 Zaira Ianniello
Zaira Ianniello Alessandro Paiardini2*
Alessandro Paiardini2* Alessandro Fatica
Alessandro Fatica- 1Department of Biology and Biotechnology “Charles Darwin,” Sapienza University of Rome, Rome, Italy
- 2Department of Biochemical Sciences “A. Rossi Fanelli,” Sapienza University of Rome, Rome, Italy
Recent studies have uncovered an important role for RNA modifications in gene expression regulation, which led to the birth of the epitranscriptomics field. It is now acknowledged that RNA modifiers play a crucial role in the control of differentiation of stem and progenitor cells and that changes in their levels are a relevant feature of different types of cancer. To date, among more than 160 different RNA chemical modifications, the more relevant in cancer biology is the reversible and dynamic N6-methylation of adenosine, yielding N6-methyladenosine (m6A). m6A is the more abundant internal modification in mRNA, regulating the expression of the latter at different levels, from maturation to translation. Here, we will describe the emerging role of m6A modification in acute myeloid leukemia (AML), which, among first, has demonstrated how mis-regulation of the m6A modifying system can contribute to the development and progression of cancer. Moreover, we will discuss how AML is paving the way to the development of new therapeutic options based on the inhibition of m6A deposition.
Introduction
Chemical modifications in eukaryotic RNAs are known from decades. However, until recent years, their role in cancer development was largely unknown. One of most studied RNA modifications with a well-define role in gene expression regulation is the N6-methyladenosine (m6A), which is present in all RNA species including mRNAs, lncRNAs, rRNAs, tRNAs, and snRNAs. Here, we will focus on the dynamic m6A modification of mRNAs. m6A is the most abundant internal modification in mRNA where it can be embedded and erased by specific proteins (1–5). m6A mark can specifically recruit reader proteins, such as the YT521-B homology (YTH) domain family of proteins, or it can produce conformational changes within local RNA structures that may indirectly affect the interaction with RNA binding protein (6–9). As a result, m6A may regulate mRNA expression at different levels by affecting splicing, nuclear export, stability and translation [reviewed in (10, 11)].
The methyltransferase-like protein 3 (METTL3, also known as MT-A70) and the methyltransferase-like protein 14 (METTL14) complex (also called MAC, m6A-METTL Complex) installs m6A in mRNAs and lncRNAs within the DRACH motif (D = A/G/U, R = A/G; H = A/C/U) while the methyltransferase-like protein 16 (METTL16) is responsible for the m6A modification in the U6 snRNA and specific mRNAs and lncRNAs containing the UACAGAGAA sequence within a specific stem-loop structure (4, 5). Notably, METTL3 is the only catalytic component of the MAC but it requires the interaction with METTL14 for RNA binding and m6A deposition. The m6A modifications present in rRNAs, tRNAs, and U2 and U4 snRNAs are installed by still unknown methyltransferases. The METTL3/METTL14 core complex is assisted by a regulatory complex (named MACOM, m6A-METTL-associated complex) composed of Wilms tumor 1-associated protein (WTAP), Vir-like m6A methyltransferase-associated (VIRMA, also known as KIAA1429), Cbl proto-oncogene like 1 (CBLL1, also known as Hakai), RNA-binding motif 15 (RBM15), and zinc finger CCCH-type containing 13 (ZC3H13) proteins (12). Interestingly, even if the METTL3/METTL14 consensus sequence can be found along all the mRNA body, m6A deposition is enriched nearby the stop codon, 3′-UTR and long internal exons. Therein, it has been suggested that MACOM is responsible for guiding the METTL3/METT14 core complex on a specific region of the mRNA. In view of its reversible nature, m6A modification can be removed by ALKBH5 (alkB homolog 5) and FTO (fat-mass and obesity associated protein) proteins. They belong to the AlkB family of the Fe(II) and α-ketoglutarate-dependent dioxygenases, which includes also DNA and histone demethylates (10).
m6A was initially identified in pioneering studies in early 1970s in mammals, and later on, in flies, plants, yeast and also RNA viruses (13–16). However, the identification and mapping of m6A modification in whole transcriptome in different cell types, states and diseases was possible only in the last years with the development of m6A specific antibodies coupled to next generation RNA sequencing technologies (17–19). We are now witnessing the dawn of a new era in cancer biology studies in which gene expression data and epigenetic status of cancer cells are integrated with epitranscriptomics analysis to acquire a better comprehension of the molecular mechanisms that drive tumorigenesis. In this context, the first experimental evidences of a direct involvement of m6A deposition in the development of cancer have been obtained in acute myeloid leukemia (AML), a devastating blood cell cancer. In this review, we describe accepted knowledge on the critical role for the m6A modifiers, erasers, and readers in AML (Figure 1). Furthermore, in view of the fact that AML represents a remarkable example of malignancy with defects in cell differentiation, we also report recent results obtained in normal hematopoietic stem cells biology. Finally, we discuss the feasibility of chemical inhibition of the writing complex as novel therapeutic option for AML patients.
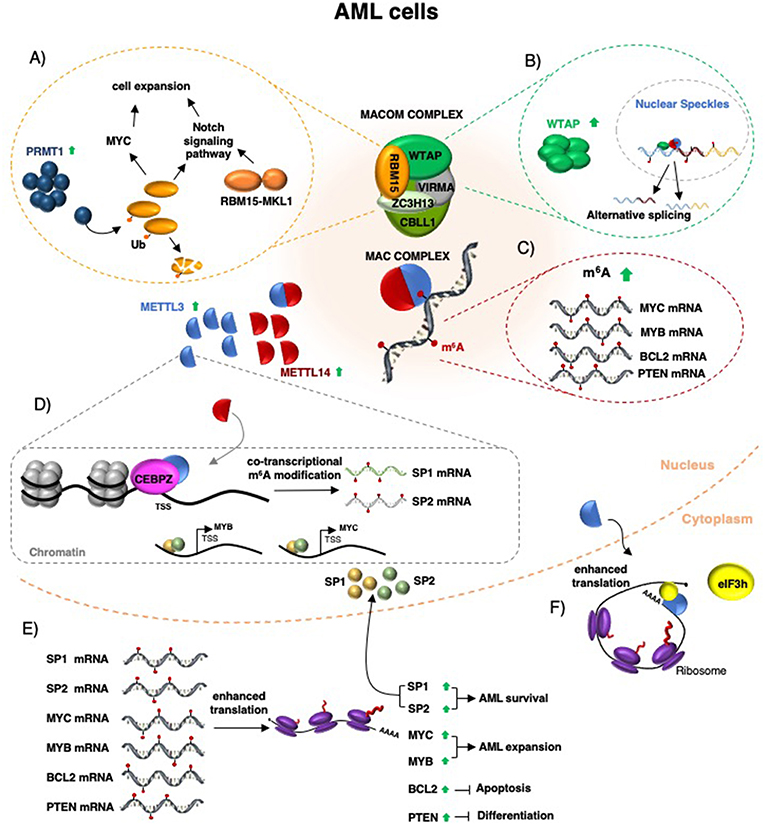
Figure 1. Schematic representation of m6A function in AML. The MACOM and MAC complex components are required for AML survival (see main text for details). (A) RBM15 protein controls cell expansion and differentiation by regulating c-Myc levels and the Notch signaling pathway. In some forms of pediatric AML, RBM15 is fused to MKL1 and induced leukemia by aberrant regulation of the Notch signaling pathway. (B) WTAP protein is upregulated in AML, localized in nuclear speckles, and regulates alternative splicing. (C) METTL3 and METTL14 upregulation in AML increases m6A methylation of specific mRNAs, including MYC, MYB, PTEN, and BCL2 mRNAs. (D) In addition, METTL3 is recruited by CEBPZ on specific promoter regions and this results in co-transcriptional m6A methylation of different mRNAs, including that one encoding for the SP1 and SP2 transcription factors. (E) Increased m6A methylation enhances mRNA translation and produced increased protein levels. (F) In AML, METTL3 mis-localizes in the cytoplasm where it can increase the translation of specific mRNAs independently from its catalytic activity by recruiting eIF3h.
