 Yi Miao
Yi Miao L. Jeffrey Medeiros
L. Jeffrey Medeiros Zijun Y. Xu-Monette
Zijun Y. Xu-Monette Jianyong Li2
Jianyong Li2 Ken H. Young
Ken H. Young- 1Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Hematology, The First Affiliated Hospital of Nanjing Medical University, Jiangsu Province Hospital, Nanjing, China
- 3Graduate School of Biomedical Sciences, The University of Texas Health Science Center at Houston, Houston, TX, United States
Diffuse large B cell lymphoma (DLBCL) is the most common type of lymphoma worldwide, representing 30–40% of non-Hodgkin lymphomas, and is clinically aggressive. Although more than half of patients with DLBCL are cured by using standard first-line immunochemotherapy, the remaining patients are refractory to the first-line therapy or relapse after complete remission and these patients require novel therapeutic approaches. Understanding the pathogenesis of DLBCL is essential for identifying therapeutic targets to tackle this disease. Cell survival dysregulation, a hallmark of cancer, is a characteristic feature of DLBCL. Intrinsic signaling aberrations, tumor microenvironment dysfunction, and viral factors can all contribute to the cell survival dysregulation in DLBCL. In recent years, several novel drugs that target abnormal cell survival pathways, have been developed and tested in clinical trials of patients with DLBCL. In this review, we discuss cell survival dysregulation, the underlying mechanisms, and how to target abnormal cell survival therapeutically in DLBCL patients.
Introduction
Diffuse large B cell lymphoma (DLBCL) is the most common type of lymphoma and represents 30–40% of all non-Hodgkin lymphomas (1). Based on gene expression profiling, two molecular subtypes of DLBCL, activated B-cell-like (ABC) subtype and germinal center B-cell-like (GCB) subtype, are identified (2, 3). Approximately 60% of patients with DLBCL can be cured by the standard immunochemotherapy regimen rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), but remaining patients are either refractory to induction immunochemotherapy or experience relapses after achieving complete response (CR) (4–6). Enormous effort has been devoted to developing new therapeutic approaches for these refractory or relapsed patients, with some success. In rituximab era, using conventional salvage immunochemotherapy with autologous transplant, only ~ 10% of refractory/relapsed DLBCL cases could be cured, whereas the remaining patients have very dismal outcome warranting development of novel therapies (7, 8). Understanding the pathogenesis of DLBCL is vital to define potential therapeutic targets and develop regimens for the treatment of DLBCL (9, 10). As is common to other types of cancer, dysregulation of cell survival or resistance to cell death also contributes to the pathogenesis of DLBCL. DLBCL lymphoma cells have evolved many strategies to resist cell death, which potentially can be therapeutically targeted. In this review, we discuss the mechanisms underlying dysregulation of cell survival in DLBCL and therapeutic options to target related pathways.
Mechanisms Underlying Dysregulation of Cell Survival in Dlbcl
B Cell Receptor (BCR) Signaling
Normal BCR Signaling
Based on dependence on antigen stimulation, BCR signaling pathways can be categorized into tonic BCR signaling or antigen-dependent BCR signaling. Tonic BCR signaling is vital for maintaining the survival of resting mature B cells, as ablation of cell surface BCR expression or inactivation of BCR result in cell death (Figure 1) (11). Tonic BCR signaling exerts its pro-survival effects via PI3K signaling, as PI3K signaling can rescue the survival of mature B cells that are deficient in BCR (12). Activated PI3K/AKT inactivates the pro-apoptotic protein BAD and stabilizes the anti-apoptotic protein MCL1, resulting in enhanced cell survival. Moreover, activated AKT phosphorylates forkhead box class O family member transcription factors (FOXOs) leading to cytosolic retention of FOXOs, impairing FOXOs-mediated transactivation of cell death genes (12). Antigen-dependent BCR signaling promotes the survival of B cells through activation of a variety of pathways. The binding of antigen causes BCR aggregation, leading to phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) of CD79A and CD79B by SRC kinases, including LYN, FYN, and B lymphocyte kinase (BLK). Phosphorylated ITAMs recruit spleen tyrosine kinase (SYK), which is then phosphorylated and activated by the SRC kinases and autophosphorylation. Activated SYK leads to phosphorylation and activation of the downstream molecules Bruton tyrosine kinase (BTK) and phospholipase Cγ2 (PLCγ2) (9). PLCγ2 hydrolyses phosphatidylinositol-4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol trisphosphate (IP3), which results in increased intracellular calcium levels. Increased calcium, in combination with DAG, triggers activation of protein kinase C β (PKCβ) and promotes assembly of the CARD11/BCL10/MALT1 (CBM) complex and NF-κB activation, providing vital pro-survival signals for B cells. In BCR signaling, LYN kinase also leads to the phosphorylation of CD19 thereby promoting recruitment recruitment of PI3K to the BCR, which then activates the PI3K/AKT pathway. Additionally, BCR signaling can also activate the RAS and mitogen-activated protein kinase (MAPK) pathway as well as the transcription factor NFAT, providing additional survival signals for B cells (9).
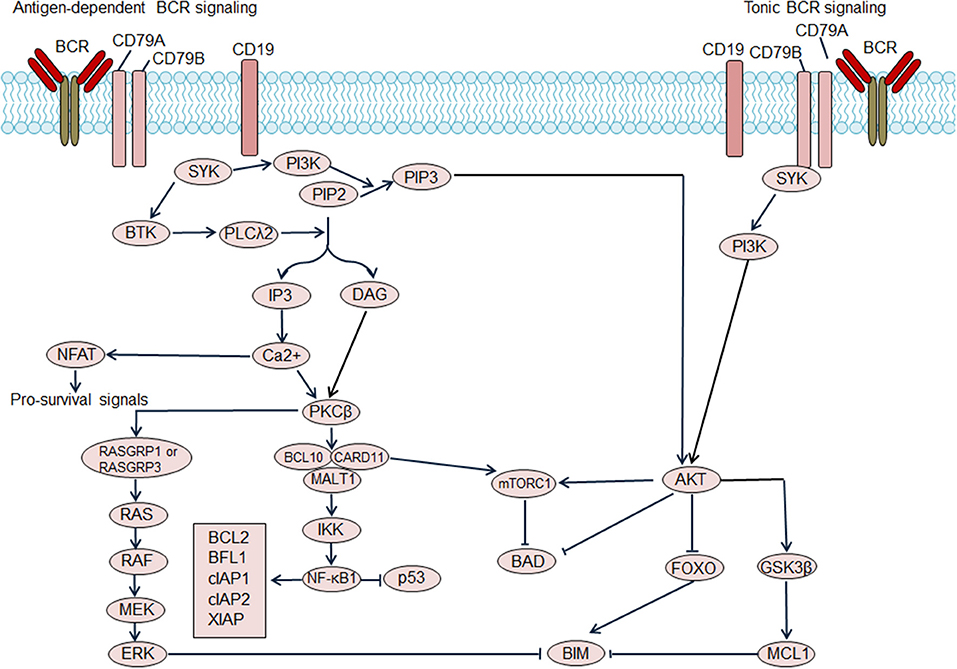
Figure 1. Regulation of B cell receptor (BCR) signaling on B cell survival. Antigen dependent BCR signaling engages several downstream pathways, which include the NF-κB pathway, PI3K/AKT/mTOR pathway, NFAT pathway, and MAPK/ERK pathway. NF-κB activation promotes transcription of the pro-survival genes including BCL2, BFL1, cellular inhibitor of apoptosis protein (cIAP) 1, cIAP2, X-linked inhibitor of apoptosis protein (XIAP) and so on. Additionally, NF-κB antagonizes the function of p53, thereby decreasing p53-mediated cell apoptosis. Activated AKT phosphorylates forkhead box class O family member transcription factors (FOXOs), suppressing the transcription of target genes including pro-apoptosis gene BIM. AKT activation also results in the activation of pro-survival MCL1. Moreover, AKT mediates the phosphorylation of the pro-apoptotic BCL2 family member BAD, and blocks BAD-induced cell apoptosis. The activation of mTOR complex 1 (mTORC1), which suppresses BAD, could be induced by AKT activation or CARD11 activation. Tonic BCR signaling triggers activation of the PI3K/AKT pathway and also promotes cell survival. BTK, Bruton tyrosine kinase; DAG, diacylglycerol; IP3, inositol trisphosphate; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKCβ, protein kinase C β; PLCλ2, phospholipase λ2; SYK, spleen tyrosine kinase.
Chronic Active BCR Signaling
ABC DLBCL cells rely on BCR signaling for survival (13). This type of BCR signaling is dependent on both proximal BCR pathway components and CARD11, similar to that in antigen activated normal B cells, therefore referred as “chronic active” BCR signaling (13). Down-regulation of chronic active BCR signaling in ABC DLBCL cells leads to inhibition of NF-κB activation as well as phosphorylation of AKT and ERK, dampening pro-survival signaling (13). Both antigen-dependent BCR activation and genetic aberrations involving the BCR signaling pathway contribute to chronic active BCR signaling activation in ABC DLBCL (13). One part of the evidence suggesting antigen-dependent BCR activation in ABC DLBCL is remarkable clustering of BCR on the ABC DLBCL cell surface, a characteristic feature of antigen dependent BCR activation in normal B cells (13). A study by Young et al. showed, in a subset of ABC DLBCL, that binding of self-antigen to BCR drove active BCR signaling and sustained survival of ABC DLBCL cells (14). Genetic aberrations affecting BCR and downstream signaling contribute to the activation of BCR signaling in ABC DLBCL. A subset of ABC DLBCL cases harbor mutations in the ITAM motif of CD79A (~3%) and CD79B (~20%) (13). Both CD79A and CD79B mutations enhance surface IgM expression, thereby increasing NF-κB activation. CD79B mutations also suppress the activity of LYN kinase, impairing LYN kinase mediated negative-feedback inhibition of BCR signaling. Consistently, inactivating LYN mutations, and LYN deletions have been identified in DLBCL, which possibly enhance activation of BCR signaling. The CARD11/BCL10/MALT1 complex is also affected by activating mutations or amplifications. CARD11 mutations, which predominantly affect the coil-coil domain, are detected in DLBCL (11–15%) including both ABC and GCB DLBCL (15, 16). These CARD11 mutations impair the inhibition of domain-mediated auto-inhibition, leading to hyper-activation of CARD11, which subsequently activates the downstream NF-κB pathway (17). A recent study showed that activated CARD11 could induce the activation of mTOR complex 1 (mTORC1), which provides additional pro-survival signals (18). With the advent of next-generation sequencing, an increasing number genetic aberrations of BCR regulators have been identified, especially negative BCR regulators, including PTPN6, PRKCD, SLA, LAPTM5, DGKZ, and MAP4K1 (16). The inactivating mutations or deletions involving these molecules release BCR signaling from inhibition, thus leading to BCR signaling activation.
