 Vivien Chan
Vivien Chan Alessandro Marro2
Alessandro Marro2 Sumit Das
Sumit Das
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol., 28 November 2018
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00567
Background: Atypical teratoid/rhabdoid tumor in adults is a relatively rare malignant neoplasm. It is characterized by the presence of rhabdoid cells in combination with loss of either the INI1 or BRG1protein from the tumor cells.
Methods: A systematic review was conducted using MEDLINE using the terms “atypical teratoid rhabdoid tumor” AND “adult.” The systematic review was supplemented with relevant articles from the references. Cases were included if the pathology was confirmed by loss of INI1 or BRG1. We included a case from our institution. The dataset was analyzed using descriptive statistics and log-rank test.
Results: A total of 50 cases from 29 articles were included in this study. The average age at diagnosis was 36.7 years. The most common locations reported are the sellar region and cerebral hemispheres (without deep gray matter involvement). Of the 50 cases, 14 were reported to show evidence of dissemination. The average overall survival was 20 months. There was a significant difference in survival between the adjuvant therapy groups (p = < 0.0001).
Conclusion: Atypical teratoid rhabdoid tumor of the central nervous system in adults is a rare neoplasm associated with a poor prognosis in a majority of patients. The treatment and clinical course are highly variable, and it remains unclear which factors impact prognosis.
Atypical teratoid/rhabdoid tumor (AT/RT) of the central nervous system is a highly malignant neoplasm (1). Although this tumor typically affects children younger than 3 years of age, it has been described in adults (1, 2). Clinical presentation can vary depending on the patient's age, location and size of tumor. In adult patients, the most common locations are cerebral hemisphere and sellar region (3). The most characteristic histological feature is the presence of rhabdoid cells; however, this alone is not sufficient for the diagnosis of AT/RT (4). The 2016 WHO Classification of Tumors of the Central Nervous System defined AT/RT by alterations of either INI1 protein (SMARCB1 gene), or rarely, BRG1 protein (SMARCA4 gene) (4).
Case reports and case series report an overall poor prognosis, with an average survival of 20 months (5). However, there are cases of adult patients with AT/RT who have survived beyond 17 years from diagnosis (6). Currently, management of AT/RT in adults is based on data extrapolated from pediatric literature (7). Little is known on the optimal management of adult patients with AT/RT (7). Given the limited number of AT/RT cases in adults and the dispersal of these cases in multiple case reports and case series, patient and tumor characteristics, overall prognosis, and impact of extent of resection and adjuvant therapy remains unclear in this patient population. In addition, other previously published systematic reviews and meta-analysis have included tumors that do not have INI1 or BRG1 alterations, resulting in an analysis of a heterogeneous population that may contain tumors that are not molecularly defined as AT/RT (7–14).
This study aimed to systematically review and analyze patient and tumor characteristics, prognosis, and impact of treatment on prognosis in adult patients with AT/RT.
The objective of this study was to report the results of a systematic review of the literature for a pooled analysis of all adult cases of AT/RT confirmed by alterations in INI1 or BRG1. This is the first systematic review to only include cases of AT/RT confirmed with either INI1 or BRG1 alterations.
From reviewing all cases of AT/RT in adult patients with confirmed genetic mutation, what are the patient and tumor characteristics and how do extent of resection and adjuvant therapy impact prognosis?
We report this systematic review in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines.
We included all articles with cases of AT/RT in adult patients with the neuropathologic diagnosis confirmed by alterations of either SMARCB1/INI1 or SMARCA4/BRG1 by immunohistochemistry and/or molecular studies. A case of ATRT in an adult patient from our institution was included in the pooled analysis.
We developed a protocol that had pre-specified objectives, eligibility criteria, data of interest, search strategy, and analyses plan.
We conducted a systematic review using MEDLINE. We restricted results to articles written in English only. There was no limitation on the date of publication; we reviewed all articles published prior to August 2018. Search terms included “Atypical teratoid rhabdoid tumor” and “adult.” References from relevant articles were used to supplement the systematic review.
Duplicate publications were removed. The title and abstracts were reviewed to identify potentially eligible articles. Full texts of potentially eligible articles were obtained and reviewed for eligibility. Data was extracted from eligible articles.
We collected data on patient demographics, tumor characteristics, survival, and treatment. A case from our institution was included in our analysis. The data was analyzed using descriptive statistics. Log-rank test was used to assess for differences in outcomes between those that received gross total resection, incomplete resection, and biopsy. Log-rank test was used to assess for differences in outcomes between those that received radiotherapy, radiotherapy and chemotherapy, and no adjuvant therapy. Kaplan-Meier curves were used to estimate the survival function.
