 Federica Barbieri1†
Federica Barbieri1† Ivan Verduci2†
Ivan Verduci2† Valentina Carlini2
Valentina Carlini2 Gianluigi Zona3,4
Gianluigi Zona3,4 Aldo Pagano4,5
Aldo Pagano4,5 Michele Mazzanti2‡
Michele Mazzanti2‡ Tullio Florio1,4*‡
Tullio Florio1,4*‡- 1Sezione di Farmacologia, Dipartimento di Medicina Interna & Centro di Eccellenza per la Ricerca Biomedica, Università di Genoa, Genoa, Italy
- 2Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano, Milan, Italy
- 3Dipartimento di Neuroscienze, Riabilitazione, Oftalmologia, Genetica e Scienze Materno-Infantili, Università di Genoa, Genoa, Italy
- 4IRCCS Ospedale Policlinico San Martino, Genoa, Italy
- 5Dipartimento di Medicina Sperimentale, Università di Genoa, Genoa, Italy
The lack of in-depth knowledge about the molecular determinants of glioblastoma (GBM) occurrence and progression, combined with few effective and BBB crossing-targeted compounds represents a major challenge for the discovery of novel and efficacious drugs for GBM. Among relevant molecular factors controlling the aggressive behavior of GBM, chloride intracellular channel 1 (CLIC1) represents an emerging prognostic and predictive biomarker, as well as a promising therapeutic target. CLIC1 is a metamorphic protein, co-existing as both soluble cytoplasmic and membrane-associated conformers, with the latter acting as chloride selective ion channel. CLIC1 is involved in several physiological cell functions and its abnormal expression triggers tumor development, favoring tumor cell proliferation, invasion, and metastasis. CLIC1 overexpression is associated with aggressive features of various human solid tumors, including GBM, in which its expression level is correlated with poor prognosis. Moreover, increasing evidence shows that modification of microglia ion channel activity, and CLIC1 in particular, contributes to the development of different neuropathological states and brain tumors. Intriguingly, CLIC1 is constitutively active within cancer stem cells (CSCs), while it seems less relevant for the survival of non-CSC GBM subpopulations and for normal cells. CSCs represent GBM development and progression driving force, being endowed with stem cell-like properties (self-renewal and differentiation), ability to survive therapies, to expand and differentiate, causing tumor recurrence. Downregulation of CLIC1 results in drastic inhibition of GBM CSC proliferation in vitro and in vivo, making the control of the activity this of channel a possible innovative pharmacological target. Recently, drugs belonging to the biguanide class (including metformin) were reported to selectively inhibit CLIC1 activity in CSCs, impairing their viability and invasiveness, but sparing normal stem cells, thus representing potential novel antitumor drugs with a safe toxicological profile. On these premises, we review the most recent insights into the biological role of CLIC1 as a potential selective pharmacological target in GBM. Moreover, we examine old and new drugs able to functionally target CLIC1 activity, discussing the challenges and potential development of CLIC1-targeted therapies.
An Introduction to Cancer Stem Cells in Human Glioblastoma
Glioblastoma (GBM) is the most aggressive and prevalent primary brain cancer in adults, characterized by morphological, cellular, and molecular heterogeneity leading to invasive growth and resistance to therapy (1). Despite the use of aggressive multimodal treatments, GBM invariably recurs, and the median overall survival time of patients is extremely poor (~15 months after diagnosis) (2). The high drug resistance and recurrence rate of GBM is mainly ascribed to a sub-population of cancer stem cells (CSCs) within the tumor mass (3). GBM CSCs (GSCs) share functional features with neural stem cells, including self-renewal and multipotency, as well as the over-activation of biochemical signaling pathways (i.e., Sonic hedgehog, Akt, and Wnt/β-catenin). On the other hand, GSCs possess distinct genetic and epigenetic alterations which sustain their in vivo tumorigenic potential: through asymmetric division GSCs give rise to all the differentiated non-tumorigenic cells forming the bulk of the tumor mass, while their stem cell-like properties provide them with inherent resistance and evasion of apoptosis (4–6).
Phenotypically, GSCs are characterized by the expression of a combination of stem cell markers (e.g., CD133, Olig2, Sox2, Nanog), although different GSC populations exist, and a unique tumor-related phenotype has not been yet identified. Several proteins contribute to the maintenance of the stem-like phenotype, the aggressiveness, and the white matter invasiveness of GSCs, including CD44, sprouty2, Notch, tGLI1, and PrP (7–11). Moreover, the microenvironment in which GSCs develop is extremely complex, harboring non-neoplastic stromal cells, mesenchymal stem cells (MSCs), endothelial cells, immune cells, and other glial cell types, organized to compose the tumor niches (12). A dynamic and reciprocal crosstalk between GSCs, GBM bulk cells and the microenvironment cells occurs in the niches, via paracrine signals, mainly mediated by chemokine systems (for ex. CXCR4/7-CXCL12) (13) or direct cell-cell interactions. This microenvironment contributes tumor progression, invasion, angiogenesis, escape from immune surveillance, drug resistance, as well as to GSC maintenance, favoring the retaining of the stem-like properties (14, 15).
GSCs sustain neovascularization via the release of pro-angiogenic factors and vascular transdifferentiation (16), and are able to secrete cytokines inducing immune suppression (17, 18). Moreover, alteration of metabolic programs (i.e., the Warburg effect) drives the aggressive phenotype of GSCs providing them biosynthetic molecules useful for rapid growth (19).
Cytotoxic drugs, such as temozolomide, might favor a mutagenic selection of treatment-resistant GSC clones, further increasing GSC genetic heterogeneity, which represents a relevant mechanism for tumor recurrence (20). In addition, GSC and non-GSC populations retain dynamic interconversion through self-differentiation and de-differentiation, respectively (21, 22). Given the capacity of GSCs to generate all the different tumor cell populations composing the tumor mass, GSC targeting agents should be used in combination with existing therapies to arrest tumor growth and improve the clinical outcome.
Overall the complex nature of GSCs makes their eradication the main therapeutic goal for GBM, but a still unsolved challenge (23). In fact, conventional antitumor drugs spare GSCs, allowing tumor re-growth. Potential innovative strategies to eradicate GSCs from tumors are directed to: (i) impair specific pathways crucial for GSC survival and functioning (i.e., Notch, Wnt, Sonic hedgehog); (ii) targeting GSC perivascular or hypoxic niches; (iii) block metabolic and/or epigenetic modifications providing GSCs with stem-like properties. However, GSCs frequently activate multiple compensatory signaling pathways, change phenotype along tumor progression, displaying genetic heterogeneity, high plasticity and diversity of stemness markers, nullifying potential effective therapies (24). The identification of the distinctive GSC Achilles heel is an urgent goal for GBM treatment, since innovative therapeutic approaches identified for other cancer types left the survival of GBM patients practically unchanged over the past decades.
Ion Channels in Cancer: CLIC1 Functional Expression and Therapeutic Potential
Ion channels are integral membrane proteins that form pores through which enable the passage of ions between cell compartments, regulating electrical excitation, cell proliferation, motility, survival, and maintaining tissue homeostasis. Structural defects or dysregulated functioning of ion channels play a pathogenic role in several human diseases including cancer. In particular, alterations of ion channel activity contribute to malignant transformation, inducing aberrant cell cycle rate, inability to activate the apoptotic program, and increased migration and invasion abilities (25). Genes encoding ion channels involved in oncogenic transformation (26) are differentially expressed in cancer and normal cells, in breast cancer (27), lung adenocarcinoma (28), and GBM (29).
While the role of plasma membrane channels has been extensively studied, less is known about intracellular ion channels. These molecules, inactive in the cytoplasm, are able to auto-insert into membranes where they act as functional integral ion channels, and have been recently recognized to regulate cell cycle, apoptosis, cell adhesion and motility (30). In this scenario, pharmacological modulation of intracellular ion channels would represent a potential innovative therapeutic option.
Among the ion channels whose aberrant expression and activity is relevant for neoplastic transformation, the chloride intracellular channel (CLIC) family recently gained particular attention. The six members of CLIC group (CLIC1-6), are present in both soluble and membrane-associated forms, displaying cell-specific expression and biological functions in mammalian tissues, not functioning as conventional chloride channels but possessing peculiar physiological roles in each different cell type (31). CLIC1 is the most widely expressed and studied channel of this family, in both physiological and pathological conditions, including brain functioning and cancer cell proliferation (32).
Overview of the Mechanisms of CLIC1 Activation and Related Physiological Functions
CLIC1 is a metamorphic protein (33) able to switch from a soluble cytoplasmic conformation to a transmembrane isoform (tmCLIC1) (34). Thus, CLIC1 exists in three different states: a monomeric soluble form, an oxidized soluble dimeric intermediate form, and an integral membrane form. The soluble monomer contains a thioredoxin-like N-domain with a glutathione binding site. The formation of the dimer is stabilized through a disulfide bond which connects two conserved cysteine residues, Cys-24 and Cys-59, which are essential for channel assembly, as the mutation of each one of them prevents the channel formation (34). Cys-24 residue is also required for the protein redox regulation, rising the hypothesis that CLIC1 membrane insertion could be controlled by reactive oxygen species (ROS) signaling (35, 36). Membrane association implies the formation of oligomeric CLIC1 complexes (37).
The ability to form the channel pore was confirmed in artificial lipid bilayers by Littler et al. (38). Membrane-associated CLIC1 exposes the N-terminal region to the extracellular space, determining the ability to activate a selective chloride conductance. Both oxidizing conditions and changes in pH levels control CLIC1 membrane insertion. In fact, CLIC1 membrane insertion is not only dependent on the level of cellular oxidation, as suggested by the observation that the dimeric intermediate form is reversible under reducing conditions, but its assembling within lipid bilayers and channel activity are also dependent on pH, being minimal at pH 7 and reaching the maximum rate at ± 2 pH units (39, 40). Mutation of two histidine residues, His-74 and His-185, impairs CLIC1 pH sensitivity, preventing membrane insertion at acidic pH 5.5 (41).
CLIC1 Signaling in Brain Function
CLIC1 is almost ubiquitously expressed in human tissues, including the central nervous system (CNS) where it is expressed in both excitable and non-excitable cells. CLIC1 is also present, in cytoplasmic conformation, in microglia, the brain intrinsic immune system (42, 43). Being chronic inflammation of the CNS during neurodegenerative disorders sustained by activated glia, tmCLIC1 was involved in the pathophysiology of Alzheimer's disease, considering that the neurodegenerative process implies an overproduction of ROS mediated by activated microglia (42, 44). This phenomenon can be reproduced in vitro stimulating microglial cells with amyloid β (Aβ) peptides: Aβ-activated microglia is characterized by high proliferative rate, production of large amount of ROS, and sustained by tmCLIC1 activity (42). Though tmCLIC1 rapidly increases in response to Aβ stimulation, it is rarely detectable in quiescent microglia cells (45). Indeed, CLIC1 downregulation in microglia by small interfering RNA or its inhibition using the channel blocker IAA94 and/or specific antibodies, prevents Aβ-induced neurotoxicity (45). Analogously, CLIC1 activity is a pre-requisite for ROS overproduction in β-amyloid-activated microglia (42). All together these findings indicate that tmCLIC1 plays a crucial role in the microglial inflammatory state characterizing the neurodegenerative processes, and support therapeutic targeting for neuroprotective strategies (44).
CLIC1 in Cancer
In the last years, growing evidence highlighted the role of CLIC1 as key factor in tumor development and progression. Working as a chloride channel, tmCLIC1 plays an essential role in tumorigenesis, controlling cell volume regulation (46), cell migration and invasion (47–49), and neoangiogenesis (50). CLIC1 is overexpressed in several human solid tumors, as compared to the surrounding normal tissue. For example, CLIC1 gene expression is significantly increased in bladder (51), in situ breast ductal (52) and ovarian (53) carcinomas, and it has been linked to oncogenic functions and poor prognosis in colorectal (48), gastric (49), hepatocellular (54), gallbladder (55), pancreatic (56), and lung carcinomas (57), and sarcomas (58). Bioinformatic analysis (cBioPortal/TCGA datasets) of CLIC1 mRNA levels in several human aggressive carcinomas (breast, colorectal, esophagus, liver, ovarian, stomach, prostate, thyroid, uterine, head & neck, and pancreas) shows that this channel is expressed at similar levels in all the different types of neoplasia, with a small increment only in colorectal, head & neck and pancreatic cancers (Figure 1). These data suggest the relevance of this channel in the development and progression of most malignant neoplasms. Moreover, CLIC1 gene is highly conserved among tumors in the various districts, with only 2% of patients carrying missense or non-sense mutations, clearly indicating that its role in tumorigenesis is more related to membrane localization and activity than to mutations.
