 Katia C. Genadry
Katia C. Genadry Silvia Pietrobono
Silvia Pietrobono Rossella Rota
Rossella Rota Corinne M. Linardic
Corinne M. Linardic- 1Division of Hematology-Oncology, Department of Pediatrics, Duke University Medical Center, Durham, NC, United States
- 2Department of Hematology-Oncology, Bambino Gesù Pediatric Hospital, IRCCS, Rome, Italy
- 3Department of Pharmacology & Cancer Biology, Duke University Medical Center, Durham, NC, United States
Soft tissue sarcomas (STSs) are an uncommon group of solid tumors that can arise throughout the human lifespan. Despite their commonality as non-bony cancers that develop from mesenchymal cell precursors, they are heterogeneous in their genetic profiles, histology, and clinical features. This has made it difficult to identify a single target or therapy specific to STSs. And while there is no one cell of origin ascribed to all STSs, the cancer stem cell (CSC) principle—that a subpopulation of tumor cells possesses stem cell-like properties underlying tumor initiation, therapeutic resistance, disease recurrence, and metastasis—predicts that ultimately it should be possible to identify a feature common to all STSs that could function as a therapeutic Achilles' heel. Here we review the published evidence for CSCs in each of the most common STSs, then focus on the methods used to study CSCs, the developmental signaling pathways usurped by CSCs, and the epigenetic alterations critical for CSC identity that may be useful for further study of STS biology. We conclude with discussion of some challenges to the field and future directions.
Introduction
Despite substantial therapeutic advances, cancer is still a significant cause of morbidity and mortality world-wide (1, 2). Solid tumors in particular show a complex mix of genomic subclones with different mutational signatures and cellular phenotypes. This “intratumoral heterogeneity” is thought to be the primary cause of therapeutic failure, followed by disease progression and relapse (3–6). To date, two opposing models have been offered to explain such tumor heterogeneity. The “stochastic” model considers it as a result of cellular natural selection (7). As such, every cell within a tumor has equivalent tumorigenic potential, and random mutations in individual tumor cells promote the selection of the fittest clone. Over time, additional advantageous mutations spawn genetically divergent subclones that independently maintain their malignant potential. By contrast, the cancer stem cell (CSC) model ascribes tumor establishment to a single, transformed, stem-like clone endowed with dysregulated and unlimited self-renewal that differentiates into less tumorigenic subclones to create a cellular “hierarchy” similar to that of normal tissue (8, 9).
CSCs, also known as “tumor-initiating cells” (TICs), were first identified in cancers of hematopoietic origin (10, 11), then in various solid tumors (12–17), where these cells show the ability to re-derive the original tumor heterogeneity when serially xenotransplanted in immunocompromised mice at very low number (8, 9). The importance of CSCs comes from their ability to constitute a small reservoir of drug-resistant cells, which overcome conventional chemotherapy due to their low rate of proliferation, thus driving tumor recurrence and metastasis (18). CSCs share several properties with non-transformed adult stem cells (SCs): multipotency, resulting in the possibility to differentiate into lineages of the embryonic layer of origin; self-renewal capacity; the ability to transition between quiescence, slow-cycling and active proliferation states; the expression of the embryonic stem cell transcription factors (TFs) OCT4, NANOG, SOX2, KLF4, and MYC; the expression of similar surface markers such as CD44, CD133, or of the intracellular enzyme aldehyde dehydrogenase (ALDH); enhanced protective mechanisms against apoptosis, DNA repair, and oxidative stress; activation of key developmental pathways; a preference for oxidative metabolism for energy production, and the expression of ATP-binding cassette (ABC) drug transporters (19). However, unlike normal SCs, CSCs are characterized by the dysregulation of those features and possess the ability to control stemness signals (5, 20).
Although the source of CSCs has been the center of investigation for many years, it remains a hotly debated topic. Reflecting the ongoing discourse, it has been suggested that the clonal evolution (stochastic) and the CSC (hierarchical) models are not necessarily mutually exclusive, as CSCs can originate not only from the malignant transformation of adult SCs (21–23) but also from mutated tumor cells that undergo a de-differentiation process in which they reacquire stem-like features in order to evolve into CSCs (24, 25). Both cell-autonomous (activation of stemness signaling pathways, epigenetic alterations, DNA damage, replicative stress) and non-cell-autonomous factors (immune system, tissue damage, signals from the microenvironment niche, therapy-induced genotoxic stress) seem to participate to the reprogramming [reviewed in Poli et al. (26)].
Soft tissue sarcomas (STSs) are uncommon malignancies of mesenchymal origin characterized by a high degree of heterogeneity in their genetic profile, histology and clinical features. They include several subtypes with onset in childhood, adolescence and even in the adult life, such as rhabdomyosarcoma (RMS), synovial sarcoma (SS), fibrosarcoma (FS), malignant peripheral nerve sheet tumor (MPNST), leiomyosarcoma (LMS), liposarcoma (LPS) and undifferentiated pleomorphic sarcoma (UPS). The prevalence of STS subtypes significantly changes from childhood (<20 years) through adolescence into adulthood representing about 5–6% of all childhood cancers and <1% of all adult malignancies (27). Various studies suggest that STSs originate from the malignant transformation of a primitive, multipotent mesenchymal stem cell (MSC), i.e., the multipotent precursor of mesodermal tissues including bone, skeletal muscle, adipose tissue, cartilage and tendon (28). It has been suggested that the same transformed MSC can give a particular subtype of STS depending on the vulnerability to subsequent mutations involving specific developmental pathways or, alternatively, in the genesis of an undifferentiated sarcoma (29, 30). However, gene expression studies showed that the signatures of several STSs were more similar to that of differentiated MSCs than of undifferentiated MSCs, suggesting that the development of STSs with distinct phenotypes and histological grades may reflect different differentiation stages of MSCs at the time of tumorigenesis initiation (31–33).
Contemporary therapies for STSs are multi-modal and include surgery, radiation and chemotherapy, although significant limitations are provided by their toxicity and partial responses. To date, the 5-year survival rate for patients with STS is about 60%, reflecting age, tumor type, stage and histologic grade, but it drops dramatically to 10–17% in high risk patients (34). Recently, STS tumor cells with stem-like properties have been identified, possibly explaining the heterogeneity that characterizes these cancers and suggesting that these cells might be responsible for relapse and metastasis.
CSC characteristics and their role in tumorigenesis has been studied and summarized for various solid tumors, including bone sarcomas (35–38). However, an overview on the role of CSCs in STS is lacking. This review summarizes the evidence for CSCs in STSs. The importance of CSC features for clinical anticancer interventions is also discussed.
Evidence for CSCs in Soft-Tissue Sarcomas
According to the type of genomic alteration, STSs can be classified into two main categories: (i) recurrent translocation-driven STSs, where reciprocal chromosomal translocations result in oncogenic fusion transcripts such as PAX3-FOXO1 in alveolar RMS (ARMS), SS18-SSX in SS, FUS-CHOP in myxoid/round-cell LPS, and (ii) non-translocation driven STSs characterized by complex genetic changes such as amplifications/deletions in various chromosomal regions as observed in embryonal RMS (ERMS), FS, LMS, LPS and MPNSTs (39). Fusion-positive STSs are characterized by cells that are morphologically and molecularly similar with the fusion oncoprotein as the major driver of the malignancy. Conversely, fusion-negative STSs show a high degree of intra-tumor heterogeneity.
Rhabdomyosarcoma (RMS)
RMS is the most common soft tissue sarcoma in children and young adults but can occur at any age (40, 41). RMS is thought to derive from myogenic precursors that lose the ability to differentiate into skeletal muscle despite the expression of the master key genes of skeletal muscle lineage (42). The two main histopathologic subtypes are ARMS and ERMS. ARMS is associated with a poorly differentiated phenotype and arises mostly in adolescents and young adults. Genetically, approximately 80% of the cases are characterized by a t(2, 13) or t(1, 13) chromosomal translocation, which generates the fusion oncoproteins PAX3-FOXO1 or PAX7-FOXO1 that work as mutant transcription factors (43, 44). ERMS is more common, usually affects children under the age of 10 years, and is for the most part associated with a favorable prognosis. Genomic landscape studies of RMS showed that ERMS has a higher mutation rate when compared to ARMS, as well as more frequent copy number variants and single nucleotide variants (45–47). Mutations identified include (among others) RAS isoforms, TP53, neurofibromin-1 (NF-1), PI3K catalytic subunit α (PIK3CA), β-catenin (CTNNB1), fibroblast growth factor receptor 4 (FGFR4), and F-box and WD repeat domain-containing 7 (FBXW7).
While the genomic homogeneity of ARMS would predict that its molecular features could be harnessed for therapeutic purposes, the PAX3-FOXO1 protein has remained therapeutically intractable (48). On the other hand, the genomic heterogeneity of ERMS highlights the challenge of finding a single target for therapeutic purposes. Using a variety of approaches, cell populations with CSC features have been reported for ERMS (49–52); the identification of ARMS CSCs has been more elusive and while a recent study showed that ARMS cells could form holoclones and spheres (53), no studies have reported in vivo functional assays for ARMS CSCs. Similar to what is observed in SS [below (54)], there is some thought that almost all PAX3-FOXO1+ ARMS tumor cells have stem cell characteristics–suggesting that ARMS is a stemness-disease, but this has yet to be demonstrated.
Synovial Sarcoma (SS)
SS is an aggressive neoplasm occurring in adolescents and young adults (aged 10 to 35 years), accounting for about 10% of all STSs (55). About 70% of cases develop metastases (56–58). SS is characterized by t(X;18)(p11;q11) (59), which generates an in-frame fusion of the synovial sarcoma translocation, chromosome 18 (SS18, also known as SYT) gene to the synovial sarcoma, X breakpoint (SSX) genes 1, 2 and, rarely, 4 (60–62). Whereas, SYT interacts with the SWI/SNF chromatin remodeling complex (63–65) to activate a host of transcriptional programs that control cell cycle, stem cell maintenance and differentiation signals (66–68), its fusion partner SSX co-localizes with the Polycomb repressor complex hampering its function (69). The resulting SS18-SSX fusion oncoprotein acts as an epigenetic modifier that is dependent upon cellular context (70), and drives SS pathogenesis (71–73). In line with fusion-driven sarcomas, SSs are genetically quiet, with no chromosomal abnormalities other than t(X;18) in the majority of cases. However, copy number gains are more common in adult patients and are typically associated with a poor outcome (74).