Tonic BCR Signaling
Absence of immobile BCR clustering on the cell surface of GCB DLBCL cells suggests lack of chronic active BCR signaling. Moreover, most GCB DLBCLs are relatively insensitive to the BCR inhibitor ibrutinib and do not show activation of NF-κB pathway, further suggesting independence of GCB DLBCL from chronic active BCR signaling (19). The study by Chen et al. suggested some DLBCL cell lines, which included GCB subtypes, displayed tonic BCR signaling, as these cell lines exhibited detectable SYK and BLNK phosphorylation without BCR crosslinking (20). Inhibition of SYK dampened tonic BCR signaling and increased cell apoptosis in BCR-dependent DLBCL cell lines, pointing to a role of tonic BCR signaling in sustaining survival of BCR-dependent DLBCL cells (20). Replacement of BCR antigen-binding regions has no impact on BCR signaling in GCB DLBCL lines, indicating that GCB DLBCL rely on tonic BCR signaling (21). The biological effect of tonic BCR signaling in GCB DLBCL is highly dependent on AKT activation, as tonic BCR signaling triggers AKT activation and forced AKT activation can rescue GCB DLBCL cells from depletion of the BCR or tonic BCR signaling mediators SYK and CD19 (21). Genetic aberrations also play a role in promoting tonic BCR signaling. PTEN deletions, which are identified in approximately 10% of DLBCL including the GCB and ABC subtypes, can result in enhanced PI3K/AKT signaling (16). Mir-17-92 targets and negatively regulates expression of PTEN protein, therefore, mir-17-92 amplification, which occurs exclusively in GCB DLBCL (~8%) (16), leads to PI3K/AKT activation. These aberrations, by activating PI3K/AKT signaling, lead to increased tonic BCR signaling.
Toll-Like Receptor Signaling and the MyD88–TLR9–BCR Supercomplex
MYD88L265P mutation, which involves the adaptor protein MYD88 in the Toll-like receptor (TLR) pathway, is found in approximately 30% of ABC DLBCL, suggesting that abnormal TLR signaling plays a role in the pathogenesis of DLBCL (22). Physiologically, following recruitment of MYD88 to TLRs, TLR4 forms a complex with interleukin-1 receptor-associated kinase (IRAK) 4, further recruiting other IRAKs (IRAK1 and IRAK2) and causing activation of downstream pathways including the NF-κB pathway (23). Inhibiting MYD88 using shRNA is toxic to ABC DLBCL cell lines suggesting that MYD88 is essential for the survival of these cell lines (24). Compared with wild-type MYD88, MYD88L265P leads to enhanced phosphorylation of IRAK, activation of NF-κB, and JAK-STAT3 signaling, and increased secretion of interleukin 6 (IL-6), interleukin 10 (IL-10), and interferon-β promoting the survival of ABC DLBCL cell lines. The study by Avbelj et al. found that MYD88L265P Toll/interleukin-1 receptor (TIR) domain can recruit endogenous wild-type MYD88 to form oligomers, which leads to hyperactivation of downstream pathway (25). Moreover, conditional expression of MYD88L265P mutation in B cells in mice leads to spontaneous development of lymphoproliferative diseases which mimic ABC DLBCL (26). These data suggest that MYD88L265P activates MYD88, promotes the survival of DLBCL cells, and contributes to the pathogenesis of ABC DLBCL. Additional conditional BCL2 overexpression in this model remarkably accelerates the development of lymphoma (26). Accordingly, in human DLBCL samples, BCL2 amplification frequently co-occur with MYD88L265P mutations in ABC DLBCL, suggesting the anti-apoptotic function of BCL2 is important for full transformation of B cells by MYD88L265P (27). This finding also provides a rationale for using BCL2 inhibitors in MYD88L265P mutated DLBCL.
Additionally, Co-occurrence of MYD88L265P and CD79B mutations is frequent in ABC DLBCL, suggesting that these two aberrations might be synergistic in driving ABC DLBCL development (27). There has been direct evidence that MYD88 and BCR cooperate in the pathogenesis of a subset of DLBCL (28). A recent study showed that MYD88, TLR9, and the BCR formed a multiprotein supercomplex (MyD88–TLR9–BCR supercomplex, the My-T-BCR supercomplex) in ibrutinib-responsive cell lines and patient samples (28). The My-T-BCR supercomplex co-localizes with mTOR on endolysosomes to drive NF-κB and mTOR signaling, both of which promote cell survival (28).
Dysregulation of Apoptosis Molecules
Dysregulation of BCL2 Family Members
The BCL2 family consists of a group of proteins that share with Bcl-2 homology (BH) domains (29). BCL2 family proteins, including anti-apoptotic and pro-apoptotic members, have a crucial role in regulating cell survival by modulating the intrinsic apoptosis pathway. Briefly, signaling including DNA damage and absence of growth factors leads to the activation of BH3-only proteins, which inactivate the pro-survival members such as BCL2, allowing activation of BAX and BAK. BAX and BAK lead to permeabilization of the outer mitochondrial membrane, releasing the pro-apoptotic cytochrome c, which activates caspases. These caspases, via their proteolytic activities, act as the direct mediators of cell apoptosis.
Dysregulation of BCL2 family members has been reported in DLBCL. BCL2, the prototype of this family, is overexpressed in 50%-53% of DLBCL (ABC DLBCL, 53–61%; GCB DLBCL, 40–44%) (30, 31). BCL2 contributes to the pathogenesis of DLBCL by promoting the survival of B cells, as BCL2-transgenic mice have B cells with extended survival and these mice spontaneously develop aggressive B cell lymphoma (32–34). Several mechanisms contribute to BCL2 overexpression in DLBCL, among which BCL2 translocation is the most common one. BCL2 translocation occurs exclusively in GCB DLBCL (~30%) (16, 35, 36), which juxtaposes BCL2 with the immunoglobulin heavy chain (IGH) enhancer, leading to increased BCL2 mRNA transcription. BCL2 copy number alterations, including gains and amplifications, are observed in ABC DLBCL and regarded as contributing factors to BCL2 overexpression (37). In normal germinal center (GC) B cells, BCL6 binds to the promoter of BCL2, and inhibits MIZ1-mediated BCL2 transcriptional activation (38). BCL2 translocations as well as mutations in BCL2 promoter regions, disrupt BCL6-mediated suppression of BCL2 transcription, thereby contributing to BCL2 overexpression (38). MicroRNA (miRNA) dysregulation also contributes to BCL2 overexpression in DLBCL (39). For instance, miR-34a, which targets BCL2, is down-regulated in DLBCL, contributing to BCL2 overexpression (39). Mutations in BCL2 coding sequence (CDS), which is located predominantly in the BH4 and flexible loop domain (FLD), also contribute to the BCL2 dysregulation by affecting the function and stability of BCL2 (40–42). The anti-apoptotic BCL2 family member MCL1 is also overexpressed in non-GCB DLBCL (~50% at protein level), which could be caused by MCL1 gains or amplification or abnormal activated STAT3 signaling (43). MCL1 possibly contributes to the pathogenesis of DLBCL by promoting cell survival, as MCL1 transgenic mice spontaneously develop DLBCL and inhibition of MCL1 in DLBCL cell lines induces cell death (43, 44). Another BCL2 family anti-apoptotic member, BCL-w, has been shown to be overexpressed in ~90% of DLBCL (60% with high expression) and BCL-w overexpression was associated with poor prognosis (45, 46). The RNA level of BCL-w is inversely correlated with that of BCL2 in DLBCL samples, pointing to a possible complementary role of BCL-w to BCL2 in the pathogenesis of DLBCL (46). Loss of BCL-w sensitized B cell to growth factor deprivation-induced cell apoptosis and suppressed MYC-induced lymphomagenesis, suggesting a crucial role of BCL-w in B cell survival and lymphoma development (45). Nevertheless, the mechanisms underlying BCL-w overexpression in DLBCL remain not well-defined. In summary, abnormalities in anti-apoptotic members of the BCL2 family, such as BCL2, MCL1, and BCL-w, lead to enhanced B cell survival and contribute to DLBCL development.
Dysregulation of Inhibitor of Apoptosis Proteins
Inhibitor of apoptosis proteins (IAPs) inhibit cell apoptosis by binding specific caspases. Dysregulation of IAPs has been reported in DLBCL. Cellular inhibitor of apoptosis protein 1 (cIAP1), cellular inhibitor of apoptosis protein 2 (cIAP2), and X-linked inhibitor of apoptosis protein (XIAP) are reported in 92, 37, and 26~55% of DLBCL, respectively (47, 48). And the overexpression of XIAP in DLBCL is significantly associated with worse outcome (48). Pharmacological inhibition of XIAP induces cell apoptosis in DLBCL cell lines, supporting the role of XIAP in maintaining cell survival of DLBCL cells (48). Survivin, another member of the inhibitor of apoptosis protein family, has been found to be overexpressed in ~60% of DLBCL (49). And in ABC DLBCL, survivin overexpression significantly predicted worse outcome (49). These pieces of evidence suggest that survivin has a pathogenic role in DLBCL.
TP53 Dysregulation
The p53 transcription factor has a crucial role in regulating cell survival. Under normal conditions, the E3 ligase MDM2 targets p53 for degradation in a negative feedback manner, thus maintaining a low expression level of p53. Additionally, MDM2 suppresses p53 mediated transcriptional activation and promotes the transportation from the nucleus to the cytoplasm (50, 51). Cellular stresses, including DNA damage, can disrupt the binding of MDM2 to p53 and increase p53 protein expression. P53 activates transcription of several genes that are involved in cell apoptosis, including BAX, PUMA, and NOXA (Figure 2). TP53 dysfunction is common in DLBCL (52) and restoration of p53 expression tumor cell induces apoptosis in an Eμ-Myc mouse model, suggesting a role of p53 dysregulation in DLBCL development (53). P53 dysregulation also contributes to chemoresistance in DLBCL, as p53 functions as an important mediator of chemotherapy induced cell death. Clinically, TP53 mutation is an independent prognostic factor of poor outcome in DLBCL patients treated with R-CHOP chemotherapy (54).