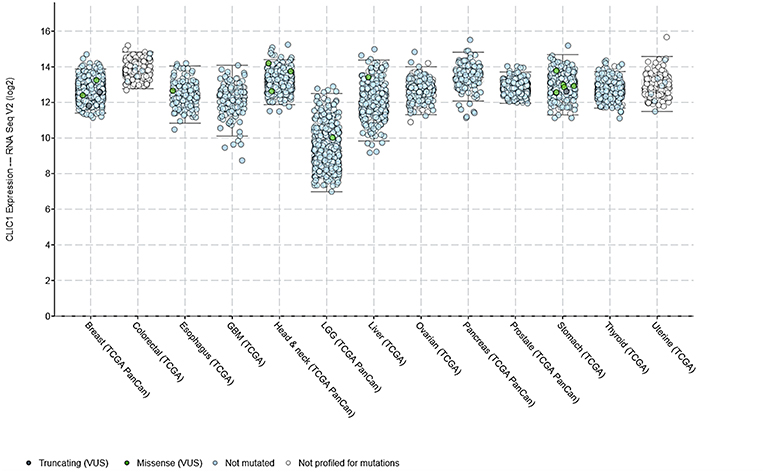
Figure 1. CLIC1 mRNA expression levels in various human carcinomas and mutations found according to the cBioPortal/TCGA datasets.
The expression and function of several ion channels are altered in GBM cells, and, within chloride channels, changes in CLIC1 gene expression have been frequently detected (51). CLIC1 mRNA and protein levels are up-regulated in GBM as compared to normal brain parenchyma. The analysis of TCGA database identified a weak correlation with tumor stage, displaying lower expression in low grade gliomas than in GBM (Figure 1), suggesting a potential role for this channel in the malignant behavior of this tumor. Similarly, CLIC1 expression levels directly correlated to GBM aggressiveness in experimental models (30). Beyond expression levels, in vitro and preclinical in vivo studies analyzing CLIC1 channel function in malignant transformation and progression, shed new light on its biological and clinical significance in tumors. CLIC1 channel activity is involved in invasion and migration through ROS-mediated MAPK pathway in colon cancer cells (46, 48), and gastric cancer cells (49, 59). Also the metastatic process has been associated to CLIC1 functioning in gallbladder and hepatocellular carcinomas (55, 60).
The abundance of tmCLIC1 expression in cancer and its high activity in all the cells with sustained proliferation rate, raised the hypothesis of an oncogenic role of CLIC1 (43, 56).
CLIC1 Role in Cell Cycle Progression of Cancer Cells
Different chloride channels are involved in cell division and specifically in the regulation of cell cycle progression, showing a functional activity restricted to a specific cell cycle phase, the G1/S transition (61, 62). In physiological conditions, CLIC1 is mostly cytoplasmic and, upon an oxidative burst, it transiently inserts into the plasma membrane. However, after persistent oxidative stress and/or alkaline cytoplasmic pH, the integral membrane channel form becomes constitutive. Oxidation and cytoplasm alkalization are hallmarks of cancer cells (63, 64) and both conditions promote G1/S cell cycle progression (65, 66). Intriguingly, high ROS production, cytoplasm alkalization, and the subsequent G1/S transition occur in the same time-window in which CLIC1 is active as ion channel (43, 67). Indeed, tmCLIC1 functional expression undergoes a well-defined timing, as shown by electrophysiology measurements, demonstrating that chloride current increases along G1/S phase progression, reaching a peak just before G1/S transition (68) (Figure 2). Fluorescence intensity analysis of tmCLIC1 by TIRF microscopy supports these results, demonstrating a different localization of the protein during the different phases of the cell cycle (67). Moreover, the inhibition of CLIC1 activity with the specific blocker IAA94 (69), or using an antibody targeting the NH2 extracellular portion of the channel, induces the accumulation of cancer cells, including GSCs, in the G1 phase with a consequent delay of cell cycle progression (68). Elevated ROS levels and alkaline pH can result from the overexpression and/or hyperactivation of NADPH oxidase and Na+/H+ exchanger 1 (NHE1), respectively, and both NADPH oxidase and NHE1 activities are impaired by targeting CLIC1 function (67). In this scenario, functional expression and activation of tmCLIC1 trigger a feed-forward mechanism which involves the activity of NAPDH oxidase and NHE1 establishing a vicious loop which generates a cellular microenvironment that favors the abnormal proliferative rate of tumor cells (67).
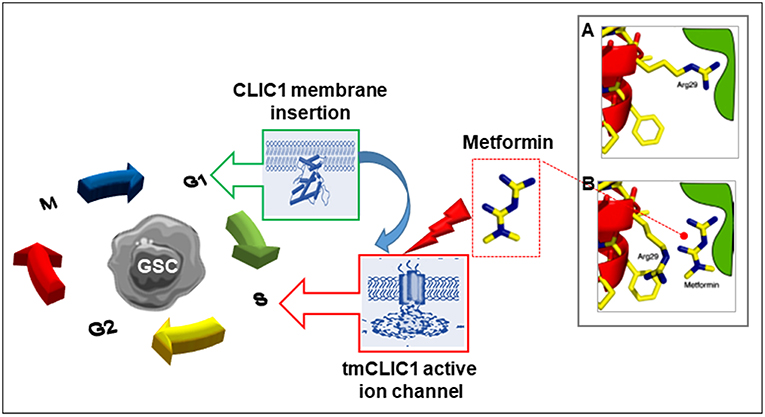
Figure 2. Scheme of the proposed mechanism by which metformin and other biguanides interacts with CLIC1 in glioblastoma stem cells. CLIC1 is a main regulator of GSC functioning once expressed into the plasma membrane and acting as chloride ion channel (tmCLIC1). CLIC1 activity promotes cell cycle progression and cell division. While in normal cells this functional expression transiently occurs during G1/S phase transition, it is constitutive in cancer cells leading to accelerated growth rate. Metformin (and other biguanides) directly interact with the extracellular portion of the active tmCLIC1, interfering with its activity and inhibiting cell cycle progression with high specificity toward GSCs, due to the high activity of CLIC1 in these cells. The insert shows a schematic representation of the putative molecular site of CLIC1 blockade by metformin: (A) In the closed state of CLIC1, the side chain of Arg29 makes an interaction that destabilizes the closed state. This facilitates the opening of the channel; (B) Metformin (and possibly other biguanides) interacts with the amino terminus of the channel, presumably in the vicinity of Arg29 side chain responsible for pore opening, stabilizing the closed state and blocking the channel activity [modified from Gritti et al. (68)].
Role of CLIC1 in Cancer Stem Cell Proliferation
The relevance of CLIC1 in tumor biology, and for GBM in particular, is further supported by the observation that tmCLIC1 is highly expressed in patient-derived GSC sub-populations (30, 68). Moreover, CLIC1-mediated chloride current is higher in GSCs isolated from neurospheres and expressing stemness markers (nestin, Sox2, Olig2), than in the differentiated GBM cell counterpart (68). Inhibition of CLIC1 activity using IAA94 (69), anti-CLIC1 N-terminus antibodies, or CLIC1 downregulation using small interfering RNA, causes a reduction of self-renewal, proliferation and in vivo tumorigenic ability of patients-derived GSCs (30, 68, 70).
This evidence strengthens the idea that CLIC1 plays a critical role in the tumorigenic potential of GBM-derived stem/progenitor cells. The differential functional expression of CLIC1 between GSCs and their differentiated counterpart could represent a possible strategy to selectively recognize and hit the tumor stem cell subset. Thus, tmCLIC1 represents a potential ideal target for antineoplastic treatments, also as chemo-sensitizing approach which, hitting CSC subpopulations, may increase tumor responsiveness to conventional anticancer therapies.
Therapeutic Potential OF CLIC1 Pharmacological Targeting in Glioblastoma
Rationale for Targeting CLIC1
To date, GBM represents the biggest challenge for cancer therapy. The main reason for the failure of GBM treatments is represented by tumor occurrence in one of the most critical area of human body, physically shielded by the skull and pharmacologically isolated by the BBB. Although GBM, as every tumor, represents a detriment from a clinical point of view, cancer cells may be considered an evolutionary successful model, being able to dynamically adapt the changing microenvironment and reprogram their own physiology setting in a new steady state. Tumor cells are in a chronic hyper-activated (allostatic) state (71) that supports their abnormal proliferative rate. A novel strategy for GBM treatment could be to hit one or more components that promote the assessment of the chronic allostatic state, restoring the physiological homeostasis (67). Several proteins, including NADPH oxidase and NHE1 exchanger, involved in the establishment of the allostatic condition (72, 73) are crucial for survival of both cancerous and normal cells, therefore limiting the possibility of their pharmacologic or genetic targeting. In this scenario, the peculiar ability of CLIC1 to change its functional localization depending on the activation state of the cells could be a compelling strategy to impair tumor cell proliferation and/or survival with a higher efficacy in CSCs (67). The possibility to selectively hit the CSC fraction could be instrumental for a more efficient activity of standard antineoplastic drugs, also considering that CLIC1 inhibition-dependent delay of G1/S phase transition might also favor microglia activity toward tumor cells, and potentiate conventional cytotoxic therapies.
Cellular and molecular steps through which ion channels, including CLIC1, support malignant cell phenotype and specifically CSC features (enhanced survival and proliferation rate, self-renewal, migration ability, and resistance to apoptosis and chemo- or radio-therapy) are still not completely defined. However, a growing bulk of evidence is currently available to be exploited in pre-clinical investigation or in medicinal chemistry studies for the identification of novel compounds able to target ion channels involved in cancer cell proliferation.
CLIC1 displays several peculiar features which render this channel an ideal pharmacological target in cancer cells. First, CLIC1 is overexpressed in several cancer types as compared to non-cancer cell counterparts; second, its activity is pivotal for cancer cell functioning; third, although in physiological conditions it is ubiquitously expressed, its chloride channel activity, absolutely dependent on its membrane insertion, is constitutive only in tumor cells and, in particular, in the CSC compartment (32). In fact, the translocation of CLIC1 to the membrane is reversible and the channel activity is transient in normal cells, with only few channels active at a given time. On the contrary, in cancer cells the specific intracellular microenvironment generated by the high production of ROS and low pH levels (74, 75) enhances CLIC1 functional expression promoting its constitutive membrane localization as an active ion channel (67). Given that tmCLIC1 is largely more abundant in GSCs than in healthy tissues (30), the possibility to specifically hit the transmembrane isoform could be a promising novel therapeutic approach for GBM. Indeed, a successful therapy should slow-down the proliferation of GBM cells, preventing relapses by inhibiting GSCs with the minimum possible systemic toxicity. However, IAA94, the only known compound able to block CLIC1 activity in vitro (69), can't be used as a potential drug for GBM due to its off-target toxicity in vivo.
Importantly, recent studies identified the well-known antidiabetic drug metformin as a compound able to impair tmCLIC1 activity (68) (Figure 2). Metformin is a generally very well tolerated type 2 diabetes (T2D) first line drug, which displays antineoplastic effects, although the molecular mechanism at the basis of this effect is still debated. Thus, understanding at a molecular level how metformin interferes with cancer cell proliferation and the role of CLIC1 in such effect might improve its repositioning as antitumor agent or, alternatively, allow the development of structural-related molecules showing higher efficacy and potency against tmCLIC1.
Development of Pharmacological Tools to Target CLIC1 Activity to Counteract Glioblastoma Cancer Stem Cell Tumorigenesis
The low clinical outcome of the available therapeutic approaches for GBM urges the identification of novel molecular targets and new molecules able to hit them. In this respect, as detailed in the previous paragraphs, CLIC1 represents a potential ideal candidate, for its relevance in GSC biology and its functional irrelevance in normal cells. If these premises are confirmed, a selective CLIC1 inhibitor should have high efficacy against tumor cells and low toxicity on the normal cell counterparts.
Unfortunately, the introduction of new molecular entities in clinics is becoming more and more difficult, due to the outraged increased costs of development and the tighter regulatory rules. Therefore, in the last years the approval of novel chemotherapeutics, with the only exception of biologicals, faces a significant slow-down. Drug repositioning, a strategy based on the identification of new disease indications and/or molecular targets for existing compounds, represents a drug discovery strategy which bypasses all the preclinical and early phase clinical trials and allows a faster, more efficient and less expensive way to bring molecules from bench to bedside. This is especially true if the studied drug has already proven good safety and tolerability profile in humans (76). Interestingly, a CLIC1 inhibitory activity was reported in some Chinese traditional medicine molecules, identified by bioinformatic strategies (77), although most attention has been dedicated to the effects of metformin a biguanide antidiabetic drug. In particular, it was shown that metformin is a powerful CLIC1 inhibitor in GSCs (78), and its repositioning as GBM drug could have a significant impact for the treatment of these patients. However, metformin represents the most studied repurposed drug in oncology in almost all human tumors and several intracellular mechanisms were proposed to mediate these effects. Thus, to show that CLIC1 inhibition is a primary target for this drug in GBM a pharmacological class effect should be demonstrated, showing that also structurally related drugs (i.e., containing a biguanide moiety) have the same biochemical mechanism (CLIC1 inhibition) and clinical effects (antitumor activity).
In the next sections we will discuss the general pharmacology of metformin (and of other biguanides), the evidence of their antiproliferative effects, and data showing that CLIC1 is one of the main molecular targets involved in the inhibition of GSC proliferation and invasiveness induced by this class of drugs, highlighting the pros and cons of their possible use for treatment of GBM.