Skeletal muscle lineage precursors (but not differentiated myocytes) have been suggested as a cell of origin for SS, since conditional expression of the human fusion gene SYT-SSX2 in Myf5-expressing murine myoblasts results in tumors with 100% penetrance (72). More recently, SYT-SSX2 forced expression in MSCs disrupted normal mesodermal differentiation, triggering a pro-neural gene signature via its recruitment to genes controlling neural lineage features (75). The authors also showed that SYT-SSX2 controlled the activation of key regulators of stem cell and lineage specification (75). Consistently, silencing of SYT–SSX induced terminal differentiation of SS cells into multiple mesenchymal lineages (osteogenic, chondrogenic and adipogenic types) (54). On the one hand, these data point to MSCs as a cell of origin of SS and suggest that deregulation of normal differentiation by SYT-SSX could constitute the basis for MSC transformation. On the other hand, they seem to also suggest that SS can develop in MSC precursors that are in a susceptible developmental stage. In the same work, Naka et al. showed that SS cell lines, similarly to SS clinical samples, contain a subpopulation of cells characterized by high levels of the pluripotency factors SOX2, OCT4, and NANOG and that exhibit in vitro self-renewal ability and in vivo tumorigenicity following xenografting (54).
Fibrosarcoma (FS)
Adult type fibrosarcoma (FS) is a malignant tumor thought to arise from fibroblasts and is characterized histologically by undifferentiated spindle cells (76). Only a few studies point to the existence of CSCs within FSs. These studies identified a subpopulation of cells characterized by increased levels of OCT3/4, NANOG, SOX2, and SOX10, possessing stem-like characteristics such as self-renewal ability, proliferation and increased chemoresistance partly conferred by the overexpression of the multidrug resistance transporter MDR1 (77, 78). There are no published studies of CSCs in infantile FS, a pediatric malignancy associated with intermediate malignant rarely metastasizing tumor characterized by the NTRK3–ETV6 translocation resulting from t(12, 15) (79).
Malignant Peripheral Nerve Sheet Tumor (MPNST)
MPNSTs account for about 5-8% of all STSs and can occur sporadically after radiotherapy or can arise in the neurofibromatosis type 1 (NF1) syndrome (80). The NF1 gene, located on the long arm of chromosome 17 (17q11.2), encodes for the 220kDa protein neurofibromin. NF1 syndrome is characterized by mutation-induced inactivation or more rarely complete germline loss of one NF1 allele that often leads to either dermal or plexiform benign neurofibromas. The latter neurofibroma subtype, arising in nerve plexuses or deep large nerves, occurs following de novo somatic mutations or inactivation of the other NF1 allele specifically in the Schwann cell lineage and can undergo malignant transformation in MPNSTs (81). Patients with NF1 can develop other types of pediatric tumors such as pheochromocytomas, RMS, LMS, and juvenile myelomonocytic leukemia (82). In addition, inactivating mutations of NF1 have been reported in adult tumors including brain, lung and ovarian cancers and in melanomas (83). Neurofibromin inhibits RAS signaling through its RAS GTPase-activating protein (GAP) domain, thus working as a tumor suppressor (84). In agreement, the RAS pathway is constitutively over-activated in MPSNTs (85). Although neurofibromin is a member of the large RAS-GAP family proteins, it is the only one linked to a tumor predisposition syndrome when mutated. However, accumulating genomic abnormalities in tumor suppressors or oncogenes have been suggested to be responsible for the progression from benign plexiform neurofibromas to MPNSTs. Loss of TP53 and CDKN2A are common in MPNSTs (86, 87). CDKN2A encodes for both p19ARF and p16INK4A and thus its inactivation can affect both p19ARF-MDM2-p53 and p16INK4A-Cyclin D-RB pathways leading to uncontrolled proliferation. Even RB1 loss can be seen in about 25% of MPNSTs (88).
Either precursors of or postnatal Schwann-derived cells, the source of myelinating glial cells of the peripheral nervous system, seem to be the cell of origin of MPNSTs (82, 89, 90). Gene expression studies showed that MPNSTs exhibit deregulation of tumor-specific gene clusters belonging to Schwann cell development regulators, including downregulation of SOX10, which promotes the specification of Schwann progenitors and their maturation and myelin production (91) and upregulation of SOX9, which is involved in neural crest stem cell survival (92, 93). Putative CSCs of MPNSTs expressing stemness genes have been recently established under specific conditions from human cell lines and primary tumors (94). These cells were characterized by high levels of the neural lineage genes NESTIN and NGFR, and the stemness markers OCT4, NOTCH4, SOX2, and SOX9 as assessed by qPCR, but also by the expression of stem surface markers by flow cytometry, and were shown to give rise to tumors resembling human MPNSTs when they were injected subcutaneously in immunodeficient mice.
Leiomyosarcoma (LMS)
LMS accounts for about one quarter of all soft tissue tumors. It is extremely rare during infancy and childhood, occurring most commonly in middle-aged individuals. LMS has a complicated histopathological classification and a different clinical behavior depending on the location within the body (95). LMS is characterized by a high degree of genomic instability, with non-recurrent aberrations at the chromosomal level. The most common regions of chromosomal loss have been identified in 10q and 13q, where the phosphatase and tensin homolog (PTEN) and the retinoblastoma 1 (RB1) tumor suppressor genes, respectively, reside (96–98). In addition, PTEN point mutations (99, 100) as well as constitutive hyperactivation of PI3K/AKT pathway (101), have been detected in LMS, suggesting that loss of PTEN might contribute to the initiation or progression of LMS. Epigenetic changes could also contribute to LMS. For instance, Roncati et al. showed that methylation-dependent silencing of CDKN2A, which is associated with decreased expression of both p16INK4A and p14ARF and with higher activity of MDM2 and CDK4/CDK6, results in LMS progression (102). Additionally, treatment with the HDAC inhibitor vorinostat in combination with a DNA demethylating agent such as decitabine allowed overcoming the resistance to cell death induction due to promoter methylation of apoptotic genes in uterine LMS (103).
Recent findings suggest a mesenchymal stem cell origin for LMS. By using Cre-Lox technology to generate murine MSC cultures knock-out for Trp53 and Rb1 alone or in combination, Rubio et al. showed that Tp53−/− mouse adult MSCs underwent in vitro transformation and developed LMS-like tumors in vivo when injected as xenografts in immunodeficient mice (104). A microRNA (miRNA) signature distinctive of MSCs includes several components of the miR-17-92 cluster, and appeared downregulated during MSC differentiation into SMCs while up-regulated in uterine LMS, supporting the hypothesis that LMS is a mesenchymal stem cell-related malignancy (32). In testicular LMS, a subpopulation of cells with stem-like characteristics was described (105). These cells showed high tumorigenic potential and the capacity to re-derive the original parental tumor in immunodeficient mice.
Liposarcoma (LPS)
LPS is also one of the most common STS, accounting for at least 20% of all adult sarcomas. The World Health Organization classifies LPS into four main subtypes: 40–45% well differentiated (WD-LPS), 15–25% dedifferentiated (DD-LPS), 30–35% myxoid/round-cell (MR-LPS) and about 5% undifferentiated high-grade pleomorphic liposarcomas (P-LPS) (106). The histological subtypes reflect both clinical behavior and prognosis (107). WD-LPS occurs most frequently in the retroperitoneum and limbs, rarely metastasizes and shows low recurrence rates. By contrast, DD-LPS is more aggressive, with a metastatic rate of 15–20% and a worse prognosis. Both WD-LPS and DD-LPS can be distinguished from other adipocytic neoplasms based on the amplification of the chromosome region 12q13-15, in which MDM2, CDK4 and SAS genes reside (108). MR-LPS is characterized by the appearance of spindle to oval-shaped cells in a myxoid stroma (109) and has a predilection for the limbs, with abdomen and bones as typical metastatic sites (110, 111). MR-LPS harbors the chromosomal translocation t(12, 16)(q13;p11) that results in a fusion gene arrangement between FUS and the C/EBP homologous protein (CHOP, also known as DDIT3 or GADD153) and appears to constitute the primary oncogenic event in MR-LPS (112). P-LPS is a rare tumor of adulthood and can be distinguished from the other subtypes by the presence of pleomorphic lipoblasts. It occurs most commonly in soft tissues of the extremities and is associated with pulmonary metastases (113).
Previously, adipose-derived stem cells (ASC) were proposed as a cell of origin of LPS (114). In support of this, Rodriguez and colleagues showed that the expression of a FUS-CHOP transgene in Trp53-deficient mouse ASCs was able to shift the tumor phenotype toward LPS-like tumors (115). Consistently, the expression of FUS-CHOP in murine MSCs resulted in the development of tumors resembling LPS with features such as intracellular lipid accumulation, presence of lipoblasts with round nuclei, and an adipocyte differentiation block (116). In the human setting, FUS-CHOP has been reported to cooperate with other oncogenic hits to block the differentiation potential of bone marrow-derived MSCs toward adipocytes, and to transform them into LPS cells resembling the myxoid subtype (117). Matushansky and colleagues linked adipocyte differentiation from human MSC to all LPS subtypes, in dependence of their maturation status (33). They propose that additional secondary mutations could lead to morphologically diverse tumors arising from the same stage of transformation (33).
Using a LPS xenograft model, Stratford et al. identified a small (0.1–1.7%) fraction of cells characterized by a stem-like phenotype (CD133+/ALDH+). This putative CSC population was able to self-renew in vitro, differentiate into mature adipocytes and be highly tumorigenic in nude mice (118). Recently, LPS-like tumor xenografts were generated in immunocompromised mice from subcutaneous injection of murine induced pluripotent stem cells (miPSCs) that were cultured in conditioned media from the Lewis lung carcinoma (LLC) cell line, which secretes tumor-derived extracellular vesicles [the resulting cells were termed miPSC-LLCev] (119). Cells derived from the LPS-like xenografts were characterized by CSC features including self-renewal ability, increased expression of Sox2, Nanog and Klf4, the capacity to generate secondary tumors resembling the original histotype of the primary ones, and to disseminate into the mesentery of abdominal cavity. When cultured as spheres, the miPSC-LLev cells were also able to differentiate into adipocytes under appropriate conditions, suggesting phenotypic heterogeneity (119).
