 Erik W. J. Mollen1,2,3†
Erik W. J. Mollen1,2,3† Jonathan Ient1†Vivianne C. G. Tjan-Heijnen1,4Liesbeth J. Boersma1,2
Jonathan Ient1†Vivianne C. G. Tjan-Heijnen1,4Liesbeth J. Boersma1,2 Lucio Miele5,6Marjolein L. Smidt1,3
Lucio Miele5,6Marjolein L. Smidt1,3 Marc A. G. G. Vooijs1,2*
Marc A. G. G. Vooijs1,2*- 1Department of Radiotherapy, GROW School for Oncology and Developmental Biology, Maastricht University, Maastricht, Netherlands
- 2Department of Radiation Oncology (MAASTRO), Maastricht University Medical Centre+, Maastricht, Netherlands
- 3Division of Medical Oncology, Department of Surgery, Maastricht University Medical Centre+, Maastricht, Netherlands
- 4Division of Medical Oncology, Department of Internal Medicine, Maastricht University Medical Centre+, Maastricht, Netherlands
- 5Department of Genetics, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 6Stanley S. Scott Cancer Center, Louisiana State University Health Sciences Center, New Orleans, LA, United States
Breast cancer is the second most common malignancy, worldwide. Treatment decisions are based on tumor stage, histological subtype, and receptor expression and include combinations of surgery, radiotherapy, and systemic treatment. These, together with earlier diagnosis, have resulted in increased survival. However, initial treatment efficacy cannot be guaranteed upfront, and these treatments may come with (long-term) serious adverse effects, negatively affecting a patient's quality of life. Gene expression-based tests can accurately estimate the risk of recurrence in early stage breast cancers. Disease recurrence correlates with treatment resistance, creating a major need to resensitize tumors to treatment. Notch signaling is frequently deregulated in cancer and is involved in treatment resistance. Preclinical research has already identified many combinatory therapeutic options where Notch involvement enhances the effectiveness of radiotherapy, chemotherapy or targeted therapies for breast cancer. However, the benefit of targeting Notch has remained clinically inconclusive. In this review, we summarize the current knowledge on targeting the Notch pathway to enhance current treatments for breast cancer and to combat treatment resistance. Furthermore, we propose mechanisms to further exploit Notch-based therapeutics in the treatment of breast cancer.
Introduction
Breast Cancer
Breast cancer is the second most common malignancy, worldwide (1). Breast cancer screening and early detection has increased, leading to better outcome. Furthermore, a number of treatment options, have improved survival (2). First line therapies include surgery, radiotherapy, and systemic treatment (including: chemotherapy, endocrine therapy and targeted therapy). Treatment options for breast cancer exist in the neo-adjuvant (prior to surgery) and adjuvant setting (after surgery).
There is strong evidence that tumors that respond to neo-adjuvant chemotherapy with a pathological complete remission (pCR) have improved long-term prognosis (3). Conversely, tumors that do not respond to neo-adjuvant chemotherapy have a higher chance of recurrence. Adjuvant therapy targets remaining (micro-metastatic) cancerous cells, thereby preventing recurrent disease.
Clinical choices for the type of systemic treatment are guided by expression of estrogen receptor (ER), progesterone receptor (PR), and Human Epidermal Growth Factor Receptor 2 (HER2) in tumor biopsies, in concurrence with TNM classification, tumor grade, and age. However, it is widely accepted that breast cancer is a heterogeneous disease, from primary tumor to metastatic sites (4). Gene expression profiling (involving hierarchical clustering) has had a significant impact on classification of breast malignancies. Molecular breast cancer subtypes revealed different clinical behaviors and retain distinct differences in biological mechanisms (Table 1)—associated with tumor aggressiveness, metastasis, and response (8, 10–18, 22). Based on gene expression profiling, the MINDACT trial has shown that tumors with genomic “low risk” features do not require chemotherapy. This included some node-positive tumors irrespective of molecular subtype (23). Similarly, the TAILORx study has recently shown that early stage, node-negative breast cancers with low or intermediate recurrence scores do not benefit from adjuvant chemotherapy and can be treated with endocrine therapy alone (24). Additionally, gene expression analysis showed a sub-classification in Triple Negative Breast Cancer (TNBC) into at least 4 molecular subgroups (12, 19, 25) with observed differences in response to chemotherapy (20), by providing more detailed information inter-tumor heterogeneity (26). The combination of histological and genetic classification of each tumor will further guide therapy selection and disease outcome (26, 27) and, ultimately, form the basis for personalized precision medicine.
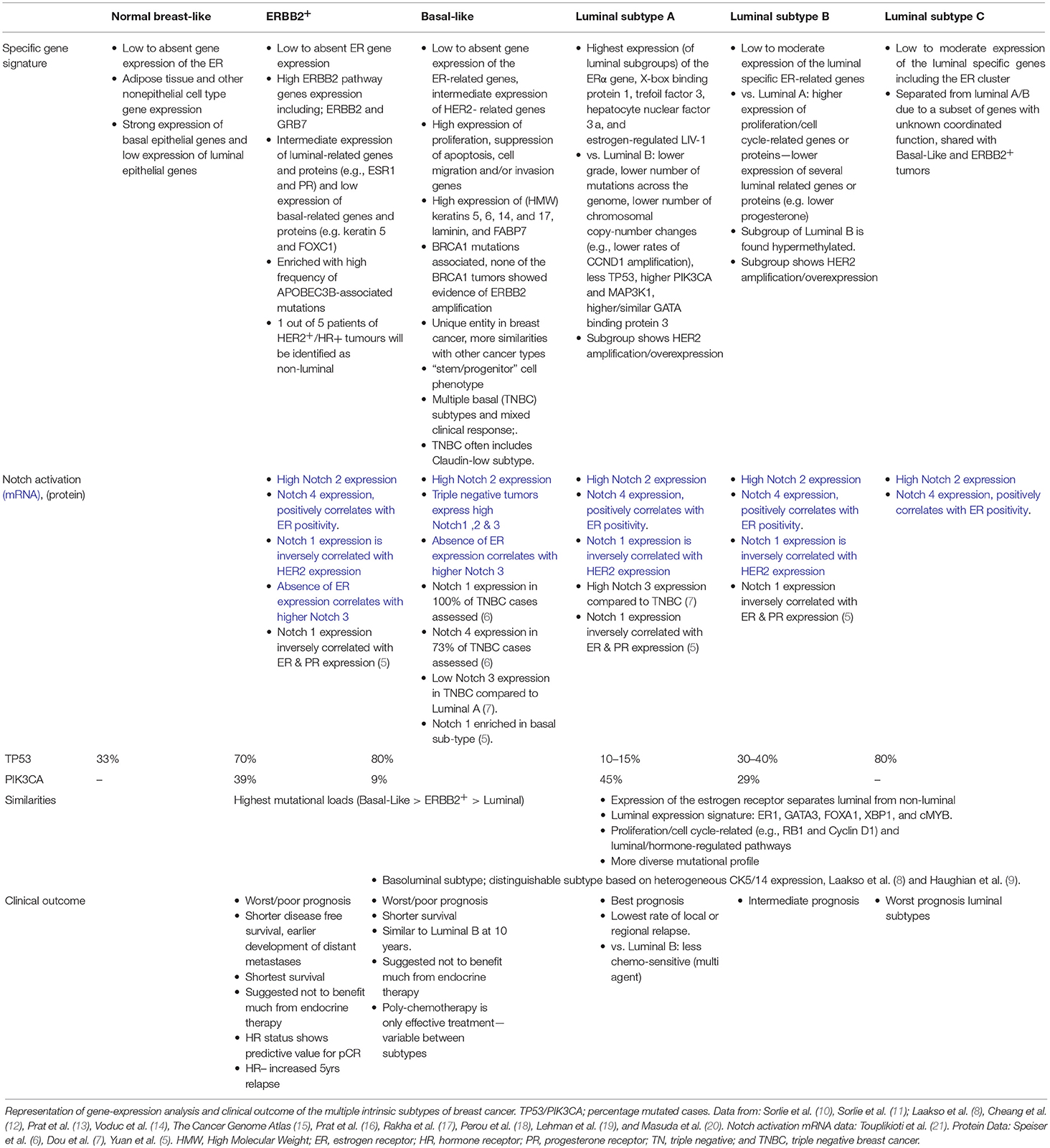
Table 1. Subtype classification of breast cancer.
Intra-Tumor Heterogeneity and Tumor Stem Cells
Regardless of clinical or molecular subtype, intra-tumor heterogeneity is a common feature of all human solid tumors (28), and is a major determinant of treatment outcome in breast cancer (15, 29). Tumor growth is thought to be driven by small populations of cancer cells with self-renewal and multi-potential properties (30), coined cancer stem cells (CSC) (31). These CSCs are involved in malignant behavior (invasion and metastasis) and resistance to treatment (32). Thus, CSCs are of high clinical importance, and targeting CSC self-renewal appears necessary for obtaining a durable response. Furthermore, intra-tumor heterogeneity can be driven by mutation or deregulation of stem cell signaling pathways such as Notch, Wnt, Shh, and others as well as through the tumor microenvironment; including nutrient-, oxygen levels, and paracrine interactions with other cell types (fibroblasts, blood vessels, and immune cells) (33). Herein, Notch has shown interesting targeting opportunities in cancer (34).
Notch
Notch Signaling
Notch signaling (Figure 1) is a cell-to-cell communication system of type I single-pass transmembrane Notch receptors (Notch 1-4) and transmembrane ligands (Delta/Jagged (JAG)). Notch receptor maturation starts in the Golgi/Endoplasmic reticulum. Glycosylation of Notch proteins in the Golgi and ER is known to play a role in the regulation of Notch activity (35). Fringe proteins can both positively and negatively regulate Notch ligands however the full scope of their roles in breast cancer are unclear (36). Furin-like convertases cleave the non-covalently associated Notch heterodimer, which is transported to the plasma membrane (Figure 2A).
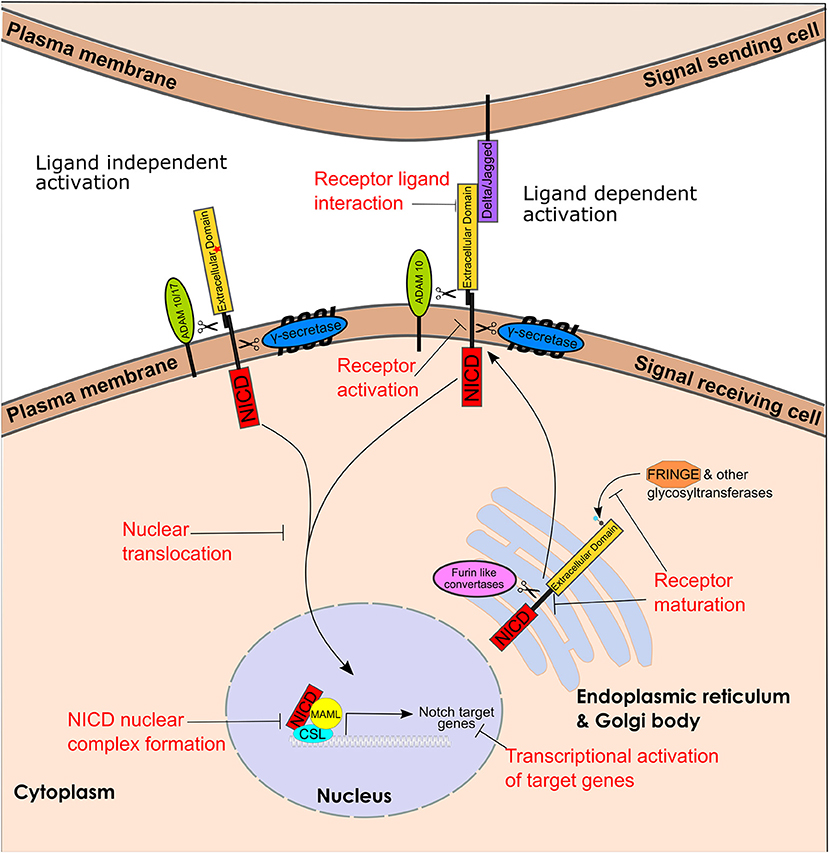
Figure 1. Notch receptor maturation, ligand dependent and independent activation pathway and targetable steps. Representation of Notch receptor maturation, ligand dependent and independent activation and the key enzymes involved. Text in red represents steps that can be targeted. The red star in Notch extracellular domain represents Notch activating mutations leading to ligand independent signaling. (CBF1, Su(H), Lag1), CSL; NICD, Notch intracellular domain and MAML, Mastermind-Like.
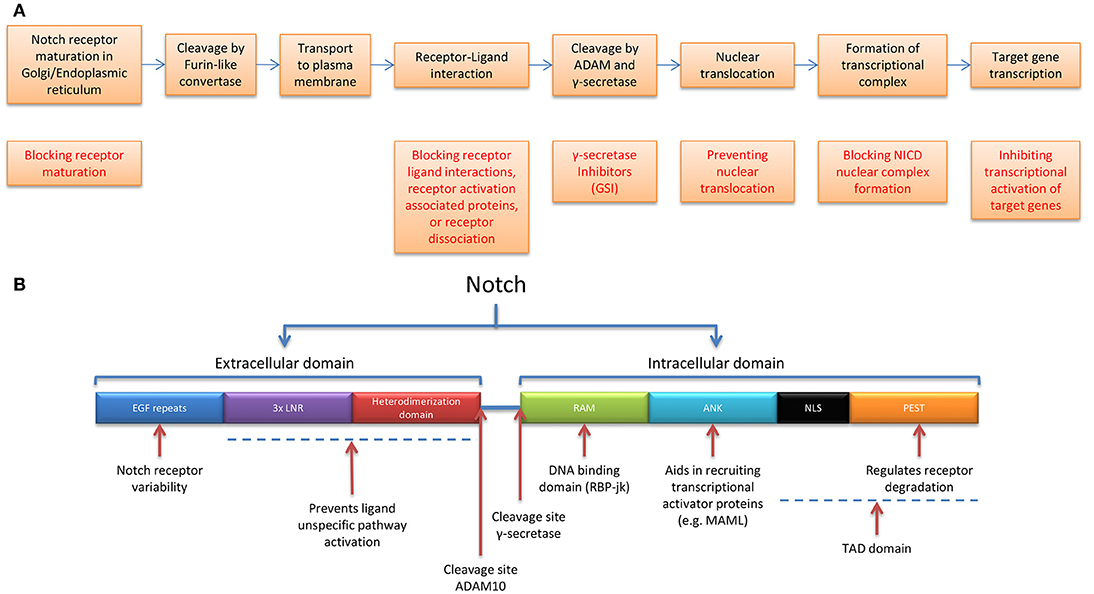
Figure 2. Notch receptor maturation and pathway activation, targetable options, and receptor functionality. (A) Stepwise representation of the process of Notch receptor maturation until receptor activation, followed by transcriptional output (not shown), and possibilities in targeting the Notch receptor pathway. (B) Notch receptor functional domains and corresponding functions ANK, Ankyrin repeats; LNR, Lin12-Notch Repeats and RAM, RBP-jk association module.
The extracellular domain consists of epidermal growth factor (EGF)-like repeats, followed by a negative regulatory region (NRR) which includes 3 LNRs (Lin12-Notch repeats) and a heterodimerization domain which prevents receptor activation in the absence of ligand(37) The intracellular portion of Notch (NICD) also contains multiple regions and domains, the RBP-jk association module (RAM), Ankyrin repeats (ANK domain), and the TAD domain—which consists of Nuclear Localization Sequences (NLS) and the PEST domain (regulates receptor degradation) (38) (Figure 2B). Notch 1-4 have relatively short lifespans and undergo degradation through the ubiquitin-proteasome and lysosomal pathways. The PEST domain contained in the NICD is likely to play a role in E3 ubiquitin mediated turnover. In fact, mutations in the PEST domain of Notch 1-3 in TNBCs, have been shown to increase Notch half-life and lead to increases in Notch downstream targets. Promisingly TNBCs with these mutations have been shown to be sensitive to GSIs (39). Similarly, alterations in the tumor suppressor and ubiquitin ligase Fbwx7/cdc4 target the PEST domain of Notch (40).
