 Ali Vaziri-Gohar
Ali Vaziri-Gohar Mahsa Zarei
Mahsa Zarei Jonathan R. Brody
Jonathan R. Brody Jordan M. Winter1,5*
Jordan M. Winter1,5*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 12 December 2018
Sec. Molecular and Cellular Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00617
This article is part of the Research Topic Cancer Ecosystems View all 19 articles
Pancreatic ductal adenocarcinoma (PDA) is a highly lethal cancer with a long-term survival rate under 10%. Available cytotoxic chemotherapies have significant side effects, and only marginal therapeutic efficacy. FDA approved drugs currently used against PDA target DNA metabolism and DNA integrity. However, alternative metabolic targets beyond DNA may prove to be much more effective. PDA cells are forced to live within a particularly severe microenvironment characterized by relative hypovascularity, hypoxia, and nutrient deprivation. Thus, PDA cells must possess biochemical flexibility in order to adapt to austere conditions. A better understanding of the metabolic dependencies required by PDA to survive and thrive within a harsh metabolic milieu could reveal specific metabolic vulnerabilities. These molecular requirements can then be targeted therapeutically, and would likely be associated with a clinically significant therapeutic window since the normal tissue is so well-perfused with an abundant nutrient supply. Recent work has uncovered a number of promising therapeutic targets in the metabolic domain, and clinicians are already translating some of these discoveries to the clinic. In this review, we highlight mitochondria metabolism, non-canonical nutrient acquisition pathways (macropinocytosis and use of pancreatic stellate cell-derived alanine), and redox homeostasis as compelling therapeutic opportunities in the metabolic domain.
Pancreatic cancer is the third leading cause of cancer-related death in the United States (1). The disease is predicted to be the second leading cause within the next decade (2). Cures are exceedingly rare, and the 5-years survival for patients with metastatic disease is just 3%. Even patients with localized PDA who undergo resection with curative intent have a 5-years survival of only 30% (1). This survival rate is by far the lowest of the common cancers, and is attributable in large part to PDAs uniquely aggressive behavior and resistance to conventional therapy (3, 4).
The majority of pancreatic cancer patients already have experienced macroscopic or microscopic spread at the time of diagnosis (5–7). Cytotoxic chemotherapeutic agents are the only approved systemic treatments for these patients, and are grouped into two separate multi-agent regimens used as standard-of-care: (1) gemcitabine and nab-paclitaxel (albumin-bound paclitaxel or Abraxane) or (2) 5-fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX). Unless researchers discover effective strategies to detect PDA earlier or preventative tactics, new ways to treat invasive PDA are desperately needed (8, 9).
Pancreatic cancer develops over many years due to the additive effects of numerous genetic changes (10). Gain-of-function mutations in KRAS at codons 12, 13, and 61 are observed in over 90% of PDAs. Loss-of-function mutations in three specific tumor suppressor genes occur in the majority of PDAs: TP53, CDKN2A, and SMAD4 (11–14). These oncogenic changes occur early in the adenoma to carcinoma progression (15, 16). The vast majority of pancreatic cancers are sporadic. Approximately 10% of patients have one or more immediate family members with a history of PDA. Germline culprits include genes that are important for DNA repair, such as BRCA2, PALB2, ATM, FANCC, and FANCG genes (11, 17).
Histologically, pancreatic tumorigenesis passes through three non-invasive, pre-malignant stages before acquiring an invasive phenotype. The premalignant stages are referred to as pancreatic intraepithelial neoplasia (PanIN 1, 2, and 3). More detailed descriptions of the genetics and pathobiology of pancreatic cancer appear elsewhere (18–20). In addition to well-characterized genetic mutations, there other molecular abnormalities common to PDA including hyperactivated growth factor signaling, epigenetic changes, dysregulated gene expression (transcriptional or post-transcriptional), and abnormal post-translational modifications (18, 21–23).
The PDA tumor microenvironment is characterized by one of the most abundant stromal compartments of any tumor type, and this feature is the principal biologic driver of the PDA metabolic program. Neoplastic epithelial cells account for roughly 10–15% of the tumor mass, while non-neoplastic elements comprise the remainder of the tumor. The stroma, or desmoplastic response, consists of an extracellular matrix and diverse cellular elements including fibroblasts, myofibroblasts, lymphatic vessels, blood vessels, pancreatic stellate cells, and immune cells (24, 25). As a result of these elements, the stroma is extremely dense, with a high interstitial fluid pressure (26). Consequently, blood vessels are compressed by biochemical forces and microscopic arteriolar-venular shunts are common events (27, 28). Compounding this significant biologically relevant perfusion challenge, microscopic vessel density is markedly reduced in PDA stroma as compared to normal pancreata (26).
PDA hypovascularity is easy to appreciate macroscopically. Transected PDAs are pale gray and necrotic (Figure 1A). On imaging, PDA appears as a hypodense tumor, easily distinguished by the well-perfused and contrast-enhancing normal pancreatic parenchyma (Figure 1B). This biologic reality accounts for the harsh, nutrient deprived, and hypoxic conditions that are hallmarks of the PDA microenvironment. Indeed, PDA survives, and thrives, in a desert!
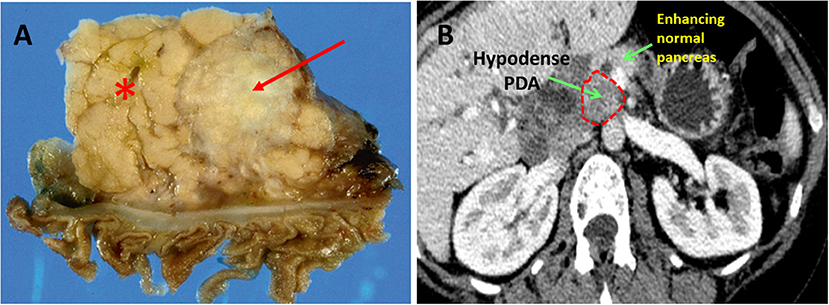
Figure 1. Pancreatic ductal adenocarcinoma. (A) Resected human PDA. The arrow identifies the characteristically pale, gray, and hypovascular pancreatic ductal adenocarcinoma. The asterisk marks pancreatic parenchyma. (B) On CT scan imaging with intravenous contrast, PDA appears hypodense (dark gray), while well-perfused normal pancreatic parenchyma shows bright enhancement due to penetration by the contrast.