Undifferentiated Pleomorphic Sarcoma (UPS)
UPS is the most common STS in the elderly. In the 1960s this malignancy was thought to arise from histiocytes and thus was named “malignant fibrous histiocytoma.” It was later proven that this tumor was not a true histiocytic malignancy and it was renamed pleomorphic fibrosarcoma, or undifferentiated high-grade pleomorphic sarcoma (UPS) (120). UPS cells have variable morphology without a hint of differentiation (121). Recently, rare gene fusions involving PRDM10 were identified in UPS tumors (122). Li et al formed pleomorphic sarcomas in immunodeficient mice out of a transformed culture of bone marrow stromal cells (30). Martinez et al were also able to engineer a model of UPS out of a mutated human bone-marrow derived MSCs, proving that MSCs are most likely the origin this sarcoma (123, 124). Rubin et al. were able to generate UPS through the loss of Tp53 in combination with Ptch1 in Myf6-expressing muscle cells. Interestingly, however, when these cells were depleted only of Trp53, they formed ERMS, suggesting a common progenitor (125). Wang et al. isolated a subpopulation of cells in UPS that exhibits stem-like properties identified as a “side population” by flow cytometry (126).
Isolation and Characterization of CSCs in STS
Successful and specific investigation of CSCs is a prerequisite for better understanding of the molecular mechanisms underlying STS initiation, progression, relapse, metastasis and resistance to therapies. Currently, evaluation of CSCs is achieved through both in vitro and in vivo approaches, including (i) generation of non-adherent “sarcosphere” cultures under serum-free conditions; (ii) flow cytometry sorting based on expression of CSC-specific surface markers; (iii) assessment of aldehyde dehydrogenase (ALDH) activity through the ALDEFLUOR (Stemcell Technologies) assay; (iv) detection of a side population identified by exclusion of Hoechst 33342 dye; (v) xenograft tumor initiation of the ability of a low number of cells in immunodeficient mice, and (vi) reprogramming of cancer cells back to pluripotency (127–129). Remarkably, since none of these methods alone is enough to identify CSCs unequivocally within a tissue, the use of several markers and properties in combination could be helpful to better define the CSC phenotype in these tumors. The stem cell markers and assays that have been used to identify, isolate and characterize potential STS CSCs are summarized in Table 1. An overview of STS CSCs including their characteristics and signaling is provided in Figure 1.

Table 1. List of soft tissue sarcoma (STS) subtypes and the stem cell markers and assays that have been used to investigate their cancer cell stemness.
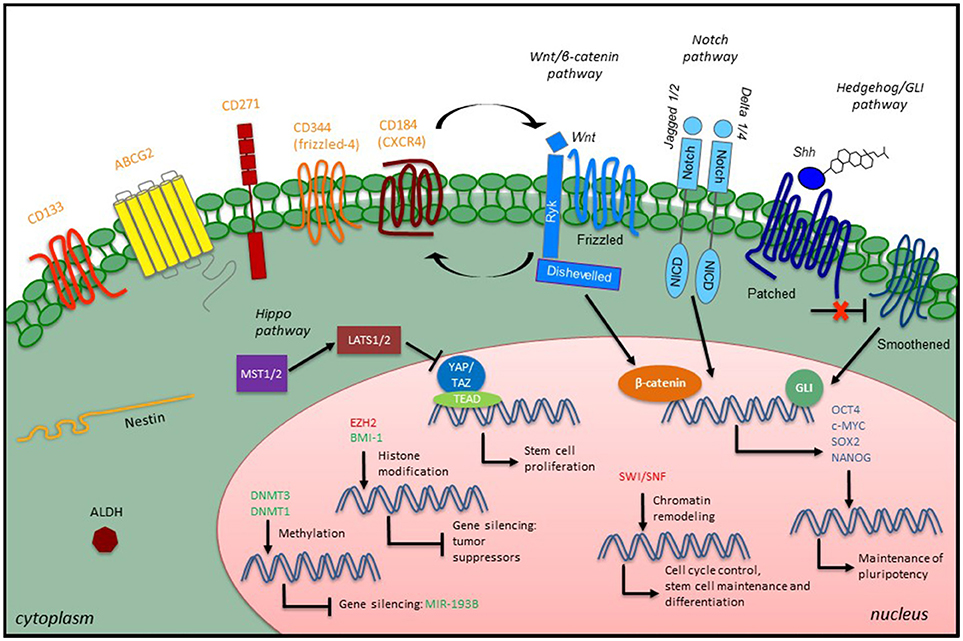
Figure 1. Overview of CSC characteristics and signaling in STS. STS CSCs express specific stem cell surface markers (orange), which have been used as CSC identifiers, along with some intracellular markers such as the intermediate filament Nestin or the enzyme ALDH. Developmental signaling pathways play a role in the CSC phenotype by promoting the expression of embryonic transcription factors (blue). Epigenetic modulators (green, confirmed modulators; red, putative modulators) also participate in the CSC phenotype through different mechanisms: maintenance of existing methylation patterns or de novo methylations at CpG islands, histone modification and chromatin remodeling.
Three-Dimensional Cell Cultures
First used by Reynolds and Weiss to isolate stem cells of neural origin (130), the 3D model enables cells to grow in all dimensions, thus mimicking the interactions between cells of interest and the microenvironment in a given tissue (131). A single-cell suspension is grown in low-density conditions to avoid cell aggregation, and in defined serum-free media supplemented with specific growth factors (epidermal growth factor, and basic fibroblast growth factor), in ultra-low attachment plates (132). In these conditions, cells can proliferate to form non-adherent, floating spheres, which in turn can be dissociated to allow secondary and tertiary sphere formation. Each sphere consists of a small percentage of self-renewing cells and a large percentage of progenitor cells at various stages of differentiation (133). In the last few years, sphere culture techniques have been employed to allow CSC enrichment in STSs, including both ERMS and ARMS (50, 53), FS (77), SS (54, 77), MPNSTs (94), LMS (105), and LPS (118, 119).
Stem Surface Markers
The first idea for CSCs isolation based on the expression of certain surface markers came with the identification of a small subset of leukemic stem cells with a CD34+CD38− phenotype. When these cells were transplanted into non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice, they were able to initiate acute myeloid leukemia with the same heterogeneity of the primary cancer, whereas the most abundant subset of CD34+CD38+ was ineffective (11). These results stimulated many researchers to isolate CSCs from heterogeneous cell populations of STS through fluorescent-activated cell sorting (FACS) of cells expressing specific stem surface markers, alone or in combination. Following is a list of cell surface markers thought to have roles in stemness.
CD133 (Prominin-1) is a glycosylated protein involved in topological organization of the cell membrane (134). CD133 has been shown to mark a subpopulation of tumor cells characterized by high levels of stemness genes, higher clonogenicity and in vitro self-renewal, and increased in vivo tumorigenicity than the CD133- subpopulation in several STS, including SS (135–137), RMS (49, 50, 52, 138) and FS (77, 78, 136). Similarly, LPS putative CSCs, prospectively isolated by FACS of the stem surface marker CD133 and of ALDH activity, were shown to produce tumors at limiting cell dilution more efficiently compared to the other sorted subpopulations (118). In MPNST, CD133+ cells were able to self-renew, yielding more spheres and proliferating faster compared to the CD133− cells, and to give rise to tumors resembling the original heterogeneous tumor when injected at low number (94). In LMS, She et al. found that CD133 expression positively correlates with tumor size, mitotic counts and histological grade in primary retroperitoneal LMS, proposing it as prognostic marker (139).
CD184 (chemokine receptor type-4, CXCR4) is a seven transmembrane chemokine receptor normally expressed on immune cells, but also on embryonic stem cells (ESCs) (140) and MSCs (141). Recently, CD184 has been identified as a SS-initiating surface marker. By using sphere formation assays, the authors enriched for a CSC subpopulation that was characterized by high levels of CXCR4. These cells exhibited higher tumor initiation potential after serially transplantations into NOD/SCID mice compared to their CXCR4− counterpart and were able to recapitulate the phenotype observed in the original tumor (142). Interestingly, CXCR4 was found highly expressed on the surface of ARMS cells, where it correlates with unfavorable primary sites, advanced stage, decreased overall survival and bone marrow involvement (143, 144), and was also used as a prognostic marker for MPNSTs, LMS, LPS and FS (145). However, the above-mentioned studies did not determine whether CD184 is associated with a CSC phenotype in these STS.
CD271 (low-affinity nerve growth factor receptor), is expressed in neural crest tissue and suggested to be a CSC surface marker in SS, FS, LMS and LPS (146).
CD344 (frizzled-4), a neuronal stem cell marker that plays important roles in vascular development of the retina and inner ear, has been shown to identify a tumor cell subpopulation with increased capacity for proliferation and sarcosphere formation and resistance to doxorubicin in LMS and SS cells (146).
ABCG2 (ATP binding cassette G2) has been used to isolate a subpopulation of CSCs with increased drug resistance in SS and FS (77, 78). ABCG2 expression has been associated with shortened survival in RMS patients (147).
Nestin is an intermediate filament protein first identified in stem cells of neuroepithelial origin. It is expressed in several cell types during development, including neural crest cells and myocytes. It was found upregulated in tumor cell spheres derived from MPNSTs compared to their corresponding adherent cells (94) and overexpressed in RMS (148).
ALDH Activity
In addition to the cell surface markers described above, a few specific intracellular enzymes and their activity can be utilized to identify CSCs. Among them, the detoxifying enzyme aldehyde dehydrogenase (ALDH), which is highly expressed in normal stem cells (149, 150), has been recently implicated in therapy resistance and tumor recurrence (151–154). Detection of ALDH activity is captured through the ALDEFLUOR assay, an enzyme-based assay thought to specifically detect the ALDH isoform ALDH1A1 (155). ALDH proteins have been used as markers of CSC identification in many STSs including ERMS (51), LPS (118), FS and SS, in which their expression correlates with higher proliferation and clonogenicity, and is associated with increased drug resistance (156).