The E3 ligase MDM2 has been shown to contribute to the degradation of Numb and through ubiquitination leading to activation of Notch in breast cancer. Treatment of MCF7 cells with drugs targeting the acidic domain of MDM2 showed a reduction in Notch signaling (41). Furthermore, ubiquitination of Notch1 by MDM2 has been shown to activate Notch rather than leading to degradation (42). MDM2 has also been shown to regulate p53 degradation through ubiquitination, which, along with its role in Notch regulation makes it an attractive target for drug discovery along with other E1-3 ligases and interacting proteins. Knockdown of the E2-conjugating enzyme UBC9 and inhibition of the E1 activating complex SAE1/UBA2 has also been shown to impair the growth of Notch1-activated breast epithelial cells (43). Pevonedistat (MLN4924), is an inhibitor of NEDD8, a ubiquitin like protein that can neddylate E3 ligases. Pevonedistat has been shown to induce apoptosis in MCF-7 & SKBR-3 cells in combination with 2-deoxyglucose (44), and to sensitize breast cancer cells to radiation in vivo (45). Bortezomib, an FDA approved proteasome inhibitor has been shown to inhibit multiple genes associated with poor prognosis in ERα breast cancer (46), however several clinical studies have shown contradictory results in advanced/metastatic breast cancer (47–49).
A number of components of post-translational modifications pathway have been implicated in regulating Notch stability including Fbw7, Itch, β-arrestin, Fe65 and Numb (50–53). Numb negative breast cancers have increased Notch signaling which can be reverted to basal levels with overexpression of Numb and visa-versa knockdown of Numb in Numb positive breast cancers leads to upregulation of Notch signaling (54). Further research into the mechanisms of Notch post-translational modifications and degradation may provide novel therapeutic targets as well as for other malignant diseases.
Signal transduction occurs through the Notch ligand on the “signal-sending cell” interacting with the Notch receptor on the “signal-receiving cell.” This interaction process involves two sequential proteolytic cleavage events – first by the ADAM10 metalloprotease which sheds the extracellular domain, leading to the release of the NICD. The γ-secretase complex is composed of 4 polytopic transmembrane proteins including a catalytic subunit the aspartyl protease presenilin (55) The sequential proteolysis activation mechanism is essentially the same for Notch1-3 receptors (56). The activation mechanism for Notch4 -although likely similar to the other Notch family members- has not been reported yet. The NICD then translocates to the nucleus where it forms a protein complex with CSL (Cbf-1/RBP-jk in mammals, Su(H) in Drosophila and Lag-1 in C. elegans) and MAML (Mastermind-like) and induces transcription of multiple Notch downstream target genes (38) (Figure 2B). Additionally, a number of non-canonical pathways have been described downstream of Notch, including transcriptional activation of ERα-dependent genes (57), and NF-kB (58), activation of the PI3K-AKT-mTOR pathway (59), and activation of mitochondrial metabolism (60, 61). Mammalian cells express four Notch receptors and five ligands in a highly tissue specific and content dependent manner (38). Activation levels of specific pathways within the global notch signaling pathway has been found to differ within mammary epithelial cells and this can lead to different phenotypic responses (62).
Notch can also be phosphorylated which can have contradictory effects depending on the number of cleavage steps it has undergone and the specific kinase involved in phosphorylation (63). Phosphorylation of Notch by glycogen synthase kinase 3 (GSK-3) can reduce Notch transcriptional activity & protein levels (64, 65) and may be a target for possible therapies. Site specific methylation of NICD1 has been shown to make it less stable than a methylation defective mutant (66) indicating other possible post-translational targets (67).
Targeting Notch in Cancer
In many solid tumors the Notch signaling pathway is deregulated or mutated (68), affecting most hallmarks of cancer (69). Notch gene expression is frequently deregulated in breast cancer (70) and shows extensive crosstalk with several major signaling pathways. Further, there is ample evidence for the diverse role of Notch signaling in tumor formation, progression, and resistance to treatment in breast cancer (71).
Due to the multi-step activation process, several Notch pathway interventions are being explored at the level of: I. blocking receptor maturation, II. receptor–ligand interactions, III. receptor activation associated proteins, IV. nuclear translocation, V. NICD nuclear complex formation, and VI. transcriptional activation of target genes (72) (Figure 2A). This includes, but is not limited to antibodies, small molecule inhibitors, and inhibitors of γ-secretase (GSI) (72, 73).
Despite the increased evidence for deregulated Notch in numerous malignancies and resensitization opportunities (34, 74–81), many clinical studies investigating Notch targeting are on hold or have been terminated. Notably, most of these trials were conducted in recurrent, heavily pre-treated chemo resistant cancers and used dose-limiting non-selective pan-Notch/GSIs. Additionally, because of a lack of biomarkers predicting outcome to Notch therapies, potential responders were thus not effectively selected [reviewed in (71)]. As a result, this has not led to effective interventions using Notch inhibitors combined with standard of care. Therefore, in this review, we have focused on the possible role of Notch in enhancing the efficacy of breast cancer treatment.
Notch and Breast Cancer
Role of Notch in breast development.
The normal mammary gland experiences a period of rapid growth and development at puberty. Thereafter, and until menopause, it undergoes cycles of expansion and regression with each estrous cycle, pregnancy, lactation, and involution (82). This homeostasis requires stem cells and their existence was first demonstrated using transplantation experiments to reconstitute a functional mammary gland in rodents (83). Dontu et al. demonstrated the presence of early progenitor/stem cells capable of differentiating along all three mammary epithelial lineages (myoepithelial, ductal-, and alveolar epithelial). Gene expression analysis revealed similarities with progenitor and stem cell associated pathways, thereby identifying mammary stem cells (MaSCs) in 3D culture systems (84). More recently, in vivo imaging has identified bi-potent basal stem cells in the mammary gland, yielding both myoepithelial and luminal cells (85) and Notch plays a role in this process (86–89). Bouras et al., have performed extensive research on the role of Notch in MaSCs. In MaSCs, Notch1 is differentially expressed between subtypes (90), and its expression is higher in the luminal type cells (90, 91). Furthermore, Notch1/3 mark the luminal progenitor cells in mammary gland development (89, 91). Downregulation of Cbf-1/RBP-jk resulted in increased proliferation of MaSCs, thereby influencing absolute stem cell numbers. However, this proliferation resulted in increased and disorganized side branching, with increased number of end buds and basal cells in these end buds. Therefore, RBP-jk downregulation regulates the formation of a more basal cell phenotype. Additionally, overexpression of the endocytic protein NUMB, a negative regulator of Notch, produced the same effects. This shows that reduced Notch signaling is important in proliferation of the basal cell population and MaSCs. Conversely, increased levels of Notch1 in the luminal cells showed that constitutive Notch activation is important for commitment to the luminal cell lineage (High Keratin8/18, Stat5, and p63 downregulation) (90). Moreover, it has been reported that Notch4 is involved in promoting stem cell renewal of mammary epithelial cells (mammospheres) in vitro (92, 93), and is involved in stem cell activity (94)–possibly through JAG1 signaling (95) and PKCa-Notch4 interaction (96). Furthermore, Notch and p63 signaling guide the establishment of basal and luminal epithelial cells (97) and PTEN/JAG1 play an important role in mammary epithelial stem cells (98).
Role of Notch in breast cancer development and metastasis.
The role of different Notch pathway components in breast cancer development has been extensively researched. Stylianou et al. showed that in many breast cancer cell lines Notch ligands, receptors, and target genes are aberrantly expressed (99). Charafe-Jauffret et al. identified a 413-gene CSC profile (including Notch2) using normal and malignant mammary tissue (100), identifying breast cancer stem cells (BCSCs) through ALDH+ (101). ALDH+ cells were capable of self-renewal, differentiation, tumor formation in mice, and showed increased metastatic potential. ALDH− cells hardly generated tumors. Results from a Meta–analysis involving 3867 patients showed that Notch1 expression positively correlates with breast cancer progression and that higher expression is associated with a transition from ductal carcinoma in situ to invasive cancer. Furthermore notch1 overexpression was correlated with significantly worse overall and recurrence-free survival. The data further suggested that Notch inhibitors may be useful in blocking early progression of ductal carcinoma in situ (5).
Aberrant activation of the Notch signaling pathway has been shown to promote an aggressive phenotype partially through NF-κB, whereas de-activation of Notch signaling abrogates this aggressive phenotype (58). Furthermore, in TNBC, tumor cells activated NF-κB upregulates Jagged-1, which stimulates Notch signaling in CSCs (102).Tumor derived Jagged1 has been shown to be an important mediator of bone metastasis in breast cancer. Jagged1 activates stromal Notch signaling which in turn induces IL-6 secretion from osteoblasts stimulating tumor growth. Notch signaling also directly stimulates maturation of osteoclasts exacerbating bone metastasis. Destruction of bone matrices releases TGF-β upregulating Jagged1 in the tumor giving a positive feedback loop. GSIs treatment in turn reduces bone metastasis by targeting stromal Notch signaling (103).
In vivo studies using TNBC and ERα+ cell lines showed an association between Notch3 expression and distant metastases which was diminished in Notch3 null cells. This finding was corroborated using TNBC cells from a patient-derived brain metastasis (104). An in vivo study using a more metastatic variant of the HER2+ MDA-MB435 isolated from in vivo brain metastasis showed activation of the Notch signaling pathway. Inhibition of Notch using the γ-secretase inhibitor DAPT or knockdown using RNAi against Notch and Jagged2 resulted in inhibition of the migratory and invasive phenotype (105). Furthermore fewer brain micrometastases were found when Notch1 was silenced in an MDA-MB-231 model (106). Breast tumor cells in the brain highly express IL-1β which leads to surrounding astrocytes expressing Jagged1 which stimulates Notch signaling in CSCs (107). Oskarsson et al. showed that breast cancer cells that metastasize to the lungs enhance their ability to survive through expression of the extracellular matrix protein tenascin C. Tenascin C is associated with aggressiveness and pulmonary metastasis and enhances stem signaling components including Notch (108).
Notch1. Notch1 is aberrantly expressed in breast cancer (99) and high Notch1/4 mRNA expression and activity are associated with worse prognosis (30). In Ductal Carcinoma in situ (DCIS), Notch1 signaling is active and associated with the development of breast cancer (109). Both Notch1 and Notch4 are identified as common sites of proviral integration in mammary mouse tumors (110, 111), and induce mammary (MMTV)-tumors when overexpressed in transgenic mice (112–114). Larger studies have shown that expression of Notch1/4 and JAG1 is associated with poor prognosis in breast cancer (115). Moreover, JAG1 expression is an independent predictor of poor outcome in node-negative disease (116) and higher NICD1 expression correlated with sentinel lymph-node positive patients (117). Notch1 levels were progressively associated with the transition from DCIS to invasive basal cancer (5).
In human breast cancer, a meta-analysis including approximately 4000 cases showed that elevated Notch signaling is associated with increased disease recurrence (118). Pathway and network analysis revealed that altered Notch1 signaling occurred in ER+/PR+/HER2+/− breast cancers (119), whereby Notch1 mutations are more prevalent in HER2− than HER2+ tumors (120). JAG1-Notch signaling leads to Cyclin-D1 induction (121), a gene that is essential for normal breast development in mice (122) and frequently deregulated or amplified in human breast cancer (123, 124). k Notch1 activating mutations/rearrangements have also been observed in TNBC (in EGF repeats and NRR) (125) and in the basal-like phenotype (116). Additionally, Notch1 promotes stem cell maintenance through c-Jun signaling (126). Further, Reedijk et al. revealed that JAG1 is an independent predictor of poor outcome in multivariate-analysis (115) with other well-known outcome predictors (nodal metastases, patient age, tumor size, node status, ER positivity, and tumor grade) (5, 115, 127). Higher NICD1 expression correlated with sentinel lymph-node positive patients—strengthening Notch1's role in the metastatic process (117).
Leong et al. provided data that JAG1 and Notch1 are involved in epithelial-mesenchymal transition (EMT) through SLUG and E-cadherin. They showed that SLUG facilitated E-cadherin repression (through Notch1 inhibition) and inhibition of HEYL blocked tumor growth and metastasis, showing JAG1-Notch1-SLUG dependency (128). Furthermore, NICD1 expression negatively correlated with E-cadherin and showed increased invasive capacity of Notch1, (129). This was also the case under hypoxia with differences observed in high/low Notch signaling cell lines (130). Mechanistically, hypoxia-induced EMT is mediated through SLUG and SNAIL (131). A JAG2-EMT relationship has been shown too (through Notch1), revealing a broader spectrum of Notch1 activation and involvement in hypoxia and metastatic potential of CSCs (132). Additionally, high Notch1 and HIF predict a worse prognosis (133). These results show that Notch1 signaling is important for EMT and downregulation of E-cadherin, ultimately creating a more invasive phenotype. Furthermore, as described above, the invasiveness of the tumor and hypoxia induced EMT requires Notch1 signaling, demonstrating a hypoxia/Notch1/EMT axis. Thus, inhibition of Notch1 can be tumor suppressive by removing the inhibition on E-cadherin expression, regardless of hypoxia.
Downregulation of JAG1 or blocking Notch with GSI in a metastatic breast cancer model (MDA-231) attenuates bone metastasis by reducing osteolysis in the bone microenvironment. Conversely, overexpression of JAG1 is sufficient to induce bone metastasis in this model (103). Others have demonstrated a role for Notch1 of tumor dormancy in the bone marrow microenvironment, instigating metastases, through a Notch1/STAT3/LIFR signaling axis (134). Furthermore, circulating tumor cells “primed” for breast cancer brain metastases have a specific gene signature (HER2+/EGFR+/HPSE+/Notch1+) (135, 136). These CTCs could either be derived from the primary tumor or from metastatic lesions. Importantly, these CTCs were EPCAM−. This would make them undetectable by the only FDA approved clinical test for CTCs, which is based on an EPCAM+ profile (136).
Notch2. Notch2 can act as a transcriptional and functional regulator of Notch1 and Notch3 (137) and has been shown to be involved in specific mammary epithelial lineages affecting luminal cellular hierarchy (138). Mutations in Notch2 show increased incidence in breast cancer, and in addition to the TCGA database new mutations have been found (139). Notch2 is positively correlated with HER2 (140), low-grade tumors and improved outcome (141), and increased apoptosis (142). In the basal subtype, JAG1 and DLL4-induced Notch2 activation under the influence of FYN/STAT5 maintained the mesenchymal-phenotype. Notch2 siRNA decreased the EMT markers VIM, SNAI1, SNAI2 (SLUG), TWIST, and ZEB1 (143). Notch2/3 inhibition (Tarextumab) decreased CSC numbers in the UM-PE13 breast cancer cells (144). Furthermore, mutations in Notch2 could facilitate development of liver metastasis (145). However, other studies showed that, Notch2 mutations do not unequivocally associate with better prognosis and therapy efficacy (144, 146).
Notch3. Expression of oncogenic Notch3 in mice leads to mammary cancer (111), and is involved in: hormone-receptor positive breast cancer (120), the proliferation of HER2− breast cancer (147) and HER2+ DCIS (148), and TNBC (149). Notch3 is involved in HER2+ DCIS through transcriptional upregulation of the Notch pathway by HER2–whereby Notch3 upregulates the formation of luminal cells and increases proliferation through Cyclin-D1, c-MYC, and AKT (148). Furthermore, Notch3 signaling has been proposed to be an important regulator of the process whereby bipotent progenitors commit to the luminal lineage (93). Additionally, evidence from nonsense and missense mutations in multiple cancers, including breast cancer, showed tumor suppressor capabilities of Notch3 through controlling of the cellular senescence pathway (150). Interestingly, no significant change in Notch1, Notch2, or Notch4 expression was observed in these studies.
In TNBC, ectopic NICD3 (over)expression facilitated the inhibition of EMT through upregulation of the HIPPO pathway and E-cadherin in a RBP-jk dependent manner, whereby knockdown of Notch3 abrogated this effect (151). Furthermore, a correlation was shown between Notch3 and p21, a well-known senescence-involved protein. A significant decrease in Notch3 was observed in primary breast cancer, compared with normal tissue, suggesting a protective mechanism against Notch3-initiated cellular senescence. Re-introduction of Notch3 resulted in growth inhibition and activation of cellular senescence, suggesting that loss of Notch3 expression facilitates senescence induction and could play a critical role in tumor progression. Notch3 silencing has recently been shown to sensitize TNBC cells to the EGFR inhibitor gefitinib by promoting EGFR tyrosine dephosphorylation and internalization (152). Notch3/4 were shown to have increased expression in low-burden metastatic cells relative to the primary tumor (153).
Notch4. The oncogenic function of Notch4 was first demonstrated by retroviral insertion in MMTV-induced mammary tumors (110). Additionally, Notch4 is highly expressed, and gain of function mutations have been identified, in mouse mammary cancer models [reviewed in (154)]. Expression of activated Notch4 in mammary epithelial cells lead to transformation (155) and rapid development of poorly differentiated adenocarcinoma in transgenic mice (110, 156). Additionally, truncated human Notch4/Int3 (activated Notch4) instigated mammary tumors (112), through transcription of RBP-jk (157) and ANK repeats (158). Interestingly, transgenic expression of Notch4 NICD caused mammary tumors in the absence of RBP-jk in mice harboring conditional knockout of RBP-jk (157). This suggests that non-canonical pathways may participate in the oncogenic activity of Notch4.