m6A Deposition is Required for AML Cells Survival
The MACOM components WTAP and RBM15 had already been involved in AML before knowing they were regulators of m6A modification. In particular, WTAP was initially identified as interactor of the Wilms' tumor gene [WT1; (20)], whose high expression in AML is associated with poor prognosis (21). Later on, WTAP protein was found upregulated in AML and to act as an oncogene (22). According to its oncogenic role, WTAP knockdown in AML cell lines and AML xenograft model decreased proliferation and induced apoptosis (22). Mechanistically, WTAP silencing in AML cell lines altered alternative splicing (22, 23) and this was in accordance with its previously reported localization in nuclear speckles (24), where the mRNA splicing reaction occurs. Notably, before knowing its role as regulator of m6A deposition, the WTAP complex was purified from human cell lines and different components of the MACOM complex were identified as interacting proteins, including VIRMA, RBM15, and CBLL1 (23). Furthermore, in agreement with its role in splicing regulation, it was also demonstrated a transient interaction of the WTAP complex with the splicing machinery (23). Notably, the METTL3 and METTL14 proteins were also identified as WTAP complex interactors in different cell lines (23). However, albeit it was known since 1990s that METTL3 was responsible for m6A modification in human mRNAs (25) and that it was implicated in splicing regulation (26, 27), the link between WTAP and m6A was initially ignored. Few years later, with the birth of epitransciptomics, the WTAP protein was identified as an important regulator of the METTL3/METTL14 complex (28).
RBM15 is a member of the SPEN (Split-end) family of proteins, a group of proteins with RNA recognition motifs that functions in transcriptional regulation, post-transcriptional processing and nuclear export of mRNAs (29, 30). RBM15 is highly expressed in the hematopoietic system (29). Notably, chromosomal translocation between RBM15 and MKL1 were identified in some forms of pediatrics AML (also referred to as Acute Megakaryoblastic Leukemia). Similar to WTAP, knockdown of RBM15 in AML cell lines inhibited cell differentiation and induced apoptosis (31, 32). In mouse, RBM15 has an important role in regulating HSCs expansion and differentiation. In particular, conditional knockout of RBM15 in the hematopoietic compartment produced a block in B cell differentiation and myeloid and megakaryocytic development (29, 33). Strikingly, it was shown that the effect of RBM15 in the hematopoietic system and AML was partly due to deregulation of the Notch signaling (31, 34) and regulation of c-MYC expression (33), which were later identified as relevant m6A targets in haematopoiesis and AML (35–38). In addition, RBM15 protein can be modified by protein arginine methyltransferase 1 (PRMT1), resulting in its ubiquitylation and degradation by proteasome (39). Overexpression of PRMT1 altered alternative splicing and blocks AML cell differentiation (39). Therein, it is very likely that PRMT1 can indirectly control m6A deposition by regulating MACOM activity through RBM15 post-translational modification.
More recently, m6A modification came into focus of AML studies (40). AML is one of the cancers with the highest expression of both METTL3 and METTL14. Both genes were found upregulated in all subtypes of AML compared to normal hematopoietic cells (37, 38, 41, 42), despite the heterogeneity of this blood cell cancer in terms of chromosomal rearrangement and gene mutations. Moreover, this correlate with higher levels of m6A modified mRNAs in AML cell compared to normal hematopoietic progenitors (37, 38, 41). Downregulation of METTL3 and METTL14 expression has been performed in cell lines derived from different human AML subtypes by RNA interference and CRISPR/Cas9 genome editing. In all cases, it has been reported an inhibition of differentiation, proliferation arrest and massive induction of apoptosis (37, 38, 41). Strikingly, the apoptotic response is specific for leukemic cells and it has not been observed in normal hematopoietic progenitors. On the other hand, overexpression of METTL3 and METTL14 promoted AML cell proliferation and the effect was abolished by a catalytic inactive form of METTL3. Despite the common results observed at the cellular level by independent studies, the molecular mechanisms identified differ (Table 1). However, it should be considered that the strong induction of apoptosis observed upon METTL3/METTL14 depletion complicates the subsequent gene expression analysis. Moreover, the techniques to study m6A level in the transcriptome are impacted by the methodology, the antibody and sometimes also influenced by culture/experimental conditions (19, 45). In particular, in a first study performed on the MOLM-13 AML cell lines, which carries the FLT3 internal tandem duplication (FLT3-ITD) that in patients is associated with a more aggressive disease, knockdown of METTL3 resulted in a m6A dependent reduction of c-MYC, BCL2 and PTEN mRNA translation while the overexpression of METTL3 produced increased protein levels of all three proteins (37). c-MYC is a well-known oncogene in leukemia, while BCL2 and PTEN are negative regulators of apoptosis and PI3K/AKT pathway, respectively. However, activation of the PI3K/AKT pathway was also observed by increasing the expression of a non-functional METTL3 indicating that it is not merely due to m6A modification (37). In addition, the translation defects of BCL2 and c-MYC mRNA are recovered after few days of METTL3 silencing despite the persistence of the proliferation arrest and apoptosis (37). Therein, this indicates that additional mechanisms might be responsible for the observed cellular phenotype.