Side Population
The side population phenotype (SP) was first defined in hematopoietic cells (157, 158). SP cells show limited intake or active extrusion of the fluorescent dye Hoechst 33342 as a consequence of increased expression of the ABC transporter ABCG2 (159–161). SP cells can be isolated by flow cytometry based on the absence of accumulation of Hoechst dye, and have been used to enrich for CSCs in various cancers, including sarcomas (162, 163). Indeed, the SP phenotype has been shown to correlate with high tumorigenicity in immunocompromised mice, the ability to repopulate both the SP and the non-SP fractions (164, 165) and resistance to therapy (166–168)-all established criteria for CSCs. In the context of STSs, Alman's group was the first to identify a SP fraction within human LMS and SS through Hoechst dye staining. The size of this SP appeared to positively correlate with the tumor grade, although it is unclear whether the SP fraction isolated in this study reflected a population of cells enriched in CSC features such as self-renewal ability and higher in vivo tumorigenicity compared to non-SP cells (169). By contrast, Sette et al. demonstrated that the subpopulation of testicular LMS stem-like cells characterized by a SP phenotype showed enhanced ability to extrude doxorubicin and high clonogenicity in limiting dilution assays (105).
Limiting Dilution in vivo
The most stringent method to define the frequency of CSCs in vivo is the limiting dilution cell transplantation assay (LDA). In this assay, tumor cells are transplanted at defined, decreasing doses into animals and tumors allowed to develop over time. At analysis, the percentage of animals that develop (or do not develop) tumors is used to determine the number of tumor cells with self-renewal capacity (170, 171). These “LDA frequencies” are calculated using a web-based tool called ELDA (extreme limiting dilution analysis), which is the first software for limiting dilution analysis that delivered useful confidence intervals for all LDA cell subpopulations, even those with no (or complete) response (172). In vivo LDA must be performed to confirm that a defined marker enriches for CSC activity, and must be done with both the positive and negative fractions. It is important to highlight that the in vitro sphere-forming assay does not constitute a surrogate for the in vivo LDA, and can only complement, rather than replace, it. By using a Trp53-null mouse model of breast cancer, Zhang and colleagues identified a cell subpopulation characterized by high levels of CD24 and CD29 using in vitro LDA and subsequent transplantation in vivo (173). However, to date LDA has been performed only for few STS tumors in vivo, and further studies are required to confirm the true nature of marker-sorted CSCs in STSs.
Reprogramming Mechanisms Potentially Involved in CSCs in STS
Similar to normal cells, in which reprogramming toward a pluripotent state can be achieved by nuclear transfer, blastocyst injection, or by applying induced pluripotent stem cell (iPSC) technology [forced expression of a specific panel of TFs such as Oct4, Sox2, Klf4, and Myc (174, 175)], human cancer cells can also be reprogrammed. Hochedlinger and colleagues showed that introduction of nuclei derived from mouse melanoma cells into enucleated oocytes induced the establishment of an ESC line from blastocysts with the potential to generate teratomas (176). Alternative methods to reprogram cancer cells include: the transfection of cancer cells with the family of micro-RNA miR-302 that is highly expressed in ES cells, thus generating pluripotent stem-like cells with both self-renewal and multipotency properties (177, 178); the use of small chemical molecules to enrich for cancer stem-like cells endowed with increased in vitro tumor-sphere formation and in vivo tumorigenic abilities (179); and the application of the iPSC technology to primary tumor cells to successfully convert several cancer types into induced-pluripotent cancer stem cells (iPCSCs) (176, 180–182). These iPCSCs resemble the ES cell state at both epigenetic and transcriptional levels and repress the reprogrammed cancer genome in the pluripotent state, constituting a live cell model for studying cancer progression (183).
Stemness Signals
It is known that adult SCs and CSCs both express key evolutionarily conserved developmental pathways such as Wnt/β-catenin, Notch, Hedgehog and Hippo, which play pivotal roles in regulating stemness and differentiation (184–189). In fact, aberrant stemness signaling has been related to tumorigenesis, as deregulation of these pathways in adult SCs can lead to unchecked cell proliferation and aberrant differentiation in a tissue-specific manner. Besides, their reactivation in bulk tumor cells plays critical roles in cancer plasticity, by inducing a CSC phenotype, to promote EMT and to enhance drug resistance (190–192). However, the final goal is the alteration of gene expression patterns through the induction of embryonic TFs such as OCT4, NANOG, SOX2, KLF4, and MYC, which are part of a transcriptional network in which they regulate each other and affect chromatin remodeling (193–195). The existence of a link between reprogramming mechanisms and the stem cell TF network is supported by the revolutionary study of Yamanaka and colleagues, showing that lineage committed cells can be reprogrammed to an induced pluripotent state after the introduction of Sox2, Oct4, Klf4, and Myc (175). As such, a stemness signature is seen more often in less differentiated cancers with worse clinical outcomes (196, 197).
Hedgehog pathway
The Hedgehog (HH) pathway is a signaling network that plays a crucial role during organogenesis in the developing embryo, mainly by modulating genes involved in stem cell fate determination (198). Similarly, it is important for the existence of CSCs, as it is believed to support the CSC phenotype by driving the expression of stemness-related genes, including Oct4, Sox2, Bmi1, and Nanog (199). However, few studies are currently available about its role in CSCs of STS. In 1996, Hahn et al described a critical role for HH pathway in ERMS (200). Later studies confirmed then its activation in ERMS, its role in promoting self-renewal of ERMS CSCs (201, 202), and its association with a poor prognosis (203). The HH pathway has been proven to be hyperactivated also in ARMS, although it appears mainly activated in translocation-negative ARMS compared to translocation-positive ones (202), and its upregulation has been associated with a poor prognosis (204). A recent study showed HH pathway activation in cells contained in CSC-enriched structures including holoclones and spheres (53). In UPS, the HH pathway has been linked to the maintenance of self-renewal and proliferation of a subpopulation of putative CSCs characterized by a SP phenotype (126).
Hippo pathway
The Hippo tumor suppressor pathway plays key roles in tissue homeostasis and repair by regulating stem cell proliferation and expansion (203). It consists of cytoplasmic kinases (MST1/2 and LATS1/2) that act as tumor suppressors by restraining the activity of the transcriptional co-activators yes-associated protein (YAP1)/transcriptional co-activator with PDZ-binding motif (TAZ, also known as WWTR1) via phosphorylation, hence preventing their nuclear localization and activation of TEA domain (TEAD) family members transcription factors. Dysregulation of the Hippo pathway has been linked to cancer development, including STSs (205–208). Besides, increased expression of YAP/TAZ has been correlated with the acquisition of cancer stem cell traits that lead to EMT, drug resistance and metastasis (209). Recent evidence points to the role of the Hippo pathway in maintaining CSCs in STS. For instance, Linardic and co-workers identified a novel NOTCH-YAP1-SOX2 circuit critical for maintaining stem cell plasticity in ERMS (210). The same group found expression of TAZ in ARMS, in which it supports stemness and promotes drug resistance (211).
Notch pathway
The Notch pathway regulates cell fate determination mainly of stem and/or progenitor cells during embryonic development of organs such as pancreas, bones, muscles, heart and the nervous system (186, 212, 213), and its role in CSC establishment and maintenance has been reported in a wide range of human cancers (214, 215). The Notch pathway was found deregulated in some STS. For instance, both ERMS and ARMS show significant upregulation of the pathway, which has been shown to affect motility and invasiveness of both RMS subtypes (216). However, its importance in RMS CSC self-renewal and differentiation has been reported only for the ERMS subtype (217). The Notch pathway was found hyperactivated also in SP cells of UPS compared to non-SP cells, pointing toward a critical role in in maintaining CSC self-renewal and proliferation (126). In SS, Notch pathway components NOTCH1, JAG1 and the transducin-like enhancer of split (TLE)-1 were found overexpressed, although any association with the CSC phenotype of SS has not been yet reported (218).
Wnt/β-catenin pathway
The Wnt/β-catenin pathway plays roles in cell fate determination, cell proliferation and migration during embryogenesis. After development, it participates in preserving homeostasis in different organs, mainly those who rely on the function of stem cells (184, 185). Wnt/β-catenin signaling is involved in MSC self-renewal and differentiation (219) and it appears aberrantly expressed in a variety of CSC settings (220), although its role as oncogenic influence is still debated since its recent association with a tumor suppressive effect (221, 222). As an example, in ERMS some researchers observed inhibition of the Wnt/β-catenin pathway (223), whereas others identified opposing roles for the canonical and non-canonical pathway in regulating ERMS self-renewal and differentiation, as the canonical pathway plays a tumor-suppressor role whereas the non-canonical pathway an oncogenic one (224). By contrast, Wnt/β-catenin pathway has not been extensively studied in ARMS, although Kephart et al. found that an inhibitor of the Wnt/β-catenin pathway secreted frizzled related protein 3 is upregulated in PAX3-FOXO1+ human ARMS cells, suggesting a tumor-suppressive role for Wnt/β-catenin signaling (225).
In SS, Barham et al. provided evidence that the fusion protein SYT-SSX2 activates Wnt/β-catenin signaling through its nuclear reprogramming function, using a combination of SS cell cultures, xenografts and a SYT-SSX2 transgenic mouse model. In this study, inhibition of the Wnt pathway with small molecule CK1α activators induced SS growth arrest and, importantly, reversed the myogenesis block induced by the fusion oncoprotein by downregulating many of the Wnt/β-catenin targets involved in the embryonic program, including pluripotency factors, differentiation blockades, embryonic lineage determinants, homeobox, and forkhead box factors (226), suggesting that targeting this pathway may a rational approach in patients with SS (227, 228).
In MPNSTs, a forward genetic screen using the Sleeping Beauty mutagen transposon approach in vivo revealed the over-activation of Wnt signaling, exemplified by overexpression and enhanced activity of β-catenin due to both decreased levels of the destruction complex and up-regulation of R-spondin2, a secreted ligand that can stimulate Wnt signaling. Data were confirmed in murine models of MPNST and in primary samples from patients, and the translational relevance was verified from the evidence that the blockade of this pathway affected the tumorigenic properties of tumor cell lines in vitro and in vivo. Notably, simultaneous inhibition of Wnt signaling and mTOR pathway, the latter over-activated in this tumor, showed synergistic effects suggesting a path to intervention (229). It has been proposed that Wnt/β-catenin signaling activation could be also triggered by an autocrine loop via the activation of the CXCR4 receptor, which is overexpressed on the surface of MPNST cells, resulting in the inactivation of GSK3β that mediates β-catenin destabilization (230).