PEST domain. The PEST domain is a degradation domain that regulates the stability of all NICDs through ubiquitination and proteasomal degradation (39). Nonsense mutations are common in T-ALL (159, 160) and have been observed in Notch1/2/3 receptors in TNBC (39). Furthermore, Notch pathway and target genes, including Notch1/3, HES1, HEY2, HES4, MYC, Cyclin-D1, and NRARP, were highly overexpressed in TNBC (39, 125). Notch mutation-activated dependency was shown using GSI, as wild type tumors showed little to no response (39).
Notch pathway-associated proteins
Fringe. Fringe is an important regulator of the Notch receptor-ligand interaction (161) through modification (glycosylation) of EGF repeats in the extracellular domains of Notch receptors (162). Fringe enzymes add N-acetyl glucosamine to fructose residues in the extracellular domains of Notch receptors. More glycosylated receptors retain high affinity for Delta ligands but have reduced affinity for Serrate/JAG ligands. Hence, loss of Fringe glycosylation enhances Notch affinity for Serrate/JAG ligands. There are three Fringe genes in mammals: Lunatic Fringe (LF), Manic Fringe (MF), and Radical Fringe (RF). In breast stem or progenitor cells, and especially the terminal end bud cap cells termed “leader cells” (163), LF is highly expressed (93). Conversely, the majority of basal tumors and a subset of claudin-low tumors show reduced LF expression (164). In MMTV-driven tumors, absence of LF exclusively caused triple negative tumors. Furthermore, deletion of LF was enough to cause Notch-driven (Notch1-4) basal-like tumors via enhanced stem/progenitor cell proliferation (163). These tumors resembled “claudin-low” (mesenchymal) subtype of TNBC. p53 loss of function in these tumors resulted in a clear EMT profile (Vimentin, TWIST, E-cadherin) (165). Cells showed increased levels of Vimentin and E-cadherin and decreased expression of cytokeratin 8/14–this coincided with decreased differentiation, increased levels of proliferation, and stem cells. Co-deletion of LF and p53 resulted in upregulated NICD3 and HES5, and downregulation of HES1. These data connect expression of LF, Notch (signaling), and p53 to impaired luminal differentiation.
In contrast to LF, MF is highly expressed in the claudin-low subtype of breast cancer and is associated with Notch4 (166). Deletion of MF shifted the tumor resemblance to a less claudin-low like, more luminal subtype—through increased levels of the luminal marker CK8 and basal marker CK14, and decreased levels of stem cell marker ALDH1. Furthermore, MF was shown to be able to regulate cancer stem cells and their migration in a spheroid model by increasing NICD1 expression and PIK3CG (encoding the g catalytic subunit of PIK3-γ) (166). These data show that Fringe is involved in a Notch-dependent manner in breast cancer with different roles observed for different Fringes (no data has been reported yet on RF)—causing a Fringe-dependent subtype switch (basal-luminal).
NUMB. NUMB is a cell fate determinant and endocytic protein that acts as a negative regulator of the Notch signaling pathway (54, 167, 168). NUMB is frequently down-regulated in breast cancer and suppresses the growth of breast cancer cells in vitro (169, 170) often involving the attenuation of the p53 tumor suppressor pathway (168). NUMB can drive Notch toward endocytic degradation. Additionally, NUMB inhibits ubiquitin ligase MDM2, which targets p53 for degradation. Hence Loss of NUMB results in a high-Notch, low p53 phenotype. Mechanistically, NUMB forms a ternary complex with MDM2 and TP53 and inhibits the activity of MDM2 (168, 171). In a cohort of breast cancer patients receiving adjuvant chemotherapy, NUMB, and indirectly Notch activation, were inversely correlated with clinical and pathological parameters indicative aggressive disease progression (168). In NUMB-deficient cells, p53 is ubiquitinated and degraded, resulting in chemoresistance and high Notch activity. MDM2 also ubiquitinates NUMB, which results in nuclear translocation and degradation (172). Thus, NUMB connects the MDM2/p53 pathway, the most frequent mutated pathway in human cancers, with Notch signaling.
MAST. In many breast cancers, gene translocations and fusions have been described. Recurrent gene arrangements involve MAST and Notch family members (Notch1/2), both showing phenotypic effects in breast cancer (e.g., greater proliferation). Notch fusions were found, almost exclusively, in ER− breast carcinomas. All the fusion transcripts retained the exons that encode for the NICD. Furthermore, higher Notch responsive transcriptional activity was seen in breast cancer cell lines carrying MAST-Notch fusions, and showed dependence on Notch signaling for proliferation and survival (173). The discovery of these Notch fusions warrants further investigation and may identify a biomarker for Notch based therapeutics.
Nicastrin. Nicastrin is an essential component of the γ-secretase complex; it encodes an integral membrane protein which associates with the catalytic subunit of γ-secretase, Presenilin (174). Nicastrin is crucial for maturation of Presenilin and cells that lack Presenilin are γ-secretase and Notch-deficient (175, 176). In breast cancer, high Nicastrin is mainly observed in the ER+ subtypes. Nicastrin expression correlates with age and tumor grade–and predicts worse tumor survival (177). Additionally, a set of 22 genes (located on chromosome 1) has been co-identified with Nicastrin amplification and breast cancer (178), however, these genes showed no clear Notch signature. Furthermore, Nicastrin seems to play a role in EMT (177, 179). Targeting of Nicastrin affects breast cancer stem cells and inhibits tumor formation in vivo (179). Inhibiting Nicastrin in TNBC, using monoclonal antibodies, showed anti-tumor activity (180). Thus, aiming at Nicastrin provides another opportunity to target the involvement of Notch in breast cancer.
These data suggest that (deregulated) Notch receptor/ligand signaling influences cell renewal in the mammary gland and reaches far beyond mammary development, as it possesses the ability to influence the pre-malignant lesions, primary tumors, the metastatic potential of tumors, and therapy resistance.
Notch Signaling in the Tumor Microenvironment
The breast microenvironment consists of a number of cell types including fibroblasts, adipocytes, endothelial and immune cells as well as extracellular matrix.
Cancer associated fibroblasts (CAFs) have been shown to induce Notch activation in breast cancer cell lines through secretion of IL-6 (181). There is also evidence supporting a role for fibroblast-derived microvesicles in endocrine resistance. Cancer-Associated-Fibroblast (CAF)-derived microvesicles, containing oncomiR-221 promoted de novo endocrine resistance—as overexpression of oncomiR-221/222 in luminal breast cancer cells reduces ER expression (182) Furthermore CAFs can promote the cancer stem cell phenotype by secreting CCL2, inducing Notch1 (183). Stromal cells including fibroblasts have also been shown to promote therapy resistance in breast cancer cells through expression of Jagged1 and exosomal transfer leading to Notch3 and STAT1 signaling in cancer cells (184). GPER signaling from both CAFs and cancer cells has been shown to upregulate Notch signaling. 17β-estradiol and GPER ligand G-1 induces γ-secretase-dependent activation of Notch1. Furthermore, the 17β-estradiol and GPER induced migration of breast cancer cells and CAFs is attenuated with GSI treatment (185).
17β-estradiol also promoted increased Jagged1 as well as Notch1 expression in MCF7 cells and was similarly found in endothelial cells. The endothelial cells formed cord-like structures in matrigel in contrast to cells expressing a dominant negative form of Notch1. 17β-estradiol treatment was also able to increase tumor microvessels in vivo, which correlated with Notch1 expression (186). Clinical data has shown higher Notch1 activation in tumor endothelial cells compared to non-malignant tissue. A correlation between the rate of NICD1-positve vs negative tumor endothelial cells was higher in patients with positive sentinel lymph nodes (117) Co-culture in vitro and in vivo has demonstrated upregulation of notch ligands in endothelial cells after contact with breast cancer cells. Proliferation and survival was significantly reduced along with a reduction in the stem-cell population when co-cultures were treated with GSI. Knockdown of Jagged1 in endothelial cells reduced the survival ability of breast cancer cells under starvation conditions. Knockdown also reduced tumor cell proliferation but did not reduce survival of knockdown epithelial cells (187). Wnt signaling is known to be up-regulated in breast cancer. Aberrant wnt signaling has been shown to give a tumorigenic phenotype to primary epithelial cells. This conversion is in part caused by up-regulation of the Notch ligands Dll1, Dll3 & Dll4 which are required for the tumorigenic phenotype (188).
Mammospheres enriched with stem/progenitor cells from node invasive breast carcinoma tissue expressed more IL-6 than matched non-neoplastic mammary glands. Il-6 was only detected in basal-like breast carcinoma tissue which contained stem cell features. Il-6 upregulated Jagged1 and lead to growth and a hypoxia-resistant/invasive phenotype through Notch3 dependent expression of CAIX (189).
Adipocytes within the tumor microenvironment secrete leptin and IL-6. Leptin and IL-6 signaling in breast cancer cells adjacent to adipocytes upregulate multiple pathways including Notch promoting a stem-like phenotype as well as epithelial-mesenchymal transition (190). Leptin is able to induce Notch 1,3 & 4 however Notch3 appears to be cell dependent. The leptin-Notch signaling axis is involved in proliferation and migration and leads to higher incidence and aggressiveness in obese patients. Leptin inhibitors were able to reduce Notch receptor, ligand and target expression (191).
Dll4 and Jag1 have opposite effects on regulating angiogenesis. Jag1 induces maturation of blood vessels, while Dll4/Notch regulates sprouting angiogenesis (192). Thus targeting Dll4 or Jag1 will have different effects. Targeting Dll4 using antibodies promotes non-productive angiogenesis (193). GSI treatment however targets both and leads to a decrease in angiogenesis (194). These differences in targeting may explain the contrasting in angiogenesis seen in pre-clinical models treated with GSI or Dll4 antibodies. In a phase I clinical trial, enoticumab, a Dll4 monoclonal antibody targeting the tumor vasculature, showed stable disease as best response in 2 of the 6 breast cancer patients enrolled. The antibody also gave a number of side effects, seen with previous Notch targeting therapeutics, as well as ventricular dysfunction and pulmonary hypertension (195).
Notch and the immune response
The role of Notch signaling in the immune response to tumors is complex and is dependent on the tumor type and microenvironment factors. Notch signaling is a key regulator of hematopoietic development and controls self-renewal, lineage commitment and terminal differentiation of the innate and adaptive immune system including B cells, T cells, myeloid cells, dendritic cells and natural killer cells (196, 197) Notch signaling, both canonical and non-canonical, also plays a role in tumor induced immuno-suppression.
It has been established that most stages of the tumor development from initiation to malignant conversion, invasion, metastasis, therapy resistance and relapse involve the inflammatory response (198). The interaction between tumor cells and immune cells in the tumor microenvironment controls the overall immune surveillance and response to therapies and patient outcome. The role of Notch signaling in the immune response to tumors is complex and is dependent on the tumor type and microenvironment factors Notch as well as regulating many aspects of the immune system regulates many components of the tumor microenvironment (199, 200).
There is a strong causal relationship between endocrine resistance and Jagged NOTCH signaling in breast cancer which promotes macrophage differentiation toward tumor-associated macrophages (TAMs), the most common immune cell found in the breast tumor microenvironment (200). TAMs can be pro or anti-inflammatory depending on micro environmental factors, which in most breast cancers develop the anti-inflammatory phenotype (200, 201). The anti-inflammatory phenotype in breast cancer plays a role in suppressing immune surveillance as well as promoting proliferation, angiogenesis and tissue remodeling (198). In a model of basal-like breast cancer, tumor cells secrete the CCL2 & IL-1β cytokines in a Notch dependent manner, which work to recruit monocytes (202). Within the tumor microenvironment monocytes differentiate into TAMs with a pro tumor phenotype supporting tumor growth and metastasis (203). TAMs also interact with cancer cells via TGFβ, promoting Jagged 1 expression, causing a feedback loop that amplifies cytokine/chemokine secretion.
Myeloid-derived suppressor cells (MDSCs) promote tumor progression through a variety of mechanisms including immune suppression and enhancing angiogenesis and metastasis. MDSCs have been shown to have lower Notch activity in conditioned media from breast cancer cell lines through an inhibitory phosphorylation of NICD by casein kinase 2, disrupting NICD/ CSL interaction (204). MDSCs in breast cancer have also been shown to induce Notch signaling in cancer cells and promote CSC capacity through IL6/STAT3 & Nitric Oxide/Notch cross talk signaling (205, 206). Cancer cells also increase Jagged-1 & Jagged-2 expression in MDSCs leading to a positive feedback loop between cancer cells, immune cells and CSCs.
Notch has been shown to be important in the regulation of Tregs, a subtype of T cells, which is important in peripheral self-tolerance and plays a role in tumor immunosuppression (207). Tregs promote evasion of immune surveillance and are linked to tumor invasiveness and poor prognosis. Notch-1-TGF-β signaling directly induces peripheral Tregs through upregulation of Foxp3 (208). Both Jagged-1 and Jagged-2 increase the generation of Tregs (209) and are highly expressed in TNBC, CSCs and treatment resistant populations (95, 132).
On the other hand CD8+ cytotoxic T cells, which have been shown to have anti-tumor function, require Notch to become activated (210), and Notch2 has been shown to be required for the anti-tumor effect of cytotoxic T lymphocytes (211). Furthermore, selective activation of the Notch pathway in hematopoietic environments enhances T-cell activation and infiltration, inhibiting tumor growth in mouse models.
Research into targeting the immune response and the tumor microenvironment is ongoing and detailed reviews strategies and treatments can be found here (212, 213). GSI treatment has been shown to reduce the numbers of TAMs, MDSCs and TRegs, however it can't be excluded that this was in part due to inhibiting tumor growth (214). More research is needed to fully elucidate the complex interplay between Notch, tumor microenvironment and the immune system in breast cancer and to develop strategies that enhance the anti-tumorigenic effect but do not suppress the anti-tumor immune response.
Notch in Breast Cancer Therapies
Radiotherapy
For breast cancer, radiotherapy is mainly implemented in the adjuvant setting and involves the targeting of remaining tumor cells, with the aim to prevent recurrence of residual disease. Gene signatures (IGKC, RGS1, ADH1B, DNALI1) in primary breast cancers predict low and high risk groups for local regional recurrence after Radiotherapy (215, 216). Generally, cancer stem cells are often radiation resistant (217, 218). Radiotherapy resistance could be intrinsic or acquired through changes in gene expression profiles and radiotherapy-resistant CSCs have been observed in breast cancer (219, 220). More specifically, BCSCs (CD44+/CD24−/low) were shown to be resistant to radiation (compared to non- CD44+/CD24−/low mono-layer cultures), and contributed to tumor recurrence after fractionated radiation. In a clinically more relevant culture system (mammospheres) higher radiation resistance was observed correlating with lower levels of ROS compared to monolayer cultures. Consistently, mammosphere cultures showed higher radiation resistance than irradiated single cell suspensions. Thus, during fractionated radiation, repopulation derives from the more resistant subpopulation of CSCs. Increased levels of Notch1/JAG1 signaling could stimulate the more resistant phenotype of CD44+/CD24−/low CSCs (219). Lagadec et al. showed that radiotherapy-exposed cancer cells have increased mammosphere formation, increased tumorigenicity, and (re)expressed stemness-related genes (transcription factors Oct4, Y-box 2, Nanog, and Klf4). Interestingly, both NICD1 and JAG1 expression were upregulated only in response to fractionated radiation (5 × 3Gy) and not after a single dose (10Gy) (221). Additionally, other research showed that a singular dose of 3Gy did upregulate NICD1 and JAG1 (222). Thus, induction of Notch pathway genes is radiation (multi)dose-dependent (222). Furthermore, targeting of Notch using siRNA (221) or GSI (222) decreased the induced BCSCs population after irradiation of non-tumorigenic cells. These data indicate that Notch is involved in the induction of radiation-induced CSCs from partially differentiated tumor cells. Recently it has been shown that, that Notch1 directly regulates the DNA damage response, through physical interaction and suppression of phosphorylation of ATM kinase (223). A plausible hypothesis is that after repeated irradiation, Notch1 could suppress apoptosis-inducing signals from the activated DNA damage response.