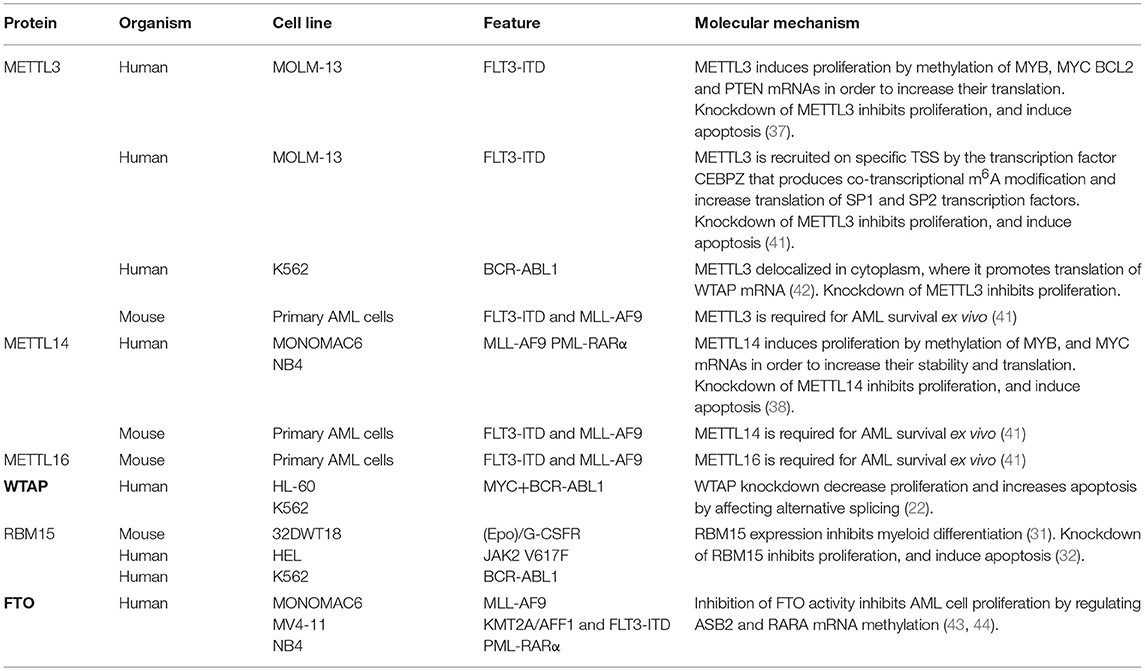
Table 1. The role of m6A regulators in leukemia.
A second study performed again in MOLM-13 cells, showed that METTL3 is recruited on specific promoter transcription start sites (TSS) by the transcription factor CCAAT enhancer binding protein zeta (CEBPZ, also known as DDIT3 and CHOP) (41). This has been indicated as a mechanism that produces co-transcriptional m6A modification on specific RNAs. Notably, CEBPZ gene was found recurrently mutated in different AML subtypes (46, 47), suggesting that this might result in altered recruitment of METTL3. Similarly, METTL14 was also found associated with several TSS but, surprisingly, METTL14 peaks do not overlap with those of METTL3 (41). As METTL14 is strictly required for METTL3 modifying activity, this suggests that the function of the two proteins on chromatin might be independent from m6A modification. Interestingly, the Saccharomyces cerevisiae METTL14 homologous protein KRF4 was also found associated to chromatin and initially described as a transcription factor (48). Two of the relevant METTL3 modified transcripts identified in MOLM-13 cells were the mRNAs encoding for the SP1 and SP2 transcription factors. SP1 and SP2 belong to the SP/FLF family, whose members have several roles in tumor development (49). Similar to what has been found for c-MYC, upon METTL3 knockdown these transcripts are translated less efficiently even if mRNA levels are not changed. Interestingly, it has also been demonstrated that SP1 and SP2 proteins directly regulate c-MYC transcription (41). Therein, in this case the regulation of c-MYC by METTL3 appears to be indirect. Notably, in MOLM-13 cells silenced for METTL3, overexpression of SP1 rescues cell growth. On the other hand, deletion of SP1 is lethal (41), thus, indicating a relevant role for SP1 in supporting METTL3 function in AML cells. However, SP1 is ubiquitously expressed, and regulates the expression of many genes within the cells. Furthermore, while SP1 knockout mice die early during embryogenesis, mESCs from SP1 knockout animals are viable and they can be induced to differentiate and form embryoid bodies (50). On the other hand, METTL3 knockout in mESCs impairs exit from self-renewal and block differentiation (51), indicating SP1 independent function. It is worth noting that some of the differences between these two studies performed on the same AML cell line might depend from the different antibodies utilized for the identification of m6A modified mRNAs. Indeed, it has been shown that the anti-m6A antibody influences the efficiency of m6A detection (19, 45).
Human AML subtypes are characterized by the presence of gene translocation that results in the expression of oncogenic fusion proteins that contribute to the differentiation block observed in AML (52). Expression of these proteins in normal mouse hematopoietic progenitor cells strongly induced expression of METTL3 and METTL14 (38). More importantly, conditional deletion of METTL14 strongly reduce the oncogenic potential of AML fusion proteins both in primary cells and in recipient mice (38). In addition, the ablation of METTL14 delayed the onset of leukemia and prolonged the survival of mice. Knock-down of METTL14 in the Mono-Mac 6 and NB4 human AML cell lines, which express the oncogenic MLL-AF9 and PML-RARα fusion proteins, respectively, strongly reduced both the mRNA stability and translation of the oncogenes MYB and c-MYC (38). However, it should be considered that the fate of m6A modified mRNAs depend on the identity of the reader protein. For instance, in contrast to YTHDF2, IGF2BPs were shown to stabilize mRNAs, including MYC (53). Moreover, ectopic overexpression of MYB and c-MYC partially counteracts the effect of METTL14 depletion on AML cell proliferation and differentiation. Notably, similar to c-MYC, also the MYB promoter is regulated by the SP1 transcription factor (54), whose levels are regulated by m6A (see above). Thus, some of the observed phenotypes might be due to indirect effects. Taken together, these results indicate that the oncogenic function of m6A writers in AML is mediated by different pathways, which include modulation of SP1, c-MYC, and MYB expression. However, not all the observed cellular phenotypes may be ascribed to the identified regulatory networks and it should be considered that simple m6A/target relationships may dictate some phenotypes and complex networks of m6A changes within the all transcriptome may underlie others.
Interestingly, a genome-wide CRISPR/Cas9 screening performed in mouse primary leukemia cells expressing both FLT3-ITD and MLL-AF9 fusion genes identified besides METTL3 and METTL14 also METTL16 as critical gene for AML survival (41). METTL16 positively regulates the expression of the human S-adenosylmethionine (SAM) synthetase MAT2A (4, 5, 55), whose expression contribute to appropriate SAM levels. SAM is the major donor of methyl transfer within the cell. Therein, METTL16 expression may indirectly regulate the activity of METTL3/MELL14 and also of many other RNA, DNA and protein methyltransferases.