Studies implicate mutant KRAS as a key driver of the desmoplastic response. Temporally, this genetic model fits since KRAS mutations arise in PanIN1 lesions when the stroma first develops. As evidence, withdrawal of mutant KRAS expression using shRNAs in genetically engineered KPC mice (autochthonous PDA mice with conditional expression of oncogenic KRAS and TP53 mutations) (16) resulted in the disappearance of the stromal compartment (29).
Oxidative phosphorylation is an energy extracting process where pyruvate enters the mitochondria matrix via pyruvate translocase and is oxidized to generate ATP, H2O, and CO2. Oxygen acts as a final electron acceptor in the electron transport chain. As a result of the associated redox reactions, an electrochemical potential is generated across the inner mitochondrial membrane, which drives a proton-motive force across the membrane, and directly results in the formation of ATP. Thus, carbon flows through the tricarboxylic acid (TCA) cycle and is oxidized to its simplest form, creating basic energy subunits with high energy phosphate bonds that drive other chemical reactions required for cell viability. Well-perfused and differentiated normal cells generate the bulk of their cellular energy through oxidative phosphorylation in the mitochondria (30). Cancer cells, on the other hand, are often oxygen-poor. This scenario theoretically poses an obstacle for effective oxidative phosphorylation. Even in the presence of sufficient oxygen, however, scientists believed for decades that cancer cells were reprogrammed.
Scientists posited that cancer cells relied on cytosolic glycolysis to produce ATP instead of oxidative phosphorylation, even though the energy yield was far less efficient (2 ATP vs. 36 ATP). A preference for aerobic glycolysis is eponymously referred to as the “Warburg's effect” (31), and there have been numerous lines of evidence that in fact reveal robust glycolytic activity in pancreatic cancer cells in certain experimental models, including patient samples. For instance, endogenous expression of many glycolytic enzymes is increased, including hexokinase 2, enolase 2, and lactate dehydrogenases (both LDHA and LDHB isoforms) (32–35). Consequently, glycolytic metabolites, including lactate, are also elevated in pancreatic cancer cells (32, 33, 36).
Generally speaking, a biochemical penchant for glycolysis can serve cancer cells in multiple ways. First, Lactic acid build-up reduces cellular and extracellular pH. This contributes to invasiveness by promoting genetic changes in PDA cells (spurring on genetic selection), impairing the anti-tumor immune response (protecting cancer cells from immune patrol), reducing adherens junctions on cancer cell membranes (facilitating detachment and metastases), and hydrolysis of extracellular proteins to encourage cell invasion (37). Just as important, enhanced glycolytic activity minimizes combustion of carbon from glucose to CO2. Instead, organic carbon is preserved, and diverted into cellular building blocks through biosynthetic pathways, such as lipid synthesis, the hexosamine biosynthesis pathway, and the pentose phosphate pathway. This reprogramming effort promotes the macromolecular synthesis needed for cell proliferation (i.e., anabolism) (30, 38, 39). As stated above for desmoplasia (and a common theme for many of the adaptive reprogramming responses described below), oncogenic KRAS drives glycolytic activity in PDA. For instance, KRAS activation leads to increased glucose uptake and elevated levels of glycolysis-associated metabolites (40, 41). In vivo models modified by an inducible KRAS extinction mechanism in pancreatic tumors corroborate these findings (29).
Additional studies show that oncogenic KRAS activity supports glycolysis by replenishing the supply of NAD+, via upregulation of NAD(P)H oxidase (42). At a regulatory level, the Pasteur effect (an increase in glucose consumption and lactic acid fermentation under low oxygen conditions) is also encouraged by certain transcription factors and related proteins (34, 43). As examples, HIF-1α and MUC1 upregulate glucose transporter 1 (GLUT1) and aldolase A, which leads to increased glucose uptake and glycolysis. MUC1 collaborates with HIF-1α for this purpose. Additionally, under hypoxic conditions, pyruvate dehydrogenase kinase 1 (PDHK1) protein expression is increased, leading to a reduction in pyruvate dehydrogenase (32, 44, 45) activity, and consequently a reduction in oxidative phosphorylation (46).
As it turns out, the classic Warburg model misses a large part of the story. Current perspectives maintain that a balance between aerobic glycolysis and oxidative phosphorylation is much more complex, and seems to be highly variable between different tumor types. Moreover, the balance is not fixed in a given tumor. Rather, the metabolic program is dynamic, and responds to ambient conditions (47). Contrary to Warburg's teachings, mitochondria in cancer are often highly functional, and aerobic respiration is even critical for cancer cell survival (48). A preponderance of new evidence shows that pancreatic cancer cells are especially dependent on mitochondrial oxidative phosphorylation under low nutrient conditions, and that mitochondrial metabolism represents a key metabolic vulnerability (47–51).
Diverse macromolecular substrates are utilized by pancreatic cancer cells for catabolic and anabolic purposes, but glucose is consistently viewed as the most important nutrient. Glucose is even likely more limiting in the microenvironment than oxygen, especially in poorly perfused PDA. Oxygen levels exist in the microenvironment around 1.5% (compared to 21% in the atmosphere and 5% in normal tissues (52). At these low levels, mitochondria still function relatively well. In fact, mitochondria perform sufficiently at oxygen levels as low as 0.5% (53, 54). Glucose concentrations frequently dip below 1 mM in poorly perfused tumors, and these levels are profoundly deleterious. Cell necrosis is the outcome at these levels, even when sufficient oxygen is present (55).
An siRNA screen of 2,752 metabolic genes revealed that mitochondrial genes encoding electron transport chain components were functionally the most important genes in cancer cell survival under low glucose conditions (51). Moreover, when 28 cell lines were evaluated for their ability to withstand low glucose conditions, resistant cell lines consistently increased oxygen consumption on demand, as compared to the most vulnerable cell lines. Further, the vulnerable cell lines exhibited higher rates of genetic mutations in mitochondria-encoded electron transport genes (51). Studies revealed that under low glucose, cell proliferation markedly decreases (49, 56, 57). All of these findings suggest a model where cancer cells that are well-adapted to harsh and unfavorable metabolic conditions prioritize the conservation of glucose, and reprogram their biology to maximize energy yields through enhanced mitochondrial respiration. This metabolic shift ensures the production of sufficient ATP to power critical cellular processes. Under austere conditions, cancer cells choose to divert carbon away from biosynthetic pathways, which minimizes unnecessary ATP utilization. Cell proliferation is deferred for a later time when energy supplies become more available, or the cells become even more adept at scavenging nutrients from a deprived microenvironment (Figure 2).