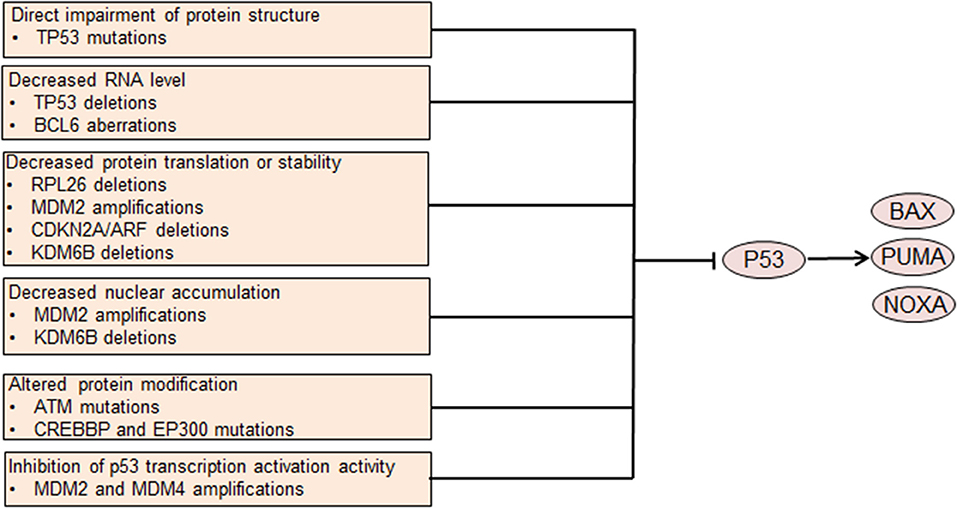
Figure 2. Genetic aberrations causing p53 dysfunction leads to dysregulated cell survival. Mutations involving the coding DNA sequence region of the TP53 gene affect the protein structure and abrogate p53 tumor suppressor function. Other aberrations lead to decreased p53 translation or stability or altered protein modification. MDM2 and MDM4 also inhibit p53-mediated transcription activation. Decreased p53 nuclear accumulation caused by MDM2 amplifications or KDM6B deletions leads to p53 dysfunction. Impaired p53 function leads to decreased transcription of pro-apoptotic genes, resulting in abnormally enhanced cell survival.
Many different mechanisms contribute to p53 dysregulation in DLBCL. Among these mechanisms, TP53 mutation is the most common one, occurring in ~20% of all DLBCL cases (16, 52). TP53 mutations in the CDS regions mostly target exons 4 through 9, frequently involving the DNA binding domain and impairing p53 mediated transcriptional transactivation (52). Heterogeneous or homogeneous TP53 deletions that lead to decreased TP53 gene dosage occur in ~ 10% of DLBCL cases (52, 55). BCL6, which is frequently dyregulated in DLBCL, binds to the TP53 promoter region and suppresses the transcription of TP53 gene (56). MDM2 is amplified in a subset of DLBCL, contributing to increased ubiquitylation and degradation of p53 as well as decreased nuclear accumulation and transcriptional activity (57). MDM4, a protein that shares structural similarity with MDM2, inhibits p53 transcriptional activity. Amplifications of MDM4 are also detected in DLBCL, possibly decreasing transcriptional activity of p53 (57). ARF, which encodes a protein that promotes MDM2 degradation and stabilizes p53, is frequently deleted in DLBCL, as a part of the CDKN2A locus (55, 58). ARF deletion contributes to the pathogenesis of DLBCL, at least partly via decreasing p53 protein level. Deletions of KDM6B and RPL26, which are located with TP53 in the 17p13.1 region, are recurrent in DLBCL (57). Under normal conditions, the demethylase KDM6B can activate ARF transcription by demethylating the p14ARF locus (59). Additionally, KDM6B directly interacts with p53 by demethylating p53 and causes accumulation of p53 in the nucleus, thereby activating the function of p53 (60). Therefore, loss of KDM6B might decrease ARF transcription and p53 stability and p53 nuclear accumulation, thereby leading to p53 dysregulation.
RPL26 can bind the 5′ UTR of TP53 and facilitate p53 translation, which increases stress-induced p53 expression (61). Thus, lack of RPL26 may decrease p53 expression by impeding p53 translation. Several other genetic aberrations may attenuate p53 protein function. ATM, which phosphorylates and activates p53, is recurrently mutated in DLBCL and may lead to impaired activation of p53 (16). Both CREBBP and EP300 can acetylate p53 and activate p53 transcriptional activity (62). Mutations in CREBBP and EP300 occur in DLBCL inactivating the acetyltransferase activity of these two proteins, thereby impairing p53 acetylation and activity (63).
Tumor Microenvironment (TME) Dysfunction
The TME consists of vasculature, immune cells, fibroblasts, signaling molecules, and the extracellular matrix. In contrast with low-grade B cell lymphomas, DLBCL is relatively less dependent on the TME. Nevertheless, there is evidence suggesting that the TME provides indispensable survival signals in at least some cases of DLBCL. Not all DLBCL cells can be cultured in vitro, suggesting that these cells might be dependent on the TME to survive (64). Furthermore, in clinical settings the presence of certain types of immune cells predicts a poorer prognosis in DLBCL patients, suggesting that these immune cells might support the survival of DLBCL tumor cells or mediate lymphoma cell drug-resistance (65–68). Immune escape definitely contributes to the survival of DLBCL cells, however, this is not the focus of our review. Here, we discuss how the TME provides pro-survival signals to tumor cells. Several types of cells in the TME can provide survival signals for DLBCL cells (Figure 3).
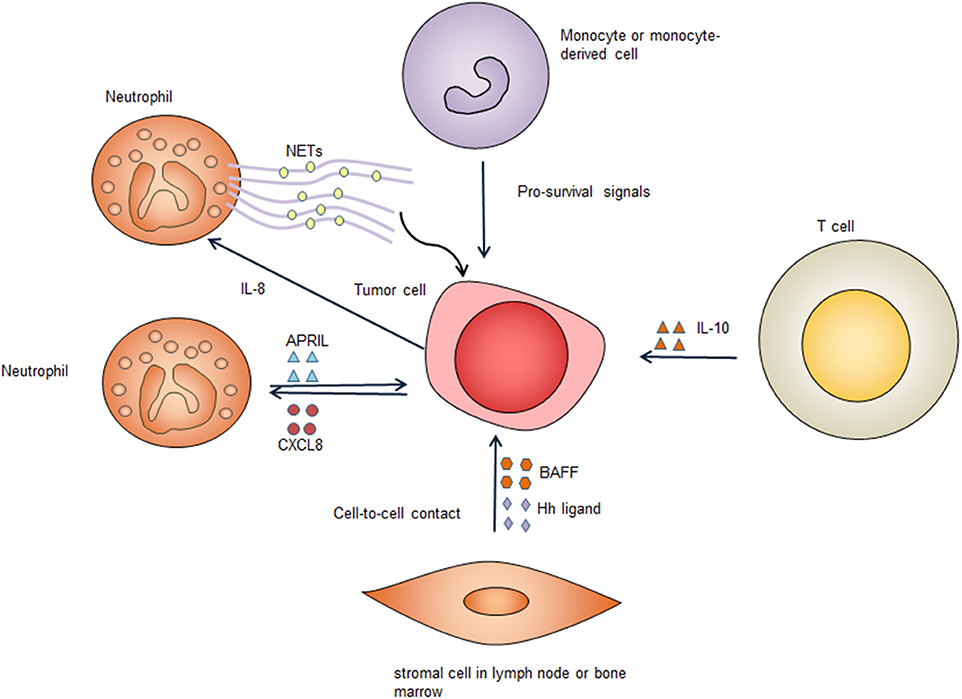
Figure 3. Survival signals from the tumor microenvironment. Immune cells and stromal cells promote tumor cell survival through cell-to-cell contact and secreting pro-survival factors. Several types of cells in the tumor microenvironment secret pro-survival factors including B-cell activating factor (BAFF), a proliferation-inducing ligand (APRIL), hedgehog (Hh) ligands, and interleukin-10 (IL-10). These factors bind to corresponding receptors on the tumor cell surface, providing important pro-survival signals to the tumor cells. Neutrophils form neutrophil extracellular traps (NETs), which activate Toll-like receptor 9 (TLR9) pathway to promote survival of diffuse large B cell tumor cells. CXCL8, C-X-C motif chemokine ligand 8; IL-8, interleukin-8.
Neutrophils
Coculture of neutrophils with DLBCL cell lines can sustain the survival of DLBCL cell lines (69). This effect is associated with decreased DLBCL cell apoptosis, suggesting that neutrophils promote the survival of DLBCL cells (69). In DLBCL, tumor-infiltrating neutrophils can secrete a proliferation-inducing ligand (APRIL) and cause overexpression of APRIL in DLBCL tissues, which could be related to poorer outcome of DLBCL patients (70). APRIL, which is a ligand of tumor necrosis factor (TNF) superfamily, can bind to B cell maturation antigen (BCMA) and the transmembrane activator and calcium modulator cyclophilin ligand interactor (TACI) and has a crucial role in regulating B cell survival (71). Mechanistically, DLBCL can produce C-X-C motif chemokine ligand 8 (CXCL8), which can recruit neutrophils expressing APRIL, thereby providing the tumor cells with pro-survival signals (72). Additionally, DLBCL derived interleukin-8 (IL-8) can induce neutrophils to form neutrophil extracellular traps (NETs) by binding to its receptor CXCR2 on neutrophils. The formed NETs directly activates TLR9 pathways in DLBCL, leading up-regulation of NF-κB, STAT3 and p38 pathways, which provide pro-survival signals (73).
