel TRBC2 binder and anti-TRBC2 CAR for TRBC2 restricted clones (20). These strategies open new opportunities for the treatment of T cell malignancies.
There is an expanding interest in using natural killer cells as source of autologous and allogeneic cellular therapy. NK cells are cytotoxic immune cells which represent our first line of defense against pathogens and malignant cells. NK cells express CD56, CD16, and CD7 but lack TCR, CD3, and CD5 expression (36). Notably, they are able to kill target cells in a non-antigen dependent fashion, do not cause GVHD and are short-lived relative to their T-cell counterparts. All these characteristics make them attractive candidates for genetic engineering with CAR molecules, especially in the context of T cell malignancies. As NK cells do not share as many common T-cell antigens, fratricide would not be expected during cellular manufacturing. Additionally, their short lifespan may prevent long-term T cell aplasia while their lack of a of TCR make them a potential source of allogeneic products.
For these reasons CAR NK cells targeting CD3, CD4, CD5, and CD7 have been explored (Table 1). Chen et al. utilized a NK-92 human cell line transduced with a 3rd generation anti-CD3 CAR and demonstrated in vitro activity against T-ALL cell lines and primary PTCL samples. Although these NK-92 CAR cells were unable to completely eradicate leukemic cells in a xenograft mouse model, improvements in vivo persistence may improve on the short lived nature of CD3CAR NK-92 cells (21). This same approach utilizing the NK-92 cell line for CAR therapy has been demonstrated for CD4 and CD5 positive lymphomas as well as with improvements in both in vivo persistence as well as activity in other disease such as T-ALL and PTCL (22, 23). Although NK-92 CARs carry promise, there is a significant safety concern regarding the use of a transformed human NK lymphoma cell line as an NK cell source. Approaches utilizing irradiated CAR NK-92 cells are being evaluated in clinical trials (NCT03081910, NCT02742727), however any approach meant to limit the persistence and expansion of an immortalized cell line in vivo will have similarly detrimental effects on CAR persistence and efficacy.
The major challenges in the development of adoptive cell therapy for T cell tumors, as mentioned above, remain fratricide, T cell aplasia and the potential for leukemic transduction or poor T cell function if used in the autologous setting. Approaches to overcome fratricide include the genetic modification and deletion of the T cell antigen in the case of long-term CAR-T cell persistence or regulated CAR-T expression. To ensure restoration of T cell immunity, transient CAR expression can be achieved incorporation of a CAR suicide gene, transient CAR expression using mRNA electroporation, or short-lived NK cell lines. Finally, given that these toxicities may be tolerable initially, CAR-T cells followed by an ablative hematopoietic stem cell transplant may allow for hematologic rescue following CAR-T mediated disease clearance. As most of the models to date have utilized normal donor human T cells for CAR manufacturing, we must also consider the underlying fitness of the starting cell product. Peripheral T-cells from patients with underlying T cell malignancy routinely demonstrate impairments in T cell function as well as reduced quantities in peripheral blood, given the extensive prior treatment burden and immune-dysregulation. Approaches utilizing allogeneic donors and gene-editing techniques to remove the endogenous TCR, or CAR products generated from autologous or allogeneic NK cells may offer creative solutions. These include the potential for multiple allogeneic sources, such as peripheral blood, umbilical cord blood, or effector cells generated from induced pluripotent stem cells (iPCSs). Regardless of cell source, target antigen, and the challenges and obstacles each approach may carry, CAR-effector cells as a treatment option for T-cell lymphomas may provide an exciting opportunity for these diseases.
IS wrote and edited the manuscript. MF and MM contributed to manuscript planning and editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Wilcox RA. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. (2016) 91:151–65. doi: 10.1002/ajh.24233
2. Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. (2009) 113:5064–73. doi: 10.1182/blood-2008-10-184168
3. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. (2005) 105:3768–85. doi: 10.1182/blood-2004-09-3502
4. Argnani L, Broccoli A, Zinzani PL. Cutaneous T-cell lymphomas: focusing on novel agents in relapsed and refractory disease. Cancer Treat Rev. (2017) 61:61–9. doi: 10.1016/j.ctrv.2017.10.007
5. Folkes AS, Feng M, Zain JM, Abdulla F, Rosen ST, Querfeld C. Targeting CD47 as a cancer therapeutic strategy: the cutaneous T-cell lymphoma experience. Curr Opin Oncol. (2018) 30:332–37. doi: 10.1097/CCO.0000000000000468
6. Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab vedotin or physician's choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet. (2017) 390:555–66. doi: 10.1016/S0140-6736(17)31266-7
7. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
8. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
9. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. (2018) 24:1499–503. doi: 10.1038/s41591-018-0201-9
10. Pinz K, Liu H, Golightly M, Jares A, Lan F, Zieve GW, et al. Preclinical targeting of human T-cell malignancies using CD4-specific chimeric antigen receptor (CAR)-engineered T cells. Leukemia. (2016) 30:701–7. doi: 10.1038/leu.2015.311
11. Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. (2015) 126:983–992. doi: 10.1182/blood-2015-02-629527
12. Mamonkin M, Mukherjee M, Srinivasan M, Sharma S, Gomes-Silva D, Mo F, et al. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol Res. (2018) 6:47–58. doi: 10.1158/2326-6066.CIR-17-0126
13. Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. (2017) 130:285–96. doi: 10.1182/blood-2017-01-761320
14. Png YT, Vinanica N, Kamiya T, Shimasaki N, Coustan-Smith E, Campana D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. (2017) 1:2348–60. doi: 10.1182/bloodadvances.2017009928
15. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. (2018) 32:1970–83. doi: 10.1038/s41375-018-0065-5
16. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. (2017) 127:3462–71. doi: 10.1172/JCI94306
17. Scarfo I, Ormhøj M, Frigault MJ, Castano AP, Lorrey S, Bouffard AA, et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood. (2018) 132:1495–506. doi: 10.1182/blood-2018-04-842708
18. Perera LP, Zhang M, Nakagawa M, Petrus MN, Maeda M, Kadin ME, et al. Chimeric antigen receptor modified T cells that target chemokine receptor CCR4 as a therapeutic modality for T-cell malignancies. Am J Hematol. (2017) 92:892–901. doi: 10.1002/ajh.24794
19. Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med. (2017) 23:1416–23. doi: 10.1038/nm.4444
20. Onuoha S, Ferrari M, Bulek A, Bughda R, Manzoor S, Srivastava S, et al. Structure Guided Engineering of Highly Specific Chimeric Antigen Receptors for the Treatment of T Cell Lymphomas. ASH Annual Meeting, San Diego, CA (2018).
21. Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. (2016) 7:56219–32. doi: 10.18632/oncotarget.11019
22. Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. (2017) 8:112783–96. doi: 10.18632/oncotarget.22626
23. Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. (2017) 31:2151–60. doi: 10.1038/leu.2017.8
24. Campana D, van Dongen JJ, Mehta A, Coustan-Smith E, Wolvers-Tettero IL, Ganeshaguru K, et al. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood. (1991) 77:1546–54.
25. Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. (1993) 82:343–62.
26. Kroesen BJ, Bakker A, van Lier RA, The HT, de Leij L. Bispecific antibody-mediated target cell-specific costimulation of resting T cells via CD5 and CD28. Cancer Res. (1995) 55:4409–15.
27. Vandenberghe P, Ceuppens JL. Immobilized anti-CD5 together with prolonged activation of protein kinase C induce interleukin 2-dependent T cell growth: evidence for signal transduction through CD5. Eur J Immunol. (1991) 21:251–9. doi: 10.1002/eji.1830210203
28. Vandenberghe P, Verwilghen J, Van Vaeck F, Ceuppens JL. Ligation of the CD5 or CD28 molecules on resting human T cells induces expression of the early activation antigen CD69 by a calcium- and tyrosine kinase-dependent mechanism. Immunology. (1993) 78:210–7.
29. Lu X, Axtell RC, Collawn JF, Gibson A, Justement LB, Raman C. AP2 adaptor complex-dependent internalization of CD5: differential regulation in T and B cells. J Immunol. (2002) 168:5612–20. doi: 10.4049/jimmunol.168.11.5612
30. Murphy K, Weaver C. Janeway's immunobiology. New York, NY: Garland Science/Taylor and Francis Group, LLC (2016). p. 904.
31. Gorczyca W. Atlas of Differential Diagnosis in Neoplastic Hematopathology. (2014). CRC Press; Taylor & Francis Group.
32. Falini B, Pileri S, Pizzolo G, Dürkop H, Flenghi L, Stirpe F, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. (1995) 85:1–14.
33. Zheng W, Medeiros LJ, Young KH, Goswami M, Powers L, Kantarjian HH, et al. CD30 expression in acute lymphoblastic leukemia as assessed by flow cytometry analysis. Leuk Lymphoma. (2014) 55:624–7. doi: 10.3109/10428194.2013.820293
34. Kim YH, Tavallaee M, Sundram U, Salva KA, Wood GS, Li S, et al. Phase II investigator-initiated study of brentuximab vedotin in mycosis fungoides and sezary syndrome with variable CD30 expression level: a multi-institution collaborative project. J Clin Oncol. (2015) 33:3750–8. doi: 10.1200/JCO.2014.60.3969
35. Duvic M, Tetzlaff MT, Gangar P, Clos AL, Sui D, Talpur R. Results of a phase II trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis. J Clin Oncol. (2015) 33:3759–65. doi: 10.1200/JCO.2014.60.3787
Keywords: T cell lymphomas/leukemias, CAR T cells, adoptive cell therapy, immunotherapy, cutaneous
Citation: Scarfò I, Frigault MJ and Maus MV (2019) CAR-Based Approaches to Cutaneous T-Cell Lymphoma. Front. Oncol. 9:259. doi: 10.3389/fonc.2019.00259
Received: 21 January 2019; Accepted: 22 March 2019;
Published: 16 April 2019.
Edited by:
Catherine Grace Chung, The Ohio State University, United StatesReviewed by:
Yona Keisari, Tel Aviv University, IsraelCopyright © 2019 Scarfò, Frigault and Maus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela V. Maus, bXZtYXVzQG1naC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.