Epigenetic Alterations
Alterations of the epigenetic machinery are considered critical for CSC formation and persistence. In both embryonic and adult SCs, epigenetic processes modulate the transcriptional programs to regulate the balance of self-renewal vs. differentiation. Thus, undifferentiated SCs express high levels of TFs OCT4, SOX2, and NANOG, which work to ensure the maintenance of self-renewal and pluripotency through their co-localization on specific regulatory regions (231, 232). During the differentiation program, genes that are associated with pluripotency and self-renewal become silenced, whereas tissue-specific genes are turned on (233). Interestingly, developmental and lineage-committed genes are often present within “bivalent” chromatin domains (promoters and enhancers) containing both repressive (H3K27me3 or H3K9me3) and permissive (H3K4me3 or H3K4me1) histone marks (234–237). These genes are repressed even if polymerase II is present on bivalent promoters/enhancers to allow rapid activation (236, 238, 239) by safeguarding differentiation. These bivalent regions appear to be enriched for binding sites for at least one of the pluripotency TFs mentioned above and can be poised for transcriptional activation or repression during both developmental and differentiation processes to respond quickly to developmental cues (238, 240–242).
Some cancer cells have bivalently marked genes that correspond to those in embryonic SCs, but a remarkable number of regions that are bivalently marked in SCs are frequently hypermethylated and thus completely silenced in cancer cells (243, 244). Local DNA hypermethylation at tumor suppressor genes or genes associated with differentiation has been shown to predispose precancerous cells to oncogenic transformation and CSC establishment (243, 244). Mechanistically, many tumor cells show aberrant activation of the DNA methyltransferases DNMT1 and DNMT3, which are involved in the maintenance of existing methylation patterns or in the de novo methylation at CpG islands, respectively. DNMT hyperactivation is also required for the maintenance of the CSC subpopulations (245).
In the context of STSs, increased expression of DNMT1 in LPS (compared to fat) results in miR-193b downregulation by promoter methylation (246). Expression of miR-193b has been linked with adipogenesis in adipose-derived SCs and with increased apoptosis in LPS cells, as miR-193b mimetics were able to inhibit LPS xenograft growth in vivo (246). However, further studies are required to clarify whether DNMT1 might play a role in the establishment of CSCs in LPS. Similarly, in ERMS DNMT3B appears to be important for the maintenance of a less differentiated phenotype, since its depletion reverses cell cancer phenotype by rescuing the myogenic program (247). In uterine LMS, treatment with the HDAC inhibitor vorinostat in combination with a DNA demethylating agent such as decitabine allowed overcoming the resistance to cell death induction due to promoter methylation of apoptotic genes (103). Given that resistance to chemotherapeutics represents an essential characteristic of CSCs, it is possible that DNA methylation might favor a CSC phenotype.
The process of CSC reprogramming has been also correlated to histone modifications. Thus, it is not surprising that epigenetic modifiers constitute the most altered genes in both solid cancers and hematological malignancies (248). For example, enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the polycomb repressive complex PRC2, has been found overexpressed in several tumors, in which it contributes to H3K27me3-mediated silencing of tumor suppressor genes, besides promoting a self-renewal transcriptional program that allows CSC expansion (249–253). Similarly, upregulation of a key subunit of the PRC1 complex, BMI1, has been shown to favor the reprogramming toward a CSC phenotype through the repression of tumor suppressor pathways in tumor-initiating cells (254, 255).
Except for BMI1, which has been reported to be highly expressed in fractions of CD133+ SS cells, characterized by increased clonogenicity, self-renewal, and in vivo tumor formation (137), no association between histone modifications and CSC phenotype has been demonstrated to date in STS. However, studies showed altered expression of the epigenetic modifier EZH2 in ERMS and SS, which has been related to the survival of cancer cells and to the maintenance of a less differentiated and more aggressive phenotype, suggesting pharmacological inhibition of EZH2 as adjuvant differentiation therapy (256–258). Also, a direct involvement of PRC2 components in the progression from neurofibroma to MPNST has been demonstrated showing that, surprisingly, EZH2 works as a tumor suppressor, and the detection of the loss of H3K27 trimethylation has entered the clinical practice to help in the diagnosis of MPNST (83).
Three well characterized ATP-dependent chromatin remodelers (SWI/SNF, ISWI, CHD) have also been also implicated in tumor initiation. Loss of the SWI/SNF complex, for instance, plays an important role in sarcomagenesis (259–261). Together, these data indicate that alterations in chromatin status may represent a key step for CSC formation and maintenance, by inducing the activation of several stemness signals in differentiated cancer cells. In SS, the SS18-SSX fusion protein has been shown to compete with SS18, a normal subunit of the SWI/SNF complex, for the assembly into the complex, thus reverting the H3K27me3-mediated repression at the Sox2 locus (262). In RMS, several lines of evidence suggested that alteration of SWI/SNF components might help to maintain tumor cells in a less-differentiated state. Indeed, the activation of the ATPase subunit of the SWI/SNF chromatin remodeling complex BRG1, for instance, provided an open chromatin conformation for the induction of myogenin in ERMS cell lines (263). Likewise, silencing of the SWI/SNF complex subunit BAF53a, which has been found hyper-expressed in primary RMS tumors compared to normal muscle, increases the expression of myogenic markers contributing to the differentiation program (264). Interestingly, CHD4 was identified as an important epigenetic co-regulator of PAX3-FOXO1 activity in ARMS. Together, both of these proteins bind to the regulatory regions of PAX3-FOXO1 target genes (265). All together, these data emphasize the necessity to address the requirement of these epigenetic modifiers in the maintenance of a stem-like phenotype in STSs.
Challenges and Future Directions
The role of CSCs in tumorigenesis and the ability to therapeutically target their vulnerabilities will continue to be important for all cancer types. However, since STSs are less common than—for example—cancers of the breast, colon, lung, prostate, and melanoma, and exhibit enormous cellular and molecular heterogeneity, information regarding stemness in STS and how to apply it therapeutically will lag. On the other hand, because of the unique characteristics of STSs, there may be unanticipated opportunities for investigation. For example, since STSs are all soft tissue cancers of mesenchymal origin, can we identify conserved CSC signatures spanning STSs that can be exploited for therapy? Since many STSs have unique signature translocations that drive their tumorigenesis, can we compare and contrast the impact of the encoded oncogenic fusion proteins on CSC stemness and identify commonalities to target? It is likely that there is not a “one-size-fits-all” CSC for STSs, and that translocation-positive STSs with their more homogeneous genomes may need to be studied separately from translocation—negative STSs in order to understand how and when the stochastic and hierarchical models of CSCs apply.
There remain many gaps in the STS CSC field. Some of these gaps are technical—for example, can we standardize our definition of CSC, and consistently apply the most stringent criteria for “stemness,” which is the ability for a small number of tumor cells to give rise to an STS, rather than rely on descriptions of stemness TF or cell surface marker expression? On the other hand, some of these gaps are conceptual. Because of the sheer number of STSs subtypes and the intrinsic complexity of a heterogeneous tumor, it has not been possible to undertake a comprehensive and systematic investigation of other forces that impact cancer cell stemness in STSs, such as the microenvironment, the CSC niche, the role of immunoediting, mechanical cellular forces, and so forth (26, 266). Bridging these knowledge gaps will take time and coordinated effort between fields including but not limited to cancer biology, bioinformatics, mathematics, bioengineering, immunology, and evolutionary biology.
Regarding future directions, two fields in particular are rapidly changing and having an immediate impact on STS biology and therapy: epigenetics and immunotherapy. Knowledge of epigenetic circuitry in both SC and CSC is increasing, and many of the involved proteins have druggable moieties (267). Can these moieties be evaluated in STS basic and preclinical studies? Once we identify these moieties, can we then understand patterns of treatment resistance, whereby one CSC epigenetic circuit might compensate for another? Knowledge and application of immunotherapy has also revolutionized the treatment of cancer at the CSC level, including via monoclonal antibodies, checkpoint modulators, and CAR-T approaches (268). For example, targeting CSC markers is currently being attempted via CAR-T cells against CD133 (clinicaltrials.gov NCT02541370, NCT03423992, NCT03473457). Caution must be maintained, however, since in some carcinoma xenograft studies even CD133− cells can initiate tumors (269). Nevertheless, with transdisciplinary approaches, we should have confidence that knowledge of STS cancer stem cell biology will also progress and lead to improve patient outcomes.
Author Contributions
CL, RR, SP, and KG: Conceived review; KG and SP: Wrote the paper; CL and RR: Edited the paper.