Pharmacology of Biguanides
Biguanides are a class of drugs whose functional group consists of two guanidines linked by a common nitrogen (Figure 3); biguanides have a broad range of medical indications spanning from the first line pharmacological approach for T2D, by metformin and its derivatives phenformin and buformin (although the latter compounds are no longer used in therapy), to antimalarial prophylaxis and therapy by proguanil, and antiviral and antimicrobial activity by moroxydine, chlorophenylbiguanide, and chlorhexidine.
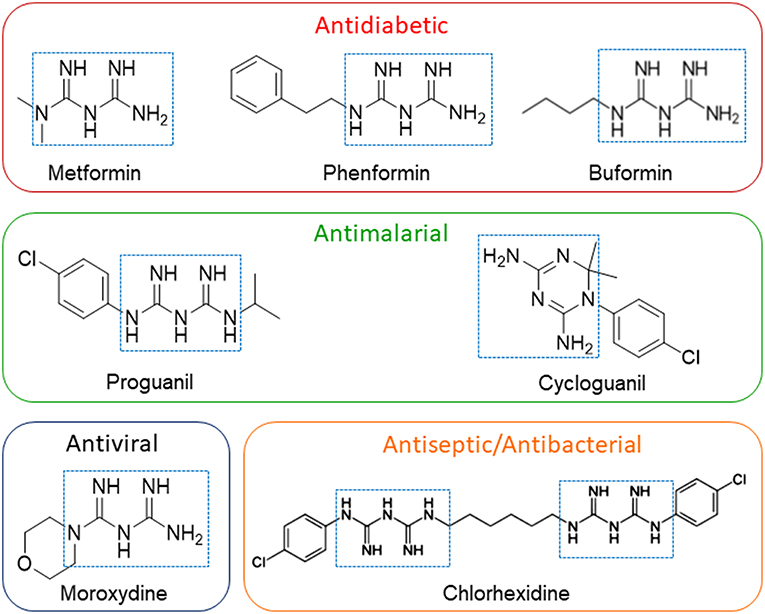
Figure 3. Structures of drugs possessing a biguanide moiety as pharmacophore. The presence of the biguanide moiety in clinically-relevant compounds, highlighting their actual clinical applications, is evidenced by the dotted squares. Figure includes compounds affecting CLIC1 activity to inhibit GSC proliferation and self-renewal.
Original interest in biguanides derived from their potential antimalarial effects, particularly by proguanil the first compound of this class used in 1950, and still a current antimalarial drug. Proguanil is a synthetic arylbiguanide acting as oral prodrug and is considered the safest antimalarial compound; in vivo it is metabolized to the active derivative cycloguanil, which contains a cyclized biguanide moiety and acts as dihydrofolate reductase and folate synthesis inhibitor within malaria parasites (79). Proguanil is used for both malaria prophylaxis and treatment, in combination with atovaquone or cloroquine (80). The observation that this drug may cause hypoglycemia as side-effect, triggered the development of the dimethylbiguanide metformin (81).
Metformin was licensed as anti-diabetic agent in the UK in 1958, but only in 1995 in the USA, due to concerns about lactic acidosis and cardiac mortality, which, however, are now considered as very rare occurrences. Among the different biguanides introduced for diabetes therapy in late 1950s, metformin shows the better safety profile and tolerability (82). Two other biguanides, phenformin (phenethyl biguanide) and buformin (N-butyl biguaninde), although more potent than metformin as hypoglycemizing agents, were discontinued in 1970s due to the same adverse events (lactic acidosis and cardiac mortality) but occurring at higher rate than observed with metformin (83). In T2D patients, glucose-lowering effect of metformin is attributed to the reduction of hepatic glycogenolysis and gluconeogenesis, enhancement of insulin receptor tyrosine kinase activity, improvement of insulin sensitivity, and reduction of enteric glucose absorption (84). Metformin also induces peripheral glucose uptake, increasing glucose transporter GLUT4 activity, and glycogen synthesis, stimulates glucagon-like-peptide-1 (GLP-1) release, and reduces lipolysis and triglyceride levels.
Moroxydine, is another heterocyclic biguanide proposed in the 1950s as anti-influenza agent. Moroxydine exhibits anti-viral activity against RNA and DNA viruses, and was originally used for prophylaxis or therapy of viral infections. Moroxydine has negligible side effects, but very little information exists on its mechanism of action (85). Despite its favorable pharmacological profile, moroxydine has been scanty investigated, and only recently it has gained new interest as potential anti-hepatitis C agent (86).
Repositioning of Metformin and Other Biguanides as Antitumor Agents
Repositioning of Metformin as Anti-tumor Agent
On the basis of several epidemiological and preclinical observations, several biguanides have been proposed to possess anti-neoplastic activity; to date, metformin is the most promising application of repositioning of a non-oncological drug as anti-cancer agent. Epidemiologic studies suggested a correlation between chronic use of metformin in T2D patients and the reduction of incidence and related mortality of several solid tumors, when compared to T2D patients treated with other classes of hypoglycemic drugs (87–89). These observations triggered a series of pre-clinical and clinical investigations in several tumor types to detail the antitumor mechanism of action of metformin and its potential efficacy as adjuvant agent in clinics (90, 91). In diabetic patients, metformin use was correlated to a reduced risk of development of different cancer types, including pancreatic cancer (92), and lung and hepatocellular carcinomas also increasing survival time (93–95). However, to date non-univocal results were reported in the many studies published. Some meta-analyses confirmed a significant association between metformin use and lower incidence of pancreatic, liver, renal, endometrial, prostate, breast, colorectal, and ovarian carcinomas, while no correlation was found in other studies (96–103). Metformin use in T2D patients with HER2-positive breast cancer was associated with a better outcome (104) and a meta-analysis in breast cancer patients reported a significant association between metformin therapy and the reduction of all-cause mortality without observing a reduction of breast cancer incidence in these subjects (105).
Repositioning of Other Biguanides
Although less investigated than metformin, also other biguanides were shown to possess anti-cancer activity. Phenformin exerts antitumor activity in preclinical models in vitro and in vivo, using ovarian cancer (106) NSCLC (107), and hepatocellular carcinoma (108) cells, and pancreatic cancer patient-derived xenografts (109); moreover, phenformin was also reported to selectively affect the CSC compartment (110). Other studies showed that the antitumor activity of phenformin in mammary cancer was dependent on the inhibition of angiogenesis, apoptosis, and epithelial-mesenchymal transition (EMT) (111–113). The anti-cancer activity of buformin in rat mammary breast cancer carcinogenesis was also reported (114, 115).
However, compared to the other biguanides, the evidence of a potential broad antitumor activity, associated with the overall safety and low cost, has opened a new horizon for repurposing of metformin in oncology (87, 116, 117).
Repurposing of Biguanides for Targeting of CSCs
Several preclinical studies reported that metformin is effective against CSC subpopulations, the key target for all antitumor pharmacological approaches, at odd with most conventional anti-neoplastic drugs which have little or no effect on CSCs (118). Selective anti-cancer properties of metformin have been described in CSC-like cultures derived from colorectal (119), gastric (120), breast (121, 122), prostate (123), pancreatic (124, 125), and ovarian (126) carcinomas and osteosarcoma (127, 128). Metformin effects on CSCs include the impairment of self-renewal and survival, decreased expression of stemness markers, the slow-down of cell cycle progression and inhibition of invasiveness. Moreover, a chemo-sensitizing activity of metformin, helping to overcome refractory CSCs to radiotherapy (129), and chemotherapeutic agents (130) has been also described. As far as GBM, metformin was reported to synergize with temozolomide (131) and reduce the acquired resistance to this alkylating agent (132).
While most of the human tumors, at least at preclinical level, are affected by metformin, this drug is almost completely harmless for normal cells. The low toxicity observed in T2D patients after chronic treatment already suggested this eventuality, but it was directly demonstrated by in vitro experiments, in which metformin concentrations able to reduce CSC viability were ineffective in normal cells, including MSCs (68, 70, 133). These data clearly suggest that a tumor-specific target should mediate metformin antitumor effects.
To date, mainly pharmaco-epidemiologic and preclinical data were at the basis of the assumption that metformin may be useful in cancer prevention or treatment. However, hundreds of clinical trials are in progress to validate this hypothesis (see www.clinicaltrials.gov). Translation from retrospective to prospective trials however, is not easy-going also in light of several biases often present in retrospective studies (134). Some preoperative or neo-adjuvant window of opportunity studies reported a decrease in the expression of Ki-67, a marker of cell proliferation, after metformin treatment in breast, prostate, and endometrial cancers (135–137), although another study found no effects in breast cancer (138). An unpublished study (NCT01620593) found a significant decrease of prostate-specific antigen (PSA) after treatment of prostate cancer patients with metformin, while, in ovarian cancer (NCT01579812) no changes in progression-free and overall survival were reported (www.clinicaltrials.gov). Only few prospective studies have been to date published, reporting that metformin provided benefit in patients in colorectal adenoma and, in association with paclitaxel, in non-small cell lung cancer patients (139, 140).
Phenformin has been evaluated in a clinical trial (NCT03026517) in combination with dabrafenib and trametinib (RAF and MEK inhibitors, respectively) in patients with BRAF-mutated melanoma, but, till now, no results are available.
Overall the available literature data about the clinical antitumor efficacy of metformin are not conclusive, possibly due to the heterogeneous composition of patient cohorts, the study design, pharmacokinetics and posology discrepancies, as well as variable responses in different cancer types (141). Thus, repositioning of metformin and, potentially, other biguanide derivatives, in oncology is still a controversial topic, and results from clinical trials that are going to be concluded in the next years in different cancer types, mainly investigating the adjuvant efficacy of metformin in association with chemo- and radio-therapy, will provide a clearer picture of its clinical impact.
Notwithstanding these unsolved problems, a huge amount of data has been produced to detail the molecular mechanism(s) of the antiproliferative activity of metformin.
Molecular Mechanisms of Metformin Antitumor Effect
Although numerous experimental studies analyzed the antiproliferative, pro-apoptotic, and anti-invasive activity of metformin, at present, the exact molecular mechanisms through which this drug exerts its antitumor activity is only partially known. In fact, most of the possible intracellular pathways involved in tumor cell proliferation have been reported to be affected by metformin treatment in different cancers. Consequently, not only metformin seems to not display tumor specificity but also its activity seems to involve a wide plethora of intracellular signaling pathways.
The classical intracellular pathway proposed as molecular target for metformin antitumor effects has been derived by the mechanism activated in the liver to control glucose release (142). Metformin affects the energetic balance interfering with the complex I enzymes within mitochondrial respiration, reducing ATP content and the ATP/ADP ratio (143). This alteration, altogether with a direct regulation via liver kinase B1 (LKB1), causes the activation of the cellular energy sensor AMP-activated protein kinase (AMPK), which in turn leads to the inhibition of mTOR (144, 145), a kinase acting as crucial mediator of tumor cell metabolism (146). AMPK, activated after metformin treatment, was reported to directly phosphorylate PD-L1 causing its endoplasmic reticulum (ER) accumulation and ER-associated protein degradation. In fact, breast cancers from metformin-treated patients exhibit reduced PD-L1 levels, which enhances cytotoxic cell immunity against cancer cells (147). In addition, metformin, lowering the plasma levels of insulin and insulin-like growth factors, might indirectly inhibit the PI3K/Akt/mTOR pathway (148). AMPK activation following metformin treatment has been described in several human cancer cell types including breast (149, 150), endometrial (151), ovarian (152), pancreatic (153, 154), lung (155), prostate (123), head and neck (156), and colon carcinomas (157), often correlating with antiproliferative effects.
However, AMPK-independent pathways have gained increasing attention. Metformin was reported to directly inhibit mTOR signaling by inactivating Rag GTPases (158), or inducing cell cycle arrest through REDD1, a negative regulator of mTOR (159). Furthermore, several other intracellular effectors were reported to be modulated by metformin treatment to reduce cell proliferation, including, among others, the VEGF/PI3K/Akt pathway (160) in prostate cancer cells, Sonic hedgehog (Shh) signaling pathway in gastric cancer cells (161), inactivation of p38 MAPK and activation of ERK3 (both effects leading to inhibition of mTORC1, in which AMPK was only partially involved) in intrahepatic cholangiocarcinoma cells (162), reversal of the activation of ERK1/2 in ovarian cancer cells (163), inhibition of CLIC1 in gallbladder cancer cells (164); metformin also counterbalanced the overactivation of Notch1/Hes1 signaling observed in colorectal cancer patients (165), and induced apoptosis via the up-regulation of adenosine A1 receptor in human colorectal cancer cells (166). Other putative mechanisms of metformin anti-tumor activity involve the reduced RANKL (167) or caveolin 1 (168) expression in breast CSCs, and HIF-1α gene expression in oral squamous cell carcinoma cell lines, which caused inhibition of cell proliferation and migration (169). Moreover, metformin antiproliferative activity was also ascribed to enhanced autophagy in cancer cells, which causes cell cycle arrest or apoptosis (170, 171), and the modulation of miRNA activity (172, 173).