Chemotherapy
Chemotherapy is an important component of standard cancer treatment and includes anthracyclines, cyclophosphamide, and taxanes. Resistance to chemotherapy is the main cause of treatment failure in 90% of the patients with metastatic cancers (224). Importantly, chemo-resistance accompanies endocrine resistance, so that ER-positive recurrent tumors that are resistant to endocrine therapy are also almost invariably chemo-resistant. One of the main underlying causes for treatment failure is intra-tumor heterogeneity, a process affected by the presence of CSCs (168, 225).
Anthracycline/Cyclophosphamide
A role of Notch in doxorubicin sensitivity and resistance has been reported by Zang et al. (226). They showed that Notch1 inhibition (RNAi) and doxorubicin treatment led to a 50 and 70% growth inhibition, and increased apoptosis, compared to chemotherapy alone–in the MCF7 and MDA-MB-231 cell lines respectively. Li et al. showed that the efficacy of doxorubicin could be increased when used in combination with a GSI (227). Additionally, chemotherapy increases the percentage of treatment resistant CD44+/CD24low breast cancer cells in patients. In tumor xenografts combination treatment with GSI and doxorubicin led to better tumor control–by reducing CD44+/CD24low population (168).
Interestingly, ALDH expression has been shown to inactivate chemotherapeutics such as doxorubicin and cyclophosphamide (228–230). In addition, Suman et al. (231) showed that Notch inhibition was effective in both ALDH− and ALDH+ cells, though ALDH− cells were more sensitive. Additionally, they showed that Notch1 downregulation (using Psoralidin) and silencing resulted in inhibition of cell viability and proliferation, and a downregulation of EMT factors SLUG and TWIST.
In ER+ cell lines (MCF7 and T47D) Notch target genes HES1 and HEY1 were induced by doxorubicin, and could be inhibited using a GSI–suggesting a Notch signaling dependent effect. Furthermore, expression of Notch was associated with expression of multi drug resistance protein 1 (MRP1), a potential predictor of chemotherapy response and clinical outcome, in a dose-dependent manner (232). Importantly, in patients treated with neoadjuvant chemotherapy (anthracyclines ± taxanes), pre-treatment NICD1 levels were very low or absent, while post-therapy NICD1 was significantly upregulated (232).
In a doxorubicin resistant engineered-cell line, MCF7-AMD, Notch3 was shown to be downregulated in chemo-resistant cells, and EMT was activated. Furthermore, in ER+ patients, low Notch3 predicted distant relapse-free survival, with Fos-related antigen 1 (Fra1) being negatively regulated by Notch3 (233).
Taxanes
The two most common used taxanes for breast cancer treatment are docetaxel and paclitaxel (234, 235). Qiu et al. showed that docetaxel treatment resulted in increased primary mammosphere formation. Notch1 inhibition increased chemotherapy efficacy in TNBC BCSCs (CD44+/CD24−/low population) in vitro and in a patient-derived xenograft breast cancer model (236). In line with this, Zhang et al. reported similar findings, using a GSI in multiple xenograft models (237). “Tumor debulking” by docetaxel resulted in an increased BCSC population, quantified using ALDH+/CD133+/CD44+. Interestingly, the CD44+/CD24−/low population was not altered, however, this might be due to differential targeting methods (Qiu et al. (236): mAb vs. Zhang et al. (237): GSI). Docetaxel-treated tumors showed increased NICD1. Combination of GSI with docetaxel showed significant improved effect compared to docetaxel alone. Mechanistically, treatment with docetaxel caused an increase in survivin (inhibitor of apoptosis) and drug transporters, which could be inhibited by GSI. Furthermore, decreased expression of NUMB was observed in docetaxel treated tumors but not after dual treatment with GSI. Docetaxel treatment increased EMT markers SNAIL, SLUG and N-cadherin, which could be blocked by Notch inhibition. These findings indicate that Notch1 is involved in the resistance mechanisms of docetaxel treated tumors and that dual treatment could block enrichment of the BCSC population and increase therapy efficacy.
Schott et al. showed a residual BCSC subpopulation to be insensitive to docetaxel alone (238). However, in tumor-derived xenografts treatment with GSI (MK-0752) reduced the BCSC population; this resulted in reduced mammosphere formation and decreased NICD and HES1 expression. A concurrent clinical study, including 30 patients with recurrent disease after anthracycline treatment, showed that repeated cycles of GSI resulted in partial response in 11 patients and evidence for a reduction in CD44+/CD24−/low and ALDH+ cells. Repeated biopsies showed an initial increase in BCSC populations until after the 1st treatment cycle, after which it declined—this is consistent with the ability of GSIs to decrease BCSCs. However, additional treatment cycles where needed to additionally reduce BCSCs and tumor burden. An additive effect of Notch inhibitors and docetaxel has been recently observed in a phase 1b trial in TNBC, whereby docetaxel and GSI (PF-03084014) showed 4 partial responses and 9 had stable disease out of 25 patients, with a manageable safety profile (by dose reduction) (239). All in all, the combination of docetaxel and Notch1 targeting showed synergy, with a manageable toxicity profile (238, 239).
In TNBC cells treated with the microtubule stabilizing agent paclitaxel, surviving breast cancers cells expressed Notch1, Sox2, Oct3/4, c-Myc, c-SRC, c-MET, Nanog, and E-cadherin, and were highly tumorigenic. Surviving cells also became resistant to the BCR-Abl/Src family kinase inhibitor dasatinib (240). In parental MDA-231 cells, dasatinib reduced NICD1 and Cyclin-D1 levels, but in paclitaxel resistant clones NICD1 levels were not affected. Dasatinib resistant MDA-231 clones were not cross-resistant to doxorubicin or docetaxel. Targeting Notch1 signaling in TNBC (using GSI) was additive to paclitaxel treatment, as Notch wildtype tumors showed no additive effect (125). These results support a protective mechanism whereby Notch1 is upregulated to protect the survival of paclitaxel-treated TNBC cells. In the TNBC UM-PE13 xenograft, blockage of DLL-4, decreasing Notch1 signaling, resulted in delayed tumor regrowth after paclitaxel treatment, with additionally decreasing the CSC frequency (241). Paclitaxel is capable of preventing breast cancer bone metastases. However, resistance emerges over time through induction of osteoblast JAG1 expression. Hence, metastatic seeding could be prevented using a JAG1 antibody (15D11). Synergistic effects (100x, compared to IgG) were observed when used in combination with paclitaxel (242).
All together, these data indicate that Notch inhibition may sensitize breast cancer to chemotherapeutics and that this involves a treatment-resistant BCSC population characterized by CD44+/CD24−/low cells. Further, chemotherapy resistant cell lines may be resensitized after treatment with Notch inhibitors.
Endocrine Therapy
In ER+ breast cancers, estrogen receptor signaling plays a pivotal role in tumor development and progression (243). Treatments that target the ER include blocking of receptor with an antagonist (e.g., selective estrogen receptor modulators such as tamoxifen or selective estrogen receptor disruptors such as fulvestrant), or depriving the tumor of estrogen (aromatase inhibitors). This mainly targets the tumor bulk, however, important implications have been made for hormone receptor-positive stem cells (244, 245). Despite similar expression of hormone receptors, some tumors are more sensitive to endocrine therapy than others, resulting in inter- and intra-patient differences. Additionally, differences in clinical outcome are observed based on breast cancer subtype (Table 1). Notably, expression of ER/PR is not universal in both tumor and metastases (246), and this does affect tumor prognosis (247). Receptor conversion and intra-tumor heterogeneity of ER expression in primary and metastatic tumors are therefore still a barrier to effective endocrine therapy. Point mutations in the ESR1 gene, encoding ERα, have been shown to arise during endocrine therapy and lead to endocrine resistance (248, 249).
Notch, Estrogen Receptor Interactions, and Therapy Sensitivity/Resistance
It has been suggested that in endocrine-resistant tumors, the ER is not the main survival pathway of breast cancer cells. Additionally, ER-targeting treatment resistance mechanisms are already in place (250, 251), and these resistance mechanisms show potential activating crosstalk with Notch (57). Endocrine resistant breast cancers show increased BCSCs numbers (9, 252) with Notch3/4 expression (94, 252, 253). Interestingly, in BCSCs paracrine EGFR and Notch signaling (under the influence of estrogen), is capable of activating estrogen signaling in ER− BCSCs (253).
Estradiol inhibits the activation of Notch1/4, causing membrane accumulation of uncleaved receptors (254), and upon estrogen deprivation or anti-estrogen drugs increased Notch signaling was observed (254). Luminal breast cancers with Notch1 remain hormone responsive (9). Hence, decreasing Notch signaling using GSI in cell lines and xenografts resulted in G2 growth arrest (254). Additionally, estrogen deprivation of luminal ER+ cells (MCF-7) inhibits tumor growth. Conversely, in the engineered HER2+ MCF-7 cell line, tamoxifen stimulated growth, even in combination with estrogen deprivation. This was accompanied by molecular crosstalk between ER and HER2 (255). Furthermore, involvement of the Akt and MAPK pathways were observed, with possible roles for Notch in this resistance (59, 256, 257). These experiments indicate that HER2 expression plays an important role in endocrine therapy resistance mechanism; however luminal cells are still dependent on estrogen receptor activation.
Interestingly, when grown orthotopically, original ER+/PR+/CK5− tumors showed an increased population of ER−/PR−/CK5+ “luminobasal cells,” this population further increased when estrogen was withdrawn, revealing receptor conversion when exposed to a new environmental niche (9). However, others have stated that this ER−/PR−/CK5+ population doesn't increase over time, is under the influence of progesterone signaling, and is capable of surviving extensive ER-targeting (258). Many Notch1 pathway genes were included in this new so-called luminobasal gene signature—involving TWIST1 and SLUG upregulation. These luminobasal cells resemble a more TNBC basal-like phenotype (CK5+) while retaining their luminal origin, expand (at higher rates) within luminal tumors when deprived of estrogen signaling due to their independence of the estrogen receptor, and showed sensitivity to Notch1 silencing. These data suggest an important link between ovarian endocrine sensitivity (both progesterone and estrogen) and Notch1, and support a luminal origin of basal-like cells (9, 258).
Elevated Notch1/3 signaling upregulates IL6 and activates the JAK/STAT pathway, however, dependent on p53/IKKa/IKKb status, and through a non-canonical mechanism. Furthermore, Notch signaling upregulation resulted in different Notch target genes in different molecular subtypes of breast cancer (basal vs. luminal B) (259). This growth promoting effect can also be instigated by fibroblasts secreting IL6, in relation with Notch3 and JAG1 (181). Pioneering research by Sansone et al. showed that Notch3-IL6 signaling is under indirect control of hypoxia and that it promotes self-renewal and survival in mammary gland stem cells (260, 261). CD133high cells express low levels of ER, but high levels of Notch3 (252), are endocrine resistant and promote metastases. This process is regulated through IL6-Notch3 signaling (261). IL6 expression could be induced either by Tamoxifen or HER2. CD133high expressing cells could be resensitized to endocrine therapy through IL6R blockade, which reduced Notch3, STAT3, and CD133. Knockdown of STAT3 resulted in reduced Notch3 mRNA levels and re-expression of ERα, without changes in CD133 expression. Notch3 thus, indirectly, plays an important role in endocrine resistance observed in metastatic breast cancer by influencing stem cell behavior (260, 261).
As described earlier fibroblast-derived microvesicles containing oncomiR-221 promoted de novo endocrine resistance—as overexpression of oncomiR-221/222 in luminal breast cancer cells reduces ER expression (182). These microvesicles were capable of blocking endocrine therapy Notch3 down regulation and causing an estrogen-independent phenotype in breast cancer cells (96, 262). This was also observed in endocrine resistant luminal breast cancers whereby blockage of Notch3 abrogated the growth of these ER-resistant cells (262).
Moreover, Notch4 is a crucial mediator of endocrine therapy resistance in models of luminal breast cancers (95, 261–263). BCSC induced by endocrine treatment are characterized by upregulation of Notch target genes [and additionally induces an EMT phenotype (263)], and endocrine resistance in BCSC is driven through JAG1/Notch4 signaling (95). This could be inhibited through targeting of Notch4 using GSI RO4929097. Notch4 inhibition reduced HES1 and HEY1 expression, reversed EMT, decreased CSC populations, thereby attenuating proliferation and invasion. Notch4 thus promotes estrogen-independent, endocrine therapy resistant growth of breast cancer cell lines (95, 263) possibly through a Notch4/STAT3/EMT regulated axis (264). Very recent evidence shows that mutations in the ligand binding domain of ERα, which occur in patients and are associated with endocrine therapy resistance, promote a stem-cell-like phenotype through activation of Notch4 (265).
Targeted Therapy (HER2)
HER2, a family member of the ERBB transmembrane receptor tyrosine kinases (ERBB1-4 or also known as EGFR and HER2-4) is a well-known target in HER2-amplified breast cancer therapy for both primary tumors (266, 267) and metastases (268–270). However, it is still unclear whether HER2+ cells are truly addicted to oncogenic HER2 signaling as other EGFR members can compensate after HER2 blockade (266). Moreover, a single copy of HER2 (in the absence of genomic amplification) can elicit an expression signature associated with HER2 dependence. Thus, HER2-non-amplified tumors may in some cases benefit from HER2 targeted therapy. Yet, such tumors are currently not being selected for treatment (267, 271). HER2+ breast cancer is mainly treated with combinations including taxane-based chemotherapy plus trastuzumab (272), pertuzumab (273), the tyrosine kinase inhibitor lapatinib (274, 275), or combinations thereof (266, 276). Many trials have shown remarkable response rates (277–281), and therefore HER2-targeted therapy is standard of care. However, intrinsic and acquired resistance may still result in relapse and progression of HER2+ disease. This resistance can occur on many levels, including activation of the downstream signaling pathways, constitutively activated HER2, and crosstalk of HER2 with other growth factor receptors such as other EGFR-members and IGF (282–285).
HER2 is a direct Notch target gene and bidirectional crosstalk between Notch and HER2 has been extensively reviewed (286). Under trastuzumab treatment, Notch activation occurs and contributes to trastuzumab resistance (284, 287). Trastuzumab-resistant cells (treated with trastuzumab for 6 months) expressed higher levels of Notch pathway genes, and this could be reversed by Notch inhibition (siRNA). GSIs decreased proliferation (288). In HER2+ xenograft experiments, GSI MK0752 alone did not affect tumor volume, while trastuzumab alone caused complete regression of tumors. However, trastuzumab-treated tumors recurred in approximately 50% of the cases. When trastuzumab was combined with GSI MK0752, complete cures were obtained with no observed recurrences. This suggests that the combination trastuzumab/GSI targeted stem-like cells responsible for recurrent disease. Notch inhibition resulted in HER2 down regulation (under the influence of Notch/RBP-jk binding sites in HER2 promotor sequences), followed by decreased mammosphere formation (286, 289).
Furthermore, Notch signaling is upregulated after treatment with lapatinib, a clinically active small molecule EGFR/HER2 inhibitor. Blockade of HER2 signaling in HER2-dependent primary tumor cells led to upregulation of Notch signaling [NICD1, HEY1, and HEY2 (266)]. The feedback signaling between these pathways was confirmed by the ability of HER2 to represses Notch signaling through HES1 and NRARP. In a HER2-inducible mouse model, Notch1 gain-of-function constructs identified Notch dependency in tumors recurring after suppression of HER2 expression in an HER2 inducible mouse model. After HER2 removal, the rate of recurrence was much higher in primary tumors that overexpressed NICD1, and this could be blocked using GSIs (118). The GSI sensitivity of these tumors suggests that other wild-type Notch paralogs (e.g., Notch3) induced by NICD1, may play a role. Moreover, a meta-analysis (17 studies, including 4,463 patients) revealed increased Notch activity in a subset of breast cancers associated with poor clinical outcomes (including basal-like tumors). These data suggest that Notch is positively associated with tumor recurrence in breast cancer patients and implicate that Notch targeting might prevent recurrent disease by targeting the dormant residual tumor cells.
Interestingly, HER2 expression can be heterogeneous both in bulk tumor cells (290) and BCSCs (291), and shows plasticity (291). ER+/HER2− and TNBC acquire a HER2+ subpopulation following therapy exposure (267, 291). Cultured BCSCs from ER+/HER2− patients retained HER2+/− subpopulations and switching between these HER2 states is dependent on environmental stimuli (291). Notch was inversely correlated with HER2 expression and HER2− cells were sensitive to Notch inhibition. HER2+ cells showed higher proliferation but were not addicted to HER2 oncogenic signaling. Following these sub-profiles, a proliferative state/niche favored the HER2+ phenotype, whereas oxidative stress or chemotherapy selected for, or initiated transition to, HER2− BCSCs. Thus, Notch might mediate a protective mechanism by functioning in the switch between proliferative and survival-prone phenotypes of HER2+/− BCSCs.