Funding
This research was supported by a St. Baldrick's Summer Student 2017 Fellowship (to KG), Ministry of Health Ricerca Finalizzata PE-2013-02355271 and AIRC 15312 grants (to RR), and V Foundation, Hyundai Hope on Wheels, and Glenn and Stacy Schiffman Pediatric Cancer Research Fund grants (to CL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the RR and CL laboratories for their helpful suggestions.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. (2016) 66:7–30. doi: 10.3322/caac.21332
2. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol Biomarkers Prev. (2016) 25:16–27. doi: 10.1158/1055-9965.EPI-15-0578
3. Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. (2017) 2017:5619472. doi: 10.1155/2017/5619472
4. Zheng S, Xin L, Liang A, Fu Y. Cancer stem cell hypothesis: a brief summary and two proposals. Cytotechnology (2013) 65:505–12. doi: 10.1007/s10616-012-9517-3
5. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. (2015) 42(Suppl. 1):S3–17. doi: 10.1053/j.seminoncol.2015.01.001
6. Zhang D, Tang DG, Rycaj K. Cancer stem cells: regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. (2018) 52:94–106. doi: 10.1016/j.semcancer.2018.05.001
7. Nowell PC. The clonal evolution of tumor cell populations. Science (1976) 194:23–8. doi: 10.1126/science.959840
8. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. (2006) 66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126
9. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer (2012) 12:133–43. doi: 10.1038/nrc3184
10. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367:645–8. doi: 10.1038/367645a0
11. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
12. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
13. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature (2007) 445:106–10. doi: 10.1038/nature05372
14. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature (2007) 445:111–5. doi: 10.1038/nature05384
15. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432:396–401. doi: 10.1038/nature03128
16. Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. (2005) 65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343
17. Perego M, Tortoreto M, Tragni G, Mariani L, Deho P, Carbone A, et al. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. J Invest Dermatol. (2010) 130:1877–86. doi: 10.1038/jid.2010.69
18. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. (2006) 355:1253–61. doi: 10.1056/NEJMra061808
19. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—What challenges do they pose? Nat Rev Drug Discov. (2014) 13:497–512. doi: 10.1038/nrd4253
20. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. (2011) 8:97–106. doi: 10.1038/nrclinonc.2010.196
21. Blanpain C. Tracing the cellular origin of cancer. Nat Cell Biol. (2013) 15:126–34. doi: 10.1038/ncb2657
22. Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, et al. Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell (2015) 28:800–14. doi: 10.1016/j.ccell.2015.10.003
23. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature (2009) 457:608–11. doi: 10.1038/nature07602
24. White AC, Lowry WE. Refining the role for adult stem cells as cancer cells of origin. Trends Cell Biol. (2015) 25:11–20. doi: 10.1016/j.tcb.2014.08.008
25. Varga J, Greten FR. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat Cell Biol. (2017) 19:1133–41. doi: 10.1038/ncb3611
26. Poli V, Fagnocchi L, Zippo A. Tumorigenic cell reprogramming and cancer plasticity: interplay between signaling, microenvironment, and epigenetics. Stem Cells Int. (2018) 2018:4598195. doi: 10.1155/2018/4598195
27. Ferrari A, Sultan I, Huang TT, Rodriguez-Galindo C, Shehadeh A, Meazza C, et al. Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer (2011) 57:943–9. doi: 10.1002/pbc.23252
28. Mohseny AB, Hogendoorn PC. Concise review: mesenchymal tumors: when stem cells go mad. Stem Cells (2011) 29:397–403. doi: 10.1002/stem.596
29. Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, et al. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. (2007) 117:3248–57. doi: 10.1172/JCI31377
30. Li Q, Hisha H, Takaki T, Adachi Y, Li M, Song C, et al. Transformation potential of bone marrow stromal cells into undifferentiated high-grade pleomorphic sarcoma. J Cancer Res Clin Oncol. (2010) 136:829–38. doi: 10.1007/s00432-009-0723-0
31. Nielsen TO, West RB, Linn SC, Alter O, Knowling MA, O'Connell JX, et al. Molecular characterisation of soft tissue tumours: a gene expression study. Lancet (2002) 359:1301–7. doi: 10.1016/S0140-6736(02)08270-3
32. Danielson LS, Menendez S, Attolini CS, Guijarro MV, Bisogna M, Wei J, et al. A differentiation-based microRNA signature identifies leiomyosarcoma as a mesenchymal stem cell-related malignancy. Am J Pathol. (2010) 177:908–17. doi: 10.2353/ajpath.2010.091150
33. Matushansky I, Hernando E, Socci ND, Matos T, Mills J, Edgar MA, et al. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. Am J Pathol. (2008) 172:1069–80. doi: 10.2353/ajpath.2008.070284
34. Casali PG, Abecassis N, Bauer S, Biagini R, Bielack S, Bonvalot S, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2018) 29 (Suppl. 4):iv51–67. doi: 10.1093/annonc/mdy096
35. Fujiwara T, Ozaki T. Overcoming therapeutic resistance of bone sarcomas: overview of the molecular mechanisms and therapeutic targets for bone sarcoma stem cells. Stem Cells Int. (2016) 2016:2603092. doi: 10.1155/2016/2603092
36. Abarrategi A, Tornin J, Martinez-Cruzado L, Hamilton A, Martinez-Campos E, Rodrigo JP, et al. Osteosarcoma: cells-of-origin, cancer stem cells, and targeted therapies. Stem Cells Int. (2016) 2016:3631764. doi: 10.1155/2016/3631764
37. Laranjeira AB, Yang SX. Therapeutic target discovery and drug development in cancer stem cells for leukemia and lymphoma: from bench to the clinic. Expert Opin Drug Discov. (2016) 11:1071–80. doi: 10.1080/17460441.2016.1236785
38. Khan MI, Czarnecka AM, Helbrecht I, Bartnik E, Lian F, Szczylik C. Current approaches in identification and isolation of human renal cell carcinoma cancer stem cells. Stem Cell Res Ther. (2015) 6:178. doi: 10.1186/s13287-015-0177-z
39. Jain S, Xu R, Prieto VG, Lee P. Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol. (2010) 3:416–28.
40. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. (2009) 27:3391–7. doi: 10.1200/JCO.2008.19.7483
41. Shern JF, Yohe ME, Khan J. Pediatric rhabdomyosarcoma. Crit Rev Oncog. (2015) 20:227–43. doi: 10.1615/CritRevOncog.2015013800
42. Saab R, Spunt SL, Skapek SX. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol. (2011) 94:197–234. doi: 10.1016/B978-0-12-380916-2.00007-3
43. Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. (1993) 3:113–7. doi: 10.1038/ng0293-113
44. Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. (1994) 54:2869–72.
45. Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell (2013) 24:710–24. doi: 10.1016/j.ccr.2013.11.002
46. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. (2014) 4:216–31. doi: 10.1158/2159-8290.CD-13-0639
47. Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. (2015) 6:7557. doi: 10.1038/ncomms8557
48. Olanich ME, Barr FG. A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma. Expert Opin Ther Targets (2013) 17:607–23. doi: 10.1517/14728222.2013.772136
49. Hirotsu M, Setoguchi T, Matsunoshita Y, Sasaki H, Nagao H, Gao H, et al. Tumour formation by single fibroblast growth factor receptor 3-positive rhabdomyosarcoma-initiating cells. Br J Cancer (2009) 101:2030–7. doi: 10.1038/sj.bjc.6605407
50. Walter D, Satheesha S, Albrecht P, Bornhauser BC, D'Alessandro V, Oesch SM, et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS ONE (2011) 6:e19506. doi: 10.1371/journal.pone.0019506
51. Nakahata K, Uehara S, Nishikawa S, Kawatsu M, Zenitani M, Oue T, et al. Aldehyde dehydrogenase 1 (ALDH1) is a potential marker for cancer stem cells in embryonal rhabdomyosarcoma. PLoS ONE (2015) 10:e0125454. doi: 10.1371/journal.pone.0125454
52. Skoda J, Nunukova A, Loja T, Zambo I, Neradil J, Mudry P, et al. Cancer stem cell markers in pediatric sarcomas: Sox2 is associated with tumorigenicity in immunodeficient mice. Tumour Biol. (2016) 37:9535–48. doi: 10.1007/s13277-016-4837-0
53. Almazan-Moga A, Zarzosa P, Vidal I, Molist C, Giralt I, Navarro N, et al. Hedgehog pathway inhibition hampers sphere and holoclone formation in rhabdomyosarcoma. Stem Cells Int. (2017) 2017:7507380. doi: 10.1155/2017/7507380
54. Naka N, Takenaka S, Araki N, Miwa T, Hashimoto N, Yoshioka K, et al. Synovial sarcoma is a stem cell malignancy. Stem Cells (2010) 28:1119–31. doi: 10.1002/stem.452
55. Herzog CE. Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol. (2005) 27:215–8. doi: 10.1097/01.mph.0000161762.53175.e4
56. Canter RJ, Qin LX, Maki RG, Brennan MF, Ladanyi M, Singer S. A synovial sarcoma-specific preoperative nomogram supports a survival benefit to ifosfamide-based chemotherapy and improves risk stratification for patients. Clin Cancer Res. (2008) 14:8191–7. doi: 10.1158/1078-0432.CCR-08-0843
57. Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the surveillance, epidemiology, and end results program, 1983 to 2005: an analysis of 1268 patients. Cancer (2009) 115:3537–47. doi: 10.1002/cncr.24424
58. Vlenterie M, Litiere S, Rizzo E, Marreaud S, Judson I, Gelderblom H, et al. Outcome of chemotherapy in advanced synovial sarcoma patients: review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur J Cancer (2016) 58:62–72. doi: 10.1016/j.ejca.2016.02.002
59. Knight JC, Reeves BR, Kearney L, Monaco AP, Lehrach H, Cooper CS. Localization of the synovial sarcoma t(X;18)(p11.2;q11.2) breakpoint by fluorescence in situ hybridization. Hum Mol Genet. (1992) 1:633–7. doi: 10.1093/hmg/1.8.633
60. Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. (1994) 7:502–8. doi: 10.1038/ng0894-502
61. Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. (1995) 14:2333–40. doi: 10.1002/j.1460-2075.1995.tb07228.x
62. Skytting B, Nilsson G, Brodin B, Xie Y, Lundeberg J, Uhlen M, et al. A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst. (1999) 91:974–5. doi: 10.1093/jnci/91.11.974
63. Thaete C, Brett D, Monaghan P, Whitehouse S, Rennie G, Rayner E, et al. Functional domains of the SYT and SYT-SSX synovial sarcoma translocation proteins and co-localization with the SNF protein BRM in the nucleus. Hum Mol Genet. (1999) 8:585–91. doi: 10.1093/hmg/8.4.585
64. Kato H, Tjernberg A, Zhang W, Krutchinsky AN, An W, Takeuchi T, et al. SYT associates with human SNF/SWI complexes and the C-terminal region of its fusion partner SSX1 targets histones. J Biol Chem. (2002) 277:5498–505. doi: 10.1074/jbc.M108702200
65. Middeljans E, Wan X, Jansen PW, Sharma V, Stunnenberg HG, Logie C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE (2012) 7:e33834. doi: 10.1371/journal.pone.0033834
66. Svejstrup JQ. Synovial sarcoma mechanisms: a series of unfortunate events. Cell (2013) 153:11–2. doi: 10.1016/j.cell.2013.03.015
67. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. (2013) 45:592–601. doi: 10.1038/ng.2628
68. Nielsen TO, Perou CM. CCR 20th anniversary commentary: the development of breast cancer molecular subtyping. Clin Cancer Res. (2015) 21:1779–81. doi: 10.1158/1078-0432.CCR-14-2552
69. Barco R, Garcia CB, Eid JE. The synovial sarcoma-associated SYT-SSX2 oncogene antagonizes the polycomb complex protein Bmi1. PLoS ONE (2009) 4:e5060. doi: 10.1371/journal.pone.0005060
70. Tamaki S, Fukuta M, Sekiguchi K, Jin Y, Nagata S, Hayakawa K, et al. SS18-SSX, the oncogenic fusion protein in synovial sarcoma, is a cellular context-dependent epigenetic modifier. PLoS ONE (2015) 10:e0142991. doi: 10.1371/journal.pone.0142991
71. Panagopoulos I, Mertens F, Isaksson M, Limon J, Gustafson P, Skytting B, et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer (2001) 31:362–72. doi: 10.1002/gcc.1155
72. Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell (2007) 11:375–88. doi: 10.1016/j.ccr.2007.01.016
73. Carmody Soni EE, Schlottman S, Erkizan HV, Uren A, Toretsky JA. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin Orthop Relat Res. (2014) 472:874–82. doi: 10.1007/s11999-013-3065-9
74. Lagarde P, Przybyl J, Brulard C, Perot G, Pierron G, Delattre O, et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol. (2013) 31:608–15. doi: 10.1200/JCO.2012.46.0147
75. Garcia CB, Shaffer CM, Alfaro MP, Smith AL, Sun J, Zhao Z, et al. Reprogramming of mesenchymal stem cells by the synovial sarcoma-associated oncogene SYT-SSX2. Oncogene (2012) 31:2323–34. doi: 10.1038/onc.2011.418
76. Augsburger D, Nelson PJ, Kalinski T, Udelnow A, Knosel T, Hofstetter M, et al. Current diagnostics and treatment of fibrosarcoma perspectives for future therapeutic targets and strategies. Oncotarget (2017) 8:104638–53. doi: 10.18632/oncotarget.20136
77. Liu WD, Zhang T, Wang CL, Meng HM, Song YW, Zhao Z, et al. Sphere-forming tumor cells possess stem-like properties in human fibrosarcoma primary tumors and cell lines. Oncol Lett. (2012) 4:1315–20. doi: 10.3892/ol.2012.940
78. Feng BH, Liu AG, Gu WG, Deng L, Cheng XG, Tong TJ, et al. CD133+ subpopulation of the HT1080 human fibrosarcoma cell line exhibits cancer stem-like characteristics. Oncol Rep. (2013) 30:815–23. doi: 10.3892/or.2013.2486
79. Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. (1998) 18:184–7. doi: 10.1038/ng0298-184
80. Mackall CL, Meltzer PS, Helman LJ. Focus on sarcomas. Cancer Cell (2002) 2:175–8. doi: 10.1016/S1535-6108(02)00132-0
81. Serra E, Rosenbaum T, Winner U, Aledo R, Ars E, Estivill X, et al. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. (2000) 9:3055–64. doi: 10.1093/hmg/9.20.3055
82. Carroll SL. The challenge of cancer genomics in rare nervous system neoplasms: malignant peripheral nerve sheath tumors as a paradigm for cross-species comparative oncogenomics. Am J Pathol. (2016) 186:464–77. doi: 10.1016/j.ajpath.2015.10.023
83. Kim A, Pratilas CA. The promise of signal transduction in genetically driven sarcomas of the nerve. Exp Neurol. (2018) 299:317–25. doi: 10.1016/j.expneurol.2017.08.014
84. Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell (1990) 62:599–608. doi: 10.1016/0092-8674(90)90024-9
85. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature (1992) 356:713–5. doi: 10.1038/356713a0
86. Legius E, Dierick H, Wu R, Hall BK, Marynen P, Cassiman JJ, et al. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer (1994) 10:250–5. doi: 10.1002/gcc.2870100405
87. Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. (1999) 155:1879–84. doi: 10.1016/S0002-9440(10)65507-1
88. Mantripragada KK, Spurlock G, Kluwe L, Chuzhanova N, Ferner RE, Frayling IM, et al. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. (2008) 14:1015–24. doi: 10.1158/1078-0432.CCR-07-1305
89. Kluwe L, Friedrich R, Mautner VF. Loss of NF1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer (1999) 24:283–5. doi: 10.1002/(SICI)1098-2264(199903)24:3<283::AID-GCC15>3.0.CO;2-K
90. Carroll SL. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. (2012) 123:321–48. doi: 10.1007/s00401-011-0928-6
91. Schreiner S, Cossais F, Fischer K, Scholz S, Bosl MR, Holtmann B, et al. Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development (2007) 134:3271–81. doi: 10.1242/dev.003350
92. Cheung M, Chaboissier MC, Mynett A, Hirst E, Schedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival, and delamination. Dev Cell (2005) 8:179–92. doi: 10.1016/j.devcel.2004.12.010
93. Miller SJ, Jessen WJ, Mehta T, Hardiman A, Sites E, Kaiser S, et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol Med. (2009) 1:236–48. doi: 10.1002/emmm.200900027
94. Spyra M, Kluwe L, Hagel C, Nguyen R, Panse J, Kurtz A, et al. Cancer stem cell-like cells derived from malignant peripheral nerve sheath tumors. PLoS ONE (2011) 6:e21099. doi: 10.1371/journal.pone.0021099
95. Jagannathan JP, Tirumani SH, Ramaiya NH. Imaging in soft tissue sarcomas: current updates. Surg Oncol Clin N Am. (2016) 25:645–75. doi: 10.1016/j.soc.2016.05.002
96. El-Rifai W, Sarlomo-Rikala M, Knuutila S, Miettinen M. DNA copy number changes in development and progression in leiomyosarcomas of soft tissues. Am J Pathol. (1998) 153:985–90. doi: 10.1016/S0002-9440(10)65640-4
97. Otano-Joos M, Mechtersheimer G, Ohl S, Wilgenbus KK, Scheurlen W, Lehnert T, et al. Detection of chromosomal imbalances in leiomyosarcoma by comparative genomic hybridization and interphase cytogenetics. Cytogenet Cell Genet. (2000) 90:86–92. doi: 10.1159/000015640
98. Hu J, Rao UN, Jasani S, Khanna V, Yaw K, Surti U. Loss of DNA copy number of 10q is associated with aggressive behavior of leiomyosarcomas: a comparative genomic hybridization study. Cancer Genet Cytogenet. (2005) 161:20–7. doi: 10.1016/j.cancergencyto.2005.01.011
99. Amant F, de la Rey M, Dorfling CM, van der Walt L, Dreyer G, Dreyer L, et al. PTEN mutations in uterine sarcomas. Gynecol Oncol. (2002) 85:165–9. doi: 10.1006/gyno.2002.6601
100. Kawaguchi K, Oda Y, Saito T, Takahira T, Yamamoto H, Tamiya S, et al. Genetic and epigenetic alterations of the PTEN gene in soft tissue sarcomas. Hum Pathol. (2005) 36:357–63. doi: 10.1016/j.humpath.2005.01.017
101. Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J, et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. (2007) 13:748–53. doi: 10.1038/nm1560
102. Roncati L, Barbolini G, Sartori G, Siopis E, Pusiol T, Maiorana A. Loss of CDKN2A promoter methylation coincides with the epigenetic transdifferentiation of uterine myosarcomatous cells. Int J Gynecol Pathol. (2016) 35:309–15. doi: 10.1097/PGP.0000000000000181
103. Frohlich LF, Mrakovcic M, Smole C, Lahiri P, Zatloukal K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE (2014) 9:e91558. doi: 10.1371/journal.pone.0091558
104. Rubio R, Garcia-Castro J, Gutierrez-Aranda I, Paramio J, Santos M, Catalina P, et al. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo . Cancer Res. (2010) 70:4185–94. doi: 10.1158/0008-5472.CAN-09-4640
105. Sette G, Salvati V, Memeo L, Fecchi K, Colarossi C, Di Matteo P, et al. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PLoS ONE (2012) 7:e46891. doi: 10.1371/journal.pone.0046891
106. Conyers R, Young S, Thomas DM. Liposarcoma: molecular genetics and therapeutics. Sarcoma (2011) 2011:483154. doi: 10.1155/2011/483154
107. Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology (2014) 64:38–52. doi: 10.1111/his.12311
108. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol. (2012) 36:462–9. doi: 10.1097/PAS.0b013e3182417330
109. Chufal SS, Chufal KS, Pant P, Rizvi G, Pandey HS, Shahi KS. Hypercellular round cell liposarcoma: a comprehensive cytomorphologic study and review of 8 cases. J Cytol. (2017) 34:78–83. doi: 10.4103/0970-9371.203574
110. Hoffman A, Ghadimi MP, Demicco EG, Creighton CJ, Torres K, Colombo C, et al. Localized and metastatic myxoid/round cell liposarcoma: clinical and molecular observations. Cancer (2013) 119:1868–77. doi: 10.1002/cncr.27847
111. Lin S, Gan Z, Han K, Yao Y, Min D. Metastasis of myxoid liposarcoma to fat-bearing areas: a case report of unusual metastatic sites and a hypothesis. Oncol Lett. (2015) 10:2543–6. doi: 10.3892/ol.2015.3585
112. Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature (1993) 363:640–4. doi: 10.1038/363640a0
113. Downes KA, Goldblum JR, Montgomery EA, Fisher C. Pleomorphic liposarcoma: a clinicopathologic analysis of 19 cases. Mod Pathol. (2001) 14:179–84. doi: 10.1038/modpathol.3880280
114. Gimble JM, Katz AJ, Bunnell BA. Adipose-derived stem cells for regenerative medicine. Circ Res. (2007) 100:1249–60. doi: 10.1161/01.RES.0000265074.83288.09