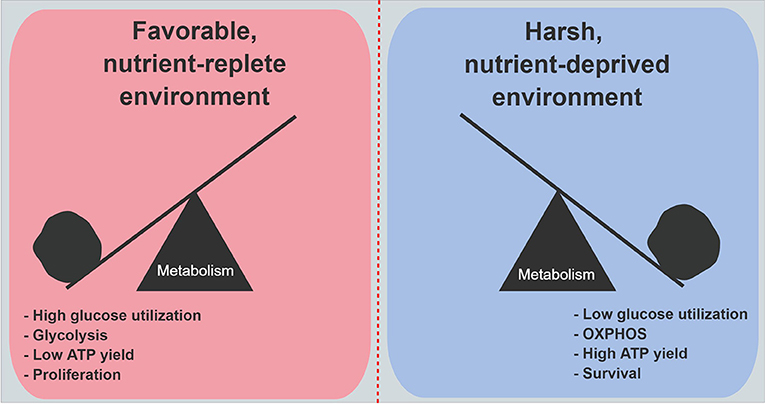
Figure 2. Metabolic features in PDA cells under nutrient abundance and deprivation. Under nutrient abundance, PDA cells have a proliferative phenotype and macromolecular synthesis is prioritized over ATP generation. Under nutrient deprivation, PDA cells have a survival phenotype, and nutrient conservation with maximal ATP generation are prioritized. OXPHOS, oxidative phosphorylation.
A number of bioenergetic observations support this model. As glucose levels decline, cellular respiration markedly increases. The mitochondrial matrix becomes more acidic as protons return to the matrix through ATP synthase, and hydroxide anions are expelled in phosphate/OH− exchangers. This supports ATP production. A burst of oxidative activity is usually observed (49). Morphologically, the matrix condenses, and cristae expand in functional and working mitochondria. Mitochondrial fusion and elongation are favored over fission events (58–60). Inhibition of an important fission-related protein, dynamin-related protein 1 (Drp1), has been shown to shift the balance from mitochondrial fission to fusion under low nutrient conditions (61).
We have recently found that an RNA-binding protein and regulator of acute survival processes, HuR (ELAVL1), plays an important role in increasing mitochondrial performance in stressed PDA cells. This occurs over a very short timescale. We observed that HuR-proficient PDA cells cultured under low glucose conditions exhibited higher rates of oxygen consumption and ATP production, as compared to isogenic HuR-deficient cancer cells (50). Additionally, HuR expression promoted mitochondrial biogenesis (Vaziri-Gohar, unpublished). Consequently, HuR-deficient PDA cells were unable to ramp up mitochondrial activity under metabolic stress, and were especially vulnerable under low glucose conditions in vitro.
Thus, it stands to reason that mitochondrial biology represents a promising therapeutic target in nutrient-deprived cancers (e.g., PDA). There are some clinical studies that support this idea in pancreatic cancer patients. A common diabetic drug, metformin, inhibits complex 1 of the electron transport chain. A meta-analysis of nine retrospective studies and two randomized studies in patients with pancreatic cancer revealed that metformin use was associated with prolonged survival (62–64). Notably, however, the two randomized, phase II studies included in the meta-analysis (both in patients with advanced PDA) were negative studies (63, 64). Prolonged survival was only observed in patients with localized PDA, suggesting that the drug may not be effective in patients with macroscopic distant disease (62). The strategy may be correct, but for improved efficacy, a more potent drug may be required.
More recently, a novel mitochondrial inhibitor was tested in a phase I study of patients with advanced pancreatic cancer. The results were extremely promising. CPI-613 is a lipoic acid analog that disrupts the activity of two mitochondrial enzymes: pyruvate dehydrogenase and α-ketoglutarate dehydrogenase (65, 66). The drug was given to 18 patients at a maximum tolerated dose of 500 mg/m2 on days 1 and 3, of a 2-weeks cycle, and in combination with modified FOLFIRINOX (67). The disease control rate, response rate, and complete response rate were 89, 61, and 17%, respectively. In contrast, the rates for FOLFIRINOX alone in a prior phase III study are 71, 32, and 0.6%, respectively (68). A registration phase III trial is planned to begin shortly as of this manuscript writing, and we are poised to test the same combination in a phase II trial of patients with locally advanced PDA at our institution. These studies are expected to be actively accruing by the start of 2019.
PDA cells also adapt to low nutrient conditions by recruiting unconventional nutrient sources or biochemical salvage pathways to meet bioenergetic demands in a nutrient-deprived microenvironment. There is a small, but important body of literature that highlights some of these biologic processes. Like mitochondrial biology, these non-canonical metabolic pathways represent novel therapeutic opportunities. We have categorized them as intracellular processes (intrinsic) or those dependent on the microenvironment (extrinsic). While some salvage pathways, like nucleotide salvage, are not directly addressed here, we have focused on ones that have received attention in the recent pancreatic cancer literature.
Autophagy is a cytoplasmic recycling process that breaks down dysfunctional organelles and unfolded proteins into their basic components for reuse by cells. Mechanistically, unwanted structures are enwrapped by a double-membrane structure called a phagophore to produce an autophagosome. When the vesicle fuses with a lysosome, the contents are enzymatically digested. Autophagy is driven by starvation-induced activation of AMPK, and is suppressed by mTORC1 in normal cells (69, 70). However, oncogenic KRAS signaling appears to regulate or induce autophagy in PDA cells (71, 72). Conceptually, autophagy functions as an effective nutrient salvage pathway for KRAS-driven PDA, especially when extrinsic nutrient sources are deficient. Cleaved LC3 is an indicator of active autophagy (70, 73) and is increased in late PanIN lesions, as well as in PDA (74). Increased autophagy markers have also been associated with worse prognosis in patients with PDA (75). At the subcellular level, inhibiting autophagy in PDA cells disrupts mitochondrial oxidative phosphorylation and exacerbates oxidative damage (74). Autophagy inhibitors like chloroquine and Bafilomycin A1 have been shown to reduce PDA growth in cell culture and pre-clinical animal models (74, 76).
NADH is an essential cofactor for enzymatic reactions in both glycolysis and respiration. The molecule provides reducing equivalents for the electron transport chain, which drives the proton-motive force to ultimately yield ATP for basic cellular functions. The regeneration of NAD+ as an upstream substrate of NADH production is, therefore, an absolute requirement PDA cell survival, particularly when mitochondrial demands escalate. Tryptophan is the principal source of NAD+ production through canonical biochemical pathways (77). However, PDA cells show increased reliance on a separate NAD+ salvage pathway. In the cancer-associated salvage pathway, nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting step in the production of the NAD+ precursor molecule nicotinamide mononucleotide (NMN). NAMPT expression was found to be elevated in PDA cell lines and tissues (78), and its expression was inversely linked to miR-206 activity (78). Based on these findings, NAMPT is recognized as another promising metabolic target against PDA. Inhibitors like FK866 and STF-118804 reduce NAD+ levels, glycolytic activity, and mitochondrial function. Importantly, these drugs reduced PDA growth both in vitro and in vivo (78–80).