Besides a direct link between Notch and HER2, Notch also interacts with downstream or parallel HER2 signaling pathways. Co-suppressing the activation of these pathways upon resistance (267) might bypass these resistance mechanisms. Alternative mechanisms to activate signaling pathways such as PI3K/AKT and/or MAPK can be triggered in response to trastuzumab or through constitutive activation of HER2. These pathways may mediate treatment resistance in selected clones. The communication between Notch and PI3k/AKT has been shown extensively in hematological cancers (59, 292) and to lesser extent in breast cancer (293). Bidirectional MAPK-Notch interactions have been described (256, 257). Additionally, when the HER2 receptor is inhibited, signaling might still occur due to dimerization with IGF1-R (285) and Notch interaction (294), possibly resulting in therapy resistance.
Discussion, Conclusion, and Future Perspectives
There is overwhelming evidence for a role of the Notch signaling pathway in breast cancer development and progression through upregulation of Notch receptors, ligands, and regulators. Overall, high Notch pathway activity is associated with more aggressive disease and poor outcomes. Only a limited number of breast cancers harbor Notch gain of function mutations, but in many breast cancers Notch is expressed, active, and crosstalks with other oncogenic pathways. Further, many studies support an important role for Notch in the response to radiation, chemotherapy, hormonal therapy, and targeted therapies. Importantly, there is compelling evidence that treatment-resistant breast cancer and other malignancies can be resensitized by Notch inhibition (77, 78, 295–297). Taken together, this provides a strong rationale for studies combining Notch inhibitors with current breast cancer treatment modalities.
However, an important and complicating feature of Notch signaling is its receptor-ligand specific and context dependent signaling in different cancer subtypes. Furthermore, the optimal timing to initiate treatment to achieve therapeutic efficacy must be carefully considered. In treatment-naïve tumors, Notch activation might not become clinically evident until treatment initiation, as a resistance mechanism triggered by treatment, or after occurrence of metastases with different mutational profiles compared to primary tumors (4). Selecting patients most likely to benefit from Notch inhibition will require molecular profiling and screening to show possible co-targeting options (298). The identification of predictive biomarkers is of paramount importance.
In this review, we have highlighted several opportunities for Notch targeting in the context of first line breast cancer treatment and resistance. Additionally, we have discussed its extensive communication with many other pathways (59, 256, 257, 292, 293), its role in recurrent disease and involvement in the metastatic process (103, 134, 136, 145), and its association with clinically relevant hallmarks in breast cancer (69).
Research in the past decade has focused on preventing or treating tumor recurrence by targeting CSCs. Multiple different stem-like cell populations have been proposed within tumors, based on the expression of CD44high/CD24−, ALDH+, CD133, CD29high/CD61+, CD49f+, and CD90 (299–302). These cells showed increased levels of therapy resistance and distinctive gene expression patterns, irrespective of their potential origin (e.g., from transformation of mammary stem cells or from de-differentiation of non-stem-like tumor cells)—as stem cell plasticity occurs within tumors (32, 303). Notch signaling plays an important role in mammary stem cells as well as breast cancer stem cells (BCSCs) (84, 92, 304)—well documented for triple-negative breast cancer (94, 109, 153, 244, 253, 304–307). Furthermore, Notch4 has been shown to maintain the BCSC population (94, 307). Notch-PTEN signaling is important in the expansion of these stem-like cells (98, 308). PTEN/PIK3CA mutations are often observed in breast cancer and loss of PTEN decreases radiation sensitivity (309). In the future, combining radiotherapy and small molecule targeting in BCSC may improve the efficacy of radiation therapy and forestall radiation resistance. However, the timing and sequencing of treatments should be carefully optimized in order to achieve maximum efficacy. Radiotherapy dose scheduling might be easily adapted from the current schedule standards (310–312).
The effects of chemo-, radio- and targeted therapy on Notch signaling require further investigation. Observations have been made for Notch and tumor vascularization under the influence of both anthracycline and taxane-based chemotherapy (313). Taxane (paclitaxel) therapy resistance coincides with the development of metastatic bone lesions, preventable by targeting JAG1 in osteoblasts (242).
BCSCs in ER+ tumors show responsiveness to hormone signaling/targeting despite often lacking ER and PR (244, 245). This may be mediated by paracrine crosstalk with ER+/PR+ bulk tumor cells. Many endocrine therapy resistance mechanisms have been revealed (314–317). This has guided research toward the development of new therapeutic regimens (318), such as CDK4/6 inhibitors (319)—which have been clinically implemented. Notch inhibition could play a significant role in combinations targeting these resistance mechanisms. For instance, Notch inhibition could reverse ER-targeted-treatment resistance and improve the efficacy of CDK4/6 inhibitors through decreasing Cyclin-D1 (121).
Notch has been shown to crosstalk with the HER2 receptor (289) and development of breast cancer metastases is affected by HER2 (268–270, 320) and progesterone (268). Interestingly, plasticity of HER2 expression has been observed in circulating tumor cells—with a distinctive role for Notch1 (291). Thus, Notch is involved in the heterogeneity and plasticity observed in HER2−/+ breast cancer, and the development of distant metastases. Combining CDK4/6 inhibitors (321, 322) and Notch inhibitors, it may be possible to simultaneously attenuate two main drivers in breast cancer, HER2 and Cyclin-D, promoting local control and preventing distant relapse.
A step forward, for individualized patient care, could be the use of patient-representative culture models, such as organoids, to capture information on individual tumor drug sensitivity ex-vivo (323). In general, organoids can provide rapid insight into individual treatment combinations and relationships between Notch signaling and breast cancer treatment (resistance), before the start of treatment. These models more closely represent individual tumors, and may enable us to rationally investigate the context-dependence of Notch signaling in each tumor. Breast cancer organoids have recently been developed, but to what extent they will be strong predictors of treatment response and their use as prospective platforms for individualized precision treatment remains to be established (324).
This review summarizes the evidence supporting the hypothesis that targeting Notch could a promising option in re-sensitizing breast cancer to current standard of care treatments (Figure 3). When biomarker quantification and patient stratification allow Notch targeting to live up to its potential, this strategy may be applicable to other cancers as well, targeted with concurrent chemo-radiation or targeted inactivation of other growth promoting pathways. However, clinical evidence in solid tumors showed that therapy timing is highly important to reach maximum effectivity (325). Thus, additional clinical and translational research will be required to determine the exact role of Notch in each disease- and treatment-specific context and fine-tune the use of Notch targeting agents to prevent or treat or acquired resistance. With the benefit of sufficient mechanistic knowledge, we propose that in some cancer patients targeting Notch can be a major part of an effective strategy to address therapy resistance.
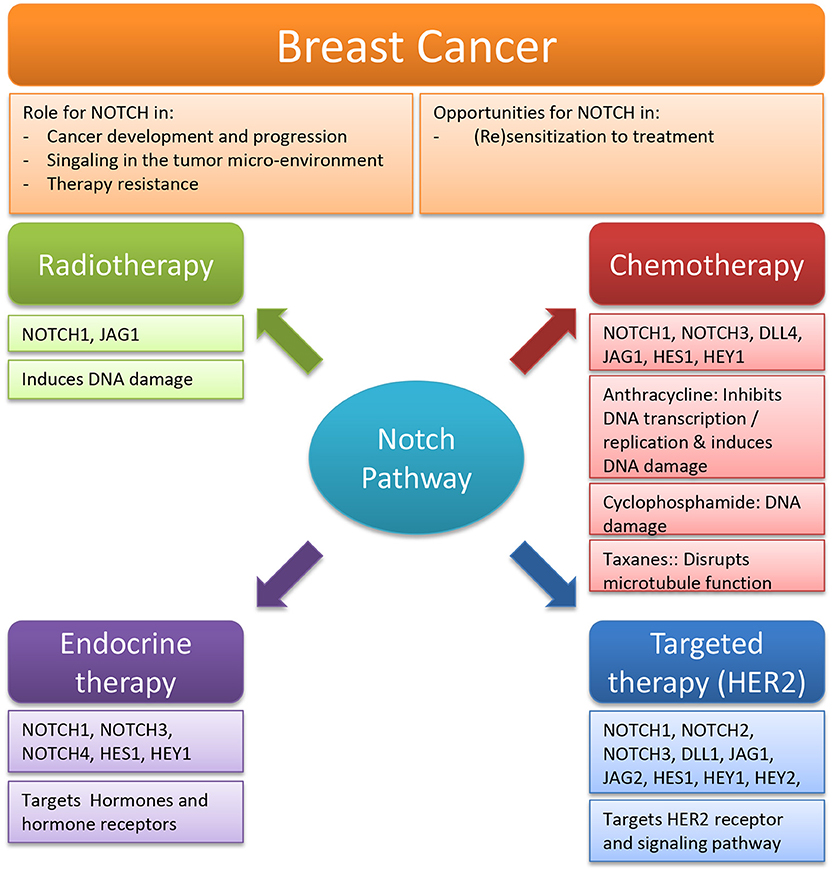
Figure 3. Overview of the role and opportunities for Notch in breast cancer therapy. Summary of the 4 fields of breast cancer therapy [radiotherapy, chemotherapy, endocrine therapy, and targeted therapy (HER2)] in which Notch targeting can play a significant role.
Author Contributions
EM, JI, MS, and MV made substantial contributions to conception and design of the review, and analysis and interpretation of articles. All authors have been involved in drafting the manuscript or revising it critically for important intellectual content. All authors have given final approval of the version to be published. EM, JI, MS, and MV have agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
JI was supported by a grant from the European Research Council (FP7/2007-2013, ERC grant no. 617060) to MV.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
(B)CSC, Breast Cancer Stem Cell; ANK, Ankyrin Repeats; CAF, Cancer Associated Fibroblast; DCIS, Ductal Carcinoma in situ; EGF, Epidermal Growth Factor; EMT, Epithelial-Mesenchymal Transition; ER, Estrogen Receptor; GSI, Y-Secretase Inhibitor; HER2, Human Epidermal Growth Factor Receptor 2; JAG, Jagged; LF, Lunatic Fringe; LNR, Lin12-Notch Repeats; LRR, Loco-Regional Recurrence; MAML, Mastermind-Like; MaSCs, Mammary Stem Cells; MDSC, Myeloid-derived suppressor cell; MET, Mesenchymal-Epithelial Transition; MF, Manic Fringe; MMTV, Mouse Mammary Tumor Virus; MRP1, Multi Drug Resistance Protein 1; NICD, Notch Intracellular Domain; NLS, Nuclear Localization Sequences; NRR, Negative Regulatory Region; pCR, Pathological Complete Remission; PR, Progesteron Receptor; RAM, RBP-Jk Association Module; RF, Radical Fringe; TAM, Tumor associate macrophage; TCGA, The Cancer Genome Atlas; TN(BC), Triple Negative (Breast Cancer).
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends–An Update. Cancer Epidemiol Biomarkers Prev. (2016) 25:16–27. doi: 10.1158/1055-9965.EPI-15-0578
3. Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, et al. Preoperative chemotherapy: updates of national surgical adjuvant breast and bowel project Protocols B-18 and B-27. J Clin Oncol. (2008) 26:778–85. doi: 10.1200/JCO.2007.15.0235
4. Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. (2017) 32:169–84 e7. doi: 10.1016/j.ccell.2017.07.005
5. Yuan X, Zhang M, Wu H, Xu H, Han N, Chu Q, et al. Expression of Notch1 correlates with breast cancer progression and prognosis. PLoS ONE (2015) 10:e0131689. doi: 10.1371/journal.pone.0131689
6. Speiser J, Foreman K, Drinka E, Godellas C, Perez C, Salhadar A, et al. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int J Surg Pathol. (2011) 20:137–43. doi: 10.1177/1066896911427035
7. Dou X-W, Liang Y-K, Lin H-Y, Wei X-L, Zhang Y-Q, Bai J-W, et al. Notch3 Maintains luminal phenotype and suppresses tumorigenesis and metastasis of breast cancer via trans-activating estrogen receptor-α. Theranostics (2017) 7:4041–56. doi: 10.7150/thno.19989
8. Laakso M, Tanner M, Nilsson J, Wiklund T, Erikstein B, Kellokumpu-Lehtinen P, et al. Basoluminal carcinoma: a new biologically and prognostically distinct entity between basal and luminal breast cancer. Clin Cancer Res. (2006) 12(14 Pt 1):4185–91. doi: 10.1158/1078-0432.CCR-06-0353
9. Haughian JM, Pinto MP, Harrell JC, Bliesner BS, Joensuu KM, Dye WW, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc Natl Acad Sci USA. (2012) 109:2742–7. doi: 10.1073/pnas.1106509108
10. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
11. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. (2003) 100:8418–23. doi: 10.1073/pnas.0932692100
12. Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. (2008) 14:1368–76. doi: 10.1158/1078-0432.CCR-07-1658
13. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. (2010) 12:R68. doi: 10.1186/bcr2635
14. Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. (2010) 28:1684–91. doi: 10.1200/JCO.2009.24.9284
15. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490:61–70. doi: 10.1038/nature11412
16. Prat A, Pineda E, Adamo B, Galvan P, Fernandez A, Gaba L, et al. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast (2015) 24(Suppl. 26):2568–81. doi: 10.1016/j.breast.2015.07.008
17. Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. (2008) 26:2568–81. doi: 10.1200/JCO.2007.13.1748
18. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406:747–52. doi: 10.1038/35021093
19. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. (2011) 121:2750–67. doi: 10.1172/JCI45014
20. Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. (2013) 19:5533–40. doi: 10.1158/1078-0432.CCR-13-0799
21. Touplikioti P. Expression of Notch receptors in primary breast cancer and correlation with pathological features. Clin Exp Pharmacol. (2012) 2:1–8. doi: 10.4172/2161-1459.1000109
22. Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. (2010) 28:3271–7. doi: 10.1200/JCO.2009.25.9820
23. Cardoso F, van't Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. (2016) 375:717–29. doi: 10.1056/NEJMoa1602253
24. Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N Engl J Med. (2018) 379:111–21. doi: 10.1056/NEJMoa1804710
25. Le Du F, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F, et al. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget (2015) 6:12890–908. doi: 10.18632/oncotarget.3849
26. Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet (2011) 378:1812–23. doi: 10.1016/S0140-6736(11)61539-0
27. Curigliano G, Burstein HJ, Winer EP, Gnant M, Dubsky P, Loibl S, et al. De-escalating and escalating treatments for early-stage breast cancer: the St. Gallen International Expert Consensus Conference on the Primary Therapy of Early Breast Cancer 2017. Ann Oncol. (2018) 28:1700–12. doi: 10.1093/annonc/mdx806
28. Tu SM, Lin SH, Logothetis CJ. Stem-cell origin of metastasis and heterogeneity in solid tumours. Lancet Oncol. (2002) 3:508–13. doi: 10.1016/S1470-2045(02)00820-3
29. Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. (2015) 12:381–94. doi: 10.1038/nrclinonc.2015.73
30. Ercan C, van Diest PJ, Vooijs M. Mammary development and breast cancer: the role of stem cells. Curr Mol Med. (2011) 11:270–85. doi: 10.2174/156652411795678007
31. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
32. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29:4741–51. doi: 10.1038/onc.2010.215
33. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature (2013) 501:346. doi: 10.1038/nature12626
34. Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. (2010) 16:3141–52. doi: 10.1158/1078-0432.CCR-09-2823
35. Takeuchi H, Haltiwanger RS. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun. (2014) 453:235–42. doi: 10.1016/j.bbrc.2014.05.115
36. Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, et al. Fringe is a glycosyltransferase that modifies Notch. Nature (2000) 406:369–75. doi: 10.1038/35019000
37. Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A Ligand-Induced Extracellular Cleavage Regulates γ-Secretase-like Proteolytic Activation of Notch1. Mol Cell (2000) 5:197–206. doi: 10.1016/S1097-2765(00)80416-5
38. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi: 10.1016/j.cell.2009.03.045
39. Wang K, Zhang Q, Li D, Ching K, Zhang C, Zheng X, et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a gamma-secretase inhibitor. Clin Cancer Res. (2015) 21:1487–96. doi: 10.1158/1078-0432.CCR-14-1348
40. O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. The Journal of experimental medicine. (2007) 204:1813–24. doi: 10.1084/jem.20070876
41. Sczaniecka M, Gladstone K, Pettersson S, McLaren L, Huart AS, Wallace M. MDM2 protein-mediated ubiquitination of NUMB protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J. Biol. Chem. (2012) 287:14052–68. doi: 10.1074/jbc.M111.303875
42. Pettersson S, Sczaniecka M, McLaren L, Russell F, Gladstone K, Hupp T, et al. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem J. (2013) 450:523–36. doi: 10.1042/BJ20121249
43. Licciardello MP, Müllner MK, Dürnberger G, Kerzendorfer C, Boidol B, Trefzer C, et al. NOTCH1 activation in breast cancer confers sensitivity to inhibition of SUMOylation. Oncogene (2015) 34:3780–90. doi: 10.1038/onc.2014.319
44. Oladghaffari M, Islamian JP, Baradaran B, Monfared AS, Farajollahi A, Shanehbandi D, et al. High Efficiency Apoptosis Induction in Breast Cancer Cell Lines by MLN4924/2DG Co-Treatment. Asian Pac J Cancer Prev. (2015) 16:5471–6. doi: 10.7314/APJCP.2015.16.13.5471