A total of 152 results were found from the MEDLINE search (Figure 1). Of the 152 results, 24 articles were deemed relevant to this study. Of these 24 articles, one was removed because the case was previously reported in another article included in this study and 8 were removed because loss of SMARCB1/INI1 or SMARCA4/BRG1 was not present or confirmed. The systematic review was supplemented by the references cited in the 24 relevant articles. From the references cited, 35 relevant articles were identified. Of the 35, 11 were removed because loss of SMARCB1/INI1or SMARCA4/BRG1 was not present or confirmed. A total of 39 articles were identified to be relevant to this study, 15 from the MEDLINE search and 24 from references cited.
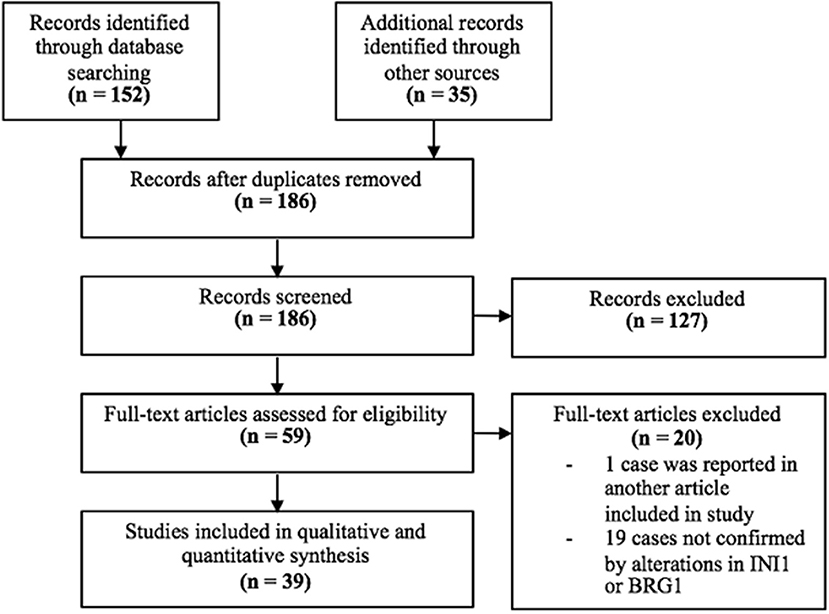
Figure 1. PRISMA Flow Diagram.
From our systematic review, we found 50 adult patients who were diagnosed with AT/RT from the 39 articles included in this study (Table 1). The Supplementary Table 1 will include all the patients included in the pooled analysis (1, 2, 4–9, 11–42). Of the 50 patients, the average age was 36.69 (SD = 15.19) years with range from 18 to 59 years. Of the 39 patients, 34 (68%) were female, 15 (30%) were male, and 1 (2%) was not identified. Signs and symptoms were related to location of the tumor. The most common symptom was headache (n = 20, 40%), followed by cranial nerve deficit (n = 17, 34%) and visual disturbance (n = 14, 28%). Average time with symptoms before surgery was 5.27 (SD = 12.72) months. The range was 0–60 months.
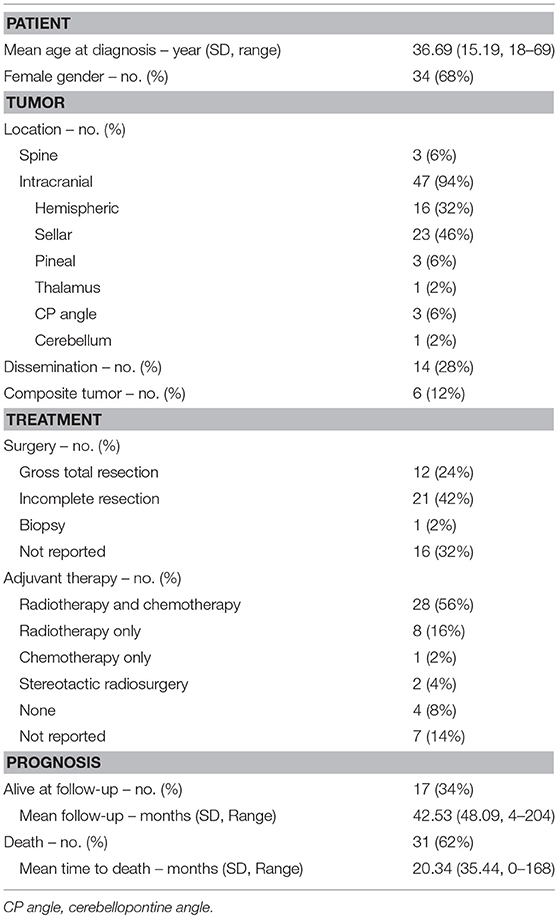
Table 1. Patient and tumor characteristics from systematic review.