Stromal Cells
Bone marrow stromal cells protect DLBCL cell lines and primary DLBCL cells through a combination of soluble factors and cell-to-cell contact. When co-cultured with mouse bone marrow stromal cells, primary human DLBCL cells have significantly increased clonogenicity and tumorigenicity (74). Bone marrow stromal cells secret B-cell activating factor (BAFF) and promote survival and drug-resistance of DLBCL cell lines. The adhesion of bone marrow stromal cells to DLBCL cells also can lead to activation of NF-κB pathway, which further up-regulates anti-apoptotic proteins c-IAPs and XIAP, leading to improved survival of lymphoma cells (75). These data suggest that bone marrow stromal cells have an important role in enhancing the survival of DLBCL tumor cells in the bone marrow microenvironment. Additionally, stromal cells can express hedgehog ligands and activate hedgehog signaling in B cell malignancies including DLBCL, promoting survival of these lymphoma cells (76). Co-culture of DLBCL cells with stromal cells results in up-regulation of BCL2, BCL-xL, and BCL2A1, which enhance cell survival (77). Inhibition of hedgehog signaling triggers apoptosis via down-regulating BCL2, suggesting the pro-survival effect provided by stromal cells is mediated, at least partly, by up-regulation of BCL2 (76). Stromal cells induced hedgehog signaling also contribute to chemoresistance by activating the transcription of adenosine triphosphate-binding cassette drug transporter ABCG2, thereby preventing DLBCL cells from cell death caused by chemotherapy (77). In addition to directly supporting the survival of DLBCL cells, stromal cells also recruits monocytes and neutrophils, which can provide additional pro-survival signals (73).
Other Types of Cells
Expansion of CD14 (+) CD169 (–) monocyte-derived cells that have dendritic cell differentiation potential is frequent in DLBCL (78). Coculture of monocytes with DLBCL cells can prolong the survival of the lymphoma cells (79). The mechanisms underlying monocyte support for DLBCL survival are less clear, and the effects are not mediated by BAFF, suggesting other factors may play a role (79). It is known that follicular helper T (Tfh) cells have a crucial role in the development of B cells. It remains to be determined if Tfh cells in the TME have a role in promoting the survival of DLBCL lymphoma cells. It has been shown that circulating CXCR5+CD4+ T cells enhance the survival of primary DLBCL cells through IL-10, indicating that these T cells may have a similar role in the TME (80).
These studies, in combination, support a role for the TME in enhancing DLBCL cell survival.
Epstein-Barr Virus (EBV) Infection
EBV infects B cells in most human hosts and is related to several types of B cell lymphoma. EBV-positive DLBCL, not otherwise specified, which accounts for ~ 10% of DLBCL (81), has been recognized as a distinct lymphoma entity. This disease usually occurs in individuals aged >50 years but can also occur in younger patients (82). Although elderly patients have an unfavorable outcome, the prognosis of young patients is excellent (82). By regulating different pathways, EBV promotes the survival of B cells and is implicated in DLBCL pathogenesis. EBV nuclear antigen 2 (EBNA2) and latent membrane protein 1 (LMP1) are expressed in 7–36% and over 90% of EBV+ DLBCL cases, respectively, suggesting type III and more frequently type II EBV latency (82). It is recognized that LMP1 is indispensable for prolonging the survival of transformed B cells (83). By mimicking the TNF-receptor superfamily member CD40, LMP1 activates the NF-κB pathway and several other pathways to promote cell proliferation and survival in transformed B cells (84, 85) (Figure 4). The cytoplasmic c-terminal tail of LMP1 has two effector domains, C-terminal activation regions 1 and 2 (CTAR1 and 2), which are essential for downstream NF-κB activation (85). CTAR1 recruits and interacts with TNF receptor associated factor (TRAF) members, thus activating downstream NF-κB, PI3K/AKT, p38 MAPK, and the ERK/MAPK pathway (86–88). CTAR2 recruits TNF receptor-associated death domain proteins to activate downstream pathways, including NF-κB, JNK, and the ERK/MAPK pathway (85, 87). Another effector domain, CTAR3, is located between CTAR1 and CTAR2 and can interact with JAK3 and activate the JAK/STAT pathway (89). Additionally, LMP1 induces BCL2 expression in infected B cells and prevents these cells from undergoing apoptosis (90). Accordingly, BCL2 is virtually always positive in EBV+ DLBCL (91). LMP1 down-regulates the expression of a sphingosine-1-phosphate (S1P) receptor S1PR2 at the transcriptional level, a molecule that inhibits PI3K/AKT pathway, contributing to PI3K/AKT pathway activation (92). EBNA2 also up-regulates the anti-apoptotic protein BFL1 (93). Some EBV miRNAs improve the survival of B cells by targeting pro-apoptotic proteins. For example, EBV-miR-Bam HI A region rightward transcript (BART)-cluster 1 and 2 and EBV-miR-BART5 target the pro-apoptotic proteins BIM and PUMA, respectively, thereby increasing the survival of infected B cells (94, 95).
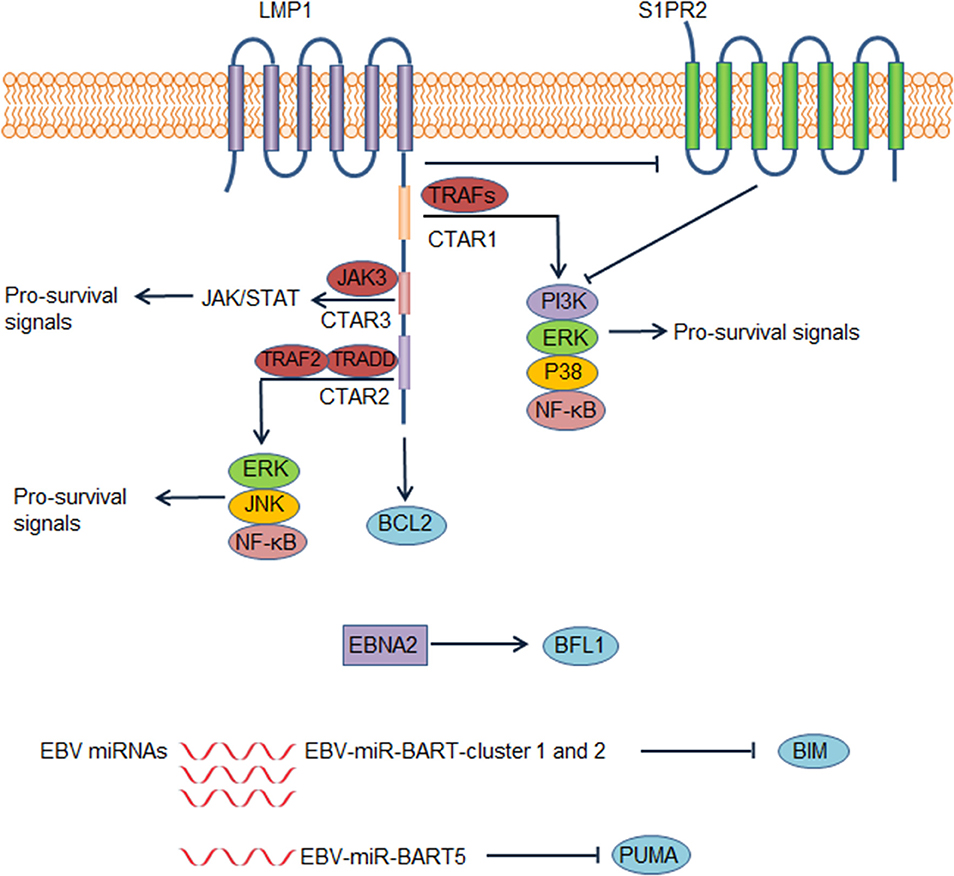
Figure 4. Epstein-Barr virus (EBV) contributes to dysregulation of survival of diffuse large B cell lymphoma (DLBCL) cells. In EBV positive DLBCL, LMP1, EBV nuclear antigen 2 (EBNA2), and several EBV microRNAs could lead to abnormal survival of DLBCL cells, contributing to the pathogenesis of EBV positive DLBCL. These factors lead to enhanced survival of DLBCL cells by increasing pro-survival signals or suppressing pro-apoptotic proteins. Abbreviations: BART, Bam HI A region rightward transcript; CTAR, C-terminal activation region; TRADD, tumor necrosis factor receptor type 1–associated death domain; TRAF, tumor necrosis factor receptor associated factor.
Therapeutic Strategies
An increasing number of novel drugs targeting aberrant cell survival through modulating different pathways have shown efficacy in preclinical studies and/or clinical trials, with some of these drugs showing moderate or significant activities (1, 96). These drugs targeting different pathways are summarized in Table 1. Ongoing trials testing some of these drugs in DLBCL have been summarized in Table 2.
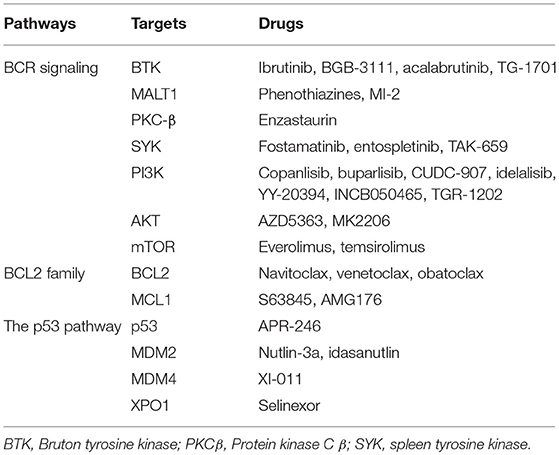
Table 1. Selected drugs targeting aberrant cell survival in diffuse B cell lymphoma.