In some tumors, including AML, METTL3 mis-localize to the cytoplasm where it can promote the translation of specific mRNAs independently from its catalytic domain (42, 56). In particular, it has been shown that METTL3 binds m6A modified regions close to the stop codon promoting mRNA circularization and, eventually, mRNA translation by interacting with the eIF3 translation initiation factor subunit eIF3h (57). In AML, higher levels of cytoplasmic METTL3 results in concomitant increase of WTAP protein expression (42). This mechanism might be relevant to increase WTAP protein levels concomitantly to the METTL3/METTL14 complex and sustain its oncogenic role in AML (42). Importantly, the binding of cytoplasmic METTL3 to mRNA occurs independently from METTL14 and it is still not clear how METTL3 would specifically recognize m6A mRNAs and, above all, how it will remain stably associated to mRNAs. In the nucleus, the RNA binding activity of METTL3 depends on the presence of a conserved cluster of positively charged residues across the METTL3/METTL14 heterodimer interface and a N-terminal Zinc finger domain in the METTL3 protein (58–61). However, as expected for a writing complex, the affinity of the METTL3/METTL14 heterodimer for RNA is very weak (61). Thus, it is very likely that METTL3 needs specific protein partners for stable mRNA binding in the cytoplasm.
By contrast with the reported oncogenic role of m6A in AML, high expression of the FTO demethylase has been also reported in AML carrying the FTL3-ITD, MLL-AF9 or PML-RARA gene translocations (43). Moreover, it was also shown that inhibition of FTO activity in the Monomac6, MV4-11, and NB4 cell lines affected AML cell proliferation capacity (43, 44). These results are in sharp contrast with what has been shown upon METTL3 and METTL14 downregulation in the same AML cell lines (see Table 1). However, in addition to m6A, it has been recently reported that FTO also demethylates N6, 2-O-dimethyladenosine (m6Am) at the 5' cap in mRNA and N1-methyladenosine (m1A) in tRNA (62). Therein, it is very likely that the observed phenotype may be m6A independent. The PCIF1 protein (also referred to as CAPAM, cap-specific adenosine methyltransferase) has been recently identified as the methyltransferase responsible for the m6Am modification at the 5' cap in mRNA (63, 64). Thus, it would be very interesting to investigate the potential impact of PCIF1 in AML.
Non-coding Transcripts as m6A Targets in AML
Despite many of the studies on m6A performed in AML focused on coding RNAs, the METTL3/METTL14 and METTL16 methyltransferases can also modify non-coding transcripts with relevant role in cancer, such as lncRNAs and circular RNAs (circRNA). For example, deletion in the mouse hematopoietic system of the X-inactive specific transcript (Xist), which controls X-dosage compensation in mammals, causes blood cancer (65). Notably, Xist contains several m6A modifications that are required for Xist-mediated transcriptional repression (66), thus suggesting that alteration of m6A levels might alter Xist function in hematopoietic cells. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1, also known as NEAT2) is another highly methylated lncRNA (17). MALAT1 is mis-regulated in several human cancers, including leukemia (67). MALAT1 is a nuclear lncRNA that interacts with splicing factors and regulates alternative splicing (68). Moreover, MALAT1 has also been shown to act as a competing endogenous RNA (ceRNA) (69) and as a scaffold for the polycomb repressive complexes 1 and 2 (PRC1 and PRC2) (70–72). Two of the m6A marks in MALAT1 affect local RNA structures and regulate the accessibility of RNA binding proteins, a mechanism referred to as m6A-riboswitch (73, 74). Therein, it is possible that the higher levels of m6A observed in AML might increase the binding of proteins, such as splicing regulators or epigenetic modifiers, that results in gene mis-regulation. Several other lncRNAs have been shown to play critical role in AML (75). Hence, alteration of their structure or expression levels by m6A modifications might influence their activity.
Another important class of m6A modified molecules is the circRNA family (76). circRNAs are covalently closed circular molecules derived from back-splicing reactions, in which the 5′ splice site of the exon joins with the 3′ splice site of an upstream intron (77). Interestingly, m6A modifications in circRNAs differ from the patters of the corresponding linear mRNAs (76) and, more importantly, m6A-modified circRNAs regulate the stability of the corresponding linear mRNA in a YTHDF2-dependent manner (76). Thus, it is likely that changes in circRNA m6A levels might have a great impact in gene expression regulation. Notably, a correlation between circRNA levels and cell proliferation has already been shown in cancer (78). Moreover, about half of AML patients carry aberrant chromosomal translocations that can produce specific fusion-circRNAs (f-circRNA) between rearranged loci (79). In particular, the PML/RARα translocation, which characterizes a subtype of AML referred to as acute promyelocytic leukemia (APL), produces oncogenic f-circRNAs that have been shown to favor leukemia progression in transgenic mouse models (79). Therein, it will be interesting to study the relationship between the m6A modifications and f-circRNA activity. circRNAs may regulate gene expression by several mechanisms, including regulators of splicing and transcription, ceRNAs and protein competitors (77). Moreover, specific circRNA can also be translated in protein by a cap-independent manner (77). This kind of translation is less efficient than cap-dependent translation, but might have an important role under stress condition and in tumor and, importantly, can be regulated by m6A modifications (10). A peptide produced from a circular form of the linc-PINT lncRNA has been recently shown to play an important role in glioblastoma tumorigenesis (80) and it is very likely that other examples of translated circRNAs with a role in cancer will follow.
m6A Role in Normal Hematopoiesis
Defects in cell differentiation are a hallmark of AML. In particular, AML is characterized by an accumulation of immature cells which fail to respond to normal regulators of differentiation within the bone marrow (52). Therein, the role of m6A RNA modification in normal hematopoiesis has been also analyzed in both purified hematopoietic stem/progenitor cells (HSPCs) and mouse model systems. In cytokine-driven differentiation of human CD34+ HSPCs purified from umbilical cord blood, METTL3/METL14 expression decreased with the progression of myeloid differentiation (37, 38). Furthermore, similar to AML cells, knockdown of METTL3 and METTL14 in CD34+ HSPCs accelerated myeloid differentiation while their overexpression stimulated proliferation and inhibited differentiation (37, 38). Notably, downregulation of METTL3 and METTL14 in purified HSPCs inhibits cell growth but does not induce massive apoptosis as in AML cells (37, 38). In contrast, conditional knockout of METTL3 in the adult mouse hematopoietic system produced an expansion of the HSCs in bone marrow without any significant alteration in mature myeloid cells production (81). Similar results were obtained with the conditional knockout of the m6A reader YTHDF2 (77, 82) but, surprisingly, not upon deletion of METTL14 (81), which is required for METTL3 function. Moreover, HSC with ablation of YTHDF2 have elevated regeneration capacity (77). The differences observed in vitro and in vivo might reflect the fact that purified cord blood cells differ from their counterparts in the bone marrow (BM). In particular, they have greater proliferative response to cytokines and are less dependent on stromal cells than the corresponding HSCPs in the BM (83). Under physiological conditions, HSPCs homeostasis is maintained by the interaction with stromal cells within the BM and the conditional system utilized for METTL3 and YTHDF2 deletion may also target the stromal cells. Thus, the expansion observed in HSCs in vivo might be also due to an alteration of the stem cell niche. However, these data indicate that m6A modification plays an important role in maintaining adult HSCs quiescence and, above all, that the inhibition of the m6A modification system is well tolerated by the normal HSCs in vivo.