When glucose is scarce, amino acids are able to fuel the tricarboxylic acid cycle through various anaplerotic reactions that involve the conversion of aspartate to oxaloacetate (aspartate transaminase), glutamate to α-ketoglutarate (glutamate dehydrogenase), or alanine to pyruvate (alanine transaminase). Biologic processes exploited by PDA cells to extract these anaplerotic substrates from the microenvironment represent additional metabolic dependencies (81), and consequently are also metabolic vulnerabilities and therapeutic targets in the context of an austere PDA microenvironment.
As with so many other adaptive responses used by PDA cells in the context of severe stress, macropinocytosis is enhanced by oncogenic KRAS (35, 82). In this cellular process, the plasma membrane envelops ambient polypeptides, such as albumin within a macropinosome. Like autophagy, the protein containing vesicle fuses with lysosomes, and proteins are proteolyzed into constituent amino acids (83). Glutamine is the most abundant amino acid released through this process. As a result of macropinocytosis, PDA cells are able to sustain the tricarboxylic acid cycle in the absence of glucose or free glutamine. Targeted inhibition of macropinocytosis with 5-(N-ethyl-N-isopropyl)amiloride (EIPA) in tissue culture models impaired PDA growth (35). In vitro studies revealed that PDA cells experienced sustained viability in the absence of essential amino acids, as long as albumin was present in the media (84). When resected PDA tissues were incubated with tetramethylrhodamine-conjugated dextran (TMR-dextran) for detection purposes, macropinosomes were clearly visualized by immunofluorescence microscopy (84).
While PDA stroma has relatively low levels of free nutrients, the stroma is, in fact, replete with non-neoplastic cell types that are potential fuel sources. Pancreatic stellate cells appear important to PDA cells toward this end. These cells are myofibroblasts that generate the extracellular matrix in the exocrine pancreas and provide a scaffold for desmoplasia in PDA. Studies indicate that pancreatic stellate cells produce alanine by autophagy. Interestingly, PDA cells appear to stimulate this process, possibly through paracrine signaling, although the inducing factor remains unknown. Free alanine excreted by pancreatic stellate cells is imported into PDA cells and converted into pyruvate by alanine transaminase. Alanine anaplerosis than supplies the tricarboxylic acid cycle to sustain it under nutrient stress (85). The authors refer to the communication and interdependence between PDA cells and pancreatic stellate cells as an example of metabolic cross-talk. It is becoming increasingly clear that multi-lineage 3-D models to study metabolic cross-talk in PDA may yield new key insights into additional metabolic vulnerabilities.
In summary, macropinocytosis and metabolic cross-talk between PDA cells and pancreatic stellate cells offer two compelling examples of how PDA cells hijack existing biologic processes or resources from the microenvironment for their own survival advantage. By recruiting these pathways, PDA cells have discovered alternative strategies to fuel the tricarboxylic acid cycle and meet their bioenergetic when the pantry is otherwise bare (Figure 3).
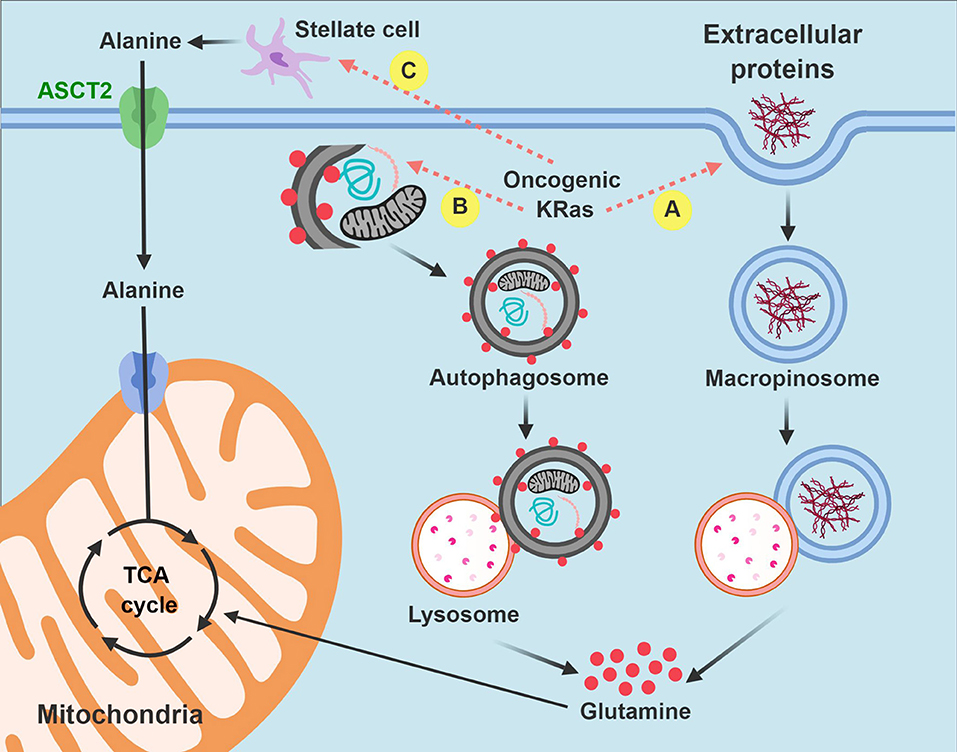
Figure 3. Intrinsic and extrinsic nutrient acquisition pathways in PDA cells. PDA cells hijack stromal elements to fuel the tricarboxylic acid (anaplerosis) and other biochemical processes when nutrients are limited. Carbon is extracted by macropinocytosis (A) and autophagy (B) after auto-digestion by lysosomes. PDA cells also stimulate pancreatic stellate cells to produce and excrete free alanine (C). Non-functional mitochondria are dark in the figure and are targeted for autophagy. Functional mitochondria are colored pink. TCA, tricarboxylic acid.
PDA cells utilize a positive feedback loop between oncogenic KRAS signaling and reactive oxygen species (ROS) to sustain tumor growth (86, 87). This interplay seems to be especially important early in the adenoma-to-carcinoma progression sequence when ROS levels are manageable. ROS stimulates other pro-growth pathways early on in cancer progression as well, like PI3K signaling (88). The generation of genetic mutations by ROS may also play a tumor-promoting role in cancer development (89). The link between ROS and early cancer progression fits with the timing of KRAS mutations, which first appear in PanIN1 lesions. Along these lines, a reduction in mitochondrial ROS using a mitochondrial antioxidant, mitoQ, actually thwarted PanIN formation in an animal model (86).