In addition, metformin downregulates ROS production of through inhibition of mitochondrial complex I (174, 175), and possesses anti-inflammatory and immunomodulatory activity, affecting energy metabolism of immune cells and stimulating CD8+ tumor infiltrating lymphocytes leading to a cytotoxic response against cancer cells (176); moreover, metformin enhances immune response in vivo in mouse melanoma model (177), and inhibits NF-κB nuclear localization and Stat3 activity in breast cancer CSCs (178).
Metformin disrupts TGFβ-mediated oncogenesis and invasiveness (179) either by direct binding (180) or by blocking autocrine TGFβ1 signaling (181). TGFβ1-dependent metastasization and invasive effects are mainly mediated by epithelial-to-mesenchymal transition (EMT). In this context metformin acts as EMT suppressor in different epithelial tumors (e.g., melanoma, colon, breast, lung, prostate, and thyroid cancer cells) (182–185). In prostate cancer, metformin represses EMT and metastasis by targeting the COX2/PGE2/STAT3 axis (186), while in breast cancer the AKT/mTOR/ZEB1 pathway was involved (187). Metformin also directly affects cancer cell metabolism interfering with glycolysis and the tricarboxylic acid cycle, decreases the production of ATP, NADH-linked respiration in cells and mitochondria, and the aspartate biosynthesis (188, 189), while induces indirect antiproliferative effects reducing hormones, cytokines and growth factor production (144, 190, 191).
Altogether, these preclinical studies, reporting metformin ability to modulate multiple, apparently unrelated mechanisms, strongly support its antitumor activity. However, the unexpected and unprecedented high number of different intracellular mechanisms regulated by a single drug in such different tumor cell types, suggests that most of these intracellular pathways could be indirectly modulated, being downstream from a common tumor-specific target directly affected by metformin.
However, it is worth to note that several unsolved issues are present in these studies. First, metformin concentrations used to cause antitumor effects in all in vitro studies here reported, largely exceed those obtained by the antidiabetic doses, and are difficult to be reached in patients. A second issue puzzling the anti-cancer use of metformin is its hydrophilic nature which limits its passive diffusion into cells (192), making necessary organic cationic transporters (OCT1, OCT2, and OCT3) for its internalization within cells (193, 194) and to cross the blood-brain barrier (195). The overexpression of these transporters is considered at the basis of the observation that metformin concentration in tissues is much higher than in plasma, and in tumors higher than in normal cells. Intratumor accumulation of metformin, induced by OCTs, has been involved in the direct antineoplastic activity of this biguanide (196). For example, in mammary tumor-bearing rats and in ovarian tumor biopsies form metformin-treated patients, metformin effects were dependent on high intratumor concentrations, which in the mammary cancer model were related to OCT2 expression (197, 198). Thus, it was suggested the possibility to potentiate metformin antiproliferative activity, obtaining clinically relevant concentrations due to the specific drug accumulation within tumors. This opportunity was demonstrated using pharmaceutic preparations and routes of administration different from the oral way (i.e., subcutaneous) allowing a topical tumor treatment (199–201).
Potential Role of Metformin and Other Biguanides as Antiproliferative Agents for Glioblastoma Stem Cells
Although in vitro studies reported the antiproliferative and proapoptotic efficacy of several drugs on GSC cultures (202–204), the same activity in patients was never reported. Thus, the lack of effective antitumoral drugs for GBM patients, pushed the testing of metformin repositioning in in vitro and in vivo GBM models (78).
Metformin reduces survival and proliferation rate not only of GBM cell lines (131, 205–207) but also of patient-derived GSC cultures (68, 70, 208–211) suggesting its efficacy to impair mechanisms involved in cancer cell stemness. This effect is time-dependent, since prolonged treatment caused significant antiproliferative effects also for relatively low concentrations (68). Importantly, metformin interference with GSC activity was further supported by the observation that, beside proliferation, several distinctive stemness features were also impaired in metformin-treated cultures, including self-renewal ability, as shown by colony-forming and spherogenesis assays (70, 132, 210, 211), migration and invasiveness (132, 210, 211) (Figure 4), and EMT (187), which is activated in GSCs to sustain GBM aggressiveness (212).
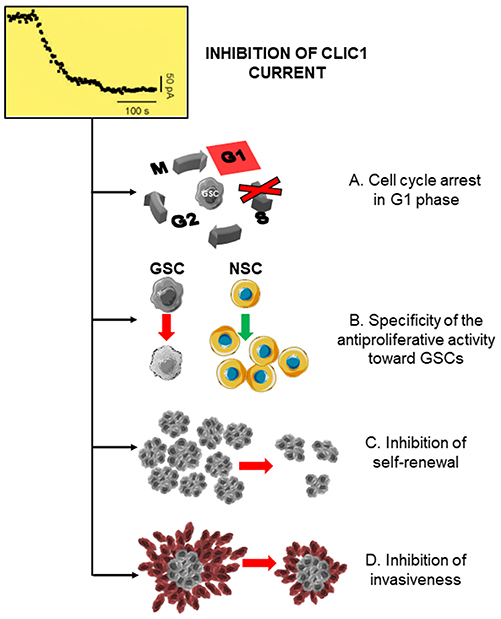
Figure 4. CLIC1 inhibition leads to the inhibition of glioblastoma stem cell proliferation, self-renewal, and invasiveness. Inhibition of CLIC1 activity by metformin and other biguanides causing a decrease in the chloride current induces several inhibitory effects in GSCs, including: (A) cell cycle arrest and cell accumulation in G1 phase, acting (B) selectively in GSCs, while sparing normal stem cells (NSC); (C) impairment of GSC self-renewal ability; and (D) inhibition of the invasive behavior.
Metformin was also tested in combination therapy with classical anti-cancer drugs, mainly the alkylating agent temozolomide, the GBM standard of care. In vitro and in vivo studies describe a potential synergism between the antiproliferative effects of metformin and temozolomide in GBM cell lines and GSCs (131, 132, 206, 207, 213). Moreover, a strong synergism between the antitumor effects of metformin and in vitro cell irradiation (5Gy), in the presence or absence of temozolomide, was also reported in GBM U87, U251, LN18, and SF767 cells (206).
Also in GBM several intracellular mechanisms mediating the antiproliferative and anti-invasive activity of metformin were reported. These include the inhibition of STAT3 (214) and Akt (70), the induction of apoptosis by increasing Bax/Bcl-2 ratio, reduced ROS production when co administered with temozolomide (213), or the downregulation of AKT-mTOR signaling pathway (207). Although some studies proposed an AMPK-dependent mechanism for the antitumor activity of metformin (208, 209, 215, 216) in other studies the inhibition of proliferation and self-renewal occurred in the absence of AMPK activation (70). Moreover, comparing the effects of metformin with a “pure” AMPK activator, the peptide A769662, which was unable to inhibit mTOR and GBM cell proliferation, it was shown that metformin suppresses GBM proliferation enhancing PRAS40–RAPTOR association to inhibit mTOR, independently of AMPK (217). Although this issue is still debated, recent data seem to confirm that the activation of AMPK and the inhibition of mTOR are not the main targets in GBM. In fact, on one hand a randomized phase II study assessing the efficacy of everolimus in combination with chemoradiation showed that mTOR inhibition does not improve GBM patients PFS (218), and, on the other, AMPK was shown to be chronically activated under cancer-associated stress conditions, to increase proliferation and survival. Moreover, AMPK inhibition reduces viability of patient-derived GBM stem cells (GSCs) (219), clearly indicating that, at least in GBM, AMPK activation cannot justify metformin antiproliferative effects and different molecular targets have to be found.
Furthermore, several studies showed that metformin activity was selectively directed against GSCs rather than differentiated glioma cells (68, 70, 211), indicating that a CSC-specific target mediates its activity.
In vivo, metformin significantly impairs GBM growth either after subcutaneous (206–208, 217) or intracranial grafting (210, 213, 220) in immunocompromised mice. These effects were obtained mainly after i.p. injection, although in one study (207) metformin was administered per os by gavage. In the cited studies, metformin induced a slow-down in tumor growth and prolonged mice survival, mainly acting in synergy with temozolomide or 2-deoxyglucose, inducing a significant effect also in temozolomide-resistant cells (213). However, it is worth to note that most studies were carried out on human GBM cell lines (mainly U87, U251) and only in few cases patient-derived GSCs were used (208, 210).
Phenformin exerts antitumor effects in GSCs overcoming resistance to temozolomide, suppressing GSC self-renewal via the reduction of the expression of stemness and mesenchymal markers, and the increase of miR-124, miR-137 and let-7 expression (221). Phenformin activity has been also analyzed as potential way to disrupt energetic/metabolic pathways sustaining GSC survival and proliferation (222, 223). In vivo, phenformin added to the drinking water, caused a significant inhibition of the growth of GBM, in an orthotopic model in which patient-derived GSCs were grafted in nude mice (221), confirming that beside metformin also other biguanides are active against GBM.
In fact, biguanides, unrelated to the antidiabetic drugs (i.e., moroxydine, and cycloguanil) were also reported to significantly reduce proliferation, self-renewal and invasiveness of GSCs, showing higher in vitro potency than metformin (211). This observation suggests that antitumor activity against the GBM stem cell-like compartment is a common feature of all biguanides. However, if this is true, all the biguanide moiety-containing molecules have to act through a common intracellular mechanism and all the other pathways proposed for metformin antitumor activity should represent tumor-specific downstream effectors dependent on a common effector which represents the direct biguanide target.
CLIC1 as Preferential Molecular Target Mediating Metformin, and Other Biguanides, Antitumor Effects in Glioblastoma Stem Cells
The controversies about the anticancer mechanisms of metformin led to overemphasize the role of AMPK in GSC antiproliferative effects, since the liver anti-hyperglycemic activity of this drug is mediated through the activation of this kinase (142). However, a growing bulk of evidence reported, in different cancer models, and in CSCs in particular, that (i) several AMPK-independent pathways are activated by metformin (70, 217); (ii) contrarily to what initially hypothesized, AMPK agonists enhance cancer cell proliferation and metabolism under metabolic stress (i.e., A-769662), while metformin and phenformin inhibit these cellular functions in an AMPK-independent manner (224); (iii) other compounds with a biguanide structure (i.e., moroxydine and cycloguanil), used with different clinical indications, and devoid of AMPK-related effects in the liver, induce the same anti-proliferative and anti-invasive activity in GSCs (211). The latter evidence strongly supports the possibility that, in GSCs, a common molecular target can be hit by all the biguanide-based compounds representing a new pharmacological class effect.
In this line of research, the observation that metformin and related compounds exert their activity on CSCs, not only in GBM (70, 208, 210, 211, 221) but also in different tumor types, such as breast cancer (121), while differentiated cells composing the tumor mass are relatively spared, clearly indicates that biguanides should interact with a CSC-specific target. In recent years, among the possible cancer-specific molecular targets for metformin, CLIC1 has been proposed to represent the main transducer of the biguanide effects in GSCs (68, 211). As detailed in the previous paragraphs, CLIC1 behaves as CSC-specific target because, although expressed in most normal and differentiated (non-stem) tumor cells, it is mainly present as inactive cytosolic monomer, with a very low activation rate (68, 211). This activation kinetics renders non-CSC subpopulations (and normal cells) relatively independent from CLIC1 for proliferation and survival. Conversely, CLIC1 is functionally expressed in GSCs, where it shows a constitutive activity with a peak at the G1-S transition (67), and its activity is absolutely necessary for GSC proliferation (Figure 2). Metformin treatment causes a significant inhibition of CLIC1 activity, measured by voltage-clamp electrophysiology experiments (Figure 4), reaching at high concentrations (5–10 mM) the same efficacy observed using IAA94. Electrophysiology experiments showed that metformin perfusion decreases the whole cell current that cannot be further reduced by the perfusion of IAA94. Current/voltage relationships show that the current amplitudes, at different membrane potentials, are superimposed, suggesting that the two drugs converge on the same molecular target (68). By single amino acid mutation experiments, metformin was also shown to directly interact with tmCLIC1 through Arg29 located within the inner side of the pore structure of the channel (68) (Figure 2). Interestingly this binding site is different from that of IAA94 identified as the external Cys24 (35) allowing a possible discrimination between the effects of the two drugs.
CLIC1 blockade directly correlates with the antiproliferative effects of metformin causing GSC arrest in the G1 phase of the cell cycle. Conversely, metformin, used at the same concentrations, was harmless for cells in which CLIC1 activity was negligible (i.e., MSCs or differentiated GBM cells) confirming the specificity of these effects for GCSs (Figure 4). Moreover, the down-regulation of CLIC1, while reducing the growth rate of GSCs (30) also diminished the antiproliferative activity of metformin, corroborating the hypothesis that, at least in these cells, CLIC1 is the main target of this biguanide (68, 211). This evidence implies that, although metformin directly or indirectly modulates different intracellular signaling, the inhibition of CLIC1 activity is sufficient and necessary to induce antiproliferative activity, at least in GSCs. Moreover, it is important to remark that the inhibition of a GSC-specific molecular target (i.e., tmCLIC1) confers metformin with high selectivity against tumor cells, while sparing normal cells, as also confirmed by the known very low toxicity observed when metformin is used as antidiabetic agent. This observation provides the molecular basis for metformin repositioning as promising novel antitumor agent, being at the same time highly effective toward tumor cells and causing low systemic toxicity (78).