Only 3 (6%) were in the spinal cord while the remaining 47 (94%) were intracranial. The most common intracranial location was the sellar region (n = 23, 46%), followed by cerebral hemispheres (n = 16, 32%). Other reported locations were pineal region (n = 3, 6%), cerebellopontine angle (n = 3, 6%), cerebellum (n = 1, 2%), and, from our case report, thalamus. Over the entire course of the disease, 14 (28%) patients experienced dissemination of disease. 2 (4%) patients were reported to have evidence of dissemination at the time of diagnosis. In 6 (12%) patients, the tumor was a composite tumor involving components of AT/RT and another primary CNS tumor.
Of the 50 cases, 12 (24%) had gross total resection, 21 (42%) had subtotal or partial resection, and1 (2%) had a biopsy. Extent of resection was not reported in 17 cases. With regards to adjuvant therapy, 28 (56%) received combined radiotherapy and chemotherapy, 8 (16%) received radiotherapy only, 1 (2%) received chemotherapy only, 2 (4%) received stereotactic radiosurgery, and 4 (8%) did not receive adjuvant therapy. Of the 36 patients that received radiotherapy, 9 received craniospinal irradiation. Of the 29 patients that receive chemotherapy, 6 received high-dose chemotherapy. Of the 28 patients who received chemotherapy and radiotherapy, 5 patients received both craniospinal irradiation and high-dose chemotherapy. Of those that received adjuvant therapy, 2 patients' death could be attributed to complications and side effects of the therapy.
Of the 50 patients, 31 (62%) had succumbed to their disease with an average time to death of 20.34 (SD = 35.44) months. The range was 0 to 168 months. 17 (34%) were alive at last follow-up with a mean follow-up of 42.53 (SD = 48.41) months and median follow-up of 28 months (range 4 to 204 months). The range was 4 to 204 months. Of the 17 patients, 11 had no evidence of recurrence at follow-up, with follow-up ranging from 4 to 54 months. Of the 50 patients, 24 patients had reported time to recurrence. The average time to recurrence was 12.4 (SD = 16.97) months and median time to recurrence was 5.5 months.
Of the 12 that had gross total resection, 5 (42%) had passed away with time to death ranging from 3 months to 14 years after surgery. Of the 21 that had incomplete resection, 12 (57%) had passed away with time to death ranging from postoperative to 2.5 years after surgery. The 1 patient that had a biopsy died 2.1 years after diagnosis. When comparing those that received gross total resection, incomplete resection, and biopsy, there was no significant difference on the log-rank test (Chi-square = 3.12, p = 0.21). The Kaplan-Meier curve is shown in Figure 2.
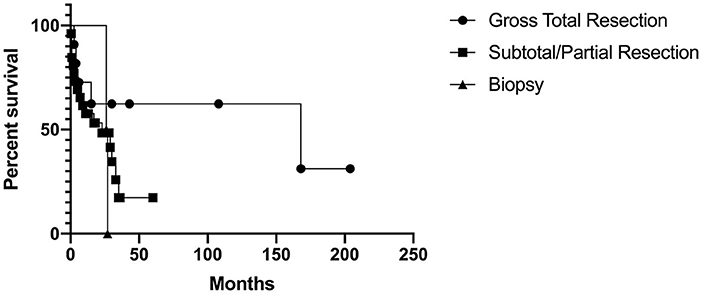
Figure 2. Kaplan-Meier curve for extent of resection.
Of the 28 patients that received combined radiotherapy and chemotherapy, 15 were alive at follow-up, ranging from 6 months to 17 years. Time to death for the remaining 13 of these 28 ranged from 3 months to 3 years after diagnosis. There were no patients alive at follow-up in the radiotherapy only, chemotherapy only, stereotactic radiosurgery only, and no adjuvant therapy groups. The 8 patients treated with radiotherapy died 2 weeks to 14 years after diagnosis. The 1 patient who received chemotherapy died 10 years after diagnosis. The two patients treated with stereotactic radiosurgery died 23 and 27 months after diagnosis. Of the 4 patients who did not receive adjuvant therapy, time to death ranged from the immediate postoperative period to 3 months after surgery. When comparing those that received radiotherapy and chemotherapy, radiotherapy only, and no adjuvant therapy, there was a significant difference in survival (Chi-square = 20.38, p = < 0.0001). Patients that received radiotherapy and chemotherapy had a significant increase in survival when compared with patients that received radiotherapy alone (Chi-square = 11.42, p = 0.0007) and patients that did not receive adjuvant therapy (Chi-square = 25.71, p = < 0.0001). There was no significant difference between The Kaplan-Meier curve is shown in Figure 3.
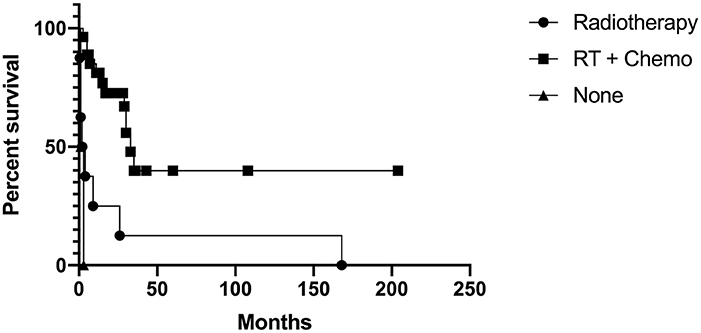
Figure 3. Kaplan-Meier curve for those that received radiotherapy and chemotherapy, radiotherapy only, and no adjuvant therapy.