However, as PDA precursor lesions advance, and invasive PDA matures, the stroma becomes more pronounced, conditions are more severe, and nutrients are in shorter supply. The nutrient-deprived stroma becomes more oxidative under these conditions. More specifically, low glucose conditions drive a surge in ROS levels (50, 90–92), principally because glucose is the main substrate for multiple NADPH-generating pathways. The pentose phosphate pathway and serine biosynthesis with one-carbon metabolism are perhaps the best-studied examples (90). Chemotherapy adds to ROS levels in the PDA microenvironment, which further compounds the oxidative perils routinely faced by these malignant cells (93). Thus, as the dangers of ROS mount, adverse consequences and toxicities associated with ROS start to outstrip any pro-survival benefits favoring tumor growth. Enhanced antioxidant defense mechanisms become paramount to PDA cells for survival (94).
KRAS-driven pancreatic cancer cells bypass the oxidative phase of the pentose phosphate pathway (29). Therefore, alternative NADPH-generating pathways are likely to be important for maintenance of cellular reductive power. There are 13 different metabolic enzymes known to directly interconvert NADP+ to NADPH, and augment the basic reducing currency in cells. These enzymes include: ME (1, 2, and 3), IDH (1 and 2), MTHFD (1 and 2), G6PD, PGD, NNT, GLUD (1 and 2), and ALDH3A1. Reducing equivalents from NADPH maintain glutathione in its reduced form. Glutathione and NADPH collaborate to biochemically prime the remainder of the antioxidant defense system, which consists of roughly 40 enzymes including superoxide dismutases, catalases, glutathione peroxidases, thioredoxins, peroxiredoxins, and glutaredoxins (95).
Son and colleagues demonstrate that malic enzyme 1 (ME1) plays an especially key role in augmenting NADPH levels in PDA. In this non-canonical NADPH biosynthesis pathway, glutamine is converted to glutamate by GLS1 in the mitochondria. Glutamate is generated and transports out of the mitochondria into the cytosol through the malate-aspartate shuttle, where it is converted to cytosolic oxaloacetate (OAA). Mitochondrial and cytosolic aspartate transaminase (GOT2 and GOT1, respectively) are required for this sequence (57). After additional oxidative reactions in the cytosol, ME1 finally yields NADPH. As seen before, oncogenic KRAS appears to influence this adaptive PDA redox program. The authors, and others identified GOT1 and GOT2 as promising therapeutic targets based on these biologic insights (57, 96).
Oncogenic KRAS also induces the transcription of the NRF2 transcription factor (97). NRF2 positively regulates antioxidant defense elements, such as genes that drive glutathione synthesis, glutathione peroxidase, glutathione reductase, glutathione transferases, thioredoxins, and several NADPH-generating enzymes (G6PD, GPD, IDH1, and ME1) (98, 99). Additionally, NAD+ kinase (NADK) was recently identified as an additional component of PDA antioxidant defense. The enzyme converts NAD+ to NADP+, and is upregulated in PDA cells, as compared to normal cells. Silencing NADK increased ROS levels, and also diminished PDA growth in cell lines and in vivo (100).
We recently reported that HuR enhances antioxidant defense through post-transcriptional stabilization of the NADPH-generating enzyme, IDH1 (50). When PDA cells are exposed to an acute oxidative stress, HuR rapidly binds to the 3′-untranslated regions of IDH1 transcripts, stabilizes the transcript, increases IDH1 protein expression and activity, augments NADPH levels, and reduces intracellular ROS (50). The whole process is executed in just a few hours, which enables PDA to respond to acute oxidative stress in short order. Genetic modulation of IDH1 with siRNAs reduced PDA survival under glucose withdrawal more than any other NADPH-generating enzyme (Vaziri-Gohar, unpublished). While IDH1 mutations are oncogenic in other cancer types, our work highlights the importance of wild-type IDH1 in PDA pathogenesis. Moreover, while IDH1 is cytosolic, enhanced reductive power related to this enzyme also appears to impact redox levels in the mitochondria [Vaziri-Gohar, unpublished, and also (101, 102)]. Figure 4 summarizes PDA adaptations that restore redox balance.
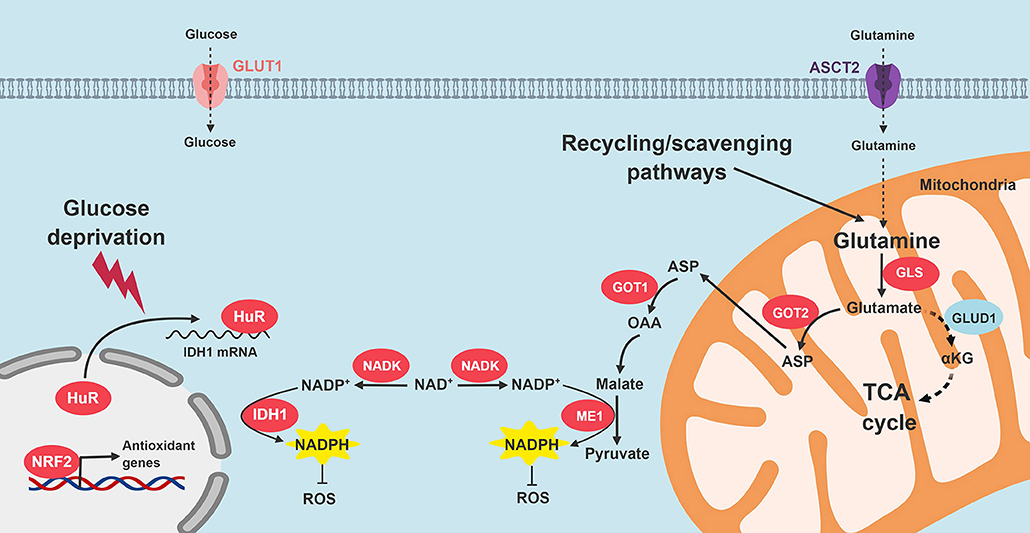
Figure 4. Signaling pathways that restore redox homeostasis in PDA. Nutrient scarcity in the PDA microenvironment augments oxidative stress. Multiple adaptive strategies are recruited by PDA cells. KRAS signaling promotes NADPH production through glutamine catabolism, followed by ME1 activity. In addition, NRF2 and NADK are upregulated. HuR cytoplasmic translocation stabilizes IDH to enhance reductive power. GLS, glutaminase; GOT, aspartate transaminase; ME1, malic enzyme 1; IDH1, isocitrate dehydrogenase 1; TCA, tricarboxylic acid; ROS, reactive oxygen species; NADK, NAD+ kinase.