A main issue in metformin-induced tmCLIC1 blockade (and in its antitumor activity, in all the tumor models analyzed) is the high drug concentration (up to 10 mM) required to induce an effect.
Thus, a potential new a therapy could be really successful if retains the efficacy and the discrimination capability among healthy and cancer cells, provided by CLIC1 localization, and the ability to block tmCLIC1, as metformin, but acting at lower doses. The search for novel, more potent tmCLIC1 inhibitors can have a big advantage whether the channel targeting ability can be shared by different structurally-related molecules representing a pharmacological class effect for biguanides. In this line, it is relevant that also phenformin and buformin, former antidiabetic biguanides, were reported to behave as anti-tumor agents (114).
The possible role of CLIC1 as molecular determinant of biguanide antitumor class effect has been recently analyzed in several patient-derived GSC cultures (211). In this study, metformin and phenformin, representative of the antidiabetic biguanides for which antitumoral activity was already proposed, were compared with two antimalarial compounds, proguanil and cycloguanil, and the antiviral compound moroxydine. All these molecules inhibited GSC proliferation, self-renewal, migration and invasion, showing a much higher potency that metformin (up to 50 fold lower IC50 than metformin observed with cycloguanil). However, proguanil effects were not specific since it was similarly toxic for GSC and normal stem cells. The direct effects of these molecules on CLIC1 activity were measured by electrophysiology experiments. All the compounds, but proguanil, exerted a significant inhibition of CLIC1-dependent ion current, acting at potency and efficacy related to the respective antiproliferative activity. The lack of efficacy of proguanil as far as CLIC1 inhibition, was proposed to be dependent on the simultaneous presence of the 1-(4-chlorophenyl) ring and the bulky 5-isopropyl group on the rigid biguanide skeleton, thus preventing the access to CLIC1 pore region.
Evidence from this study strengthen the hypothesis that molecules containing a biguanide moiety are potent CLIC1 inhibitors and, consequently, drugs able to selectively interfere with GSC proliferation, migration and self-renewal. Importantly, higher potency than metformin on both cell proliferation and CLIC1 activity inhibition was also demonstrated by these biguanides, suggesting that more easily reachable concentrations of these drugs could be similarly active as the high doses of metformin. Although all these drugs have known limitation for chronic use in patients with GBM, these data demonstrated that CLIC1 inhibition is not only a pharmacological property of metformin, but it may represent a class effect endowed of all the compounds containing a biguanide structure. The relevance of this information resides in the possibility to develop novel biguanide containing drugs, which retain the safety profile of metformin but endowed with increased efficacy and potency toward GSCs.
Conclusions and Future Presepectives
At odd with most malignant tumors, therapeutic perspective for GBM did not significantly progress in the last decades. In this context, GSCs play a central role in drug resistance, being still extremely elusive as far biological features and pharmacological sensitivity. However, the recent identification that CLIC1 activity is necessary for GSC proliferation, self-renewal and invasiveness, while it is dispensable for most non-transformed normal cell populations, opened new perspectives in the potential development of new therapeutics for this still incurable tumor. This observation found new strength after the recent report that metformin is an efficient inhibitor of CLIC1 activity, although with low potency (IC50: 10–30 mM) (211). These data are extremely relevant due to the strong interest in metformin repositioning as antitumoral agent. Several epidemiological, preclinical, and, more recently, some clinical trials are addressing the efficacy of this biguanide in basically all the human tumor types. Conversely, pharmacokinetic and even pharmacodynamic issues are still unsolved to better translate this information in a clinical setting. In particular, as far as GBM is concerned, the main intracellular mechanism associated to metformin antiproliferative activity, the activation of AMPK and the consequent mTOR inhibition, had to be discarded since AMPK was discovered to promote GSC proliferation. Thus, metformin antiproliferative activity has to depend on a completely different mechanism from its glucose-lowering effects. Among all the reported intracellular pathways affected by metformin in tumor cells, the inhibition of CLIC1 activity is of particular interest since it is GSC-specific (thus its targeting does not affect normal cell viability), in line with the low toxicity of the drug when chronically used in T2D patients. Moreover, this effect was directly evaluated by electrophysiology measurement preventing the possibility of effects mediated indirectly by other biochemical regulators. This observation supports that, in GSCs, the inhibition of CLIC1 is a common effect different drugs containing a biguanide structure.
In conclusion, the inhibition of CLIC1 is a novel and unexpected biguanide class effect, which could be used to develop novel drugs with a strong efficacy against GSCs. In fact, although all the biguanides to date tested as inhibitory of CLIC1 activity in GSCs are not completely satisfactory as far as pharmacokinetics and long term tolerability, we believe that this information might pave the way for the identification of novel structurally-related molecules, which in future will provide a better clinical outcome for GBM.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the Italian Association for Cancer Research (AIRC) # IG2015 16713, and by Fondazione Giovanni Celeghin (#2018).
References
1. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. (2013) 110:4009–14. doi: 10.1073/pnas.1219747110
2. Stupp R, Hegi ME, Mason WP, Van Den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
3. Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. (2011) 8:482–5. doi: 10.1016/j.stem.2011.04.013
4. Campos B, Gal Z, Baader A, Schneider T, Sliwinski C, Gassel K, et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J Pathol. (2014) 234:23–33. doi: 10.1002/path.4366
5. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
6. Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature. (2017) 549:227–32. doi: 10.1038/nature23666
7. Corsaro A, Bajetto A, Thellung S, Begani G, Villa V, Nizzari M, et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget. (2016) 7:38638–57. doi: 10.18632/oncotarget.9575
8. Nishikawa M, Inoue A, Ohnishi T, Kohno S, Ohue S, Matsumoto S, et al. Significance of glioma stem-like cells in the tumor periphery that express high levels of CD44 in tumor invasion, early progression, and poor prognosis in glioblastoma. Stem Cells Int. (2018) 2018:5387041. doi: 10.1155/2018/5387041
9. Park JW, Wollmann G, Urbiola C, Fogli B, Florio T, Geley S, et al. Sprouty2 enhances the tumorigenic potential of glioblastoma cells. Neuro Oncol. (2018) 20:1044–54. doi: 10.1093/neuonc/noy028
10. Rimkus TK, Carpenter RL, Sirkisoon S, Zhu D, Pasche BC, Chan MD, et al. Truncated glioma-associated oncogene homolog 1 (tGLI1) mediates mesenchymal glioblastoma via transcriptional activation of CD44. Cancer Res. (2018) 78:2589–600. doi: 10.1158/0008-5472.CAN-17-2933
11. Wang J, Xu SL, Duan JJ, Yi L, Guo YF, Shi Y, et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci. (2019) 22:91–105. doi: 10.1038/s41593-018-0285-z
12. Roos A, Ding Z, Loftus JC, Tran NL. Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Front Oncol. 7:120. doi: 10.3389/fonc.2017.00120
13. Wurth R, Bajetto A, Harrison JK, Barbieri F, Florio T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front Cell Neurosci. 8:144. doi: 10.3389/fncel.2014.00144
14. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. (2015) 2:152–63. doi: 10.1016/j.gendis.2015.02.001
15. Bajetto A, Pattarozzi A, Corsaro A, Barbieri F, Daga A, Bosio A, et al. Different effects of human umbilical cord mesenchymal stem cells on glioblastoma stem cells by direct cell interaction or via released soluble factors. Front Cell Neurosci. 11:312. doi: 10.3389/fncel.2017.00312
16. Guelfi S, Duffau H, Bauchet L, Rothhut B, Hugnot JP. Vascular transdifferentiation in the CNS: a focus on neural and glioblastoma stem-like cells. Stem Cells Int. (2016) 2016:2759403. doi: 10.1155/2016/2759403
17. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. (2010) 9:67–78. doi: 10.1158/1535-7163.MCT-09-0734
18. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
19. Thomas TM, Yu JS. Metabolic regulation of glioma stem-like cells in the tumor micro-environment. Cancer Lett. (2017) 408:174–81. doi: 10.1016/j.canlet.2017.07.014
20. Wang D, Berglund A, Kenchappa RS, Forsyth PA, Mule JJ, Etame AB. BIRC3 is a novel driver of therapeutic resistance in Glioblastoma. Sci Rep. 6:21710. doi: 10.1038/srep21710
22. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. (2012) 10:717–28. doi: 10.1016/j.stem.2012.05.007
23. Florio T, Barbieri F. The status of the art of human malignant glioma management: the promising role of targeting tumor-initiating cells. Drug Discov Today. (2012) 17:1103–10. doi: 10.1016/j.drudis.2012.06.001
24. Makena MR, Ranjan A, Thirumala V, Reddy AP. Cancer stem cells: road to therapeutic resistance and strategies to overcome resistance. Biochim Biophys Acta Mol Basis Dis. (2018). doi: 10.1016/j.bbadis.2018.11.015. [Epub ahead of print].
25. Fraser SP, Pardo LA. Ion channels: functional expression and therapeutic potential in cancer. colloquium on ion channels and cancer. EMBO Rep. (2008) 9:512–5. doi: 10.1038/embor.2008.75
26. Prevarskaya N, Skryma R, Shuba Y. Ion channels and the hallmarks of cancer. Trends Mol Med. (2010) 16:107–21. doi: 10.1016/j.molmed.2010.01.005
27. Ko JH, Ko EA, Gu W, Lim I, Bang H, Zhou T. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol Cancer 12:106. doi: 10.1186/1476-4598-12-106
28. Ko JH, Gu W, Lim I, Bang H, Ko EA, Zhou T. Ion channel gene expression in lung adenocarcinoma: potential role in prognosis and diagnosis. PLoS ONE 9:e86569. doi: 10.1371/journal.pone.0086569
29. Wang R, Gurguis CI, Gu W, Ko EA, Lim I, Bang H, et al. Ion channel gene expression predicts survival in glioma patients. Sci Rep. 5:11593. doi: 10.1038/srep11593
30. Setti M, Savalli N, Osti D, Richichi C, Angelini M, Brescia P, et al. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J Natl Cancer Inst. (2013) 105:1644–55. doi: 10.1093/jnci/djt278
31. Argenzio E, Moolenaar WH. Emerging biological roles of Cl- intracellular channel proteins. J Cell Sci. (2016) 129:4165–74. doi: 10.1242/jcs.189795
32. Peretti M, Angelini M, Savalli N, Florio T, Yuspa SH, Mazzanti M. Chloride channels in cancer: focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim Biophys Acta. (2015) 1848:2523–31. doi: 10.1016/j.bbamem.2014.12.012
33. Murzin AG. Biochemistry. Metamor. Proteins. Science. (2008) 320:1725–6. doi: 10.1126/science.1158868
34. Littler DR, Harrop SJ, Goodchild SC, Phang JM, Mynott AV, Jiang L, et al. The enigma of the CLIC proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett. (2010) 584:2093–101. doi: 10.1016/j.febslet.2010.01.027
35. Harrop S. J., Demaere M. Z., Fairlie W. D., Reztsova T., Valenzuela S. M., Mazzanti M., et al. (2001). Crystal structure of a soluble form of the intracellular chloride ion channel CLIC1 (NCC27) at 1.4-A resolution. J Biol Chem. 276, 44993–45000. doi: 10.1074/jbc.M107804200
36. Goodchild SC, Howell MW, Littler DR, Mandyam RA, Sale KL, Mazzanti M, et al. Metamorphic response of the CLIC1 chloride intracellular ion channel protein upon membrane interaction. Biochemistry. (2010) 49:5278–89. doi: 10.1021/bi100111c
37. Hare JE, Goodchild SC, Breit SN, Curmi PM, Brown LJ. Interaction of human chloride intracellular channel protein 1 (CLIC1) with lipid bilayers: a fluorescence study. Biochemistry. (2016) 55:3825–33. doi: 10.1021/acs.biochem.6b00080
38. Littler DR, Harrop SJ, Fairlie WD, Brown LJ, Pankhurst GJ, Pankhurst S, et al. The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J Biol Chem. (2004) 279:9298–305. doi: 10.1074/jbc.M308444200
39. Tulk BM, Kapadia S, Edwards JC. CLIC1 inserts from the aqueous phase into phospholipid membranes, where it functions as an anion channel. Am J Physiol Cell Physiol. (2002) 282:C1103–1112. doi: 10.1152/ajpcell.00402.2001
40. Warton K, Tonini R, Fairlie WD, Matthews JM, Valenzuela SM, Qiu MR, et al. Recombinant CLIC1 (NCC27) assembles in lipid bilayers via a pH-dependent two-state process to form chloride ion channels with identical characteristics to those observed in Chinese hamster ovary cells expressing CLIC1. J Biol Chem. (2002) 277:26003–11. doi: 10.1074/jbc.M203666200
41. Achilonu I, Fanucchi S, Cross M, Fernandes M, Dirr HW. Role of individual histidines in the pH-dependent global stability of human chloride intracellular channel 1. Biochemistry. (2012) 51:995–1004. doi: 10.1021/bi201541w
42. Milton RH, Abeti R, Averaimo S, Debiasi S, Vitellaro L, Jiang L, et al. CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J Neurosci. (2008) 28:11488–99. doi: 10.1523/JNEUROSCI.2431-08.2008
43. Averaimo S, Milton RH, Duchen MR, Mazzanti M. Chloride intracellular channel 1 (CLIC1): Sensor and effector during oxidative stress. FEBS Lett. (2010) 584:2076–84. doi: 10.1016/j.febslet.2010.02.073
44. Skaper SD. Ion channels on microglia: therapeutic targets for neuroprotection. CNS Neurol Disord Drug Targets. (2011) 10:44–56. doi: 10.2174/187152711794488638
45. Novarino G, Fabrizi C, Tonini R, Denti MA, Malchiodi-Albedi F, Lauro GM, et al. Involvement of the intracellular ion channel CLIC1 in microglia-mediated beta-amyloid-induced neurotoxicity. J Neurosci. (2004) 24:5322–30. doi: 10.1523/JNEUROSCI.1170-04.2004
46. Wang P, Zhang C, Yu P, Tang B, Liu T, Cui H, et al. Regulation of colon cancer cell migration and invasion by CLIC1-mediated RVD. Mol Cell Biochem. (2012) 365:313–21. doi: 10.1007/s11010-012-1271-5
47. Tian Y, Guan Y, Jia Y, Meng Q, Yang J. Chloride intracellular channel 1 regulates prostate cancer cell proliferation and migration through the MAPK/ERK pathway. Cancer Biother Radiopharm. (2014) 29:339–44. doi: 10.1089/cbr.2014.1666
48. Wang P, Zeng Y, Liu T, Zhang C, Yu PW, Hao YX, et al. Chloride intracellular channel 1 regulates colon cancer cell migration and invasion through ROS/ERK pathway. World J Gastroenterol. (2014) 20:2071–8. doi: 10.3748/wjg.v20.i8.2071
49. Zhao W, Lu M, Zhang Q. Chloride intracellular channel 1 regulates migration and invasion in gastric cancer by triggering the ROS-mediated p38 MAPK signaling pathway. Mol Med Rep. (2015) 12:8041–7. doi: 10.3892/mmr.2015.4459
50. Tung JJ, Kitajewski J. Chloride intracellular channel 1 functions in endothelial cell growth and migration. J Angiogenes Res. 2:23. doi: 10.1186/2040-2384-2-23
51. Biasiotta A, D'arcangelo D, Passarelli F, Nicodemi EM, Facchiano A. Ion channels expression and function are strongly modified in solid tumors and vascular malformations. J Transl Med. (2016) 14:285. doi: 10.1186/s12967-016-1038-y
52. Wulfkuhle JD, Sgroi DC, Krutzsch H, Mclean K, Mcgarvey K, Knowlton M, et al. Proteomics of human breast ductal carcinoma in situ. Cancer Res. (2002) 62:6740–9.