The studies included in this systematic review were case reports and case series. The risk of bias could not be assessed using the Cochrane Collaboration's tool for assessing risk of bias.
In accordance with the 2016 WHO Classification of Tumors of the Central Nervous System, we included only cases with confirmed mutation of INI1. Approximately 20–25% of cases have homozygous deletions of INI1 (43, 44). The majority have a mutation in one allele with a second allele lost due to monosomy 22, deletion of 22q11.2, or an acquired copy number neutral loss heterozygosity (43, 44). Reported mutation hotspots are exons 5 and 9 (5, 43). Our review did not reveal any cases of confirmed BRG1 alteration.
Ostrom et al. reported the most common location for adults to be the cerebral hemisphere, followed by the sellar region (3). In contrast, our review of published cases found the most common location to be the sellar region (46%), followed by cerebral hemisphere (32%). It has previously been suggested that sellar AT/RT represent a different subgroup given its distinct clinicopathological and genetic features (12, 5). Sellar AT/RTs were reported to be found almost exclusively in females and not in the pediatric population (5, 15). Our study found 21 of 23 cases of sellar AT/RT to occur in female patients, suggesting, although much less common, they can occur in males as well. In contrast, male-female ratio for non-sellar AT/RTs in our study were roughly even (46% female, 54% male). Sellar AT/RTs also seem to have a higher prevalence of biallelic INI1 alterations compared to AT/RT in other locations (5). A recent study, however, argues that sellar AT/RT does not form its own molecular subgroup, but can be clustered into the ATRT-MYC subgroup, one of the three recently described molecular subgroups (16, 45). However, AT/RT in the sellar location appears to have a different mutational spectrum than other tumors in the ATRT-MYC subgroup. Cases of AT/RT concurrent with other central nervous system tumors, such as pleomorphic xanthoastrocytoma (PXA), glioma, ependymoma, and prolactinoma have also been described (13, 17–21). Nobusawa et al. reported a case of a tumor with biphasic appearance consistent with histological features of ependymoma and AT/RT (17). C11 ORF95-RELA fusion, a genetic marker of supratentorial ependymomas, was present in both ependymoma and AT/RT components. However, only the AT/RT component had loss of nuclear expression of INI1. Similarly, Chacko et al. reported a composite tumor with features of AT/RT and PXA (18). The PXA component was GFAP positive. Loss of INI1 was seen in the AT/RT component, but retained in the PXA component. It is hypothesized that a postclonal inactivation of INI1 exists in a subset of the original tumor cells, leading to a composite tumor with features of AT/RT and another central nervous system tumor (17–21). The clinical significance of a concurrent CNS tumor with ATRT is unclear given the paucity of data in the literature.
The propensity for AT/RT to spread through the subarachnoid space is well known (6). In our study, only 2 patients were reported to have evidence of dissemination at the time of diagnosis. However, this value is likely an underestimation since most patients did not receive whole neuraxis imaging at the time of diagnosis. We found 28% of patients had dissemination outside of the original tumor site over the course of their disease. This figure is consistent with previously reported values (46–48). Dissemination is usually within the central nervous system; however, distant metastasis to the lung has been described (22). Even with gross total resection and adjuvant therapy, there are cases of distant metastasis at time of recurrence (19, 23–25). In our pooled analysis, of the 12 cases with gross total resection, we found 4 with dissemination. Although dissemination has been typically associated with poor prognosis, overall outcome still seems to be highly variable with some patients doing well with whole neuraxis radiotherapy and chemotherapy despite presence of dissemination at the time of diagnosis (46). In our study, we found variable outcomes in those that received whole neuraxis radiotherapy, as well as those that received high-dose chemotherapy. In a large majority of the cases that received whole neuraxis radiotherapy or high-dose chemotherapy, these measures were undertaken after discovery of rapid recurrence or new metastatic lesions. Therefore, it is difficult to draw conclusions on the impact of whole neuraxis radiation and high-dose chemotherapy on the course of the disease.