45. Yang D, Tan M, Wang G, Sun Y. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS ONE (2012) 7:e34079-e. doi: 10.1371/journal.pone.0034079
46. Thaler S, Thiede G, Hengstler JG, Schad A, Schmidt M, Sleeman JP. The proteasome inhibitor Bortezomib (Velcade) as potential inhibitor of estrogen receptor-positive breast cancer. Int J Cancer. (2015) 137:686–97. doi: 10.1002/ijc.29404
47. Engel RH, Brown JA, Von Roenn JH, O'Regan RM, Bergan R, Badve S, et al. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest. (2007) 25:733–7. doi: 10.1080/07357900701506573
48. Schmid P, Kühnhardt D, Kiewe P, Lehenbauer-Dehm S, Schippinger W, Greil R, et al. A phase I/II study of bortezomib and capecitabine in patients with metastatic breast cancer previously treated with taxanes and/or anthracyclines. Ann Oncol.. (2008) 19:871–6. doi: 10.1093/annonc/mdm569
49. Trinh XB, Sas L, Van Laere SJ, Prové A, Deleu I, Rasschaert M, et al. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol Reports (2012) 27:657–63. doi: 10.3892/or.2011.1562
50. Brou C. Intracellular trafficking of Notch receptors and ligands. Exper Cell Res. (2009) 315:1549–55. doi: 10.1016/j.yexcr.2008.09.010
51. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nature Rev Cancer. (2008) 8:83–93. doi: 10.1038/nrc2290
52. Chastagner P, Israël A, Brou C. AIP4/Itch regulates notch receptor degradation in the absence of ligand. PLoS ONE (2008) 3:e2735. doi: 10.1371/journal.pone.0002735
53. Kim MY, Mo JS, Ann EJ, Yoon JH, Park HS. Dual regulation of Notch1 signaling pathway by adaptor protein Fe65. J Biol Chem. (2012) 287:4690–701. doi: 10.1074/jbc.M111.289637
54. Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. (2004) 167:215–21. doi: 10.1083/jcb.200406140
55. Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, et al. The role of presenilin cofactors in the γ-secretase complex. Nature (2003) 422:438. doi: 10.1038/nature01506
56. Groot AJ, Habets R, Yahyanejad S, Hodin CM, Reiss K, Saftig P, et al. Regulated Proteolysis of NOTCH2 and NOTCH3 Receptors by ADAM10 and Presenilins. Mol Cell Biol. (2014) 34:2822. doi: 10.1128/MCB.00206-14
57. Hao L, Rizzo P, Osipo C, Pannuti A, Wyatt D, Cheung LW, et al. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene (2010) 29:201–13. doi: 10.1038/onc.2009.323
58. Li L, Zhao F, Lu J, Li T, Yang H, Wu C, et al. Notch-1 Signaling Promotes the Malignant Features of Human Breast Cancer through NF-κB Activation. PLoS ONE (2014) 9:e95912. doi: 10.1371/journal.pone.009591
59. Hales EC, Taub JW, Matherly LH. New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of gamma-secretase inhibitor resistant T-cell acute lymphoblastic leukemia. Cell Signal. (2014) 26:149–61. doi: 10.1016/j.cellsig.2013.09.021
60. Landor SKJ, Mutvei AP, Mamaeva V, Jin S, Busk M, Borra R, et al. Hypo- and hyperactivated Notch signaling induce a glycolytic switch through distinct mechanisms. Proc Natl Acad Sci USA. (2011) 108:18814. doi: 10.1073/pnas.1104943108
61. Perumalsamy LR, Nagala M, Sarin A. Notch-activated signaling cascade interacts with mitochondrial remodeling proteins to regulate cell survival. Proc Natl. Acad Sci USA. (2010) 107:6882. doi: 10.1073/pnas.0910060107
62. Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl. Acad Sci USA. (2010) 107:5012–7. doi: 10.1073/pnas.1000896107
63. Flores AN, McDermott N, Meunier A, Marignol L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer. Nat Rev Urol. (2014) 11:499–507. doi: 10.1038/nrurol.2014.195
64. Espinosa L, Inglés-Esteve J, Aguilera C, Bigas A. Phosphorylation by Glycogen Synthase Kinase-3β Down-regulates Notch Activity, a Link for Notch and Wnt Pathways. J Biol Chem. (2003) 278:32227–35. doi: 10.1074/jbc.M304001200
65. Jin YH, Kim H, Oh M, Ki H, Kim K. Regulation of Notch1/NICD and Hes1 expressions by GSK-3α/β. Mol Cells (2009) 27:15–9. doi: 10.1007/s10059-009-0001-7
66. Hein K, Mittler G, Cizelsky W, Kühl M, Ferrante F, Liefke R, et al. Site-specific methylation of Notch1 controls the amplitude and duration of the Notch1 response. Sci Signal. (2015) 8:ra30. doi: 10.1126/scisignal.2005892
67. Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. (2004) 16:509–20. doi: 10.1016/j.molcel.2004.10.014
68. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. (2011) 11:338–51. doi: 10.1038/nrc3035
69. Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annu Rev Pathol. (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
70. Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta (2011) 1815:197–213. doi: 10.1016/j.bbcan.2010.12.002
71. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
72. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. (2014) 13:357–78. doi: 10.1038/nrd4252
73. Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, et al. γ-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. (2017) 9:950. doi: 10.15252/emmm.201607265
74. Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. (2013) 6:1249–59. doi: 10.2147/OTT.S36162
75. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. (2013) 341:41–5. doi: 10.1016/j.canlet.2013.08.027
76. Kim SJ, Lee HW, Baek JH, Cho YH, Kang HG, Jeong JS, et al. Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene (2016) 35:251–60. doi: 10.1038/onc.2015.80
77. McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci USA. (2012) 109:E2939–48. doi: 10.1073/pnas.1206400109
78. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. (2012) 22:373–88. doi: 10.1016/j.ccr.2012.07.016
79. Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, et al. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. (2012) 113:1501–13. doi: 10.1002/jcb.24019
80. Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta (2010) 1806:258–67. doi: 10.1016/j.bbcan.2010.06.001
81. Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. (2011) 31:1105–13.
82. Russo J, Russo IH. Development of the human breast. Maturitas (2004) 49:2–15. doi: 10.1016/j.maturitas.2004.04.011
83. Kordon EC, Smith GH. An entire functional mammary gland may comprise the progeny from a single cell. Development (1998) 125:1921–30.
84. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. (2003) 17:1253–70. doi: 10.1101/gad.1061803
85. Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature (2014) 506:322–7. doi: 10.1038/nature12948
86. Tiede B, Kang Y. From milk to malignancy: the role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. (2011) 21:245–57. doi: 10.1038/cr.2011.11
87. Junankar S, Baker LA, Roden DL, Nair R, Elsworth B, Gallego-Ortega D, et al. ID4 controls mammary stem cells and marks breast cancers with a stem cell-like phenotype. Nat Commun. (2015) 6:6548. doi: 10.1038/ncomms7548
88. Pamarthy S, Mao L, Katara GK, Fleetwood S, Kulshreshta A, Gilman-Sachs A, et al. The V-ATPase a2 isoform controls mammary gland development through Notch and TGF-beta signaling. Cell Death Dis. (2016) 7:e2443. doi: 10.1038/cddis.2016.347
89. Rodilla V, Dasti A, Huyghe M, Lafkas D, Laurent C, Reyal F, et al. Luminal progenitors restrict their lineage potential during mammary gland development. PLoS Biol. (2015) 13:e1002069. doi: 10.1371/journal.pbio.1002069
90. Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell (2008) 3:429–41. doi: 10.1016/j.stem.2008.08.001
91. Lafkas D, Rodilla V, Huyghe M, Mourao L, Kiaris H, Fre S. Notch3 marks clonogenic mammary luminal progenitor cells in vivo. J Cell Biol. (2013) 203:47. doi: 10.1083/jcb.201307046
92. Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. (2004) 6:R605–15. doi: 10.1186/bcr920
93. Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. (2008) 3:109–18. doi: 10.1016/j.stem.2008.05.018
94. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. (2010) 70:709–18. doi: 10.1158/0008-5472.CAN-09-1681
95. Simoes BM, O'Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. (2015) 12:1968–77. doi: 10.1016/j.celrep.2015.08.050
96. Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, et al. Crosstalk between PKCalpha and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis (2013) 2:e60. doi: 10.1038/oncsis.2013.26
97. Yalcin-Ozuysal O, Fiche M, Guitierrez M, Wagner KU, Raffoul W, Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. (2010) 17:1600–12. doi: 10.1038/cdd.2010.37
98. Sizemore GM, Balakrishnan S, Hammer AM, Thies KA, Trimboli AJ, Wallace JA, et al. Stromal PTEN inhibits the expansion of mammary epithelial stem cells through Jagged-1. Oncogene (2017) 36:6658. doi: 10.1038/onc.2017.347
99. Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. (2006) 66:1517–25. doi: 10.1158/0008-5472.CAN-05-3054
100. Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. (2009) 69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741
101. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
102. Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J-i. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nature Commun. (2013) 4:2299. doi: 10.1038/ncomms3299
103. Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell (2011) 19:192–205. doi: 10.1016/j.ccr.2010.12.022
104. Leontovich AA, Jalalirad M, Salisbury JL, Mills L, Haddox C, Schroeder M, et al. NOTCH3 expression is linked to breast cancer seeding and distant metastasis. Breast Cancer Res. (2018) 20:105. doi: 10.1186/s13058-018-1020-0
105. Nam D-H, Jeon H-M, Kim S, Kim MH, Lee Y-J, Lee MS, et al. Activation of Notch Signaling in a Xenograft Model of Brain Metastasis. Clin Cancer Res. (2008) 14:4059–66. doi: 10.1158/1078-0432.CCR-07-4039
106. McGowan PM, Simedrea C, Ribot EJ, Foster PJ, Palmieri D, Steeg PS, et al. Notch1 Inhibition Alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol Cancer Res. (2011) 9:834. doi: 10.1158/1541-7786.MCR-10-0457
107. Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. (2013) 5:384. doi: 10.1002/emmm.201201623
108. Oskarsson T, Acharyya S, Zhang XHF, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Med. (2011) 17:867. doi: 10.1038/nm.2379
109. Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. (2007) 99:616–27. doi: 10.1093/jnci/djk133
110. Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene (1997) 14:1883–90. doi: 10.1038/sj.onc.1201035
111. Dievart A, Beaulieu N, Jolicoeur P. Involvement of Notch1 in the development of mouse mammary tumors. Oncogene (1999) 18:5973–81. doi: 10.1038/sj.onc.1202991
112. Raafat A, Bargo S, Anver MR, Callahan R. Mammary development and tumorigenesis in mice expressing a truncated human Notch4/Int3 intracellular domain (h-Int3sh). Oncogene (2004) 23:9401–7. doi: 10.1038/sj.onc.1208187
113. Kiaris H, Politi K, Grimm LM, Szabolcs M, Fisher P, Efstratiadis A, et al. Modulation of Notch Signaling Elicits Signature Tumors and Inhibits Hras1-Induced Oncogenesis in the Mouse Mammary Epithelium. Am J Pathol. (2004) 165:695–705. doi: 10.1016/S0002-9440(10)63333-0
114. Hu C, Diévart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of Activated Murine Notch1 and Notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol. (2006) 168:973–90. doi: 10.2353/ajpath.2006.050416
115. Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. (2005) 65:8530–7. doi: 10.1158/0008-5472.CAN-05-1069
116. Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, et al. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast Cancer Res Treat. (2008) 111:439–48. doi: 10.1007/s10549-007-9805-3
117. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell (2017) 31:355–67. doi: 10.1016/j.ccell.2017.01.007
118. Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, Sterner CJ, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest. (2015) 125:2484–96. doi: 10.1172/JCI74883
119. Dai X, Li Y, Bai Z, Tang XQ. Molecular portraits revealing the heterogeneity of breast tumor subtypes defined using immunohistochemistry markers. Sci Rep. (2015) 5:14499. doi: 10.1038/srep14499
120. Yi Z, Ma F, Li C, Chen R, Yuan L, Sun X, et al. Landscape of somatic mutations in different subtypes of advanced breast cancer with circulating tumor DNA analysis. Sci Rep. (2017) 7:5995. doi: 10.1038/s41598-017-06327-4
121. Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, et al. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res Treat. (2010) 123:113–24. doi: 10.1007/s10549-009-0621-9
122. Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell (1995) 82:621–30. doi: 10.1016/0092-8674(95)90034-9
123. Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. (1994) 54:1812–7.
124. Ling H, Sylvestre JR, Jolicoeur P. Notch1-induced mammary tumor development is cyclin D1-dependent and correlates with expansion of pre-malignant multipotent duct-limited progenitors. Oncogene (2010) 29:4543–54. doi: 10.1038/onc.2010.186
125. Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. (2014) 4:1154–67. doi: 10.1158/2159-8290.CD-13-0830
126. Xie X, Kaoud TS, Edupuganti R, Zhang T, Kogawa T, Zhao Y, et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene (2017) 36:2599–608. doi: 10.1038/onc.2016.417
127. Dickson BC, Mulligan AM, Zhang H, Lockwood G, O'Malley FP, Egan SE, et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. (2007) 20:685–93. doi: 10.1038/modpathol.3800785
128. Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. (2007) 204:2935–48. doi: 10.1084/jem.20071082
129. Bolos V, Mira E, Martinez-Poveda B, Luxan G, Canamero M, Martinez AC, et al. Notch activation stimulates migration of breast cancer cells and promotes tumor growth. Breast Cancer Res. (2013) 15:R54. doi: 10.1186/bcr3447
130. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. (2008) 105:6392–7. doi: 10.1073/pnas.0802047105
131. Chen J, Imanaka N, Chen J, Griffin JD. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br J Cancer. (2010) 102:351–60. doi: 10.1038/sj.bjc.6605486
132. Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, Liu W, et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene (2011) 30:4075–86. doi: 10.1038/onc.2011.122
133. Ercan C, Vermeulen JF, Hoefnagel L, Bult P, van der Groep P, van der Wall E, et al. HIF-1α and NOTCH signaling in ductal and lobular carcinomas of the breast. Cell Oncol. (2012) 35:435–42. doi: 10.1007/s13402-012-0102-8
134. Johnson RW, Finger EC, Olcina MM, Vilalta M, Aguilera T, Miao Y, et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat Cell Biol. (2016) 18:1078–89. doi: 10.1038/ncb3408
135. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. (2013) 5:180ra48. doi: 10.1126/scitranslmed.3005109
136. Boral D, Vishnoi M, Liu HN, Yin W, Sprouse ML, Scamardo A, et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun. (2017) 8:196. doi: 10.1038/s41467-017-00196-1
137. Shimizu K, Chiba S, Saito T, Kumano K, Hamada Y, Hirai H. Functional diversity among Notch1, Notch2, and Notch3 receptors. Biochem Biophys Res Commun. (2002) 291:775–9. doi: 10.1006/bbrc.2002.6528
138. Sale S, Lafkas D, Artavanis-Tsakonas S. Notch2 genetic fate mapping reveals two previously unrecognized mammary epithelial lineages. Nat Cell Biol. (2013) 15:451–60. doi: 10.1038/ncb2725
139. Lee JH, Zhao XM, Yoon I, Lee JY, Kwon NH, Wang YY, et al. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov. (2016) 2:16025. doi: 10.1038/celldisc.2016.25
140. Florena AM, Tripodo C, Guarnotta C, Ingrao S, Porcasi R, Martorana A, et al. Associations between Notch-2, Akt-1 and HER2/neu expression in invasive human breast cancer: a tissue microarray immunophenotypic analysis on 98 patients. Pathobiology (2007) 74:317–22. doi: 10.1159/000110024
141. Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. (2004) 14:779–86. doi: 10.3892/ijmm.14.5.779
142. O'Neill CF, Urs S, Cinelli C, Lincoln A, Nadeau RJ, Leon R, et al. Notch2 signaling induces apoptosis and inhibits human MDA-MB-231 xenograft growth. Am J Pathol. (2007) 171:1023–36. doi: 10.2353/ajpath.2007.061029
143. Lee G-H, Yoo K-C, An Y, Lee H-J, Lee M, Uddin N, et al. FYN promotes mesenchymal phenotypes of basal type breast cancer cells through STAT5/NOTCH2 signaling node. Oncogene (2018) 37:1857–68. doi: 10.1038/s41388-017-0114-y
144. Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. (2015) 21:2084-95. doi: 10.1158/1078-0432.CCR-14-2808
145. Anjanappa M, Hao Y, Simpson ER, Bhat-Nakshatri P, Nelson JB, Tersey SA, et al. A system for detecting high impact-low frequency mutations in primary tumors and metastases. Oncogene (2018) 37:185–96. doi: 10.1038/onc.2017.32211.