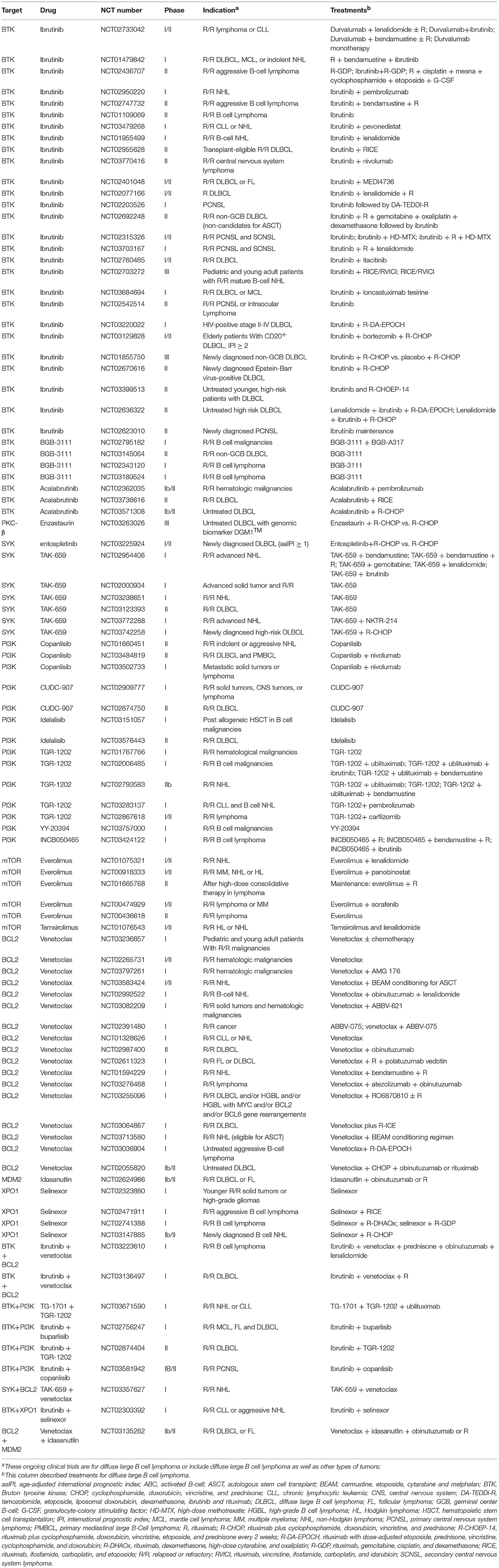
Table 2. Ongoing trials with drugs targeting cell survival in diffuse large B cell lymphoma.
Targeting the BCR Pathway
The BCR pathway has emerged as a crucial target for treating DLBCL patients. Several inhibitors that target molecules in the BCR signaling pathway have shown efficacy in preclinical studies and/or clinical trials. BTK is an important target for suppressing BCR signaling and inhibiting BTK impairs the survival of ABC DLBCL cell lines. The BTK inhibitor ibrutinib has shown significant efficacy in relapsed/refractory ABC-DLBCL patients, especially in those tumors with concurrent MYD88L265P and CD79B mutations (97). The combination of ibrutinib with immunochemotherapy has achieved promising responses in newly-diagnosed DLBCL or refractory/relapsed DLBCL patients (98, 99). Other BTK inhibitors, including BGB-3111 and acalabrutinib, are now being tested in DLBCL. MALT1, which acts as an adaptor protein to activate the downstream NF-kB pathway, has emerged as a potential therapeutic target (100). Several small molecule MALT1 inhibitors, which inhibit the protease activity of MALT1, have shown remarkable anti-tumor effects on ABC DLBCL cells in vitro and in vivo (101, 102). As a key signaling hub downstream of BCR signaling, PKC-β has been an attractive target for treating DLBCL and the PKC-β inhibitor enzastaurin has been tested in refractory/relapsed or newly-diagnosed DLBCL patients. For refractory/relapsed DLBCL, only a small proportion of patients responded to single agent enzastaurin (103). Additionally, incorporating enzastaurin into induction chemotherapy in newly diagnosed DLBCL patients or using enzastaurin as maintenance after CR in high-risk DLBCL did not provide significant survival benefits (104, 105). SYK dependent tonic BCR signaling is indispensable for the survival of DLBCL cell lines, which provides a rationale for targeting SYK in the treatment of DLBCL (20). The selective SYK inhibitor fostamatinib has shown significant activity in refractory/relapsed DLBCL, with 5 of 23 patients achieving a response in a phase 1/2 study (106). Nonetheless, a phase 2 study including 68 refractory/relapsed DLBCL patients showed that only 3% of patients achieved a response (107).
The PI3K/AKT pathway is involved in the downstream of both chronic and tonic BCR signaling. Therefore, inhibiting PI3K/AKT pathway can be used as a strategy to suppress cell survival in both ABC DLBCL and GCB DLBCL. In ABC-DBCL cell lines with CD79B mutations, PI3K inhibition decreases NF-κB activity and impairs the survival of affected DLBCL cell lines (108). Additionally, in GCB DLBCL cell lines, pharmacologic PI3K inhibition selectively impairs survival of PTEN-deficient cell lines (109). A pan-class I PI3k inhibitor, copanlisib, has been tested in refractory/relapsed DLBCL patients as a single agent in phase 1 and 2 clinical trials (110, 111). In the phase 2 trial, the overall response rate (ORR) was 25% (10 of 40) with 5 CRs. The ORR was 13.6% with 1 CR in GCB-type patients and 37.5% with 4 CRs (25%) in ABC-type patients, suggesting that ABC-type had a better response to copanlisib (111). In an expanded phase 1 trial of CUDC-907, a dual inhibitor of histone deacetylase (HDAC) and PI3K, 11 of 30 evaluable patients had an objective response including 5 CR (112). AKT inhibitors showed activities in DLBCL in preclinical studies as well (113–115). The AKT inhibitor AZD5363 was effective in PTEN-deficient DLBCL irrespective of cell-of-origin subtype (115). Another AKT inhibitor, MK2206, also showed significant activity in preclinical DLBCL models, however, in a phase 2 trial, none of 9 evaluable patients treated with MK2206 had a response (116). Activation of PI3K/AKT contributes to activation of mTOR signaling, which promotes the survival of DLBCLs. In addition to PI3K/AKT activation, mTOR could also be driven by activated CARD11 or the My-T-BCR supercomplex (28). Inhibition of mTOR also could be a strategy to suppress the survival of DLBCL cells. The mTOR inhibitors everolimus and temsirolimus have shown significant single-agent activity in refractory/relapsed DLBCL patients, providing additional salvage therapeutic options (117, 118). Given the pathogenic role of the My-T-BCR supercomplex, combination of mTOR inhibitors with BCR inhibitors could be used in DLBCL depending on the My-T-BCR supercomplex (28), and BTK inhibitor ibrutinib and mTOR inhibitor are synergistic in suppressing the growth the xenografts of TMD8, which is dependent on the My-T-BCR supercomplex, supporting combinational use of BTK inhibitors and mTOR inhibitors in My-T-BCR supercomplex positive DLBCLs (28).
Inhibiting the BCL2 Family Members
The role of BCL2 in DLBCL biology makes it a compelling target for treating DLBCL. Navitoclax (ABT-263), which inhibits both BCL2 and BCL-xL, has shown promising efficacy in B cell malignancies. Nonetheless, the thrombocytopenia caused by inhibiting BCL-xL limits the use of navitoclax. Venetoclax (ABT-199), which is a selective BCL2 inhibitor, has potent antitumor activity but spares platelets. In a phase 1 study which included 34 cases of refractory/relapsed DLBCL, 6 patients achieved objective response with 4 patients achieving CR (119). There are some clinical trials currently exploring the efficacy of combining venetoclax with other regimens or drugs in refractory/relapsed or newly-diagnosed DLBCL. MCL1 also can be targeted in DLBCL. Knockdown of MCL1 induces cell apoptosis in MCL1-positive DLBCL cell lines (43). Further, BCL2 inhibition in DLBCL cell lines results in up-regulation of MCL1 expression, possibly interfering with the therapeutic effect of venetoclax (120). Venetoclax and drugs targeting MCL1 displayed a synergistic effect in inducing apoptosis in preclinical models of high-risk DLBCL (120). A highly potent and selective inhibitor of MCL1, S63845 (MIK665), has shown significant anti-tumor activity in several DLBCL cell lines and Eμ-Myc lymphoma cells transplanted mouse models (121). One clinical trial (NCT02992483) is currently investigating the preliminary activity of S63845 in refractory/relapsed multiple myeloma or lymphoma including MYC positive DLBCL. Additionally, the pan-BCL2 inhibitor, obatoclax, which inhibits BCL2, BCL-xL, and MCL1, induces apoptotic cell death in DLBCL cell lines including MCL1 positive ones (43, 122).
Targeting Aberrant p53 Pathway
Targeting an aberrant p53 pathway could be used potentially to restore the function of p53 and trigger cell death. One low-molecular-weight compound, APR-246, is capable of restoring the transcriptional activation function of mutant p53, thus inducing apoptosis in human cancer cells (123). The safety and activity of APR-246 has been tested in refractory/relapsed hematological malignancies including 3 non-Hodgkin lymphoma cases in a phase 1 clinical trial (124). In some patients, tumor cells exhibited upregulation of p53 targeted genes and increase apoptosis. Nonetheless, the efficacy of APR-246 in DLBCL remains to be determined. Disrupting the p53-MDM2 interaction impairs MDM2-mediated p53 degradation, thereby increasing p53 stability and expression. Nutlin-3a disrupts the p53-MDM2 interaction and activates p53, thereby up-regulating the pro-apoptotic proteins BAX and PUMA and inducing apoptosis in DLBCL cell lines with t(14;18)(q32;q21) (125). Idasanutlin, a potent and selective MDM2 antagonist, when combined with obinutuzumab and venetoclax, showed significant antitumor activity in a xenograft model of DLBCL (126). This three-drug combination remarkably improved the tumor-free survival of mice. Idasanutlin in combination with rituximab and venetoclax is currently investigated in refractory/relapsed DLBCL patients (NCT03135262). One MDM4 inhibitor, XI-011, induced apoptosis in breast cancer cells by activating p53 (127). The efficacy of MDM4 inhibitors remains to be determined in DLBCL cells. The transcriptional activation activity of p53 depends on its nuclear localization. XPO1 mediates the nuclear export of proteins including p53. Selinexor, an inhibitor of XPO1, inhibits the nuclear export of p53 and restores p53 nuclear localization (128). In a phase 1 trial, 13 of 41 DLBCL patients treated with single agent selinexor had an objective response, with 4 CR (128).