There are clinical differences and similarities between the pediatric population and the adult population. In the pediatric population, there is a higher incidence in males. In a national retrospective study, 62% of reported pediatric cases occurred in males (49). This is contrast to our study which report 32% of the patients were male. In the same study, half of the patients had tumors in the infratentorial location. Whereas, in adult patients the most common locations are sellar and hemispheric. AT/RT carries a poor prognosis in both the adult and pediatric populations. In our study, we report an average survival of 20 months. This is consistent with findings reported in other studies (5, 50, 51). This is comparable to the reported median survival of 13.5 to 16.8 months in the pediatric population (49, 50). In both adult and pediatric populations, the prognosis can be highly variable. In the study by Hilden et al. on pediatric patients with AT/RT, 33% had no evidence of disease at follow-up, with a follow-up range of 9.5–96 months (50). Similarly, in our study 22% had no evidence of disease at last follow-up, with follow-up ranging from 4 to 60 months. Median time to recurrence in the pediatric population was the same as the reported value for adults in this study (49). In the pediatric population, 38% had metastatic disease during the course of their disease (49). This is comparable to the 28% reported in this study. Interestingly, in both pediatric and adult populations metastatic disease was not prognostic.
Radiological findings of AT/RT in adult patients are similar to those reported in pediatric cases (10, 52–55). A study by Kanoto et al. on adult AT/RT found 91% were hyperattenuated on CT, 80% were hypointense on T1-weighted imaging, 72% were mixed-intensity on T2-weighted imaging, 100% had restricted diffusion, 70% had heterogeneous enhancement, 56% had cyst/necrosis, 42% had hemorrhage and 33% had calcifications (56). The high density on CT and diffusion restriction on MRI is suggestive of high cell density (56, 57). The ADC value and CT density are similar to lymphoma, germinoma, and what was previously termed primitive neuroectodermal tumor (58–60).
From our analysis, there was no difference in survival between those that received gross total resection, incomplete resection, and biopsy. Based on our analysis, it remains unclear whether extent of resection impacts survival. Current adjuvant therapy treatment for AT/RT has been extrapolated from pediatric literature, which has shown poor response to adjuvant therapy (7, 47, 48). In our analysis, 15 of 16 patients that were alive at follow-up received chemotherapy and radiation; it was unknown whether the remaining patient received chemotherapy or radiotherapy. There were no patients alive at follow-up in the radiotherapy only, chemotherapy only, stereotactic radiosurgery (SRS) only, and no adjuvant therapy groups. In the no adjuvant therapy group, time to death ranged from postoperative to 3 months. Given the small number of patients in the chemotherapy only and radiosurgery only group and the large range in time to death in the radiotherapy only group (2 weeks to 14 years), it remains unclear how adjuvant therapy impacts survival. In our analysis, we found patients who received chemotherapy and radiotherapy had increased survival. However, survival associated with adjuvant therapy may be impacted by confounding factors. Most of the patients received chemotherapy and radiation unless the patient refused therapy, was too functionally impaired, or the patient succumbed due to rapidly progressive disease. The confounding factor in this context may therefore be the health and/or functional status of patients as only otherwise healthy patients or patients who were less functionally impaired received both chemotherapy and radiation while others may have only received chemotherapy only, radiotherapy only, radiosurgery only, or no adjuvant therapy.
One of the limitations of this study is the limited ability to draw robust conclusions due to the small study population and confounding factors. It is difficult to conclude from this study whether extent of resection or adjuvant therapy provides a real survival benefit or whether there were confounding factors present. Another limitation is the amount of data available in case series and case reports. Although most case series and case reports contain a large amount of specific detail such as patient factors, tumor characteristics, pathological characteristics, treatment, and prognosis, the content of the data is inconsistent from one case series/report to another, which prevented more rigorous statistical analysis, such as regression analysis, and clinically relevant analysis, such as event-free survival, from being done.
AT/RT in adults is a rare malignant neoplasm of the central nervous system diagnosed based on alterations of either INI1 or BRG1. The prognosis remains poor, with an average survival of less than 2 years. There is however a subset of patients who seem to have a much longer survival after repeat resections and adjuvant therapy. Although the sellar region and cerebral hemispheres tend to be the most common locations for AT/RT in adults, our systematic review demonstrate that AT/RT can also occur in less well known locations. From our systematic review, extent of resection may not be an important factor in prognosis, but adjuvant therapy may have an impact on prognosis. However, it is difficult to draw conclusions given the small population and confounding factors. Case reports and systematic reviews of rare malignant neoplasms remain an important component of literature in oncology as it provides information that may reveal clinicopathological patterns and factors that impact prognosis.
All datasets generated for this study are included in the manuscript and the supplementary files.