146. Kim SH, Hahm ER, Arlotti JA, Samanta SK, Moura MB, Thorne SH, et al. Withaferin A inhibits in vivo growth of breast cancer cells accelerated by Notch2 knockdown. Breast Cancer Res Treat. (2016) 157:41–54. doi: 10.1007/s10549-016-3795-y
147. Yamaguchi N, Oyama T, Ito E, Satoh H, Azuma S, Hayashi M, et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res. (2008) 68:1881–8. doi: 10.1158/0008-5472.CAN-07-1597
148. Pradeep CR, Kostler WJ, Lauriola M, Granit RZ, Zhang F, Jacob-Hirsch J, et al. Modeling ductal carcinoma in situ: a HER2-Notch3 collaboration enables luminal filling. Oncogene (2012) 31:907–17. doi: 10.1038/onc.2011.279
149. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene (2010) 29:2013–23. doi: 10.1038/onc.2009.489
150. Cui H, Kong Y, Xu M, Zhang H. Notch3 functions as a tumor suppressor by controlling cellular senescence. Cancer Res. (2013) 73:3451–9. doi: 10.1158/0008-5472.CAN-12-3902
151. Zhang X, Liu X, Luo J, Xiao W, Ye X, Chen M, et al. Notch3 inhibits epithelial-mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis (2016) 5:e269. doi: 10.1038/oncsis.2016.67
152. Diluvio G, Del Gaudio F, Giuli MV, Franciosa G, Giuliani E, Palermo R, et al. NOTCH3 inactivation increases triple negative breast cancer sensitivity to gefitinib by promoting EGFR tyrosine dephosphorylation and its intracellular arrest. Oncogenesis (2018) 7:42. doi: 10.1038/s41389-018-0051-9
153. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature (2015) 526:131–5. doi: 10.1038/nature15260
154. Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Semin Cancer Biol. (2004) 14:341–7. doi: 10.1016/j.semcancer.2004.04.013
155. Robbins J, Blondel BJ, Gallahan D, Callahan R. Mouse mammary tumor gene int-3: a member of the notch gene family transforms mammary epithelial cells. J Virol. (1992) 66:2594–9.
156. Jhappan C, Gallahan D, Stahle C, Chu E, Smith GH, Merlino G, et al. Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. (1992) 6:345–55. doi: 10.1101/gad.6.3.345
157. Raafat A, Lawson S, Bargo S, Klauzinska M, Strizzi L, Goldhar AS, et al. Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene (2009) 28:219–30. doi: 10.1038/onc.2008.379
158. Raafat A, Bargo S, McCurdy D, Callahan R. The ANK repeats of Notch-4/Int3 activate NF-kappaB canonical pathway in the absence of Rbpj and causes mammary tumorigenesis. Sci Rep. (2017) 7:13690. doi: 10.1038/s41598-017-13989-7
159. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science (2004) 306:269. doi: 10.1126/science.1102160
160. Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. (2008) 8:380–90. doi: 10.1038/nri2304
161. Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates Notch-ligand interactions. Nature (1997) 387:908–12. doi: 10.1038/43191
162. Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol. (2003) 4:786–97. doi: 10.1038/nrm1228
163. Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell (2012) 21:626–41. doi: 10.1016/j.ccr.2012.03.041
164. Zhang S, Chung WC, Miele L, Xu K. Targeting Met and Notch in the Lfng-deficient, Met-amplified triple-negative breast cancer. Cancer Biol Ther. (2014) 15:633–42. doi: 10.4161/cbt.28180
165. Chung WC, Zhang S, Challagundla L, Zhou Y, Xu K. Lunatic Fringe and p53 Cooperatively Suppress Mesenchymal Stem-Like Breast Cancer. Neoplasia (2017) 19:885–95. doi: 10.1016/j.neo.2017.08.006
166. Zhang S, Chung WC, Wu G, Egan SE, Miele L, Xu K. Manic fringe promotes a claudin-low breast cancer phenotype through notch-mediated PIK3CG induction. Cancer Res. (2015) 75:1936–43. doi: 10.1158/0008-5472.CAN-14-3303
167. Westhoff B, Colaluca IN, D'Ario G, Donzelli M, Tosoni D, Volorio S, et al. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci USA. (2009) 106:22293–8. doi: 10.1073/pnas.0907781106
168. Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, Viale G, et al. NUMB controls p53 tumour suppressor activity. Nature (2008) 451:76–80. doi: 10.1038/nature06412
169. Tosoni D, Pambianco S, Ekalle Soppo B, Zecchini S, Bertalot G, Pruneri G, et al. Pre-clinical validation of a selective anti-cancer stem cell therapy for Numb-deficient human breast cancers. EMBO Mol Med. (2017) 9:655–71. doi: 10.15252/emmm.201606940
170. Pece S, Confalonieri S, P RR, Di Fiore PP. NUMB-ing down cancer by more than just a NOTCH. Biochim Biophys Acta. (2011) 1815:26-43. doi: 10.1016/j.bbcan.2010.10.001
171. Juven-Gershon T, Shifman O, Unger T, Elkeles A, Haupt Y, Oren M. The Mdm2 oncoprotein interacts with the cell fate regulator Numb. Mol Cell Biol. (1998) 18:3974–82. doi: 10.1128/MCB.18.7.3974
172. Yogosawa S, Miyauchi Y, Honda R, Tanaka H, Yasuda H. Mammalian Numb is a target protein of Mdm2, ubiquitin ligase. Biochem Biophys Res Commun. (2003) 302:869–72. doi: 10.1016/S0006-291X(03)00282-1
173. Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. (2011) 17:1646–51. doi: 10.1038/nm.2580
174. Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature (2000) 407:48. doi: 10.1038/35024009
175. Yu G, Chen F, Nishimura M, Steiner H, Tandon A, Kawarai T, et al. Mutation of conserved aspartates affect maturation of both aspartate-mutant and endogenous presenilin 1 and presenilin 2 complexes. J Biol Chem. (2000) 275:27348–53. doi: 10.1074/jbc.M002982200
176. Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature (1999) 398:513. doi: 10.1038/19077
177. Filipović A, Gronau JH, Green AR, Wang J, Vallath S, Shao D, et al. Biological and clinical implications of nicastrin expression in invasive breast cancer. Breast Cancer Res Treat. (2011) 125:43–53. doi: 10.1007/s10549-010-0823-1
178. Sarajlić A, Filipović A, Janjić V, Coombes RC, PrŽulj N. The role of genes co-amplified with nicastrin in breast invasive carcinoma. Breast Cancer Res Treat. (2014) 143:393–401. doi: 10.1007/s10549-013-2805-6
179. Lombardo Y, Filipović A, Molyneux G, Periyasamy M, Giamas G, Hu Y, et al. Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci USA. (2012) 109:16558. doi: 10.1073/pnas.1206268109
180. Filipović A, Lombardo Y, Fronato M, Abrahams J, Aboagye E, Nguyen Q-D, et al. Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res Treat. (2014) 148:455–62. doi: 10.1007/s10549-014-3119-z
181. Studebaker AW, Storci G, Werbeck JL, Sansone P, Sasser AK, Tavolari S, et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. (2008) 68:9087–95. doi: 10.1158/0008-5472.CAN-08-0400
182. Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C, et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene (2011) 30:1082–97. doi: 10.1038/onc.2010.487
183. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates crosstalk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 72, 2768–79. doi: 10.1158/0008-5472.CAN-11-3567
184. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. (2014). Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell (2012) 159:499–513. doi: 10.1016/j.cell.2014.09.051
185. Pupo M, Pisano A, Abonante S, Maggiolini M, Musti AM. GPER activates Notch signaling in breast cancer cells and cancer-associated fibroblasts (CAFs). Int J Biochem Cell Biol. (2014) 46:56–67. doi: 10.1016/j.biocel.2013.11.011
186. Soares R, Balogh G, Guo S, Gärtner F, Russo J, Schmitt F. Evidence for the Notch Signaling Pathway on the Role of Estrogen in Angiogenesis. Molecular Endocrinology. (2004) 18:2333-43. doi: 10.1210/me.2003-0362
187. Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Rafii S, et al. Endothelial cells provide a Notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE (2014) 9:e112424. doi: 10.1371/journal.pone.0112424
188. Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K, et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc Natl. Acad Sci USA. (2006) 103:3799. doi: 10.1073/pnas.0600065103
189. Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Investig. (2007) 117:3988–4002. doi: 10.1172/JCI32533
190. Wolfson B, Eades G, Zhou Q. Adipocyte activation of cancer stem cell signaling in breast cancer. World J Biol Chem. (2015) 6:39–47. doi: 10.4331/wjbc.v6.i2.39
191. Battle M, Gillespie C, Quarshie A, Lanier V, Harmon T, Wilson K, et al. Obesity Induced a Leptin-Notch Signaling Axis in Breast Cancer. Int J Cancer J Int du Cancer (2014) 134:1605–16. doi: 10.1002/ijc.28496
192. Pedrosa AR, Trindade A, Fernandes AC, Carvalho C, Gigante J, Tavares AT, et al. Endothelial jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler Thrombos Vasc Biol. (2015) 35:1134–46. doi: 10.1161/ATVBAHA.114.304741
193. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature (2006) 444:1032–7. doi: 10.1038/nature05355
194. Paris D, Quadros A, Patel N, DelleDonne A, Humphrey J, Mullan M. Inhibition of angiogenesis and tumor growth by β and γ-secretase inhibitors. Eur J Pharmacol. (2005) 514:1–15. doi: 10.1016/j.ejphar.2005.02.050
195. Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, et al. A phase I first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res. (2015) 21:2695–703. doi: 10.1158/1078-0432.CCR-14-2797
196. Janghorban M, Xin L, Rosen JM, Zhang XHF. Notch signaling as a regulator of the tumor immune response: to target or not to target? Front Immunol. (2018) 9:1649. doi: 10.3389/fimmu.2018.01649
197. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nature Rev Immunol. (2013) 13:427–37. doi: 10.1038/nri3445
198. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
199. Grazioli P, Felli MP, Screpanti I, Campese AF. The mazy case of Notch and immunoregulatory cells. J leukocyte Biol. (2017) 102:361–8. doi: 10.1189/jlb.1VMR1216-505R
200. Liu H, Wang J, Zhang M, Xuan Q, Wang Z, Lian X, et al. Jagged1 promotes aromatase inhibitor resistance by modulating tumor-associated macrophage differentiation in breast cancer patients. Breast Cancer Res Treat. (2017) 166:95–107. doi: 10.1007/s10549-017-4394-2
201. Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. (2015) 17:101. doi: 10.1186/s13058-015-0621-0
202. Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, Murakami K, et al. Notch Shapes the Innate Immunophenotype in Breast Cancer. Cancer Disc. (2017) 7:1320–35. doi: 10.1158/2159-8290.CD-17-0037
203. Biswas SK, Allavena P, Mantovani A. Tumor-associated macrophages: functional diversity, clinical significance, and open questions. Semin Immunopathol. (2013) 35:585–600. doi: 10.1007/s00281-013-0367-7
204. Cheng P, Kumar V, Liu H, Youn J-I, Fishman M, Sherman S, et al. Effects of Notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res. (2014) 74:141 LP-52. doi: 10.1158/0008-5472.CAN-13-1686
205. Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nature Cell Biol. (2016) 18:632–44. doi: 10.1038/ncb3355
206. Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-Derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. (2016) 76:3156–65. doi: 10.1158/0008-5472.CAN-15-2528
207. Shevach EM. Biological functions of regulatory T cells. Adv Immunol. (2011) 112:137–76. doi: 10.1016/B978-0-12-387827-4.00004-8
208. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood (2008) 112:1813–21. doi: 10.1182/blood-2008-03-144980
209. Kared H, Adle-Biassette H, Foïs E, Masson A, Bach J-F, Chatenoud L, et al. Jagged2-expressing hematopoietic progenitors promote regulatory T cell expansion in the periphery through notch signaling. Immunity (2006) 25:823–34. doi: 10.1016/j.immuni.2006.09.008
210. Cho OH, Shin HM, Miele L, Golde TE, Fauq A, Minter LM, et al. Notch Regulates Cytolytic Effector Function in CD8+ T Cells. J Immunol. (2009) 182:3380 LP-9. doi: 10.4049/jimmunol.0802598
211. Sugimoto K, Maekawa Y, Kitamura A, Nishida J, Koyanagi A, Yagita H, et al. Notch2 signaling is required for potent antitumor immunity in vivo. J Immunol. (2010) 184:4673–8. doi: 10.4049/jimmunol.0903661
212. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. (2018) 34:536–48. doi: 10.1016/j.ccell.2018.07.009
213. Nakasone ES, Hurvitz SA, McCann KE. Harnessing the immune system in the battle against breast cancer. Drugs Context. (2018) 7:212520. doi: 10.7573/dic.212520
214. Mao L, Zhao Z-L, Yu G-T, Wu L, Deng W-W, Li Y-C, et al. γ-Secretase inhibitor reduces immunosuppressive cells and enhances tumour immunity in head and neck squamous cell carcinoma. Int J Cancer (2017) 142:999–1009. doi: 10.1002/ijc.31115
215. Speers C, Zhao S, Liu M, Bartelink H, Pierce LJ, Feng FY. Development and Validation of a Novel Radiosensitivity Signature in Human Breast Cancer. Clin Cancer Res. (2015) 21:3667. doi: 10.1158/1078-0432.CCR-14-2898
216. Tramm T, Mohammed H, Myhre S, Kyndi M, Alsner J, Børresen-Dale A-L, et al. Development and Validation of a Gene Profile Predicting Benefit of Postmastectomy Radiotherapy in Patients with High-Risk Breast Cancer: a study of gene expression in the DBCG82bc Cohort. Clin Cancer Res. (2014) 20:5272. doi: 10.1158/1078-0432.CCR-14-0458
217. Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: the 4 R's of radiobiology revisited. Stem Cells (2010) 28:639–48. doi: 10.1002/stem.318
218. Rich JN. Cancer stem cells in radiation resistance. Cancer Res. (2007) 67:8980–4. doi: 10.1158/0008-5472.CAN-07-0895
219. Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. (2006) 98:1777–85. doi: 10.1093/jnci/djj495
220. Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. (2007) 104:618–23. doi: 10.1073/pnas.0606599104
221. Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. (2012) 30:833–44. doi: 10.1002/stem.1058
222. Lagadec C, Vlashi E, Alhiyari Y, Phillips TM, Dratver MB, Pajonk F. Radiation-Induced Notch Signaling in Breast Cancer Stem Cells. Int J Radiat Oncol Biol Phys. (2013) 87:609–18. doi: 10.1016/j.ijrobp.2013.06.2064
223. Vermezovic J, Adamowicz M, Santarpia L, Rustighi A, Forcato M, Lucano C, et al. Notch is a direct negative regulator of the DNA-damage response. Nature Struc Mol Biol. (2015) 22:417. doi: 10.1038/nsmb.3013
224. Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. (2005) 205:275–92. doi: 10.1002/path.1706
225. Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. (2013) 2:3. doi: 10.1186/2001-1326-2-3
226. Zang S, Chen F, Dai J, Guo D, Tse W, Qu X, et al. RNAi-mediated knockdown of Notch-1 leads to cell growth inhibition and enhanced chemosensitivity in human breast cancer. Oncol Rep. (2010) 23:893–9. doi: 10.3892/or_00000712
227. Li ZL, Chen C, Yang Y, Wang C, Yang T, Yang X, et al. Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int J Clin Exp Pathol. (2015) 8:4378–87.