Strategies for Using Novel Drugs to Target Cell Death Pathway in DLBCL
Combination of novel drugs can improve therapeutic effects thorough additive effects or synergistic effects. By using a high-throughput platform to test compounds, Mathews et al. found that ibrutinib was able to cooperate with other drugs including inhibitors of PI3K pathway (agents targeting the PI3K catalytic subunit, AKT, and the mTORC1 complex), BCL2 inhibitors, and cytotoxic chemotherapeutic drugs in killing ABC DLBCL cells (129). This provides a rationale for use of combination of ibrutinib and other drugs. Furthermore, ibrutinib-resistant TMD8 cells (ABC DLBCL) had elevated BCL2 expression and were vulnerable to BCL2 inhibition; samples from ABC DLBCL patients who had poorer responses to ibrutinib also exhibited higher BCL2 expression (130). These findings support the combination of ibrutinib and BCL2 inhibitors in the treatment of ABC DLBCL (130).
The prior treatments, age and performance status need to be taken into consideration when using novel drugs in the treatment of relapsed/refractory DLBCL. It needs to be pointed out that, the ORR is low in relapsed/refractory DLBCL treated with novel drug monotherapies (97, 119). Even for patients who have achieved responses, the responses are not durable. In relapsed/refractory DLBCL patients receiving single-agent ibrutinib, the median response duration was only 4.83 months in ABC DLBCL, where ibrutinib shows significantly better activity, although patients achieving CR had a relatively long remission (97). Several strategies could possibly be used to apply novel agents in the treatment of DLBCL. In relapsed/refractory DLBCL, combining novel drugs or combining novel drugs with conventional salvage immunochemotherapies could be used to produce higher response rates; for instance, in a phase 1 trial, in 20 patients receiving ibrutinib in combination with R-ICE, 18 patients had a response, with 11 achieving CR (99). In this setting, autologous stem cell transplantation could be used as consolidation therapy in responding patients who are eligible for autologous stem cell transplantation. In the phase 1 trial of venetoclax in relapsed/refractory DLBCL, one DLBCL patient, who proceeded to allogeneic stem cell transplantation (SCT) after achieving CR with venetoclax, achieved durable remission (119). This suggests allogeneic SCT could possibly be used to as a consolidative treatment for patients who have achieved remission with novel drugs. More importantly, the chimeric antigen receptor (CAR) T-cell therapy produced significantly higher CR rates and durable remissions in relapsed/refractory DLBCL (131, 132). Therefore, these novel drugs could be used as bridging therapies to the CAR T-cell therapy or in combination with the CAR T-cell therapy. Additional efforts have been made to improve the outcome of DLBCL patients by incorporating novel drugs into R-CHOP induction in the first-line setting (Table 2). A recent study showed that the addition of ibrutinib to R-CHOP significantly improved the outcome of non-GCB DLBCL patients younger than 65 years (133).
Conclusion
Cell survival dysregulation represents a hallmark of DLBCL. Disruptions of multiple cellular pathways, including BCR signaling, the BCL2 pathway, the p53 pathway and so on, contribute to dysregulated cell survival in DLBCL. Tumor microenvironment dysfunction as well as EBV infection are also involved in promoting the survival of DLBCL cells. Several novel drugs that are capable of targeting abnormal cell survival pathways have been developed and show promising efficacy in DLBCL preclinical models and patients trials. However, until now, of the drugs discussed above, ibrutinib is the only one that has shown clinical benefit in both relapsed/refractory and untreated DLBCL patients (ABC/non-GCB subtype) (97, 133). The success of ibrutinib suggests that ABC cases, especially those with My-T-BCR supercomplex expression, are highly dependent on the BCR signaling (28). This also highlights the importance of BTK molecule in the biology of these DLBCL cases. The failure of some drugs in clinical trials could be attributed to, but not to restricted to the following reasons. DLBCL is a heterogeneous disease (16), different cases may depend on different pathways for cell survival; therefore, when a drug is tested in a group of DLBCL cases, only some of them achieve a response, highlighting the importance of identifying reliable predictors of response. Some cases can rapidly develop resistance mechanisms to a novel drug. Understanding these resistance mechanisms is helpful for developing novel strategies in the treatment of DLBCL. Further understanding of cell survival dysregulation in DLBCL will help to identify more potential targets to provide novel therapeutic options for DLBCL patients in the future.
Author Contributions
YM conceptualized and wrote the manuscript and created the figures. ZX-M and KHY contributed to the conception and writing. ZX-M and LM revised the manuscript. JL contributed to the conception and critically revised the manuscript. All authors have read and approved the final manuscript.
Funding
This study is supported by The University of Texas MD Anderson Cancer Center Lymphoma Moonshot Program, Institutional Research Grant Award, Hagemeister Lymphoma Foundation, and the University Cancer Foundation via the Sister institution network Fund at The University of Texas MD Anderson Cancer.
Conflict of Interest Statement
KHY receives research support from Roche Molecular System, Gilead Sciences Pharmaceutical, Seattle Genetics, Dai Sanyo Pharmaceutical, Adaptive Biotechnology, Incyte Pharmaceutical, and HTG Molecular Diagnostics.
The funders played no role in the study design, the collection, analysis or interpretation of data, the writing of this paper or the decision to submit it for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Dunleavy K, Erdmann T, Lenz G. Targeting the B-cell receptor pathway in diffuse large B-cell lymphoma. Cancer Treat Rev. (2018) 65:41–6. doi: 10.1016/j.ctrv.2018.01.002
2. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. (2000) 403:503–11. doi: 10.1038/35000501
3. Gutierrez-Garcia G, Cardesa-Salzmann T, Climent F, Gonzalez-Barca E, Mercadal S, Mate JL, et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood. (2011) 117:4836–43. doi: 10.1182/blood-2010-12-322362
4. Pfreundschuh M, Kuhnt E, Trumper L, Osterborg A, Trneny M, Shepherd L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. (2011) 12:1013–22. doi: 10.1016/S1470–2045(11)70235–2
5. Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Ferme C, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol. (2005) 23:4117–26. doi: 10.1200/JCO.2005.09.131
6. Pfreundschuh M, Schubert J, Ziepert M, Schmits R, Mohren M, Lengfelder E, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60). Lancet Oncol. (2008) 9:105–16. doi: 10.1016/S1470–2045(08)70002–0
7. Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. (2010) 28:4184–90. doi: 10.1200/JCO.2010.28.1618
8. Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematol Am Soc Hematol Educ Program (2011) 2011:498–505. doi: 10.1182/asheducation-2011.1.498
9. Lenz G. Insights into the molecular pathogenesis of activated B-cell-like diffuse large B-Cell lymphoma and its therapeutic implications. Cancers. (2015) 7:811–22. doi: 10.3390/cancers7020812
10. Frick M, Dorken B, Lenz G. New insights into the biology of molecular subtypes of diffuse large B-cell lymphoma and Burkitt lymphoma. Best Pract Res Clin Haematol. (2012) 25:3–12. doi: 10.1016/j.beha.2012.01.003
11. Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. (2004) 117:787–800. doi: 10.1016/j.cell.2004.05.014
12. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. (2009) 139:573–86. doi: 10.1016/j.cell.2009.08.041
13. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. (2010) 463:88–92. doi: 10.1038/nature08638
14. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proc Natl Acad Sci USA. (2015) 112:13447–54. doi: 10.1073/pnas.1514944112
15. Bohers E, Mareschal S, Bouzelfen A, Marchand V, Ruminy P, Maingonnat C, et al. Targetable activating mutations are very frequent in GCB and ABC diffuse large B-cell lymphoma. Genes Chromosomes Cancer. (2014) 53:144–53. doi: 10.1002/gcc.22126
16. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
17. Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry. (2010) 49:8240–50. doi: 10.1021/bi101052d
18. Wray-Dutra MN, Chawla R, Thomas KR, Seymour BJ, Arkatkar T, Sommer KM, et al. Activated CARD11 accelerates germinal center kinetics, promoting mTORC1 and terminal differentiation. J Exp Med. (2018) 215:2445–61. doi: 10.1084/jem.20180230
19. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. (2001) 194:1861–74. doi: 10.1084/jem.194.12.1861
20. Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. (2008) 111:2230–7. doi: 10.1182/blood-2007–07-100115
21. Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood. (2017) 130:995–1006. doi: 10.1182/blood-2016–10-747303
22. Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, et al. MYD88 L265P mutation in lymphoid malignancies. Cancer Res. (2018) 78:2457–62. doi: 10.1158/0008–5472.CAN-18–0215
23. Monlish DA, Bhatt ST, Schuettpelz LG. The role of toll-like receptors in hematopoietic malignancies. Front Immunol. (2016) 7:390. doi: 10.3389/fimmu.2016.00390
24. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
25. Avbelj M, Wolz OO, Fekonja O, Bencina M, Repic M, Mavri J, et al. Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood. (2014) 124:3896–904. doi: 10.1182/blood-2014–05-573188
26. Knittel G, Liedgens P, Korovkina D, Seeger JM, Al-Baldawi Y, Al-Maarri M, et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood. (2016) 127:2732–41. doi: 10.1182/blood-2015–11-684183
27. Wang JQ, Jeelall YS, Humburg P, Batchelor EL, Kaya SM, Yoo HM, et al. Synergistic cooperation and crosstalk between MYD88(L265P) and mutations that dysregulate CD79B and surface IgM. J Exp Med. (2017) 214:2759–76. doi: 10.1084/jem.20161454
28. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. (2018) 560:387–91. doi: 10.1038/s41586–018-0290–0
29. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. (2002) 2:647–56. doi: 10.1038/nrc883
30. Hu S, Xu-Monette ZY, Tzankov A, Green T, Wu L, Balasubramanyam A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP consortium program. Blood. (2013) 121:4021–31; quiz 250. doi: 10.1182/blood-2012–10-460063
31. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. (2012) 30:3452–9. doi: 10.1200/JCO.2011.41.0985
32. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature. (1991) 349:254–6. doi: 10.1038/349254a0
33. McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. (1989) 57:79–88. doi: 10.1016/0092–8674(89)90174–8