VC contributed to design of the study, systematic review for creation of dataset, statistical analysis, and writing the first draft of the manuscript. AM and SD contributed to writing the first draft of the manuscript. LS, JF, and SD contributed to conception of the study. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00567/full#supplementary-material
1. Horiguchi H, Nakata S, Nobusawa S, Uyama S, Miyamoto T, Ueta H, et al. Adult-onset atypical teratoid/rhabdoid tumor featuring long spindle cells with nuclear palisading and perivascular pseudorosettes. Neuropathology (2017) 37:52–7. doi: 10.1111/neup.12317
2. Samaras V, Stamatelli A, Samaras E, Stergiou I, Konstantopoulou P, Varsos V, et al. Atypical teratoid/rhabdoid tumor of the central nervous system in an 18-year-old patient. Clin Neuropathol. (2009) 28:1. doi: 10.5414/NPP28001
3. Ostrom QT, Chen YM, de Blank P, Ondracek A, Farah P, Gittleman H, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro-Oncol. (2014) 16:1392–99. doi: 10.1093/neuonc/nou090
4. Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
5. Nakata S, Nobusawa S, Hirose T, Ito S, Inoshita N, Ichi S, et al. Sellar atypical teratoid/rhabdoid tumor (AT/RT). Am J Surg Pathol. (2017) 41:932–40. doi: 10.1097/PAS.0000000000000845
6. Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neuro-Oncol. (2008) 88:321–30. doi: 10.1007/s11060-008-9571-z
7. Shonka NA, Armstrong TS, Prabhu SS, Childress A, Choi S, Langford LA, et al. Atypical teratoid/rhabdoid tumors in adults: a case report and treatment-focused review. J Clin Med Res. (2011) 3:85. doi: 10.4021/jocmr535w
8. Sinha P, Ahmad M, Varghese A, Parekh T, Ismail A, Chakrabarty A, et al. Atypical teratoid rhabdoid tumor of the spine: report of a case and literature review. Eur Spine J. (2015) 24:472–84. doi: 10.1007/s00586-014-3445-1
9. Almalki MH, Alrogi A, Al-Rabie A, Al-Dandan S, Altwairgi A, Orz Y. Atypical teratoid/rhabdoid tumor of the sellar region in an adult with long survival: case report and review of the literature. J Clin Med Res. (2017) 9:216. doi: 10.14740/jocmr2922w
10. Arrazola J, Pedrosa I, Mendez R, Saldana C, Scheithauer BW, Martinez A. Primary malignant rhabdoid tumor of the brain in an adult. Neuroradiology (2000) 42:363–7. doi: 10.1007/s002340050900
11. Dardis C, Yeo J, Milton K, Ashby LS, Smith KA, Mehta S, et al. Atypical teratoid rhabdoid tumor: two case reports and an analysis of adult cases with implications for pathophysiology and treatment. Front Neurol. (2017) 8:247. doi: 10.3389/fneur.2017.00247
12. Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim SH, et al. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat. (2014) 2:108–13. doi: 10.14791/btrt.2014.2.2.108
13. Barresi V, Lionti S, Raso A, Esposito F, Cannavò S, Angileri FF. Pituitary atypical teratoid rhabdoid tumor in a patient with prolactinoma: a unique description. Neuropathology (2018) 38:260–7. doi: 10.1111/neup.12440
14. Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: a case report and review of the literature. Surg Neurol Int. (2014) 5. 75. doi: 10.4103/2152-7806.133105
15. Nishikawa A, Ogiwara T, Nagm A, San K, Okada M, Chiba A, et al. Atypical teratoid/rhabdoid tumor of the sellar region in adult women: is it a sex-related disease? J Clin Neurosci. (2018) 49:16–21. doi: 10.1016/j.jocn.2017.12.010
16. Johann PD, Bens S, Oyen F, Wagener R, Giannini C, Perry A, et al. Sellar Region Atypical Teratoid/Rhabdoid Tumors (ATRT) in adults display DNA methylation profiles of the ATRT-MYC subgroup. Am J. Surg Pathol. (2018) 42:506–11. doi: 10.1097/PAS.0000000000001023
17. Nobusawa S, Nakata S, Hirato J, Kawashima T, Sato K, Fujimaki H, et al. Atypical teratoid/rhabdoid tumor in the sella turcica of an elderly female with a distinct vascular pattern and genetic alterations. Virchows Archiv. (2016) 469:711–5. doi: 10.1007/s00428-016-2017-7
18. Chacko G, Chacko AG, Dunham CP, Judkins AR, Biegel JA, Perry A. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neuro-Oncol. (2007) 84:217–22. doi: 10.1007/s11060-007-9361-z
19. Yamamoto J, Takahashi M, Nakano Y, Soejima Y, Saito T, Akiba D, et al. Rapid progression of rhabdoid components of a composite high-grade glioma and rhabdoid tumor in the occipital lobe of an adult. Brain Tumor Pathol. (2012) 29:113–20. doi: 10.1007/s10014-011-0069-6
20. Kleinschmidt-DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO. Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 but not claudin 6. Am J Surg Pathol. (2010) 34:341–54. doi: 10.1097/PAS.0b013e3181ce107b
21. Wyatt-Ashmead J, Kleinschmidt-DeMasters BK, Hill DA, Mierau GW, McGavran L, Thompson SJ, et al. Rhabdoid glioblastoma. Clin. Neuropathol. (2001) 20:248–55.