53. Yu W, Cui R, Qu H, Liu C, Deng H, Zhang Z. Expression and prognostic value of CLIC1 in epithelial ovarian cancer. Exp Ther Med. (2018) 15:4943–9. doi: 10.3892/etm.2018.6000
54. Wei X, Li J, Xie H, Wang H, Wang J, Zhang X, et al. Chloride intracellular channel 1 participates in migration and invasion of hepatocellular carcinoma by targeting maspin. J Gastroenterol Hepatol. (2015) 30:208–16. doi: 10.1111/jgh.12668
55. Wang JW, Peng SY, Li JT, Wang Y, Zhang ZP, Cheng Y, et al. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett. (2009) 281:71–81. doi: 10.1016/j.canlet.2009.02.020
56. Lu J, Dong Q, Zhang B, Wang X, Ye B, Zhang F, et al. Chloride intracellular channel 1 (CLIC1) is activated and functions as an oncogene in pancreatic cancer. Med Oncol. (2015) 32:616. doi: 10.1007/s12032-015-0616-9
57. Wang W, Xu X, Wang W, Shao W, Li L, Yin W, et al. The expression and clinical significance of CLIC1 and HSP27 in lung adenocarcinoma. Tumour Biol. (2011) 32:1199–208. doi: 10.1007/s13277-011-0223-0
58. Murray E., Hernychova L., Scigelova M., Ho J., Nekulova M., O'neill J. R., et al. (2014). Quantitative proteomic profiling of pleomorphic human sarcoma identifies CLIC1 as a dominant pro-oncogenic receptor expressed in diverse sarcoma types. J Proteome Res. 13, 2543–2559. doi: 10.1021/pr4010713
59. Li BP, Mao YT, Wang Z, Chen YY, Wang Y, Zhai CY, et al. CLIC1 Promotes the Progression of Gastric Cancer by Regulating the MAPK/AKT Pathways. Cell Physiol Biochem. (2018) 46:907–24. doi: 10.1159/000488822
60. Li RK, Zhang J, Zhang YH, Li ML, Wang M, Tang JW. Chloride intracellular channel 1 is an important factor in the lymphatic metastasis of hepatocarcinoma. Biomed Pharmacother. (2012) 66:167–72. doi: 10.1016/j.biopha.2011.10.002
61. Shiozaki A, Otsuji E, Marunaka Y. Intracellular chloride regulates the G(1)/S cell cycle progression in gastric cancer cells. World J Gastrointest Oncol. (2011) 3:119–22. doi: 10.4251/wjgo.v3.i8.119
62. He YM, Zhang ZL, Liu QY, Xiao YS, Wei L, Xi C, et al. Effect of CLIC1 gene silencing on proliferation, migration, invasion and apoptosis of human gallbladder cancer cells. J Cell Mol Med. (2018) 22:2569–79. doi: 10.1111/jcmm.13499
63. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. (2010) 44:479–96. doi: 10.3109/10715761003667554
64. Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 7:e2253. doi: 10.1038/cddis.2016.105
65. Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. (2004) 337:1–13. doi: 10.1016/j.gene.2004.04.032
66. Verbon EH, Post JA, Boonstra J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene. (2012) 511:1–6. doi: 10.1016/j.gene.2012.08.038
67. Peretti M, Raciti FM, Carlini V, Verduci I, Sertic S, Barozzi S, et al. Mutual influence of ROS, pH and CLIC1 membrane protein in the regulation of G1/S phase progression in human glioblastoma stem cells. Mol Cancer Ther. (2018) 17:2451–61. doi: 10.1158/1535-7163.MCT-17-1223
68. Gritti M, Wurth R, Angelini M, Barbieri F, Peretti M, Pizzi E, et al. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget. (2014) 5:11252–68. doi: 10.18632/oncotarget.2617
69. Valenzuela S. M., Mazzanti M., Tonini R., Qiu M. R., Warton K., Musgrove E. A., et al. (2000). The nuclear chloride ion channel NCC27 is involved in regulation of the cell cycle. J Physiol. 529 (Pt 3), 541–552. doi: 10.1111/j.1469-7793.2000.00541.x
70. Wurth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: a role for metformin-induced inhibition of Akt. Cell Cycle. (2013) 12:145–56. doi: 10.4161/cc.23050
71. Mcewen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. (2003) 43:2–15. doi: 10.1016/S0018-506X(02)00024-7
72. Shono T, Yokoyama N, Uesaka T, Kuroda J, Takeya R, Yamasaki T, et al. Enhanced expression of NADPH oxidase Nox4 in human gliomas and its roles in cell proliferation and survival. Int J Cancer. (2008) 123:787–92. doi: 10.1002/ijc.23569
73. Di Cristofori A, Ferrero S, Bertolini I, Gaudioso G, Russo MV, Berno V, et al. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget. (2015) 6:17514–31. doi: 10.18632/oncotarget.4239
74. Mclean LA, Roscoe J, Jorgensen NK, Gorin FA, Cala PM. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol. (2000) 278:C676–688. doi: 10.1152/ajpcell.2000.278.4.C676
75. Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC. Redox control of the cell cycle in health and disease. Antioxid Redox Signal. (2009) 11:2985–3011. doi: 10.1089/ars.2009.2513
76. Wurth R, Thellung S, Bajetto A, Mazzanti M, Florio T, Barbieri F. Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today. (2016) 21:190–9. doi: 10.1016/j.drudis.2015.09.017
77. Wang W, Wan M, Liao D, Peng G, Xu X, Yin W, et al. Identification of potent chloride intracellular channel protein 1 inhibitors from traditional chinese medicine through structure-based virtual screening and molecular dynamics analysis. Biomed Res Int. (2017) 2017:4751780. doi: 10.1155/2017/4751780
78. Wurth R, Barbieri F, Florio T. New molecules and old drugs as emerging approaches to selectively target human glioblastoma cancer stem cells. Biomed Res Int. (2014) 2014:126586. doi: 10.1155/2014/126586
79. Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. (2007) 446:88–91. doi: 10.1038/nature05572
80. Wattanagoon Y, Taylor RB, Moody RR, Ochekpe NA, Looareesuwan S, White NJ. Single dose pharmacokinetics of proguanil and its metabolites in healthy subjects. Br J Clin Pharmacol. (1987) 24:775–80. doi: 10.1111/j.1365-2125.1987.tb03245.x
81. Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American diabetes association and the European association for the study of diabetes. Diabetes Care. (2009) 32:193–203. doi: 10.2337/dc08-9025
82. Bosi E. Metformin–the gold standard in type 2 diabetes: what does the evidence tell us? Diabetes Obes Metab. (2009) 11:3–8. doi: 10.1111/j.1463-1326.2008.01031.x
83. Lalau JD. Lactic acidosis induced by metformin: incidence, management and prevention. Drug Saf. (2010) 33:727–40. doi: 10.2165/11536790-000000000-00000
84. Sirtori CR, Pasik C. Re-evaluation of a biguanide, metformin: mechanism of action and tolerability. Pharmacol Res. (1994) 30:187–228. doi: 10.1016/1043-6618(94)80104-5