34. Katsumata M, Siegel RM, Louie DC, Miyashita T, Tsujimoto Y, Nowell PC, et al. Differential effects of Bcl-2 on T and B cells in transgenic mice. Proc Natl Acad Sci USA. (1992) 89:11376–80. doi: 10.1073/pnas.89.23.11376
35. Staiger AM, Ziepert M, Horn H, Scott DW, Barth TFE, Bernd HW, et al. Clinical impact of the cell-of-origin classification and the MYC/ BCL2 dual expresser status in diffuse large b-cell lymphoma treated within prospective clinical trials of the german high-grade non-hodgkin's lymphoma study group. J Clin Oncol. (2017) 35:2515–26. doi: 10.1200/JCO.2016.70.3660
36. Visco C, Tzankov A, Xu-Monette ZY, Miranda RN, Tai YC, Li Y, et al. Patients with diffuse large B-cell lymphoma of germinal center origin with BCL2 translocations have poor outcome, irrespective of MYC status: a report from an International DLBCL rituximab-CHOP Consortium Program Study. Haematologica. (2013) 98:255–63. doi: 10.3324/haematol.2012.066209
37. Kusumoto S, Kobayashi Y, Sekiguchi N, Tanimoto K, Onishi Y, Yokota Y, et al. Diffuse large B-cell lymphoma with extra Bcl-2 gene signals detected by FISH analysis is associated with a “non-germinal center phenotype”. Am J Surg Pathol. (2005) 29:1067–73. doi: 10.1097/01.pas.0000167362.06148.95
38. Saito M, Novak U, Piovan E, Basso K, Sumazin P, Schneider C, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. (2009) 106:11294–9. doi: 10.1073/pnas.0903854106
39. Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia. (2015) 29:1004–17. doi: 10.1038/leu.2014.351
40. Deng X, Gao F, Flagg T, Anderson J, May WS. Bcl2's flexible loop domain regulates p53 binding and survival. Mol Cell Biol. (2006) 26:4421–34. doi: 10.1128/MCB.01647–05
41. Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. (2012) 26:1383–90. doi: 10.1038/leu.2011.378
42. Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. (2012) 19:295–309. doi: 10.1038/cdd.2011.97
43. Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia. (2013) 27:1381–90. doi: 10.1038/leu.2012.367
44. Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood. (2001) 97:3902–9. doi: 10.1182/blood.V97.12.3902
45. Adams CM, Kim AS, Mitra R, Choi JK, Gong JZ, Eischen CM. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J Clin Invest. (2017) 127:635–50. doi: 10.1172/JCI89486
46. Adams CM, Mitra R, Gong JZ, Eischen CM. Non-Hodgkin and hodgkin lymphomas select for overexpression of BCLW. Clin Cancer Res. (2017) 23:7119–29. doi: 10.1158/1078–0432.CCR-17–1144
47. Akyurek N, Ren Y, Rassidakis GZ, Schlette EJ, Medeiros LJ. Expression of inhibitor of apoptosis proteins in B-cell non-Hodgkin and Hodgkin lymphomas. Cancer. (2006) 107:1844–51. doi: 10.1002/cncr.22219
48. Hussain AR, Uddin S, Ahmed M, Bu R, Ahmed SO, Abubaker J, et al. Prognostic significance of XIAP expression in DLBCL and effect of its inhibition on AKT signalling. J Pathol. (2010) 222:180–90. doi: 10.1002/path.2747
49. Liu Z, Xu-Monette ZY, Cao X, Manyam GC, Wang X, Tzankov A, et al. Prognostic and biological significance of survivin expression in patients with diffuse large B-cell lymphoma treated with rituximab-CHOP therapy. Modern Pathol. (2015) 28:1297–314. doi: 10.1038/modpathol.2015.94
50. Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. (1993) 362:857–60. doi: 10.1038/362857a0
51. Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. (1999) 55:96–107. doi: 10.1007/s000180050273
52. Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an international DLBCL Rituximab-CHOP consortium program Study. Blood. (2012) 120:3986–96. doi: 10.1182/blood-2012–05-433334
53. Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. (2006) 127:1323–34. doi: 10.1016/j.cell.2006.12.007
54. Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J Cancer. (2017) 141:1381–8. doi: 10.1002/ijc.30838
55. Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P, et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood. (2010) 116:1092–104. doi: 10.1182/blood-2009-10-247122
56. Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. (2004) 432:635–9. doi: 10.1038/nature03147
57. Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell. (2012) 22:359–72. doi: 10.1016/j.ccr.2012.07.014
58. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. (1998) 92:725–34. doi: 10.1016/S0092-8674(00)81401-4
59. Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenfuhr M, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. (2009) 23:1177–82. doi: 10.1101/gad.511109
60. Sola S, Xavier JM, Santos DM, Aranha MM, Morgado AL, Jepsen K, et al. p53 interaction with JMJD3 results in its nuclear distribution during mouse neural stem cell differentiation. PLoS ONE. (2011) 6:e18421. doi: 10.1371/journal.pone.0018421
61. Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. (2005) 123:49–63. doi: 10.1016/j.cell.2005.07.034
62. Grossman SR. p300/CBP/p53 interaction and regulation of the p53 response. Eur J Biochem. (2001) 268:2773–8. doi: 10.1046/j.1432–1327.2001.02226.x
63. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. (2011) 471:189–95. doi: 10.1038/nature09730
64. Tweeddale ME, Lim B, Jamal N, Robinson J, Zalcberg J, Lockwood G, et al. The presence of clonogenic cells in high-grade malignant lymphoma: a prognostic factor. Blood. (1987) 69:1307–14.
65. Yhim HY, Kim JA, Ko SH, Park Y, Yim E, Kim HS, et al. The prognostic significance of CD11b(+)CX3CR1(+) monocytes in patients with newly diagnosed diffuse large B-cell lymphoma. Oncotarget. (2017) 8:92289–99. doi: 10.18632/oncotarget.21241
66. Mitteldorf C, Berisha A, Pfaltz MC, Broekaert SMC, Schon MP, Kerl K, et al. Tumor Microenvironment and checkpoint molecules in primary cutaneous diffuse large b-cell lymphoma-new therapeutic targets. Am J Surg Pathol. (2017) 41:998–1004. doi: 10.1097/PAS.0000000000000851
67. Nam SJ, Go H, Paik JH, Kim TM, Heo DS, Kim CW, et al. An increase of M2 macrophages predicts poor prognosis in patients with diffuse large B-cell lymphoma treated with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone. Leuk Lymphoma. (2014) 55:2466–76. doi: 10.3109/10428194.2013.879713
68. Cai QC, Liao H, Lin SX, Xia Y, Wang XX, Gao Y, et al. High expression of tumor-infiltrating macrophages correlates with poor prognosis in patients with diffuse large B-cell lymphoma. Med Oncol. (2012) 29:2317–22. doi: 10.1007/s12032–011-0123–6
69. Gregoire M, Guilloton F, Pangault C, Mourcin F, Sok P, Latour M, et al. Neutrophils trigger a NF-kappaB dependent polarization of tumor-supportive stromal cells in germinal center B-cell lymphomas. Oncotarget. (2015) 6:16471–87. doi: 10.18632/oncotarget.4106
70. Schwaller J, Schneider P, Mhawech-Fauceglia P, McKee T, Myit S, Matthes T, et al. Neutrophil-derived APRIL concentrated in tumor lesions by proteoglycans correlates with human B-cell lymphoma aggressiveness. Blood. (2007) 109:331–8. doi: 10.1073/blood-2006–02-001800
71. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. (2009) 9:491–502. doi: 10.1038/nri2572
72. Manfroi B, McKee T, Mayol JF, Tabruyn S, Moret S, Villiers C, et al. CXCL-8/IL8 produced by diffuse large b-cell lymphomas recruits neutrophils expressing a proliferation-inducing ligand APRIL. Cancer Res. (2017) 77:1097–107. doi: 10.1158/0008–5472.CAN-16–0786
73. Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil extracellular traps induced by IL-8 promote diffuse large B cell lymphoma progression via the TLR9 signaling. Clin Cancer Res. (2018). doi: 10.1158/1078-0432.CCR-18-1226. [Epub ahead of print].
74. Lin Z, Chen B, Wu T, Xu X. Highly Tumorigenic diffuse large b cell lymphoma cells are produced by coculture with stromal cells. Acta Haematol. (2018) 139:201–16. doi: 10.1159/000488385
75. Lwin T, Hazlehurst LA, Li Z, Dessureault S, Sotomayor E, Moscinski LC, et al. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-kappaB (RelB/p52) in non-Hodgkin's lymphoma cells. Leukemia. (2007) 21:1521–31. doi: 10.1038/sj.leu.2404723
76. Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. (2007) 13:944–51. doi: 10.1038/nm1614
77. Singh RR, Kunkalla K, Qu C, Schlette E, Neelapu SS, Samaniego F, et al. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. (2011) 30:4874–86. doi: 10.1038/onc.2011.195
78. Marmey B, Boix C, Barbaroux JB, Dieu-Nosjean MC, Diebold J, Audouin J, et al. CD14 and CD169 expression in human lymph nodes and spleen: specific expansion of CD14+CD169- monocyte-derived cells in diffuse large B-cell lymphomas. Hum Pathol. (2006) 37:68–77. doi: 10.1016/j.humpath.2005.09.016
79. Mueller CG, Boix C, Kwan WH, Daussy C, Fournier E, Fridman WH, et al. Critical role of monocytes to support normal B cell and diffuse large B cell lymphoma survival and proliferation. J Leukoc Biol. (2007) 82:567–75. doi: 10.1189/jlb.0706481
80. Cha Z, Qian G, Zang Y, Gu H, Huang Y, Zhu L, et al. Circulating CXCR5+CD4+ T cells assist in the survival and growth of primary diffuse large B cell lymphoma cells through interleukin 10 pathway. Exp Cell Res. (2017) 350:154–60. doi: 10.1016/j.yexcr.2016.11.017
81. Healy JA, Dave SS. The role of EBV in the pathogenesis of diffuse large b cell lymphoma. Curr Top Microbiol Immunol. (2015) 390:315–37. doi: 10.1007/978–3-319–22822-8_13
82. Nicolae A, Pittaluga S, Abdullah S, Steinberg SM, Pham TA, Davies-Hill T, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. (2015) 126:863–72. doi: 10.1182/blood-2015–02-630632
83. Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci USA. (1993) 90:9150–4. doi: 10.1073/pnas.90.19.9150
84. Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. (1999) 286:300–3. doi: 10.1126/science.286.5438.300
85. Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Semin Cancer Biol. (2001) 11:435–44. doi: 10.1006/scbi.2001.0410
86. Mainou BA, Everly DN Jr, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene. (2005) 24:6917–24. doi: 10.1038/sj.onc.1208846
87. Dawson CW, Laverick L, Morris MA, Tramoutanis G, Young LS. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J Virol. (2008) 82:3654–64. doi: 10.1128/JVI.01888–07
88. Morris MA, Dawson CW, Young LS. Role of the Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the pathogenesis of nasopharyngeal carcinoma. Future Oncol. (2009) 5:811–25. doi: 10.2217/fon.09.53
89. Higuchi M, Kieff E, Izumi KM. The Epstein-Barr virus latent membrane protein 1 putative Janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J Virol. (2002) 76:455–9. doi: 10.1128/JVI.76.1.455–459.2002
90. Henderson S, Rowe M, Gregory C, Croom-Carter D, Wang F, Longnecker R, et al. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell. (1991) 65:1107–15. doi: 10.1016/0092–8674(91)90007-L
91. Ok CY, Papathomas TG, Medeiros LJ, Young KH. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. (2013) 122:328–40. doi: 10.1182/blood-2013–03-489708
92. Vockerodt M, Vrzalikova K, Ibrahim M, Nagy E, Margielewska S, Hollows R, et al. Regulation of S1PR2 by the EBV oncogene LMP1 in aggressive ABC subtype diffuse large B cell lymphoma. J Pathol. (2019). doi: 10.1002/path.5237. [Epub ahead of print].