While the molecular determinants of aggressive PDA biology have not been definitively determined, it seems plausible that the adaptive mechanisms used by PDA cells to overcome the harsh metabolic milieu also contribute to the aggressive and chemotherapy-resistant phenotype responsible for poor patient outcomes. PDA's transformation toward a seemingly invincible state is akin to a runner training at high altitudes or a cactus surviving in a desert. These performance enhanced cells simply cannot be eradicated by conventional DNA targeting agents (i.e., chemotherapy) currently in use. A better understanding of the metabolic dependencies needed to survive harsh conditions will likely uncover metabolic vulnerabilities, and these alternative therapeutic strategies are not likely to be as critical to well-perfused normal cells.
In this review, we highlighted mutant KRAS as an important player in adaptive metabolic reprogramming to the PDA microenvironment. However, new targets or strategies have also come to light, including NRF2, HuR, mitochondrial biology, non-canonical nutrient acquisition processes (autophagy, macropinocytosis, alanine uptake), and antioxidant defense (NADPH-generating enzymes like ME1 and IDH1) (Table 1). There is a growing interest in exploiting new insights into cancer metabolism with good reason. A number of clinical trials targeting metabolic pathways in patients with PDA have been completed or are underway (Table 2).
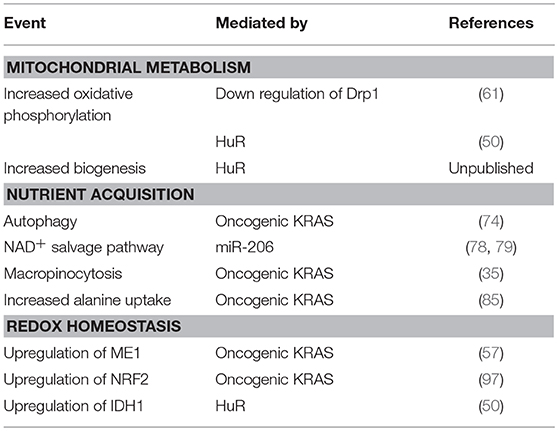
Table 1. Metabolic dependencies in PDA.
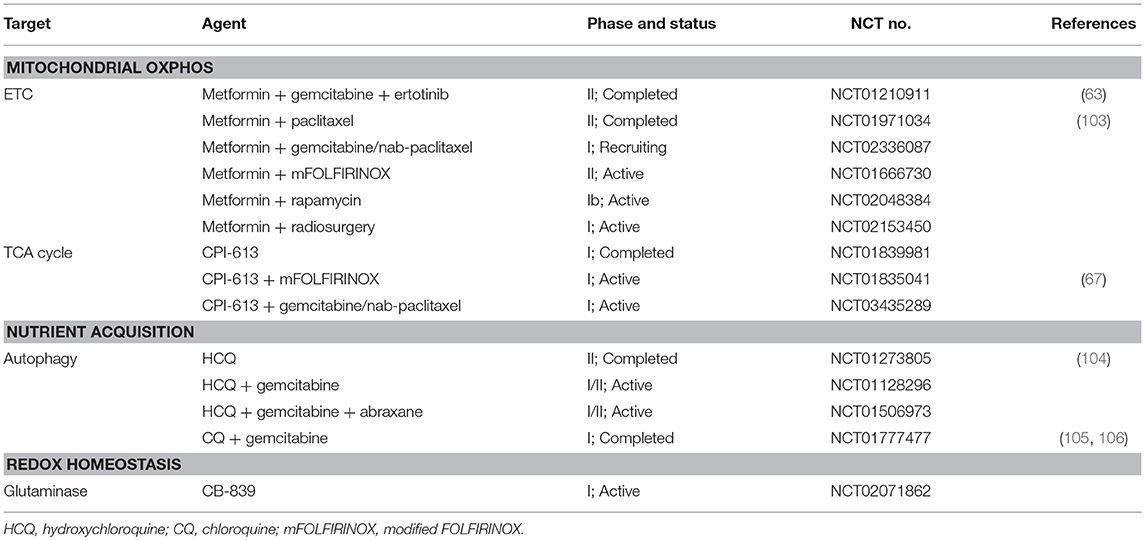
Table 2. Clinical trials targeting key steps of PDA metabolism.
AV-G and JW wrote the manuscript and prepared the figures. MZ and JB critically reviewed the manuscript. AV-G and JW approved the final manuscript.