228. Mirkes PE, Ellison A, Little SA. Role of aldehyde dehydrogenase (ALDH) in the detoxication of cyclophosphamide (CP) in rat embryos. Adv Exp Med Biol. (1991) 284:85-95. doi: 10.1007/978-1-4684-5901-2_11
229. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. (2010) 28:4006–12. doi: 10.1200/JCO.2009.27.5388
230. Chuthapisith S, Eremin J, El-Sheemey M, Eremin O. Breast cancer chemoresistance: emerging importance of cancer stem cells. Surg Oncol. (2010) 19:27–32. doi: 10.1016/j.suronc.2009.01.004
231. Suman S, Das TP, Damodaran C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br J Cancer (2013) 109:2587–96. doi: 10.1038/bjc.2013.642
232. Kim B, Stephen SL, Hanby AM, Horgan K, Perry SL, Richardson J, et al. Chemotherapy induces Notch1-dependent MRP1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer (2015) 15:634. doi: 10.1186/s12885-015-1625-y
233. Gu X, Lu C, He D, Lu Y, Jin J, Liu D, et al. Notch3 negatively regulates chemoresistance in breast cancers. Tumour Biol. (2016) 37:15825–33. doi: 10.1007/s13277-016-5412-4
234. de Weger VA, Beijnen JH, Schellens JH. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel–a review. Anticancer Drugs (2014) 25:488–94. doi: 10.1097/CAD.0000000000000093
235. Gradishar WJ. Taxanes for the treatment of metastatic breast cancer. Breast Cancer (Auckl). (2012) 6:159–71. doi: 10.4137/BCBCR.S8205
236. Qiu M, Peng Q, Jiang I, Carroll C, Han G, Rymer I, et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. (2013) 328:261–70. doi: 10.1016/j.canlet.2012.09.023
237. Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, et al. Synergistic effect of the gamma-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. (2013) 2:233–42. doi: 10.5966/sctm.2012-0096
238. Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. (2013) 19:1512–24. doi: 10.1158/1078-0432.CCR-11-3326
239. Locatelli MA, Aftimos P, Dees EC, LoRusso PM, Pegram MD, Awada A, et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget (2017) 8:2320–8. doi: 10.18632/oncotarget.13727
240. Jeong YJ, Kang JS, Lee SI, So DM, Yun J, Baek JY, et al. Breast cancer cells evade paclitaxel-induced cell death by developing resistance to dasatinib. Oncol Lett. (2016) 12:2153–8. doi: 10.3892/ol.2016.4852
241. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. (2009) 5:168–77. doi: 10.1016/j.stem.2009.05.019
242. Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell (2017) 32:731–47 e6. doi: 10.1016/j.ccell.2017.11.002
243. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology (2003) 144:4562–74. doi: 10.1210/en.2003-0567
244. Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature (2010) 465:803–7. doi: 10.1038/nature09091
245. Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature (2010) 465:798–802. doi: 10.1038/nature09027
246. Hoefnagel LD, van de Vijver MJ, van Slooten HJ, Wesseling P, Wesseling J, Westenend PJ, et al. Receptor conversion in distant breast cancer metastases. Breast Cancer Res. (2010) 12:R75. doi: 10.1186/bcr2645
247. Hoefnagel LD, Moelans CB, Meijer SL, van Slooten HJ, Wesseling P, Wesseling J, et al. Prognostic value of estrogen receptor alpha and progesterone receptor conversion in distant breast cancer metastases. Cancer (2012) 118:4929–35. doi: 10.1002/cncr.27518
248. Fuqua SAW, Gu G, Rechoum Y. Estrogen Receptor (ER) Mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat. (2014) 144:11–9. doi: 10.1007/s10549-014-2847-4
249. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations as a mechanism for acquired endocrine resistance in breast cancer. Nature Rev Clin Oncol. (2015) 12:573–83. doi: 10.1038/nrclinonc.2015.117
250. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer (2009) 9:631–43. doi: 10.1038/nrc2713
251. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
252. Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. (2017) 77:1927–41. doi: 10.1158/0008-5472.CAN-16-2129
253. Harrison H, Simoes BM, Rogerson L, Howell SJ, Landberg G, Clarke RB. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. (2013) 15:R21. doi: 10.1186/bcr3396
254. Rizzo P, Miao H, D'Souza G, Osipo C, Song LL, Yun J, et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. (2008) 68:5226–35. doi: 10.1158/0008-5472.CAN-07-5744
255. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. (2004) 96:926–35. doi: 10.1093/jnci/djh166
256. Izrailit J, Berman HK, Datti A, Wrana JL, Reedijk M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGFbeta pathways as fundamental Notch regulators in breast cancer. Proc Natl Acad Sci USA. (2013) 110:1714–9. doi: 10.1073/pnas.1214014110
257. Mittal S, Subramanyam D, Dey D, Kumar RV, Rangarajan A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol Cancer. (2009) 8:128. doi: 10.1186/1476-4598-8-128
258. Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA. (2008) 105:5774–9. doi: 10.1073/pnas.0706216105
259. Jin S, Mutvei AP, Chivukula IV, Andersson ER, Ramskold D, Sandberg R, et al. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKalpha/IKKbeta. Oncogene (2013) 32:4892–902. doi: 10.1038/onc.2012.517
260. Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, et al. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells (2007) 25:807–15. doi: 10.1634/stemcells.2006-0442
261. Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun. (2016) 7:10442. doi: 10.1038/ncomms10442
262. Magnani L, Stoeck A, Zhang X, Lanczky A, Mirabella AC, Wang TL, et al. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci USA. (2013) 110:E1490–9. doi: 10.1073/pnas.1219992110
263. Lombardo Y, Faronato M, Filipovic A, Vircillo V, Magnani L, Coombes RC. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. Breast Cancer Res. (2014) 16:R62. doi: 10.1186/bcr3675
264. Bui QT, Im JH, Jeong SB, Kim YM, Lim SC, Kim B, et al. Essential role of Notch4/STAT3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human breast cancer. Cancer Lett. (2017) 390:115–25. doi: 10.1016/j.canlet.2017.01.014
265. Gelsomino L, Panza S, Giordano C, Barone I, Gu G, Spina E, et al. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Letts. (2018) 428:12–20. doi: 10.1016/j.canlet.2018.04.023
266. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. (2011) 9:16–32. doi: 10.1038/nrclinonc.2011.177
267. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
268. Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature (2016) 540:552–8. doi: 10.1038/nature20785
269. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature (2016) 540:588–92. doi: 10.1038/nature20609
270. Schwartz RS, Erban JK. Timing of metastasis in breast cancer. N Engl J Med. (2017) 376:2486–8. doi: 10.1056/NEJMcibr1701388
271. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell (2015) 163:506–19. doi: 10.1016/j.cell.2015.09.033
272. Balduzzi S, Mantarro S, Guarneri V, Tagliabue L, Pistotti V, Moja L, et al. Trastuzumab-containing regimens for metastatic breast cancer. Cochrane Database Syst Rev. (2014) 12:CD006242. doi: 10.1002/14651858.CD006242.pub2
273. Boix-Perales H, Borregaard J, Jensen KB, Ersboll J, Galluzzo S, Giuliani R, et al. The European Medicines Agency Review of Pertuzumab for the treatment of adult patients with HER2-positive metastatic or locally recurrent unresectable breast cancer: summary of the scientific assessment of the committee for medicinal products for human use. Oncologist. (2014) 19:766–73. doi: 10.1634/theoncologist.2013-0348
274. Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. (2010) 28:1124–30. doi: 10.1200/JCO.2008.21.4437
275. Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. (2006) 66:1630–9. doi: 10.1158/0008-5472.CAN-05-1182
276. Kumler I, Tuxen MK, Nielsen DL. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev. (2014) 40:259–70. doi: 10.1016/j.ctrv.2013.09.002
277. Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. (2012) 13:25–32. doi: 10.1016/S1470-2045(11)70336-9
278. Gianni L, Pienkowski T, Im YH, Tseng LM, Liu MC, Lluch A, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. (2016) 17:791–800. doi: 10.1016/S1470-2045(16)00163-7
279. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet (2012) 379:633–40. doi: 10.1016/S0140-6736(11)61847-3
280. de Azambuja E, Holmes AP, Piccart-Gebhart M, Holmes E, Di Cosimo S, Swaby RF, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): survival outcomes of a randomised, open-label, multicentre, phase 3 trial and their association with pathological complete response. Lancet Oncol. (2014) 15:1137–46. doi: 10.1016/S1470-2045(14)70320-1
281. Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. (2010) 28:1138–44. doi: 10.1200/JCO.2009.24.2024
282. Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. (2011) 11:263–75. doi: 10.1586/era.10.226
283. Koninki K, Barok M, Tanner M, Staff S, Pitkanen J, Hemmila P, et al. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. (2010) 294:211–9. doi: 10.1016/j.canlet.2010.02.002
284. Mehta K, Osipo C. Trastuzumab resistance: role for Notch signaling. ScientificWorldJournal (2009) 9:1438–48. doi: 10.1100/tsw.2009.166
285. Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. (2010) 70:1204–14. doi: 10.1158/0008-5472.CAN-09-3321
286. Baker AT, Zlobin A, Osipo C. Notch-EGFR/HER2 Bidirectional Crosstalk in Breast Cancer. Front Oncol. (2014) 4:360. doi: 10.3389/fonc.2014.00360
287. Pandya K, Meeke K, Clementz AG, Rogowski A, Roberts J, Miele L, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer (2011) 105:796–806. doi: 10.1038/bjc.2011.321
288. Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene (2008) 27:5019–32. doi: 10.1038/onc.2008.149
289. Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. (2009) 15:1845–7. doi: 10.1158/1078-0432.CCR-08-3087
290. Seol H, Lee HJ, Choi Y, Lee HE, Kim YJ, Kim JH, et al. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Mod Pathol. (2012) 25:938–48. doi: 10.1038/modpathol.2012.36
291. Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature (2016) 537:102–6. doi: 10.1038/nature19328
292. Guo D, Ye J, Dai J, Li L, Chen F, Ma D, et al. Notch-1 regulates Akt signaling pathway and the expression of cell cycle regulatory proteins cyclin D1, CDK2 and p21 in T-ALL cell lines. Leuk Res. (2009) 33:678–85. doi: 10.1016/j.leukres.2008.10.026
293. Ramakrishnan G, Davaakhuu G, Chung WC, Zhu H, Rana A, Filipovic A, et al. AKT and 14-3-3 regulate Notch4 nuclear localization. Sci Rep. (2015) 5:8782. doi: 10.1038/srep08782
294. Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, et al. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene (2010) 29:2488–98. doi: 10.1038/onc.2010.7
295. Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. (2008) 111:2220–9. doi: 10.1182/blood-2007-07-102632
296. Yao J, Qian C. Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol. (2010) 27:1017–22. doi: 10.1007/s12032-009-9326-5
297. Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, et al. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. (2009) 69:573–82. doi: 10.1158/0008-5472.CAN-08-2088
298. McDonald ER, 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, et al. Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell (2017) 170:577–92 e10. doi: 10.1016/j.cell.2017.07.005
299. Yeo SK, Guan JL. Breast cancer: multiple subtypes within a tumor? Trends Cancer (2017) 3:753–60. doi: 10.1016/j.trecan.2017.09.001
300. Gomez-Miragaya J, Palafox M, Pare L, Yoldi G, Ferrer I, Vila S, et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem Cell Reports (2017) 8:1392–407. doi: 10.1016/j.stemcr.2017.03.026
301. Dall GV, Vieusseux JL, Korach KS, Arao Y, Hewitt SC, Hamilton KJ, et al. SCA-1 Labels a Subset of Estrogen-Responsive Bipotential Repopulating Cells within the CD24+ CD49fhi Mammary Stem Cell-Enriched Compartment. Stem Cell Reports (2017) 8:417–31. doi: 10.1016/j.stemcr.2016.12.022
302. Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signaling from monocytes and macrophages. Nature Cell Biol. (2014) 16:1105–17. doi: 10.1038/ncb3041
303. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
304. Farnie G, Clarke RB. Mammary stem cells and breast cancer–role of Notch signalling. Stem Cell Rev. (2007) 3:169–75. doi: 10.1007/s12015-007-0023-5
305. Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. (2016) 76:440–52. doi: 10.1158/0008-5472.CAN-15-1640-T
306. Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in gamma-secretase inhibitor drug responses. EMBO Mol Med. (2013) 5:1502–22. doi: 10.1002/emmm.201302558
307. D'Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther. (2015) 14:779–87. doi: 10.1158/1535-7163.MCT-14-0228
308. Zhou W, Wang G, Guo S. Regulation of angiogenesis via Notch signaling in breast cancer and cancer stem cells. Biochim Biophys Acta (2013) 1836:304–20. doi: 10.1016/j.bbcan.2013.10.003
309. Perez-Tenorio G, Alkhori L, Olsson B, Waltersson MA, Nordenskjold B, Rutqvist LE, et al. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin Cancer Res. (2007) 13:3577–84. doi: 10.1158/1078-0432.CCR-06-1609
310. Whelan TJ, Pignol JP, Levine MN, Julian JA, MacKenzie R, Parpia S, et al. Long-term results of hypofractionated radiation therapy for breast cancer. N Engl J Med. (2010) 362:513–20. doi: 10.1056/NEJMoa0906260
311. Yarnold J, Bentzen SM, Coles C, Haviland J. Hypofractionated whole-breast radiotherapy for women with early breast cancer: myths and realities. Int J Radiat Oncol Biol Phys. (2011) 79:1–9. doi: 10.1016/j.ijrobp.2010.08.035
312. Smith BD, Bentzen SM, Correa CR, Hahn CA, Hardenbergh PH, Ibbott GS, et al. Fractionation for whole breast irradiation: an American Society for Radiation Oncology (ASTRO) evidence-based guideline. Int J Radiat Oncol Biol Phys. (2011) 81:59–68. doi: 10.1016/j.ijrobp.2010.04.042
313. Zhang P, He D, Chen Z, Pan Q, Du F, Zang X, et al. Chemotherapy enhances tumor vascularization via Notch signaling-mediated formation of tumor-derived endothelium in breast cancer. Biochem Pharmacol. (2016) 118:18–30. doi: 10.1016/j.bcp.2016.08.008
314. Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell (2017) 65:1122–35 e5. doi: 10.1016/j.molcel.2017.02.008
315. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. (2013) 45:1446–51. doi: 10.1038/ng.2823
316. Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer (2015) 15:261–75. doi: 10.1038/nrc3920
317. Hart CD, Migliaccio I, Malorni L, Guarducci C, Biganzoli L, Di Leo A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat Rev Clin Oncol. (2015) 12:541–52. doi: 10.1038/nrclinonc.2015.99
318. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. (2013) 10:143–53. doi: 10.1038/nrclinonc.2013.10
319. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417–30. doi: 10.1038/nrclinonc.2016.26
320. Sidaway P. Breast cancer: brain metastasis detectable in CTCs. Nat Rev Clin Oncol. (2017) 14:588. doi: 10.1038/nrclinonc.2017.138
321. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell (2016) 29:255–69. doi: 10.1016/j.ccell.2016.02.006
322. Malumbres M. CDK4/6 Inhibitors resTORe therapeutic sensitivity in HER(2)(+) breast cancer. Cancer Cell (2016) 29:243–4. doi: 10.1016/j.ccell.2016.02.016
323. Aberle MR, Burkhart RA, Tiriac H, Olde Damink SWM, Dejong CHC, Tuveson DA, et al. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br J Surg. (2018) 105:e48–60. doi: 10.1002/bjs.10726
324. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living biobank of breast cancer organoids captures disease heterogeneity. Cell (2018) 172:373–86 e10. doi: 10.1016/j.cell.2017.11.010
Keywords: breast cancer, Notch, personalized precision treatment, treatment resistance, resensitisation
Citation: Mollen EWJ, Ient J, Tjan-Heijnen VCG, Boersma LJ, Miele L, Smidt ML and Vooijs MAGG (2018) Moving Breast Cancer Therapy up a Notch. Front. Oncol. 8:518. doi: 10.3389/fonc.2018.00518
Received: 18 July 2018; Accepted: 22 October 2018;
Published: 20 November 2018.
Edited by:
Giorgio Seano, Institut Curie, FranceReviewed by:
Gino B. Ferraro, Harvard Medical School, United StatesIsabella Screpanti, Università degli Studi di Roma La Sapienza, Italy
Copyright © 2018 Mollen, Ient, Tjan-Heijnen, Boersma, Miele, Smidt and Vooijs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc A. G. G. Vooijs, bWFyYy52b29panNAbWFhc3RyaWNodHVuaXZlcnNpdHkubmw=
†These authors have contributed equally to this work