22. Moretti C, Lupoi D, Spasaro F, Chioma L, Di Giacinto P, Colicchia M, et al. Sella turcica atypical teratoid/rhabdoid tumor complicated with lung metastasis in an adult female. Clin Med Insights: Case Rep. (2013) 6:177–82. doi: 10.4137/CCRep.S12834
23. Schweizer Y, Meszaros Z, Jones DT, Koelsche C, Boudalil M, Fiesel P, et al. Molecular transition of an adult low-grade brain tumor to an atypical teratoid/rhabdoid tumor over a time-course of 14 years. J Neuropatho Exp Neurol. (2017) 76:655–64. doi: 10.1093/jnen/nlx044
24. Wang X, Liu X, Lin Z, Chen Y, Wang P, Zhang S. Atypical teratoid/rhabdoid tumor (AT/RT) arising from the acoustic nerve in a young adult: a case report and a review of literature. Medicine (2015) 94:e439. doi: 10.1097/MD.0000000000000439
25. Biswas S, Wood M, Joshi A, Bown N, Strain L, Martinsson T, et al. Exome sequencing of an adult pituitary atypical teratoid rhabdoid tumor. Front Oncol. (2015) 5:236. doi: 10.3389/fonc.2015.00236
26. Larrán-Escandón L, Mateo-Gavira I, Vilchez-López FJ, Gómez CE, Aguilar DM. Pituitary apoplexy as presentation of atypical teratoid/rhabdoid tumor in an adult. Endocrinol Nutr. (2016) 63:364. doi: 10.1016/j.endonu.2016.05.004
27. Chi SN, Zimmerman MA, Yao X, Cohen KJ, Burger P, Biegel JA, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. (2009) 27:385. doi: 10.1200/JCO.2008.18.7724
28. Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumor in sella turcica in an adult. Acta Neurochirurgica (2008) 150:491–6. doi: 10.1007/s00701-008-1500-y
29. Takahashi K, Nishihara H, Katoh M, Yoshinaga T, Mahabir R, Kanno H, et al. A case of atypical teratoid/rhabdoid tumor in an adult, with long survival. Brain Tumor Pathol. (2011) 28:71–6. doi: 10.1007/s10014-010-0008-y
30. Bruch LA, Hill DA, Cai DX, Levy BK, Dehner LP, Perry A. A role for fluorescence in situ hybridization detection of chromosome 22q dosage in distinguishing atypical teratoid/rhabdoid tumors from medulloblastoma/central primitive neuroectodermal tumors. Human Pathol. (2001) 32:156–62. doi: 10.1053/hupa.2001.21572
31. Takei H, Adesina AM, Mehta V, Powell SZ, Langford LA. Atypical teratoid/rhabdoid tumor of the pineal region in an adult: case report. J Neurosurg. (2010) 113:374–9. doi: 10.3171/2009.10.JNS09964
32. Chou SQH, Lo SSM, Wong HN, Lau P, Chan C, Tang K, et al. Atypical teratoid/rhabdoid tumor in the sella turcica of a female adult. Hong Kong J Radiol. (2013) 16:65–8. doi: 10.12809/hkjr1311034
33. Ingold B, Moschopulos M, Hutter G, Seeger H, Röthlisberger B, Landolt H, et al. Abdominal seeding of an atypical teratoid/rhabdoid tumor of the pineal gland along a ventriculoperitoneal shunt catheter. Acta Neuropathol. (2006) 111:56–9. doi: 10.1007/s00401-005-1112-7
34. Raisanen J, Hatanpaa KJ, Mickey BE. White III CL. Atypical teratoid/rhabdoid tumor: cytology and differential diagnosis in adults. Diagn Cytopathol. (2004) 31:60–63. doi: 10.1002/dc.20064
35. Roy S, Mallik C, Maiti S, Chaudhuri T. Temporal lobe atypical teratoid/rhabdoid tumor in a 24-year old adult female. South Asian J Cancer (2013) 2:210. doi: 10.4103/2278-330X.119909
36. Schneiderhan TM, Beseoglu K, Bergmann M, Neubauer U, Macht S, Hänggi D, et al. Sellar atypical teratoid/rhabdoid tumors in adults. Neuropathol Appl Neurobiol. (2011) 37:326–9. doi: 10.1111/j.1365-2990.2010.01111.x
37. Lev I, Fan X, Yu R. Sellar atypical teratoid/rhabdoid tumor: any preoperative diagnostic clues? AACE Clin Case Rep. (2015) 1:e2–7. doi: 10.4158/EP14337.CR
38. Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. (2005) 15:23–8. doi: 10.1111/j.1750-3639.2005.tb00096.x
39. Slemp SN, Martin SE, Zhang S, Ulbright TM, Cheng L, Hattab EM. Atypical teratoid/rhabdoid tumor in an adult with disseminated mediastinal germ cell tumor. Neuropathol Appl Neurobiol. (2014) 40:789–93. doi: 10.1111/nan.12081
40. Zarovnaya EL, Pallatroni HF, Hug EB, Ball PA, Cromwell LD, Pipas JM, et al. Atypical teratoid/rhabdoid tumor of the spine in an adult: case report and review of the literature. J Neuro-Oncol. (2007) 84:49–55. doi: 10.1007/s11060-007-9339-x
41. Yu F, Chiang F, Bazan III C. Atypical teratoid/rhabdoid tumor arising from the trigeminal nerve in an adult. The neuroradiol J. (2016) 29:447–9. doi: 10.1177/1971400916654319
42. Kuge A, Sato S, Sakurada K, Takemura S, Kayama T. Atypical teratoid rhabdoid tumor located in the pineal region following prophylactic irradiation for acute lymphoblastic leukemia. Brain Tumor Pathol. (2012) 29:177–81. doi: 10.1007/s10014-011-0075-8
43. Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumors. Neurosurg Focus (2006) 20:1–7.