85. Sheppard S. Moroxydine: the story of a mislaid antiviral. Acta Derm Venereol Suppl. (1994) 183:1–9.
86. Magri A, Reilly R, Scalacci N, Radi M, Hunter M, Ripoll M, et al. Rethinking the old antiviral drug moroxydine: discovery of novel analogues as anti-hepatitis C virus (HCV) agents. Bioorg Med Chem Lett. (2015) 25:5372–6. doi: 10.1016/j.bmcl.2015.09.029
87. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. (2005) 330:1304–5. doi: 10.1136/bmj.38415.708634.F7
88. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. (2009) 52:1766–77. doi: 10.1007/s00125-009-1440-6
89. Pierotti MA, Berrino F, Gariboldi M, Melani C, Mogavero A, Negri T, et al. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. (2013) 32:1475–87. doi: 10.1038/onc.2012.181
90. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. (2012) 48:R31–43. doi: 10.1530/JME-12-0007
91. Pollak M. Metformin and pancreatic cancer: a clue requiring investigation. Clin Cancer Res. (2012) 18:2723–5. doi: 10.1158/1078-0432.CCR-12-0694
92. Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. (2009) 137:482–8. doi: 10.1053/j.gastro.2009.04.013
93. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. (2009) 32:1620–5. doi: 10.2337/dc08-2175
94. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. (2010) 30:750–8. doi: 10.1111/j.1478-3231.2010.02223.x
95. Sadeghi N, Abbruzzese JL, Yeung SC, Hassan M, Li D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin Cancer Res. (2012) 18:2905–12. doi: 10.1158/1078-0432.CCR-11-2994
96. Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res. (2010) 3:1451–61. doi: 10.1158/1940-6207.CAPR-10-0157
97. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE 7:e33411. doi: 10.1371/journal.pone.0033411
98. Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PLoS ONE 8:e71583. doi: 10.1371/journal.pone.0071583
99. Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, Decensi A, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res. (2014) 7:867–85. doi: 10.1158/1940-6207.CAPR-13-0424
100. Li Y, Hu L, Xia Q, Yuan Y, Mi Y. The impact of metformin use on survival in kidney cancer patients with diabetes: a meta-analysis. Int Urol Nephrol. (2017) 49:975–81. doi: 10.1007/s11255-017-1548-4
101. Ma S, Zheng Y, Xiao Y, Zhou P, Tan H. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine 96:e6888. doi: 10.1097/MD.0000000000006888
102. Meireles CG, Pereira SA, Valadares LP, Rego DF, Simeoni LA, Guerra ENS, et al. Effects of metformin on endometrial cancer: systematic review and meta-analysis. Gynecol Oncol. (2017) 147:167–80. doi: 10.1016/j.ygyno.2017.07.120
103. Wang SB, Lei KJ, Liu JP, Jia YM. Continuous use of metformin can improve survival in type 2 diabetic patients with ovarian cancer: a retrospective study. Medicine 96:e7605. doi: 10.1097/MD.0000000000007605
104. He X, Esteva FJ, Ensor J, Hortobagyi GN, Lee MH, Yeung SC. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol. (2012) 23:1771–80. doi: 10.1093/annonc/mdr534
105. Yang T, Yang Y, Liu S. Association between metformin therapy and breast cancer incidence and mortality: evidence from a meta-analysis. J Breast Cancer. (2015) 18:264–70. doi: 10.4048/jbc.2015.18.3.264
106. Jackson AL, Sun W, Kilgore J, Guo H, Fang Z, Yin Y, et al. Phenformin has anti-tumorigenic effects in human ovarian cancer cells and in an orthotopic mouse model of serous ovarian cancer. Oncotarget. (2017) 8:100113–27. doi: 10.18632/oncotarget.22012
107. Kang JH, Lee SH, Lee JS, Nam B, Seong TW, Son J, et al. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget. (2016) 7:49397–410. doi: 10.18632/oncotarget.10354
108. Veiga SR, Ge X, Mercer CA, Hernandez-Alvarez MI, Thomas HE, Hernandez-Losa J, et al. Phenformin-induced mitochondrial dysfunction sensitizes hepatocellular carcinoma for dual inhibition of mTOR. Clin Cancer Res. (2018) 24:3767–80. doi: 10.1158/1078-0432.CCR-18-0177
109. Rajeshkumar NV, Yabuuchi S, Pai SG, De Oliveira E, Kamphorst JJ, Rabinowitz JD, et al. Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res. (2017) 23:5639–47. doi: 10.1158/1078-0432.CCR-17-1115
110. Petrachi T, Romagnani A, Albini A, Longo C, Argenziano G, Grisendi G, et al. Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget. (2017) 8:6914–28. doi: 10.18632/oncotarget.14321
111. Liu Z, Ren L, Liu C, Xia T, Zha X, Wang S. Phenformin induces cell cycle change, apoptosis, and mesenchymal-epithelial transition and regulates the AMPK/mTOR/p70s6k and MAPK/ERK pathways in breast cancer cells. PLoS ONE 10:e0131207. doi: 10.1371/journal.pone.0131207
112. Orecchioni S, Reggiani F, Talarico G, Mancuso P, Calleri A, Gregato G, et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer. (2015) 136:E534–544. doi: 10.1002/ijc.29193
113. Guo Z, Zhao M, Howard EW, Zhao Q, Parris AB, Ma Z, et al. Phenformin inhibits growth and epithelial-mesenchymal transition of ErbB2-overexpressing breast cancer cells through targeting the IGF1R pathway. Oncotarget. (2017) 8:60342–57. doi: 10.18632/oncotarget.19466
114. Zhu Z, Jiang W, Thompson MD, Echeverria D, Mcginley JN, Thompson HJ. Effects of metformin, buformin, and phenformin on the post-initiation stage of chemically induced mammary carcinogenesis in the rat. Cancer Prev Res. (2015) 8:518–27. doi: 10.1158/1940-6207.CAPR-14-0121
115. Parris AB, Zhao Q, Howard EW, Zhao M, Ma Z, Yang X. Buformin inhibits the stemness of erbB-2-overexpressing breast cancer cells and premalignant mammary tissues of MMTV-erbB-2 transgenic mice. J Exp Clin Cancer Res. 36:28. doi: 10.1186/s13046-017-0498-0
116. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. (2012) 2:778–90. doi: 10.1158/2159-8290.CD-12-0263
117. Costa D, Gigoni A, Wurth R, Cancedda R, Florio T, Pagano A. Metformin inhibition of neuroblastoma cell proliferation is differently modulated by cell differentiation induced by retinoic acid or overexpression of NDM29 non-coding RNA. Cancer Cell Int. 14:59. doi: 10.1186/1475-2867-14-59
118. Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev. (2012) 38:589–98. doi: 10.1016/j.ctrv.2012.03.003
119. Zhang Y, Guan M, Zheng Z, Zhang Q, Gao F, Xue Y. Effects of metformin on CD133+ colorectal cancer cells in diabetic patients. PLoS ONE 8:e81264. doi: 10.1371/journal.pone.0081264
120. Courtois S, Duran RV, Giraud J, Sifre E, Izotte J, Megraud F, et al. Metformin targets gastric cancer stem cells. Eur J Cancer. (2017) 84:193–201. doi: 10.1016/j.ejca.2017.07.020
121. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. (2009) 69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994
122. Barbieri F, Thellung S, Ratto A, Carra E, Marini V, Fucile C, et al. In vitro and in vivo antiproliferative activity of metformin on stem-like cells isolated from spontaneous canine mammary carcinomas: translational implications for human tumors. BMC Cancer. (2015) 15:228. doi: 10.1186/s12885-015-1235-8
123. Mayer MJ, Klotz LH, Venkateswaran V. Metformin and prostate cancer stem cells: a novel therapeutic target. Prostate Cancer Prostatic Dis. (2015) 18:303–9. doi: 10.1038/pcan.2015.35
124. Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 8:e76518. doi: 10.1371/journal.pone.0076518
125. Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol. (2013) 6:649–59. doi: 10.1593/tlo.13556
126. Zhang R, Zhang P, Wang H, Hou D, Li W, Xiao G, et al. Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of CD44(+)CD117(+) ovarian cancer stem cells. Stem Cell Res Ther. 6:262. doi: 10.1186/s13287-015-0249-0
127. Gatti M, Solari A, Pattarozzi A, Campanella C, Thellung S, Maniscalco L, et al. In vitro and in vivo characterization of stem-like cells from canine osteosarcoma and assessment of drug sensitivity. Exp Cell Res. (2018) 363:48–64. doi: 10.1016/j.yexcr.2018.01.002
128. Paiva-Oliveira DI, Martins-Neves SR, Abrunhosa AJ, Fontes-Ribeiro C, Gomes CMF. Therapeutic potential of the metabolic modulator Metformin on osteosarcoma cancer stem-like cells. Cancer Chemother Pharmacol. (2018) 81:49–63. doi: 10.1007/s00280-017-3467-6
129. Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. (2012) 2:362. doi: 10.1038/srep00362
130. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. (2011) 71:3196–201. doi: 10.1158/0008-5472.CAN-10-3471
131. Yu Z, Zhao G, Li P, Li Y, Zhou G, Chen Y, et al. Temozolomide in combination with metformin act synergistically to inhibit proliferation and expansion of glioma stem-like cells. Oncol Lett. (2016) 11:2792–800. doi: 10.3892/ol.2016.4315
132. Yang SH, Li S, Lu G, Xue H, Kim DH, Zhu JJ, et al. Metformin treatment reduces temozolomide resistance of glioblastoma cells. Oncotarget. (2016) 7:78787–803. doi: 10.18632/oncotarget.12859
133. Ahn MJ, Cho GW. Metformin promotes neuronal differentiation and neurite outgrowth through AMPK activation in human bone marrow-mesenchymal stem cells. Biotechnol Appl Biochem. (2017) 64:836–42. doi: 10.1002/bab.1584
134. Suissa S. Metformin to treat cancer: misstep in translational research from observational studies. Epidemiology. (2017) 28:455–8. doi: 10.1097/EDE.0000000000000634
135. Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. (2012) 135:821–30. doi: 10.1007/s10549-012-2223-1
136. Joshua AM, Zannella VE, Downes MR, Bowes B, Hersey K, Koritzinsky M, et al. A pilot 'window of opportunity' neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis. (2014) 17:252–8. doi: 10.1038/pcan.2014.20
137. Schuler KM, Rambally BS, Difurio MJ, Sampey BP, Gehrig PA, Makowski L, et al. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. (2015) 4:161–73. doi: 10.1002/cam4.353
138. Kalinsky K, Crew KD, Refice S, Xiao T, Wang A, Feldman SM, et al. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Invest. (2014) 32:150–7. doi: 10.3109/07357907.2014.889706
139. Higurashi T, Hosono K, Takahashi H, Komiya Y, Umezawa S, Sakai E, et al. Metformin for chemoprevention of metachronous colorectal adenoma or polyps in post-polypectomy patients without diabetes: a multicentre double-blind, placebo-controlled, randomised phase 3 trial. Lancet Oncol. (2016) 17:475–83. doi: 10.1016/S1470-2045(15)00565-3
140. Marrone KA, Zhou X, Forde PM, Purtell M, Brahmer JR, Hann CL, et al. A randomized phase II study of metformin plus paclitaxel/carboplatin/bevacizumab in patients with chemotherapy-naive advanced or metastatic nonsquamous non-small cell lung cancer. Oncologist. (2018) 23:859–65. doi: 10.1634/theoncologist.2017-0465
141. Farmer RE, Ford D, Forbes HJ, Chaturvedi N, Kaplan R, Smeeth L, et al. Metformin and cancer in type 2 diabetes: a systematic review and comprehensive bias evaluation. Int J Epidemiol. (2017) 46:728–44. doi: 10.1093/ije/dyx046
142. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. (2017) 60:1577–85. doi: 10.1007/s00125-017-4342-z
143. Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I. Metformin as an anticancer agent. Trends Pharmacol Sci. (2018) 39:867–78. doi: 10.1016/j.tips.2018.07.006
144. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
145. Adak T, Samadi A, Unal AZ, Sabuncuoglu S. A reappraisal on metformin. Regul Toxicol Pharmacol. (2018) 92:324–32. doi: 10.1016/j.yrtph.2017.12.023
146. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. (2011) 12:21–35. doi: 10.1038/nrm3025
147. Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 71:e607. doi: 10.1016/j.molcel.2018.07.030
148. Abo-Elmatty DM, Ahmed EA, Tawfik MK, Helmy SA. Metformin enhancing the antitumor efficacy of carboplatin against Ehrlich solid carcinoma grown in diabetic mice: effect on IGF-1 and tumoral expression of IGF-1 receptors. Int Immunopharmacol. (2017) 44:72–86. doi: 10.1016/j.intimp.2017.01.002
149. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. (2006) 66:10269–73. doi: 10.1158/0008-5472.CAN-06-1500
150. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The antidiabetic drug metformin: a pharmaceutical AMPK activator to overcome breast cancer resistance to HER2 inhibitors while decreasing risk of cardiomyopathy. Ann Oncol. (2009) 20:592–5. doi: 10.1093/annonc/mdn758
151. Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation–implications for a novel treatment strategy. Gynecol Oncol. (2010) 116:92–8. doi: 10.1016/j.ygyno.2009.09.024
152. Rattan R, Giri S, Hartmann LC, Shridhar V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J Cell Mol Med. (2011) 15:166–78. doi: 10.1111/j.1582-4934.2009.00954.x
153. Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. (2009) 69:6539–45. doi: 10.1158/0008-5472.CAN-09-0418
154. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 13:235. doi: 10.1186/1471-2407-13-235
155. Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. (2013) 108:2021–32. doi: 10.1038/bjc.2013.187
156. Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. (2012) 11:1374–82. doi: 10.4161/cc.19798
157. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, Deberardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. (2007) 67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447
158. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
159. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. (2011) 71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769