This work was supported by American Cancer Society Mentored Research Scholar Grant-14-019-01-CDD (JW), R01 CA212600 (JB and JW), and 1R37CA227865 (JW and JB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ALDH3A1, aldehyde dehydrogenases; ASCT2, alanine-serine-cysteine transporter 2; dCTP, deoxycytidine triphosphate; Drp1, dynamin-related protein 1; EIPA, 5-(N-ethyl-N-isopropyl)amiloride; G6PD, glucose-6-phosphate dehydrogenase; GLUD, glutamate dehydrogenase; GLUT1, glucose transporter 1; HIF-1α, hypoxia-inducible factor-1a; IDH, isocitrate dehydrogenase; LDH, lactate dehydrogenase; ME, malic enzyme; MTHFD, methylenetetrahydrofolate dehydrogenase; NAD+, oxidized nicotinamide adenine dinucleotide; NADK, NAD+ kinase; NADP+, oxidized nicotinamide adenine dinucleotide phosphate; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NAMPT, nicotinamide phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NNT, nicotinamide nucleotide transhydrogenase; NRF2, nuclear factor erythroid 2 related factor 2; OAA, oxaloacetate; OXPHOS, oxidative phosphorylation; PanIN, pancreatic intraepithelial neoplasia; PDA, pancreatic ductal adenocarcinoma; PDHK1, pyruvate dehydrogenase kinase 1; PGD, phosphogluconate dehydrogenase; PPP, pentose phosphate pathway; ROS, reactive oxygen species; shRNA, short hairpin RNA; TCA, tricarboxylic acid.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Morgan G, Ward R, Barton M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin Oncol (R Coll Radiol). (2004) 16:549–60. doi: 10.1016/j.clon.2004.06.007
4. Van Cutsem E, Verslype C, Grusenmeyer PA. Lessons learned in the management of advanced pancreatic cancer. J Clin Oncol. (2007) 25:1949–52. doi: 10.1200/JCO.2006.09.4664
5. Ren C, Chen H, Han C, Jin G, Wang D, Tang D. Detection and molecular analysis of circulating tumor cells for early diagnosis of pancreatic cancer. Med Hypotheses (2013) 80:833–6. doi: 10.1016/j.mehy.2013.03.027
7. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet (2011) 378:607–20. doi: 10.1016/S0140-6736(10)62307-0
8. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet (2004) 363:1049–57. doi: 10.1016/S0140-6736(04)15841-8
9. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. (2013) 369:1691–703. doi: 10.1056/NEJMoa1304369
10. Yachida S, Iacobuzio-Donahue CA. Evolution and dynamics of pancreatic cancer progression. Oncogene (2013) 32:5253–60. doi: 10.1038/onc.2013.29
11. Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. (2016) 6:166–75. doi: 10.1158/2159-8290.CD-15-0402
12. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature (1999) 400:464–8. doi: 10.1038/22780
13. Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. (2006) 103:5947–52. doi: 10.1073/pnas.0601273103
14. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. (2002) 19:607–14. doi: 10.1002/humu.10081
15. Tuveson DA, Hingorani SR. Ductal pancreatic cancer in humans and mice. Cold Spring Harb Symp Quant Biol. (2005) 70:65–72. doi: 10.1101/sqb.2005.70.040
16. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
17. Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell (2002) 2:25–8. doi: 10.1016/S1535-6108(02)00093-4
18. Winter JM, Maitra A, Yeo CJ. Genetics and pathology of pancreatic cancer. HPB (2006) 8:324–36. doi: 10.1080/13651820600804203
19. Yabar CS, Winter JM. Pancreatic cancer: a review. Gastroenterol Clin North Am. (2016) 45:429–45. doi: 10.1016/j.gtc.2016.04.003
20. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. (2006) 20:1218–49. doi: 10.1101/gad.1415606
21. Murthy D, Attri KS, Singh PK. Phosphoinositide 3-kinase signaling pathway in pancreatic ductal adenocarcinoma progression, pathogenesis, and therapeutics. Front Physiol. (2018) 9:335. doi: 10.3389/fphys.2018.00335
22. Ndlovu R, Deng LC, Wu J, Li XK, Zhang JS. Fibroblast growth factor 10 in pancreas development and pancreatic cancer. Front Genet. (2018) 9:482. doi: 10.3389/fgene.2018.00482
23. Sato N, Goggins M. The role of epigenetic alterations in pancreatic cancer. J Hepatobiliary Pancreat Surg. (2006) 13:286–95. doi: 10.1007/s00534-005-1057-1
24. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. (2012) 18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114
25. Erkan M, Reiser-Erkan C, Michalski CW, Kleeff J. Tumor microenvironment and progression of pancreatic cancer. Exp Oncol. (2010) 32:128–31.
26. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
27. Xie K, Huang S. Regulation of cancer metastasis by stress pathways. Clin Exp Metastasis (2003) 20:31–43. doi: 10.1023/A:1022590402748
28. Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science (1988) 240:177–84.
29. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149:656–70. doi: 10.1016/j.cell.2012.01.058
30. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
31. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30.
32. Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezène P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci USA. (2013) 110:3919–24. doi: 10.1073/pnas.1219555110
33. Mikuriya K, Kuramitsu Y, Ryozawa S, Fujimoto M, Mori S, Oka M, et al. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int J Oncol. (2007) 30:849–55. doi: 10.3892/ijo.30.4.849
34. Akakura N, Kobayashi M, Horiuchi I, Suzuki A, Wang J, Chen J, et al. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. (2001) 61:6548–54.
35. Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature (2013) 497:633–7. doi: 10.1038/nature12138
36. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. (2014) 20:4370–80. doi: 10.1158/1078-0432.CCR-14-0186
37. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci. (2016) 73:1333–48. doi: 10.1007/s00018-015-2098-5
38. Hume DA, Weidemann MJ. Role and regulation of glucose metabolism in proliferating cells. J Natl Cancer Inst. (1979) 62:3–8.
39. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
40. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, et al. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. (1999) 274:20281–6.
41. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. (2013) 9:712. doi: 10.1038/msb.2013.65
42. Ju HQ, Ying H, Tian T, Ling J, Fu J, Lu Y, et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat Commun. (2017) 8:14437. doi: 10.1038/ncomms14437
43. Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA. (2012) 109:13787–92. doi: 10.1073/pnas.1203339109
44. Brahimi-Horn MC, Chiche J, Pouysségur J. Hypoxia signalling controls metabolic demand. Curr Opin Cell Biol. (2007) 19:223–9. doi: 10.1016/j.ceb.2007.02.003
45. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) 3:177–85. doi: 10.1016/j.cmet.2006.02.002
46. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
47. Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma? Biochim Biophys Acta (2011) 1807:552–61. doi: 10.1016/j.bbabio.2010.10.012
48. Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (Review). Oncol Lett. (2012) 4:1151–7. doi: 10.3892/ol.2012.928
49. Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. (2004) 64:985–93. doi: 10.1158/0008-5472.CAN-03-1101
50. Zarei M, Lal S, Parker SJ, Nevler A, Vaziri-Gohar A, Dukleska K, et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. (2017) 77:4460–71. doi: 10.1158/0008-5472.CAN-17-0015
51. Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature (2014) 508:108–12. doi: 10.1038/nature13110
52. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol. (2014) 87:20130676. doi: 10.1259/bjr.20130676
53. Rumsey WL, Schlosser C, Nuutinen EM, Robiolio M, Wilson DF. Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J Biol Chem. (1990) 265:15392–402.
54. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. (2015) 11:9–15. doi: 10.1038/nchembio.1712
55. Schroeder T, Yuan H, Viglianti BL, Peltz C, Asopa S, Vujaskovic Z, et al. Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Res. (2005) 65:5163–71. doi: 10.1158/0008-5472.CAN-04-3900
56. Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. (2013) 10:1312–23. doi: 10.4161/rna.25274
57. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496:101–5. doi: 10.1038/nature12040
58. Gao AW, Cantó C, Houtkooper RH. Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med. (2014) 6:580–9. doi: 10.1002/emmm.201303782
59. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
60. Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes (2009) 58:2303–15. doi: 10.2337/db07-1781
61. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. (2011) 108:10190–5. doi: 10.1073/pnas.1107402108
62. Li X, Li T, Liu Z, Gou S, Wang C. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep. (2017) 7:5825. doi: 10.1038/s41598-017-06207-x
63. Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. (2015) 16:839–47. doi: 10.1016/S1470-2045(15)00027-3
64. Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R, et al. (Ir)relevance of metformin treatment in patients with metastatic pancreatic cancer: an open-label, randomized phase II trial. Clin Cancer Res. (2016) 22:1076–85. doi: 10.1158/1078-0432.CCR-15-1722
65. Lycan TW, Pardee TS, Petty WJ, Bonomi M, Alistar A, Lamar ZS, et al. A phase II clinical trial of CPI-613 in patients with relapsed or refractory small cell lung carcinoma. PLoS ONE (2016) 11:e0164244. doi: 10.1371/journal.pone.0164244
66. Egawa Y, Saigo C, Kito Y, Moriki T, Takeuchi T. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS ONE (2018) 13:e0198940. doi: 10.1371/journal.pone.0198940
67. Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. (2017) 18:770–8. doi: 10.1016/S1470-2045(17)30314-5
68. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
69. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. (2009) 43:67–93. doi: 10.1146/annurev-genet-102808-114910
70. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell (2010) 140:313–26. doi: 10.1016/j.cell.2010.01.028
71. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. (2010) 12:814–22. doi: 10.1038/ncb0910-814
72. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer (2012) 12:401–10. doi: 10.1038/nrc3262
73. Gohar AV, Cao R, Jenkins P, Li W, Houston JP, Houston KD. Subcellular localization-dependent changes in EGFP fluorescence lifetime measured by time-resolved flow cytometry. Biomed Opt Express. (2013) 4:1390–400. doi: 10.1364/BOE.4.001390
74. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. (2011) 25:717–29. doi: 10.1101/gad.2016111
75. Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. (2008) 99:1813–9. doi: 10.1111/j.1349-7006.2008.00893.x
76. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. (2011) 25:460–70. doi: 10.1101/gad.2016311
77. Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol. (1999) 73:135–82, xi.
78. Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. (2016) 379:1–11. doi: 10.1016/j.canlet.2016.05.024
79. Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. (2014) 20:120–30. doi: 10.1158/1078-0432.CCR-13-0150
80. Espindola-Netto JM, Chini CCS, Tarragó M, Wang E, Dutta S, Pal K, et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget (2017) 8:85054–67. doi: 10.18632/oncotarget.18841
81. Sidaway P. Pancreatic cancer: pancreatic cancer cells digest extracellular protein. Nat Rev Clin Oncol. (2017) 14:138. doi: 10.1038/nrclinonc.2017.3
82. Perera RM, Bardeesy N. Pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov. (2015) 5:1247–61. doi: 10.1158/2159-8290.CD-15-0671
83. Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science (1986) 233:1061–8.
84. Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. (2015) 75:544–53. doi: 10.1158/0008-5472.CAN-14-2211
85. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536:479–83. doi: 10.1038/nature19084
86. Liou GY, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, et al. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. (2016) 14:2325–36. doi: 10.1016/j.celrep.2016.02.029
87. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. (2010) 107:8788–93. doi: 10.1073/pnas.1003428107
88. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. (2014) 2:17. doi: 10.1186/2049-3002-2-17
89. Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. (1994) 54(7 Suppl.):1890s−4s.
90. Khramtsov VV, Gillies RJ. Janus-faced tumor microenvironment and redox. Antioxid Redox Signal. (2014) 21:723–9. doi: 10.1089/ars.2014.5864
91. Ahmad IM, Aykin-Burns N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, et al. Mitochondrial O- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem. (2005) 280:4254–63. doi: 10.1074/jbc.M411662200
92. Chio IIC, Tuveson DA. ROS in cancer: the burning question. Trends Mol Med. (2017) 23:411–29. doi: 10.1016/j.molmed.2017.03.004
93. Sangeetha P, Das UN, Koratkar R, Suryaprabha P. Increase in free radical generation and lipid peroxidation following chemotherapy in patients with cancer. Free Rad Biol Med. (1990) 8:15–9.
94. Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res. (2000) 60:6201–7.
95. Policastro LL, Ibanez IL, Notcovich C, Duran HA, Podhajcer OL. The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal. (2013) 19:854–95. doi: 10.1089/ars.2011.4367
96. Yang S, Hwang S, Kim M, Seo SB, Lee JH, Jeong SM. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. (2018) 9:55. doi: 10.1038/s41419-017-0089-1
97. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature (2011) 475:106–9. doi: 10.1038/nature10189
98. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. (2014) 39:199–218. doi: 10.1016/j.tibs.2014.02.002
99. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. (2017) 29:1727–45. doi: 10.1089/ars.2017.7342
100. Tsang YH, Dogruluk T, Tedeschi PM, Wardwell-Ozgo J, Lu H, Espitia M, et al. Functional annotation of rare gene aberration drivers of pancreatic cancer. Nat Commun. (2016) 7:10500. doi: 10.1038/ncomms10500
101. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. (2012) 48:158–67. doi: 10.1016/j.molcel.2012.09.025
102. Ciccarese F, Ciminale V. Escaping death: mitochondrial redox homeostasis in cancer cells. Front Oncol. (2017) 7:117. doi: 10.3389/fonc.2017.00117
103. Braghiroli MI, de Celis Ferrari AC, Pfiffer TE, Alex AK, Nebuloni D, Carneiro AS, et al. Phase II trial of metformin and paclitaxel for patients with gemcitabine-refractory advanced adenocarcinoma of the pancreas. Ecancermedicalscience (2015) 9:563. doi: 10.3332/ecancer.2015.563
104. Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist (2014) 19:637–8. doi: 10.1634/theoncologist.2014-0086
105. Balic A, Sørensen MD, Trabulo SM, Sainz B, Cioffi M, Vieira CR, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. (2014) 13:1758–71. doi: 10.1158/1535-7163.MCT-13-0948
Keywords: pancreatic cancer, metabolism, redox homeostasis, metabolic dependencies, targeting metabolism
Citation: Vaziri-Gohar A, Zarei M, Brody JR and Winter JM (2018) Metabolic Dependencies in Pancreatic Cancer. Front. Oncol. 8:617. doi: 10.3389/fonc.2018.00617
Received: 11 October 2018; Accepted: 29 November 2018;
Published: 12 December 2018.
Edited by:
Ramon Bartrons, University of Barcelona, SpainReviewed by:
Vincenzo Ciminale, Università degli Studi di Padova, ItalyCopyright © 2018 Vaziri-Gohar, Zarei, Brody and Winter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordan M. Winter, am9yZGFuLndpbnRlckBVSGhvc3BpdGFscy5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.