m6A as an Anticancer Drug Target
Targeting m6A modification writers, erasers and readers by small molecules has been frequently hailed as a potential treatment for several kinds of cancer. Inhibitors targeting 2-oxoglutarate (2OG) and iron-dependent oxygenases [e.g., ALKBH5 (2) and FTO (84)], belonging to the 2OG-dependent nucleic acid oxygenase (NAOX) family and suppressing m6A modification demethylation of RNA, have been extensively discussed in a recent review (85). Here, we will focus on therapeutic strategies and small molecules targeting the METTL3/METTL14 complex and discuss their potential applications in cancer treatment.
Although thus far no inhibitors of METTL3/METTl14 have been reported in the literature, other than the reaction product SAH and the general nucleoside analog Sinefungin (86), the recent availability of high-resolution crystal structures for METTL3/METTL14 complexes [(58–60); (Figure 2)] provides a basis for structure-guided drug design, as the latter can be exploited by computational tools for the rational design of novel inhibitors (87). Current structural information for METTL3 and METTl14 and the potential druggability of these targets are therefore discussed.
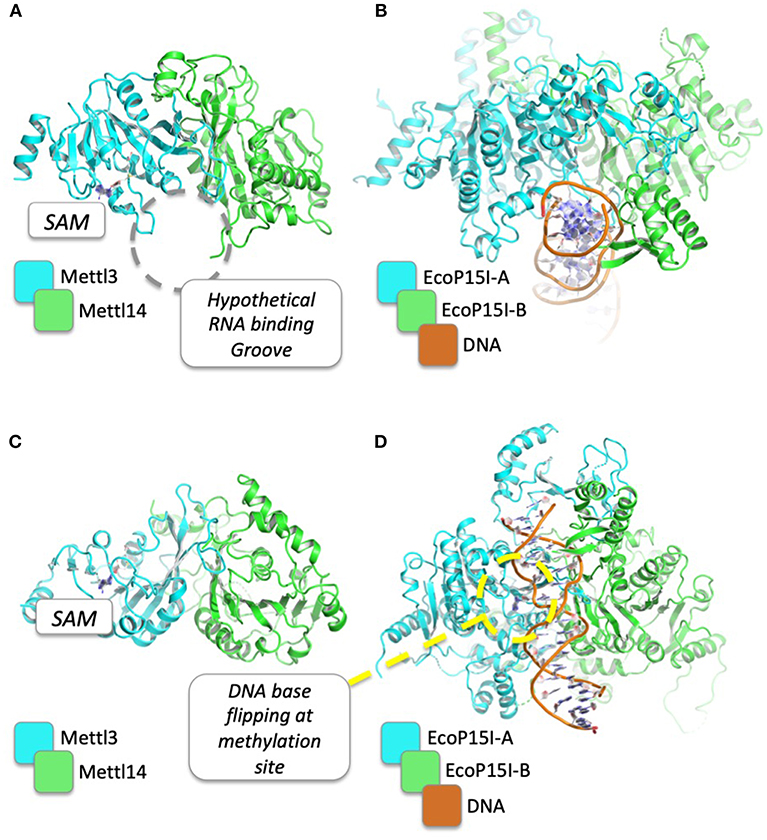
Figure 2. Structural comparison between METTL3/METTL14 complex and EcoP15I in complex with its DNA substrate. PDB codes for METTL3/METTL14 and EcoP15I are 5IL1 and 4ZCF, respectively. The hypothetical binding groove of RNA, just beside the SAM binding site, is shown (A) and compared to the DNA binding mode of EcoP15I (B). In (C,D), the structures are rotated by 90°, to show the DNA base flipping that is supposed to be required for DNA/RNA methylation of N6 of adenosine.
The crystal structures of METTL3–METTL14 complex show that both proteins belong to the class I methyltransferase family, the largest homologous group of SAM-dependent methyltransferases. The latter is characterized by a Rossman fold catalytic domain and several conserved sequence motifs (88, 89), and comprises most tRNA, cap, and m6A methyltransferases, as well as DNA methyltransferases (DNMTs), arginine methyltransferases (PRMTs), and some histone-lysine N-methyltransferases (90). METTL3 and METTL14 form an asymmetric heterodimer in which only the former is able to bind SAM and carry out the catalytic methyltransferase reaction. Structural analysis and mutagenesis indicate that both proteins are involved in RNA binding, although a complex with RNA has yet to be determined. However, because METTL3 and METTL14 are both members of the SAM-dependent methyltransferase superfamily, structural insights on RNA substrates binding mechanism can come from the comparisons with the three-dimensional structures between members of the same family. Indeed, as shown in Table 2, METTL3 shows a high structural similarity with other DNA methyltransferases (e.g., Adenine Specific DNA Methyltransferases), for which the dimeric structure in complex with the DNA substrate for methylation is already known [PDB: 1G38; (91)]. On the ground of these homology-based, functional and structural similarities, several conclusions can be drawn on the hypothetical mechanism that is responsible for m6A of mRNAs by METTL3/METTL14. Intriguingly, both METTL3 and METTL14 contain sequence motifs characteristic of amino-methyltransferases, including the “DPPW” motif that is equivalent of the “DPPY” sequence in motif IV (92). The linear order of these motifs and structural comparison with other amino-methyltransferases (e.g., EcoP15I, PDB: 4ZCF) suggests that one methyltransferase (i.e., METTL3) plays a more dominant role in adenine methylation, while the other one (i.e., METTL14) plays a more central role in recognition of the surrounding RNA secondary structure (93) (Figure 2). Moreover, a mechanism can be envisaged where, as in the case of other amino-methyltransferases and restriction endonucleases (92), the target adenosine base to be methylated is “flipped” outside the nucleic acid double helix and positioned inside the active site cleft, facing SAM and stabilized with stacking interactions with the Tryptophan residue of the conserved “DPPW” motif (88–90).