93. Price AM, Dai J, Bazot Q, Patel L, Nikitin PA, Djavadian R, et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife. (2017) 6:e22509. doi: 10.7554/eLife.22509
94. Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. (2008) 205:2551–60. doi: 10.1084/jem.20072581
95. Marquitz AR, Mathur A, Nam CS, Raab-Traub N. The epstein-barr virus BART microRNAs target the pro-apoptotic protein Bim. Virology. (2011) 412:392–400. doi: 10.1016/j.virol.2011.01.028
96. Majchrzak A, Witkowska M, Smolewski P. Inhibition of the PI3K/Akt/mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules. (2014) 19:14304–15. doi: 10.3390/molecules190914304
97. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. (2015) 21:922–6. doi: 10.1038/nm.3884
98. Younes A, Thieblemont C, Morschhauser F, Flinn I, Friedberg JW, Amorim S, et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol. (2014) 15:1019–26. doi: 10.1016/S1470–2045(14)70311–0
99. Sauter CS, Matasar MJ, Schoder H, Devlin SM, Drullinsky P, Gerecitano J, et al. A phase 1 study of ibrutinib in combination with R-ICE in patients with relapsed or primary refractory DLBCL. Blood. (2018) 131:1805–8. doi: 10.1182/blood-2017–08-802561
100. Krappmann D. Attacking MALT1 for ABC-DLBCL therapy. Oncotarget. (2012) 3:1489–90. doi: 10.18632/oncotarget.794
101. Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. (2012) 22:825–37. doi: 10.1016/j.ccr.2012.11.002
102. Fontan L, Yang C, Kabaleeswaran V, Volpon L, Osborne MJ, Beltran E, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. (2012) 22:812–24. doi: 10.1016/j.ccr.2012.11.003
103. Robertson MJ, Kahl BS, Vose JM, de Vos S, Laughlin M, Flynn PJ, et al. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. (2007) 25:1741–6. doi: 10.1200/JCO.2006.09.3146
104. Crump M, Leppa S, Fayad L, Lee JJ, Di Rocco A, Ogura M, et al. Randomized, double-blind, phase III trial of enzastaurin versus placebo in patients achieving remission after first-line therapy for high-risk diffuse large b-cell lymphoma. J Clin Oncol. (2016) 34:2484–92. doi: 10.1200/JCO.2015.65.7171
105. Hainsworth JD, Arrowsmith ER, McCleod M, Hsi ED, Hamid O, Shi P, et al. A randomized, phase 2 study of R-CHOP plus enzastaurin vs R-CHOP in patients with intermediate- or high-risk diffuse large B-cell lymphoma. Leuk Lymphoma. (2016) 57:216–8. doi: 10.3109/10428194.2015.1045898
106. Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. (2010) 115:2578–85. doi: 10.1182/blood-2009–08-236471
107. Flinn IW, Bartlett NL, Blum KA, Ardeshna KM, LaCasce AS, Flowers CR, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Eur J Cancer. (2016) 54:11–7. doi: 10.1016/j.ejca.2015.10.005
108. Kloo B, Nagel D, Pfeifer M, Grau M, Duwel M, Vincendeau M, et al. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. (2011) 108:272–7. doi: 10.1073/pnas.1008969108
109. Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. (2013) 110:12420–5. doi: 10.1073/pnas.1305656110
110. Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80–6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin's lymphomas. Ann Oncol. (2016) 27:1928–40. doi: 10.1093/annonc/mdw282
111. Lenz G, Hawkes E, Verhoef G, Haioun C, Thye LS, Heo DS et al. Phase II study of single-agent copanlisib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). J Clin Oncol. (2017) 35(Suppl. 15):7536. doi: 10.1200/JCO.2017.35.15_suppl.7536
112. Oki Y, Kelly KR, Flinn I, Patel MR, Gharavi R, Ma A, et al. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica. (2017) 102:1923–30. doi: 10.3324/haematol.2017.172882
113. Petrich AM, Leshchenko V, Kuo PY, Xia B, Thirukonda VK, Ulahannan N, et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clin Cancer Res. (2012) 18:2534–44. doi: 10.1158/1078–0432.CCR-11–1407
114. Wang J, Xu-Monette ZY, Jabbar KJ, Shen Q, Manyam GC, Tzankov A, et al. AKT Hyperactivation and the Potential of AKT-Targeted Therapy in Diffuse Large B-Cell Lymphoma. Am J Pathol. (2017) 187:1700–16. doi: 10.1016/j.ajpath.2017.04.009
115. Erdmann T, Klener P, Lynch JT, Grau M, Vockova P, Molinsky J, et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood. (2017) 130:310–22. doi: 10.1182/blood-2016–12-758599
116. Oki Y, Fanale M, Romaguera J, Fayad L, Fowler N, Copeland A, et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br J Haematol. (2015) 171:463–70. doi: 10.1111/bjh.13603
117. Smith SM, van Besien K, Karrison T, Dancey J, McLaughlin P, Younes A, et al. Temsirolimus has activity in non-mantle cell non-Hodgkin's lymphoma subtypes: the University of Chicago phase II consortium. J Clin Oncol. (2010) 28:4740–6. doi: 10.1200/JCO.2010.29.2813
118. Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. (2011) 25:341–7. doi: 10.1038/leu.2010.226
119. Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I First-in-human study of venetoclax in patients with relapsed or refractory non-hodgkin lymphoma. J Clin Oncol. (2017) 35:826–33. doi: 10.1200/JCO.2016.70.4320
120. Li L, Pongtornpipat P, Tiutan T, Kendrick SL, Park S, Persky DO, et al. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia. (2015) 29:1702–12. doi: 10.1038/leu.2015.99
121. Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, et al. Humanized Mcl-1 mice enable accurate pre-clinical evaluation of MCL-1 inhibitors destined for clinical use. Blood. (2018). 132:1573–83. doi: 10.1182/blood-2018-06-859405
122. Brem EA, Thudium K, Khubchandani S, Tsai PC, Olejniczak SH, Bhat S, et al. Distinct cellular and therapeutic effects of obatoclax in rituximab-sensitive and -resistant lymphomas. Br J Haematol. (2011) 153:599–611. doi: 10.1111/j.1365–2141.2011.08669.x
123. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. (2002) 8:282–8. doi: 10.1038/nm0302–282
124. Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. (2012) 30:3633–9. doi: 10.1200/JCO.2011.40.7783
125. Drakos E, Singh RR, Rassidakis GZ, Schlette E, Li J, Claret FX, et al. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14;18)(q32;q21). Leukemia. (2011) 25:856–67. doi: 10.1038/leu.2011.28
126. Herting F, Friess T, Umana P, Middleton S, Klein C. Chemotherapy-free, triple combination of obinutuzumab, venetoclax and idasanutlin: antitumor activity in xenograft models of non-Hodgkin lymphoma. Leuk Lymphoma. (2018) 59:1482–5. doi: 10.1080/10428194.2017.1376740
127. Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med. (2011) 17:71–8. doi: 10.1038/nm.2282
128. Kuruvilla J, Savona M, Baz R, Mau-Sorensen PM, Gabrail N, Garzon R, et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. (2017) 129:3175–83. doi: 10.1182/blood-2016–11-750174
129. Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. (2014) 111:2349–54. doi: 10.1073/pnas.1311846111
130. Kuo HP, Ezell SA, Schweighofer KJ, Cheung LWK, Hsieh S, Apatira M, et al. Combination of Ibrutinib and ABT-199 in diffuse large b-cell lymphoma and follicular lymphoma. Mol Cancer Ther. (2017) 16:1246–56. doi: 10.1158/1535–7163.MCT-16–0555
131. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
132. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
133. Younes A, Sehn LH, Johnson P, Zinzani PL, Hong XN, Zhu J, et al. A Global, Randomized, Placebo-Controlled, Phase 3 Study of Ibrutinib Plus Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone (RCHOP) in Patients with Previously Untreated Non-Germinal Center B-Cell-like (GCB) Diffuse Large B-Cell Lymphoma (DLBCL). Blood. (2018) 132 (Suppl. 1):784. doi: 10.1182/blood-2018–99-112988
Keywords: DLBCL, cell survival, apoptosis, BCR signaling, BCL2, p53, TME, EBV
Citation: Miao Y, Medeiros LJ, Xu-Monette ZY, Li J and Young KH (2019) Dysregulation of Cell Survival in Diffuse Large B Cell Lymphoma: Mechanisms and Therapeutic Targets. Front. Oncol. 9:107. doi: 10.3389/fonc.2019.00107
Received: 09 October 2018; Accepted: 05 February 2019;
Published: 01 March 2019.
Edited by:
Martine Amiot, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Narendranath Epperla, The Ohio State University, United StatesMichael Diamantidis, University Hospital of Larissa, Greece
Gaetan Jego, Université de Bourgogne, France
Copyright © 2019 Miao, Medeiros, Xu-Monette, Li and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken H. Young, a2h5b3VuZ0BtZGFuZGVyc29uLm9yZw==