160. Sun S, Gong F, Liu P, Miao Q. Metformin combined with quercetin synergistically repressed prostate cancer cells via inhibition of VEGF/PI3K/Akt signaling pathway. Gene. (2018) 664:50–7. doi: 10.1016/j.gene.2018.04.045
161. Song Z, Wei B, Lu C, Huang X, Li P, Chen L. Metformin suppresses the expression of Sonic hedgehog in gastric cancer cells. Mol Med Rep. (2017) 15:1909–15. doi: 10.3892/mmr.2017.6205
162. Ling S, Xie H, Yang F, Shan Q, Dai H, Zhuo J, et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: roles of p38 MAPK, ERK3, and mTORC1. J Hematol Oncol. 10:59. doi: 10.1186/s13045-017-0424-0
163. Dang JH, Jin ZJ, Liu XJ, Hu D, Wang J, Luo Y, et al. Metformin in combination with cisplatin inhibits cell viability and induces apoptosis of human ovarian cancer cells by inactivating ERK 1/2. Oncol Lett. (2017) 14:7557–64. doi: 10.3892/ol.2017.7176
164. Liu Y, Wang Z, Li M, Ye Y, Xu Y, Zhang Y, et al. Chloride intracellular channel 1 regulates the antineoplastic effects of metformin in gallbladder cancer cells. Cancer Sci. (2017) 108:1240–52. doi: 10.1111/cas.13248
165. Yang B, Huang CZ, Yu T, Zhou SN, Liu Q, Liu GJ, et al. Metformin depresses overactivated Notch1/Hes1 signaling in colorectal cancer patients with type 2 diabetes mellitus. Anticancer Drugs. (2017) 28:531–9. doi: 10.1097/CAD.0000000000000483
166. Lan B, Zhang J, Zhang P, Zhang W, Yang S, Lu D, et al. Metformin suppresses CRC growth by inducing apoptosis via ADORA1. Front Biosci. (2017) 22:248–57. doi: 10.2741/4484
167. Cuyas E, Martin-Castillo B, Bosch-Barrera J, Menendez JA. Metformin inhibits RANKL and sensitizes cancer stem cells to denosumab. Cell Cycle. (2017) 16:1022–8. doi: 10.1080/15384101.2017.1310353
168. Chung YC, Chang CM, Wei WC, Chang TW, Chang KJ, Chao WT. Metformin-induced caveolin-1 expression promotes T-DM1 drug efficacy in breast cancer cells. Sci Rep. 8:3930. doi: 10.1038/s41598-018-22250-8
169. Guimaraes TA, Farias LC, Santos ES, De Carvalho Fraga CA, Orsini LA, De Freitas Teles L, et al. Metformin increases PDH and suppresses HIF-1alpha under hypoxic conditions and induces cell death in oral squamous cell carcinoma. Oncotarget. (2016) 7:55057–68. doi: 10.18632/oncotarget.10842
170. Wang C, Liu C, Gao K, Zhao H, Zhou Z, Shen Z, et al. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem Biophys Res Commun. (2016) 477:534–40. doi: 10.1016/j.bbrc.2016.05.148
171. Li K, Zhang TT, Hua F, Hu ZW. Metformin reduces TRIB3 expression and restores autophagy flux: an alternative antitumor action. Autophagy. (2018) 14:1278–9. doi: 10.1080/15548627.2018.1460022
172. Zhou JY, Xu B, Li L. A new role for an old drug: metformin targets micrornas in treating diabetes and cancer. Drug Dev Res. (2015) 76:263–9. doi: 10.1002/ddr.21265
173. Bridgeman SC, Ellison GC, Melton PE, Newsholme P, Mamotte CDS. Epigenetic effects of metformin: from molecular mechanisms to clinical implications. Diabetes Obes Metab. (2018) 20:1553–62. doi: 10.1111/dom.13262
174. Li D. Metformin as an antitumor agent in cancer prevention and treatment. J Diabetes. (2011) 3:320–7. doi: 10.1111/j.1753-0407.2011.00119.x
175. Mogavero A, Maiorana MV, Zanutto S, Varinelli L, Bozzi F, Belfiore A, et al. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci Rep. 7:15992. doi: 10.1038/s41598-017-16149-z
176. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci USA. (2015) 112:1809–14. doi: 10.1073/pnas.1417636112
177. Pereira FV, Melo ACL, Low JS, De Castro IA, Braga TT, Almeida DC, et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget. (2018) 9:25808–25. doi: 10.18632/oncotarget.25380
178. Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci USA. (2013) 110:972–7. doi: 10.1073/pnas.1221055110
179. Wahdan-Alaswad R, Harrell JC, Fan Z, Edgerton SM, Liu B, Thor AD. Metformin attenuates transforming growth factor beta (TGF-beta) mediated oncogenesis in mesenchymal stem-like/claudin-low triple negative breast cancer. Cell Cycle. (2016) 15:1046–59. doi: 10.1080/15384101.2016.1152432
180. Xiao H, Zhang J, Xu Z, Feng Y, Zhang M, Liu J, et al. Metformin is a novel suppressor for transforming growth factor (TGF)-beta1. Sci Rep. 6:28597. doi: 10.1038/srep28597
181. Duan W, Qian W, Zhou C, Cao J, Qin T, Xiao Y, et al. Metformin suppresses the invasive ability of pancreatic cancer cells by blocking autocrine TGFbeta1 signaling. Oncol Rep. (2018) 40:1495–502. doi: 10.3892/or.2018.6518
182. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. (2010) 9:4461–8. doi: 10.4161/cc.9.22.14048
183. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. (2010) 9:3807–14. doi: 10.4161/cc.9.18.13131
184. Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther. (2013) 12:1605–15. doi: 10.1158/1535-7163.MCT-12-1226-T
185. Zhang J, Shen C, Wang L, Ma Q, Xia P, Qi M, et al. Metformin inhibits epithelial-mesenchymal transition in prostate cancer cells: involvement of the tumor suppressor miR30a and its target gene SOX4. Biochem Biophys Res Commun. (2014) 452:746–52. doi: 10.1016/j.bbrc.2014.08.154
186. Tong D, Liu Q, Liu G, Xu J, Lan W, Jiang Y, et al. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. (2017) 389:23–32. doi: 10.1016/j.canlet.2016.12.031
187. Song Y, Chen Y, Li Y, Lyu X, Cui J, Cheng Y, et al. Metformin inhibits TGF-beta1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget. (2018) 9:7023–35. doi: 10.18632/oncotarget.23317
188. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
189. Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol. (2018) 20:782–8. doi: 10.1038/s41556-018-0125-0
190. Vella S, Conaldi PG, Florio T, Pagano A. PPAR Gamma in neuroblastoma: the translational perspectives of hypoglycemic drugs. PPAR Res. (2016) 2016:3038164. doi: 10.1155/2016/3038164
191. Zhu M, Zhang Q, Wang X, Kang L, Yang Y, Liu Y, et al. Metformin potentiates anti-tumor effect of resveratrol on pancreatic cancer by down-regulation of VEGF-B signaling pathway. Oncotarget. (2016) 7:84190–200. doi: 10.18632/oncotarget.12391
192. Detaille D, Guigas B, Leverve X, Wiernsperger N, Devos P. Obligatory role of membrane events in the regulatory effect of metformin on the respiratory chain function. Biochem Pharmacol. (2002) 63:1259–72. doi: 10.1016/S0006-2952(02)00858-4
193. Higgins JW, Bedwell DW, Zamek-Gliszczynski MJ. Ablation of both organic cation transporter (OCT)1 and OCT2 alters metformin pharmacokinetics but has no effect on tissue drug exposure and pharmacodynamics. Drug Metab Dispos. (2012) 40:1170–7. doi: 10.1124/dmd.112.044875
194. Lee N, Duan H, Hebert MF, Liang CJ, Rice KM, Wang J. Taste of a pill: organic cation transporter-3 (OCT3) mediates metformin accumulation and secretion in salivary glands. J Biol Chem. (2014) 289:27055–64. doi: 10.1074/jbc.M114.570564
195. Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep. (2010) 62:956–65. doi: 10.1016/S1734-1140(10)70357-1
196. Segal ED, Yasmeen A, Beauchamp MC, Rosenblatt J, Pollak M, Gotlieb WH. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem Biophys Res Commun. (2011) 414:694–9. doi: 10.1016/j.bbrc.2011.09.134
197. Checkley LA, Rudolph MC, Wellberg EA, Giles ED, Wahdan-Alaswad RS, Houck JA, et al. Metformin accumulation correlates with organic cation transporter 2 protein expression and predicts mammary tumor regression in vivo. Cancer Prev Res. (2017) 10:198–207. doi: 10.1158/1940-6207.CAPR-16-0211-T
198. Liu X, Romero IL, Litchfield LM, Lengyel E, Locasale JW. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. (2016) 24:728–39. doi: 10.1016/j.cmet.2016.09.005
199. Baldassari S, Solari A, Zuccari G, Drava G, Pastorino S, Fucile C, et al. Development of an injectable slow-release metformin formulation and evaluation of its potential antitumor effects. Sci Rep. 8:3929. doi: 10.1038/s41598-018-22054-w
200. Berstein LM. Metformin: not only per os. Expert Rev Endocrinol Metab. (2018) 13:63–5. doi: 10.1080/17446651.2018.1431123
201. Maniar K, Singh V, Chakrabarti A, Bhattacharyya R, Banerjee D. High dose targeted delivery on cancer sites and the importance of short-chain fatty acids for metformin's action: Two crucial aspects of the wonder drug. Regul Toxicol Pharmacol. (2018) 97:15–6. doi: 10.1016/j.yrtph.2018.05.013
202. Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A, et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J Biol Chem. (2009) 284:7138–48. doi: 10.1074/jbc.M807111200
203. Carra E, Barbieri F, Marubbi D, Pattarozzi A, Favoni RE, Florio T, et al. Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures. Cell Cycle. (2013) 12:491–500. doi: 10.4161/cc.23372
204. Angeletti F, Fossati G, Pattarozzi A, Wurth R, Solari A, Daga A, et al. Inhibition of the autophagy pathway synergistically potentiates the cytotoxic activity of givinostat (ITF2357) on human glioblastoma cancer stem cells. Front Mol Neurosci. 9:107. doi: 10.3389/fnmol.2016.00107
205. Mouhieddine TH, Nokkari A, Itani MM, Chamaa F, Bahmad H, Monzer A, et al. Metformin and ara-a effectively suppress brain cancer by targeting cancer stem/progenitor cells. Front Neurosci. 9:442. doi: 10.3389/fnins.2015.00442
206. Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarie A, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 10:e0123721. doi: 10.1371/journal.pone.0123721
207. Yu Z, Zhao G, Xie G, Zhao L, Chen Y, Yu H, et al. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget. (2015) 6:32930–43. doi: 10.18632/oncotarget.5405
208. Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. (2012) 1:811–24. doi: 10.5966/sctm.2012-0058
209. Seliger C, Meyer AL, Renner K, Leidgens V, Moeckel S, Jachnik B, et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-beta2. Cell Cycle. (2016) 15:1755–66. doi: 10.1080/15384101.2016.1186316
210. Kim EH, Lee JH, Oh Y, Koh I, Shim JK, Park J, et al. Inhibition of glioblastoma tumorspheres by combined treatment with 2-deoxyglucose and metformin. Neuro Oncol. (2017) 19:197–207. doi: 10.1093/neuonc/now174
211. Barbieri F., Wurth R., Pattarozzi A., Verduci I., Mazzola C., Tonelli M., et al. Inhibition of chloride intracellular channel 1 (CLIC1) as biguanide class-effect to impair human glioblastoma stem cell viability Front Pharmacol. (2018) 9:899. doi: 10.3389/fphar.2018.00899
212. Kubelt C, Hattermann K, Sebens S, Mehdorn HM, Held-Feindt J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int J Oncol. (2015) 46:2515–25. doi: 10.3892/ijo.2015.2944
213. Valtorta S, Dico AL, Raccagni I, Gaglio D, Belloli S, Politi LS, et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget. (2017) 8:113090–104. doi: 10.18632/oncotarget.23028
214. Leidgens V, Proske J, Rauer L, Moeckel S, Renner K, Bogdahn U, et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget. (2017) 8:8250–63. doi: 10.18632/oncotarget.14159
215. Isakovic A, Harhaji L, Stevanovic D, Markovic Z, Sumarac-Dumanovic M, Starcevic V, et al. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci. (2007) 64:1290–302. doi: 10.1007/s00018-007-7080-4
216. Adeberg S, Bernhardt D, Harrabi SB, Nicolay NH, Horner-Rieber J, Konig L, et al. Metformin enhanced in vitro radiosensitivity associates with G2/M cell cycle arrest and Elevated Adenosine-5'-monophosphate-activated protein kinase levels in glioblastoma. Radiol Oncol. (2017) 51:431–7. doi: 10.1515/raon-2017-0042
217. Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci USA. (2014) 111:E435–444. doi: 10.1073/pnas.1311121111
218. Chinnaiyan P, Won M, Wen PY, Rojiani AM, Werner-Wasik M, Shih HA, et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: results of NRG Oncology RTOG 0913. Neuro Oncol. (2018) 20:666–73. doi: 10.1093/neuonc/nox209
219. Chhipa RR, Fan Q, Anderson J, Muraleedharan R, Huang Y, Ciraolo G, et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat Cell Biol. (2018) 20:823–35. doi: 10.1038/s41556-018-0126-z
220. Lee JE, Lim JH, Hong YK, Yang SH. High-dose metformin plus temozolomide shows increased anti-tumor effects in glioblastoma in vitro and in vivo compared with monotherapy. Cancer Res Treat. (2018) 50:1331–42 doi: 10.4143/crt.2017.466
221. Jiang W, Finniss S, Cazacu S, Xiang C, Brodie Z, Mikkelsen T, et al. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget. (2016) 7:56456–70. doi: 10.18632/oncotarget.10919
222. Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. (2015) 75:171–80. doi: 10.1158/0008-5472.CAN-14-2260
223. Park J, Shim JK, Kang JH, Choi J, Chang JH, Kim SY, et al. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro Oncol. (2018) 20:954–65. doi: 10.1093/neuonc/nox243
Keywords: glioblastoma, cancer stem cells, CLIC1, biguanide, metformin
Citation: Barbieri F, Verduci I, Carlini V, Zona G, Pagano A, Mazzanti M and Florio T (2019) Repurposed Biguanide Drugs in Glioblastoma Exert Antiproliferative Effects via the Inhibition of Intracellular Chloride Channel 1 Activity. Front. Oncol. 9:135. doi: 10.3389/fonc.2019.00135
Received: 30 August 2018; Accepted: 14 February 2019;
Published: 13 March 2019.
Edited by:
Zhi Sheng, Virginia Tech, United StatesCopyright © 2019 Barbieri, Verduci, Carlini, Zona, Pagano, Mazzanti and Florio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tullio Florio, dHVsbGlvLmZsb3Jpb0B1bmlnZS5pdA==
†These authors share joint first authorship
‡These authors have contributed equally to this work and are senior authors