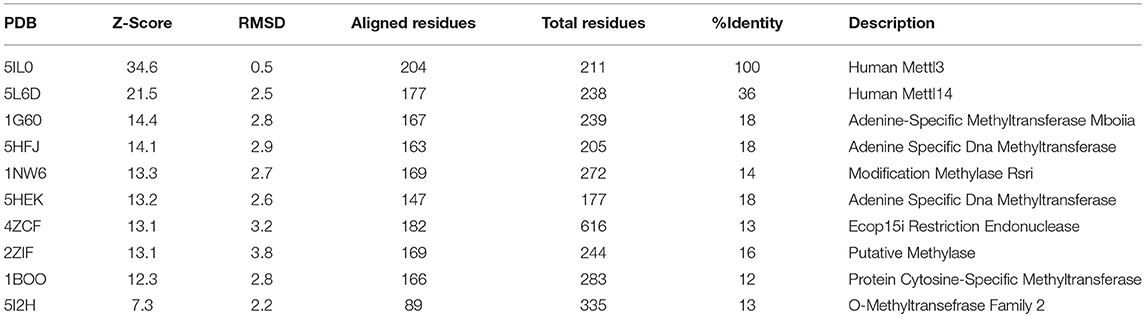
Table 2. First 10 results of a DALI search in PDB25 for structural similarities among METTL3 homologous proteins.
As previously mentioned, in spite of the fact that no inhibitors of METTL3/METTl14 have been found yet, it is reasonable to expect that such a goal is achievable, given that potent and selective inhibitors have been found for the closely related members of the class I methyltransferase family, e.g., protein lysine methyltransferases (PKMTs), protein arginine methyltransferases PRMTs and DNMTs.
The methyl-donating SAM cofactor and methyl-accepting adenosine substrate bind at distinct sites of METTL3 (71). These binding pockets are common to the Rossmann fold enzymes of class I family and are connected by a narrow channel in the protein core. Therefore, in conceiving potential METTL3/METTL14 inhibitors, nucleosides, and SAM analogs could be considered, or bisubstrates ligand mimicking both (Figure 3). For example, Azacytidine (Vidaza) and Decitabine (Dacogen) are nucleoside analogs targeting DNMTs and being approved for clinical use in hematological malignancies (94). Unfortunately, these drugs display poor bioavailability and toxicity. By contrast, small-molecule inhibitors binding within the SAM pocket have shown good pharmacological properties and oral bioavailability and are currently under clinical investigation as cancer therapeutics mainly against PKMT and PRMT protein families (95–97). Despite the fact that these SAM mimicking inhibitors share the same cofactor-binding site, side chains lining the SAM-binding cleft are usually not conserved. As in the case of protein kinases, therefore, such structural diversity could be exploited, at least in principle, to achieve highly selective inhibition (98). For example, the methylthioadenosine endogenous compound is a highly specific PRMT5 inhibitor (99). However, structure-based design of bisubstrate inhibitors holds great promise for far higher selectivity, compared to SAM and nucleoside analogs. Recently, (100) designed DNMT3A and DNMT1 bisubstrate inhibitors by linking together SAM and the deoxycytidine substrate. This approach resulted in quinazoline–quinoline derivatives as potent inhibitors, some showing also isoform selectivity. The most potent inhibitors induced demethylation of CDKN2A promoter in colon carcinoma HCT116 cells and its reactivation after 7 days of treatment. In this study, the authors highlighted the importance of the nature and rigidity of the linker between the two moieties for optimal inhibition, an issue that should be taken into account also in designing potential bisubstrate inhibitors of METTL3/METTL14.
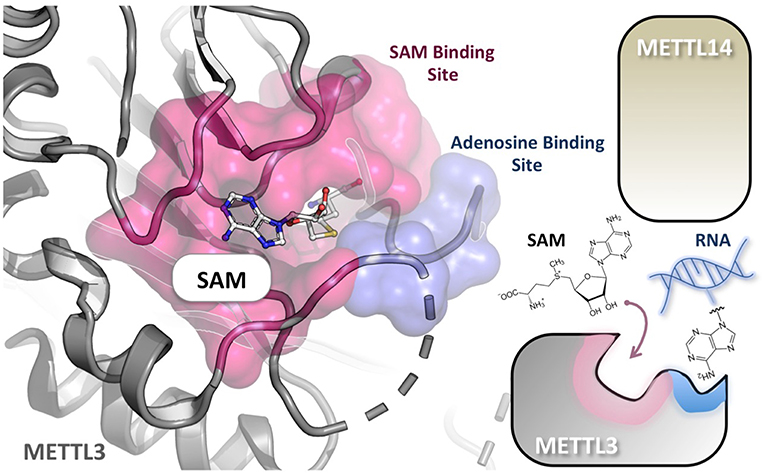
Figure 3. Substrate RNA and cofactor (SAM) of METTL3/METTL14 bind at distinct sites. The crystal structure of the METTL3 monomer in complex with SAM (PDB: 5IL1), shows that the adenosine-binding site (cyan) and the cofactor-binding pocket (purple) are connected by a narrow channel in the protein core. METTL14 is not directly involved in reaction but is probably necessary for RNA binding. A nucleotide base flipping-mechanism is also proposed.
In conclusion, the high structural diversity of these compounds pinpoints the significant range of inhibition strategies that can be conceived to target the class I methyltransferases. Although the structural details of the various members of this family are unique, the success stories of drug design for several enzymes belonging to this family portends the likely achievement of discovering potent and selective inhibitors of METTL3/METTL14.
Author Contributions
AF and AP wrote the manuscript. ZI revised and edited manuscript and prepared figures.
Funding
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) IG 17352 to AF, AIRC MFAG 20447 to AP and Progetti Ateneo Sapienza University of Rome to AF and AP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. (2011) 7:885–7. doi: 10.1038/nchembio.687
2. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. (2013) 49:18–29. doi: 10.1016/j.molcel.2012.10.015
3. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
4. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. (2017) 169:824–35.e14. doi: 10.1016/j.cell.2017.05.003
5. Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. (2017) 18:2004–14. doi: 10.15252/embr.201744940
6. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
7. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 16:1388–99. doi: 10.1016/j.cell.2015.05.014
8. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. (2015) 518:560–4. doi: 10.1038/nature14234
9. Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol. (2017) 24:870–8. doi: 10.1038/nsmb.3462
10. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. (2017) 1:31–42. doi: 10.1038/nrm.2016.132
11. Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. (2018) 28:113–27. doi: 10.1016/j.tcb.2017.10.001
12. Lence T, Paolantoni C, Worpenberg L, Roignant JY. Mechanistic insights into m(6)A RNA enzymes. Biochim Biophys Acta Gene Regul. Mech. (2018) S1874–9399:30224. doi: 10.1016/j.bbagrm.2018.10.014
13. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Natl Acad Sci USA. (1974) 71:3971–5. doi: 10.1073/pnas.71.10.3971
14. Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. (1974) 1:37–42. doi: 10.1016/0092-8674(74)90153-6
15. Adams JM, Cory S. Modified nucleosides and bizarre 5'-termini in mouse myeloma mRNA. Nature. (1975) 255:28–33. doi: 10.1038/255028a0
16. Lavi S, Shatkin AJ. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc Natl Acad Sci USA. (1975) 72:2012–6. doi: 10.1073/pnas.72.6.2012
17. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. (2012) 485:201–6. doi: 10.1038/nature11112
18. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. (2012) 149:1635–46. doi: 10.1016/j.cell.2012.05.003
19. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. (2015) 12:767–72. doi: 10.1038/nmeth.3453
20. Little NA, Hastie ND, Davies RC. Identification of WTAP, a novel Wilms' tumour 1-associating protein. Hum Mol Genet. (2000) 9:2231–9. doi: 10.1093/oxfordjournals.hmg.a018914
21. Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. (2007) 21:868–76. doi: 10.1038/sj.leu.2404624
22. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. (2014) 28:1171–4. doi: 10.1038/leu.2014.16
23. Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, et al. Identification of Wilms' tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. (2013) 288:33292–302. doi: 10.1074/jbc.M113.500397
24. Little NA, Hastie ND, Davies RC. Identification of WTAP, a novel Wilms' tumour 1-associating protein. Hum Mol Genet. (2000) 9:2231–9.
25. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. (1997) 3:1233–47.
26. Kane SE, Beemon K. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol. (1985) 5:2298–306. doi: 10.1128/MCB.5.9.2298
27. Carroll SM, Narayan P, Rottman FM. N6-methyladenosine residues in an intron-specific region of prolactin pre-mRNA. Mol Cell Biol. (1990) 10:4456–65. doi: 10.1128/MCB.10.9.4456
28. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. (2014) 24:177–89. doi: 10.1038/cr.2014.3
29. Raffel GD, Mercher T, Shigematsu H, Williams IR, Cullen DE, Akashi K, et al. (Rbm15) has pleiotropic roles in hematopoietic development. Proc Natl Acad Sci USA. (2007) 104:6001–6. doi: 10.1073/pnas.0609041104
30. Hu M, Yang Y, Ji Z, Luo J. RBM15 functions in blood diseases. Curr Cancer Drug Targets. (2016) 16:579–85. doi: 10.2174/1568009616666160112105706
31. Ma X, Renda MJ, Wang L, Cheng EC, Niu C, Morris SW, et al. Rbm15 modulates Notch-induced transcriptional activation and affects myeloid differentiation. Mol Cell Biol. (2007) 27:3056–64. doi: 10.1128/MCB.01339-06
32. Yang Y, Wang S, Zhang Y, Zhu X. Biological effects of decreasing RBM15 on chronic myelogenous leukemia cells. Leuk Lymphoma. (2012) 53:2237–44. doi: 10.3109/10428194.2012.684350
33. Niu C, Zhang J, Breslin P, Onciu M, Ma Z, Morris SW. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood. (2009) 114:2087–96. doi: 10.1182/blood-2009-01-197921
34. Mercher T, Raffel GD, Moore SA, Cornejo MG, Baudry-Bluteau D, Cagnard N, et al. The OTT-MAL fusion oncogene activates RBPJ-mediated transcription and induces acute megakaryoblastic leukemia in a knockin mouse model. J Clin Invest. (2009) 119:852–64. doi: 10.1172/JCI35901
35. Lv J, Zhang Y, Gao S, Zhang C, Chen Y, Li W, et al. Endothelial-specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. (2018) 28:249–52. doi: 10.1038/cr.2017.143
36. Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature. (2017) 549:273–6. doi: 10.1038/nature23883
37. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. (2017) 23:1369–76. doi: 10.1038/nm.4416
38. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. (2018) 22:191–205.e9. doi: 10.1016/j.stem.2017.11.016
39. Zhang L, Tran NT, Su H, Wang R, Lu Y, Tang H, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife. (2015) 4:e07938. doi: 10.7554/eLife.07938
40. Ianniello Z, Fatica A. N6-methyladenosine role in acute myeloid leukaemia. Int J Mol Sci. (2018) 19:E2345. doi: 10.3390/ijms19082345
41. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
42. Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. (2018) 9:796. doi: 10.1038/s41419-018-0843-z
43. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
44. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. (2018) 172:90–105.e23. doi: 10.1016/j.cell.2017.11.031
45. Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, et al. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. (2018) 16:e2006092. doi: 10.1371/journal.pbio.2006092
46. Herold T, Metzeler KH, Vosberg S, Hartmann L, Röllig C, Stölzel F, et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood. (2014) 124:1304–11. doi: 10.1182/blood-2013-12-540716
47. Musialik E, Bujko M, Kober P, Grygorowicz MA, Libura M, Przestrzelska M, et al. Comparison of promoter DNA methylation and expression levels of genes encoding CCAAT/enhancer binding proteins in AML patients. Leuk Res. (2014) 38:850–6. doi: 10.1016/j.leukres.2014.04.013
48. Lahav R, Gammie A, Tavazoie S, Rose MD. Role of transcription factor Kar4 in regulating downstream events in the Saccharomyces cerevisiae pheromone response pathway. Mol Cell Biol. (2007) 27:818–29. doi: 10.1128/MCB.00439-06
49. Black AR, Black JD, Azizkhan-Clifford J. Sp1 and krüppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. (2001) 188:143–60. doi: 10.1002/jcp.1111
50. Marin M, Karism A, Visser P, Grosveld F, Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. (1997) 89:619–28. doi: 10.1016/S0092-8674(00)80243-3
51. Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. (2014) 15:707–19. doi: 10.1016/j.stem.2014.09.019
52. Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. (2003) 3:89–101. doi: 10.1038/nrc989
53. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
54. Facchinetti V, Lopa R, Spreafico F, Bolognese F, Mantovani R, Tavner F, et al. Isolation and characterization of the human A-myb promoter: regulation by NF-Y and Sp1. Oncogene. (2000) 19:3931–40. doi: 10.1038/sj.onc.1203730
55. Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, et al. S-adenosylmethionine synthesis is regulated by selective N6-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. (2017) 21:3354–63. doi: 10.1016/j.celrep.2017.11.092
56. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
57. Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. (2018) 561:556–60. doi: 10.1038/s41586-018-0538-8
58. Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. (2016) 63:306–17. doi: 10.1016/j.molcel.2016.05.041
59. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. (2016) 534:575–8. doi: 10.1038/nature18298
60. Sledz P, Jinek M. Structural insights into the molecular mechanism of the m(6)A writer complex. Elife. (2016) 5:e18434. doi: 10.7554/eLife.18434
61. Huang J, Dong X, Gong Z, Qin LY, Yang S, Zhu YL, et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell. (2018) 10:272–84. doi: 10.1007/s13238-018-0518-7
62. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. (2018) 71:973–85.e5. doi: 10.1016/j.molcel.2018.08.011
63. Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science. (2019) 363:eaav0080. doi: 10.1126/science.aav0080
64. Sun H, Zhang M, Li K, Bai D, Yi C. Cap-specific, terminal N(6)-methylation by a mammalian m(6)Am methyltransferase. Cell Res. (2019) 29:80–2. doi: 10.1038/s41422-018-0117-4
65. Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. (2013) 152:727–42. doi: 10.1016/j.cell.2013.01.034
66. Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. (2016) 537:369–73. doi: 10.1038/nature19342
67. Amodio N, Raimondi L, Juli G, Stamato MA, Caracciolo D, Tagliaferri P, et al. MALAT1: a druggable long non-coding RNA for targeted anti-cancer approaches. J Hematol Oncol. (2018) 11:63. doi: 10.1186/s13045-018-0606-4
68. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. (2010) 39:925–38. doi: 10.1016/j.molcel.2010.08.011
69. Luan W, Li L, Shi Y, Bu X, Xia Y, Wang J, et al. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget. (2016) 7:63901–12. doi: 10.18632/oncotarget.11564
70. Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, et al. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. (2011) 147:773–88. doi: 10.1016/j.cell.2011.08.054
71. Wang W, Zhu Y, Li S, Chen X, Jiang G, Shen Z, et al. Long noncoding RNA MALAT1 promotes malignant development of esophageal squamous cell carcinoma by targeting beta-catenin via Ezh2. Oncotarget. (2016) 7:25668–82. doi: 10.18632/oncotarget.8257
72. Stamato MA, Juli G, Romeo E, Ronchetti D, Arbitrio M, Caracciolo D, et al. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget. (2017) 8:106527–37. doi: 10.18632/oncotarget.22507
73. Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. (2015) 519:486–90. doi: 10.1038/nature14263
74. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. (2017) 45:6051–63. doi: 10.1093/nar/gkx141
75. Morlando M, Ballarino M, Fatica A. Long non-coding RNAs: new players in hematopoiesis and leukemia. Front Med. (2015) 2:23. doi: 10.3389/fmed.2015.00023
76. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. (2017) 20:2262–76. doi: 10.1016/j.celrep.2017.08.027
77. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. (2018) 71:428–42. doi: 10.1016/j.molcel.2018.06.034
78. Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, et al. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. (2015) 5:8057. doi: 10.1038/srep08057
79. Guarnerio J, Bezzi M, Jeong JC, Paffenholz SV, Berry K, Naldini MM, et al. Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell. (2016) 165:289–302. doi: 10.1016/j.cell.2016.03.020
80. Zhang M, Zhao K, Xu X, Yang Y, Yan S, Wei P, et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat Commun. (2018) 9:4475. doi: 10.1038/s41467-018-06862-2
81. Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, et al. Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. (2018) 28:952–4. doi: 10.1038/s41422-018-0062-2
82. Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, et al. Loss of YTHDF2-mediated m(6)A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. (2018) 28:1035–8. doi: 10.1038/s41422-018-0082-y
83. Hordyjewska A, Popiolek L, Horecka A. Characteristics of hematopoietic stem cells of umbilical cord blood. Cytotechnology. (2015) 67:387–96. doi: 10.1007/s10616-014-9796-y
84. Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. (2007) 318:1469–72. doi: 10.1126/science.1151710
85. Niu Y, Wan A, Lin Z, Lu X, Wan G. N (6)-Methyladenosine modification: a novel pharmacological target for anti-cancer drug development. Acta Pharm Sin B. (2018) 8:833–43. doi: 10.1016/j.apsb.2018.06.001
86. Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. (2011) 78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x
87. Leelananda SP, Lindert S. Computational methods in drug discovery. Beilstein J Org Chem. (2016) 12:2694–718. doi: 10.3762/bjoc.12.267
88. Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. (2003) 28:329–35. doi: 10.1016/S0968-0004(03)00090-2
89. Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol. (1995) 253:618–32. doi: 10.1006/jmbi.1995.0577
90. Boriack-Sjodin PA, Ribich S, Copeland RA. RNA-modifying proteins as anticancer drug targets. Nat Rev Drug Discov. (2018) 17:435–53. doi: 10.1038/nrd.2018.71
91. Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol. (2001) 8:121–5. doi: 10.1038/84104
92. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol. (2002) 55:431–44. doi: 10.1007/s00239-002-2339-8
93. Gupta YK, Chan SH, Xu SY, Aggarwal AK. Structural basis of asymmetric DNA methylation and ATP-triggered long-range diffusion by EcoP15I. Nat Commun. (2015) 6:7363. doi: 10.1038/ncomms8363
94. Howell PM, Liu Z, Khong HT. Demethylating agents in the treatment of cancer. Pharmaceuticals (Basel). (2010) 3:2022–44. doi: 10.3390/ph3072022
95. Cao F, Townsend EC., Karatas H, Xu J, Li L, Lee S, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. (2014) 53:247–61. doi: 10.1016/j.molcel.2013.12.001
96. Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. (2015) 11:432–7. doi: 10.1038/nchembio.1810
97. Chen C, Zhu H, Stauffer F, Caravatti G, Vollmer S, Machauer R, et al. Discovery of novel Dot1L inhibitors through a structure-based fragmentation approach. ACS Med Chem Lett. (2016) 7:735–40. doi: 10.1021/acsmedchemlett.6b00167
98. Campagna-Slater V, Mok MW, Nguyen KT, Feher M, Najmanovich R, Schapira M. Structural chemistry of the histone methyltransferases cofactor binding site. J Chem Inf Model. (2011) 51:612–23. doi: 10.1021/ci100479z
99. Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. (2016) 351:1214–8. doi: 10.1126/science.aad5214
Keywords: m6A, RNA, METTL3, METTL14, AML, leukemia, epitranscriptomics
Citation: Ianniello Z, Paiardini A and Fatica A (2019) N6-Methyladenosine (m6A): A Promising New Molecular Target in Acute Myeloid Leukemia. Front. Oncol. 9:251. doi: 10.3389/fonc.2019.00251
Received: 10 January 2019; Accepted: 19 March 2019;
Published: 09 April 2019.
Edited by:
Sheng F. Cai, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Jean-Yves Roignant, Institute of Molecular Biology, GermanyRicardo Soto-Rifo, Universidad de Chile, Chile
Copyright © 2019 Ianniello, Paiardini and Fatica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Paiardini, YWxlc3NhbmRyby5wYWlhcmRpbmlAdW5pcm9tYTEuaXQ=
Alessandro Fatica, YWxlc3NhbmRyby5mYXRpY2FAdW5pcm9tYTEuaXQ=