44. Geller JI, Roth JJ, Biegel JA. Biology and treatment of rhabdoid tumor. Crit Rev Oncogene. (2015) 20:199–216 doi: 10.1615/CritRevOncog.2015013566
45. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell (2016) 29:379–93. doi: 10.1016/j.ccell.2016.02.001
46. Kawaguchi T, Kumabe T, Watanabe M, Tominaga T. Atypical teratoid/rhabdoid tumor with leptomeningeal dissemination in an adult. Acta Neurochirur. (2004) 146:1033–8. doi: 10.1007/s00701-004-0313-5
47. Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a pediatric oncology group study. Am J Surg Pathol. (1998) 22:1083–92. doi: 10.1097/00000478-199809000-00007
48. Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. (1996) 85:56–65. doi: 10.3171/jns.1996.85.1.0056
49. Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zelcer S, Wilson B, et al. Central nervous system atypical teratoid rhabdoid tumours: the canadian paediatric brain tumour consortium experience. Eur J Cancer. (2012) 48:353–9. doi: 10.1016/j.ejca.2011.09.005
50. Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. (2004) 22:2877–84. doi: 10.1200/JCO.2004.07.073
51. Dho YS, Kim SK, Cheon JE, Park SH, Wang KC, Lee JY, et al. Investigation of the location of atypical teratoid/rhabdoid tumor. Child's Nervous Syst. (2015) 31:1305–11. doi: 10.1007/s00381-015-2739-x
52. Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA. Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. Am J Neuroradiol. (2006) 27:962–71.
53. Warmuth-Metz M, Bison B, Dannemann-Stern E, Kortmann R, Rutkowski S, Pietsch T. CT and MR imaging in atypical teratoid/rhabdoid tumors of the central nervous system. Neuroradiology (2008) 50:447–52. doi: 10.1007/s00234-008-0369-7
54. Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M. Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int. (1999) 49:1114–8. doi: 10.1046/j.1440-1827.1999.00988.x
55. Ashraf R, Bentley RC, Awan AN, McLendon RE, Ragozzino MW. Implantation metastasis of primary malignant rhabdoid tumor of the brain in an adult (one case report). Med Pediatr Oncol. (1997) 28:223–7.
56. Kanoto M, Toyoguchi Y, Hosoya T, Kuchiki M, Sugai Y. Radiological image features of the atypical teratoid/rhabdoid tumor in adults: a systematic review. Clin Neuroradiol. (2015) 25:55–60. doi: 10.1007/s00062-013-0282-2
57. Han L, Qiu Y, Xie C, Zhang J, Lv X, Xiong W, et al. Atypical teratoid/rhabdoid tumors in adult patients: CT and MR imaging features. Am J Neuroradiol. (2011) 32:103–8. doi: 10.3174/ajnr.A2361
58. Yamashita Y, Kumabe T, Higano S, Watanabe M, Tominaga T. Minimum apparent diffusion coefficient is significantly correlated with cellularity in medulloblastomas. Neurol Res. (2009) 31:940–6. doi: 10.1179/174313209X382520
59. Douglas-Akinwande AC, Ying J, Momin Z, Mourad A, Hattab EM. Diffusion-weighted imaging characteristics of primary central nervous system germinoma with histopathologic correlation: a retrospective study. Acad Radiol. (2009) 16:1356–65. doi: 10.1016/j.acra.2009.05.004
Keywords: Atypical teratoid rhabdoid tumor, rhabdoid tumor, adult, systematic review, CNS tumor, brain tumor
Citation: Chan V, Marro A, Findlay JM, Schmitt LM and Das S (2018) A Systematic Review of Atypical Teratoid Rhabdoid Tumor in Adults. Front. Oncol. 8:567. doi: 10.3389/fonc.2018.00567
Received: 25 September 2018; Accepted: 13 November 2018;
Published: 28 November 2018.
Edited by:
Gordon Li, Stanford University, United StatesReviewed by:
Carsten Friedrich, University of Rostock, GermanyCopyright © 2018 Chan, Marro, Findlay, Schmitt and Das. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vivien Chan, dml2aWVua2FAdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.