115. Rodriguez R, Rubio R, Gutierrez-Aranda I, Melen GJ, Elosua C, Garcia-Castro J, et al. FUS-CHOP fusion protein expression coupled to p53 deficiency induces liposarcoma in mouse but not in human adipose-derived mesenchymal stem/stromal cells. Stem Cells (2011) 29:179–92. doi: 10.1002/stem.571
116. Perez-Losada J, Pintado B, Gutierrez-Adan A, Flores T, Banares-Gonzalez B, del Campo JC, et al. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene (2000) 19:2413–22. doi: 10.1038/sj.onc.1203572
117. Rodriguez R, Tornin J, Suarez C, Astudillo A, Rubio R, Yauk C, et al. Expression of FUS-CHOP fusion protein in immortalized/transformed human mesenchymal stem cells drives mixoid liposarcoma formation. Stem Cells (2013) 31:2061–72. doi: 10.1002/stem.1472
118. Stratford EW, Castro R, Wennerstrom A, Holm R, Munthe E, Lauvrak S, et al. Liposarcoma cells with aldefluor and CD133 activity have a cancer stem cell potential. Clin Sarcoma Res. (2011) 1:8. doi: 10.1186/2045-3329-1-8
119. Yan T, Mizutani A, Chen L, Takaki M, Hiramoto Y, Matsuda S, et al. Characterization of cancer stem-like cells derived from mouse induced pluripotent stem cells transformed by tumor-derived extracellular vesicles. J Cancer (2014) 5:572–84. doi: 10.7150/jca.8865
120. Dei Tos AP. Classification of pleomorphic sarcomas: where are we now? Histopathology (2006) 48:51–62. doi: 10.1111/j.1365-2559.2005.02289.x
122. Hofvander J, Tayebwa J, Nilsson J, Magnusson L, Brosjo O, Larsson O, et al. Recurrent PRDM10 gene fusions in undifferentiated pleomorphic sarcoma. Clin Cancer Res. (2015) 21:864–9. doi: 10.1158/1078-0432.CCR-14-2399
123. Funes JM, Quintero M, Henderson S, Martinez D, Qureshi U, Westwood C, et al. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA. (2007) 104:6223–8. doi: 10.1073/pnas.0700690104
124. Rodriguez R, Rosu-Myles M, Arauzo-Bravo M, Horrillo A, Pan Q, Gonzalez-Rey E, et al. Human bone marrow stromal cells lose immunosuppressive and anti-inflammatory properties upon oncogenic transformation. Stem Cell Rep. (2014) 3:606–19. doi: 10.1016/j.stemcr.2014.08.005
125. Rubin BP, Nishijo K, Chen HI, Yi X, Schuetze DP, Pal R, et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell (2011) 19:177–91. doi: 10.1016/j.ccr.2010.12.023
126. Wang CY, Wei Q, Han I, Sato S, Ghanbari-Azarnier R, Whetstone H, et al. Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res. (2012) 72:1013–22. doi: 10.1158/0008-5472.CAN-11-2531
127. Greve B, Kelsch R, Spaniol K, Eich HT, Gotte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry A (2012) 81:284–93. doi: 10.1002/cyto.a.22022
128. Tirino V, Desiderio V, Paino F, Papaccio G, De Rosa M. Methods for cancer stem cell detection and isolation. Methods Mol Biol. (2012) 879:513–29. doi: 10.1007/978-1-61779-815-3_32
129. Sancho-Martinez I, Izpisua Belmonte JC. Reprogramming strategies for the establishment of novel human cancer models. Cell Cycle (2016) 15:2393–7. doi: 10.1080/15384101.2016.1196305
130. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (1992) 255:1707–10. doi: 10.1126/science.1553558
131. Bielecka ZF, Maliszewska-Olejniczak K, Safir IJ, Szczylik C, Czarnecka AM. Three-dimensional cell culture model utilization in cancer stem cell research. Biol Rev Camb Philos Soc. (2017) 92:1505–20. doi: 10.1111/brv.12293
132. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia (2015) 17:1–15. doi: 10.1016/j.neo.2014.12.004
133. Suslov ON, Kukekov VG, Ignatova TN, Steindler DA. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Natl Acad Sci USA. (2002) 99:14506–11. doi: 10.1073/pnas.212525299
134. Irollo E, Pirozzi G. CD133: to be or not to be, is this the real question? Am J Transl Res. (2013) 5:563–81.
135. Terry J, Nielsen T. Expression of CD133 in synovial sarcoma. Appl Immunohistochem Mol Morphol. (2010) 18:159–65. doi: 10.1097/PAI.0b013e3181b77451
136. Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, Fazioli F, et al. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. (2011) 25:2022–30. doi: 10.1096/fj.10-179036
137. Liu A, Feng B, Gu W, Cheng X, Tong T, Zhang H, et al. The CD133+ subpopulation of the SW982 human synovial sarcoma cell line exhibits cancer stem-like characteristics. Int J Oncol. (2013) 42:1399–407. doi: 10.3892/ijo.2013.1826
138. Sana J, Zambo I, Skoda J, Neradil J, Chlapek P, Hermanova M, et al. CD133 expression and identification of CD133/nestin positive cells in rhabdomyosarcomas and rhabdomyosarcoma cell lines. Anal Cell Pathol (Amst) (2011) 34:303–18. doi: 10.1155/2011/939457
139. She Y, Liang J, Qiu Y, Liu N, Huang X, Zhao X, et al. [Expression of CD133 in primary retroperitoneal leiomyosarcoma and its relationship with Ki-67]. Zhonghua Yi Xue Za Zhi (2014) 94:1241–4. doi: 10.3760/cma.j.issn.0376-2491.2014.16.011
140. Ratajczak MZ, Zuba-Surma E, Kucia M, Reca R, Wojakowski W, Ratajczak J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia (2006) 20:1915–24. doi: 10.1038/sj.leu.2404357
141. Wynn RF, Hart CA, Corradi-Perini C, O'Neill L, Evans CA, Wraith JE, et al. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood (2004) 104:2643–5. doi: 10.1182/blood-2004-02-0526
142. Kimura T, Wang L, Tabu K, Tsuda M, Tanino M, Maekawa A, et al. Identification and analysis of CXCR4-positive synovial sarcoma-initiating cells. Oncogene (2016) 35:3932–43. doi: 10.1038/onc.2015.461
143. Strahm B, Durbin AD, Sexsmith E, Malkin D. The CXCR4-SDF1alpha axis is a critical mediator of rhabdomyosarcoma metastatic signaling induced by bone marrow stroma. Clin Exp Metastasis (2008) 25:1–10. doi: 10.1007/s10585-007-9094-6
144. Diomedi-Camassei F, McDowell HP, De Ioris MA, Uccini S, Altavista P, Raschella G, et al. Clinical significance of CXC chemokine receptor-4 and c-Met in childhood rhabdomyosarcoma. Clin Cancer Res. (2008) 14:4119–27. doi: 10.1158/1078-0432.CCR-07-4446
145. Oda Y, Tateishi N, Matono H, Matsuura S, Yamamaoto H, Tamiya S, et al. Chemokine receptor CXCR4 expression is correlated with VEGF expression and poor survival in soft-tissue sarcoma. Int J Cancer (2009) 124:1852–9. doi: 10.1002/ijc.24128
146. Wirths S, Malenke E, Kluba T, Rieger S, Muller MR, Schleicher S, et al. Shared cell surface marker expression in mesenchymal stem cells and adult sarcomas. Stem Cells Transl Med. (2013) 2:53–60. doi: 10.5966/sctm.2012-0055
147. Zambo I, Hermanova M, Zapletalova D, Skoda J, Mudry P, Kyr M, et al. Expression of nestin, CD133 and ABCG2 in relation to the clinical outcome in pediatric sarcomas. Cancer Biomark (2016) 17:107–16. doi: 10.3233/CBM-160623
148. Murphy AJ, Viero S, Ho M, Thorner PS. Diagnostic utility of nestin expression in pediatric tumors in the region of the kidney. Appl Immunohistochem Mol Morphol. (2009) 17:517–23. doi: 10.1097/PAI.0b013e3181a3259e
149. Armstrong L, Stojkovic M, Dimmick I, Ahmad S, Stojkovic P, Hole N, et al. Phenotypic characterization of murine primitive hematopoietic progenitor cells isolated on basis of aldehyde dehydrogenase activity. Stem Cells (2004) 22:1142–51. doi: 10.1634/stemcells.2004-0170
150. Hess DA, Meyerrose TE, Wirthlin L, Craft TP, Herrbrich PE, Creer MH, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood (2004) 104:1648–55. doi: 10.1182/blood-2004-02-0448
151. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res. (2009) 15:4234–41. doi: 10.1158/1078-0432.CCR-08-1479
152. Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat. (2012) 133:75–87. doi: 10.1007/s10549-011-1692-y
153. Ajani JA, Wang X, Song S, Suzuki A, Taketa T, Sudo K, et al. ALDH-1 expression levels predict response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol Oncol. (2014) 8:142–9. doi: 10.1016/j.molonc.2013.10.007
154. Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde dehydrogenase is regulated by beta-catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. (2015) 75:1482–94. doi: 10.1158/0008-5472.CAN-14-1924
155. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
156. Lohberger B, Rinner B, Stuendl N, Absenger M, Liegl-Atzwanger B, Walzer SM, et al. Aldehyde dehydrogenase 1, a potential marker for cancer stem cells in human sarcoma. PLoS ONE (2012) 7:e43664. doi: 10.1371/journal.pone.0043664
157. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. (1996) 183:1797–806. doi: 10.1084/jem.183.4.1797
158. Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, et al. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. (1997) 3:1337–45. doi: 10.1038/nm1297-1337
159. Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. (2004) 1:27–42. doi: 10.2174/1567201043480036
160. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. (2004) 101:14228–33. doi: 10.1073/pnas.0400067101
161. Christgen M, Ballmaier M, Lehmann U, Kreipe H. Detection of putative cancer stem cells of the side population phenotype in human tumor cell cultures. Methods Mol Biol. (2012) 878:201–15. doi: 10.1007/978-1-61779-854-2_13
162. Murase M, Kano M, Tsukahara T, Takahashi A, Torigoe T, Kawaguchi S, et al. Side population cells have the characteristics of cancer stem-like cells/cancer-initiating cells in bone sarcomas. Br J Cancer (2009) 101:1425–32. doi: 10.1038/sj.bjc.6605330
163. Yang M, Zhang R, Yan M, Ye Z, Liang W, Luo Z. Detection and characterization of side population in Ewing's sarcoma SK-ES-1 cells in vitro. Biochem Biophys Res Commun. (2010) 391:1062–6. doi: 10.1016/j.bbrc.2009.12.020
164. Boesch M, Zeimet AG, Reimer D, Schmidt S, Gastl G, Parson W, et al. The side population of ovarian cancer cells defines a heterogeneous compartment exhibiting stem cell characteristics. Oncotarget (2014) 5:7027–39. doi: 10.18632/oncotarget.2053
165. Boesch M, Zeimet AG, Fiegl H, Wolf B, Huber J, Klocker H, et al. High prevalence of side population in human cancer cell lines. Oncoscience (2016) 3:85–7. doi: 10.18632/oncoscience.300
166. Fukuda K, Saikawa Y, Ohashi M, Kumagai K, Kitajima M, Okano H, et al. Tumor initiating potential of side population cells in human gastric cancer. Int J Oncol. (2009) 34:1201–7. doi: 10.3892/ijo_00000248
167. Loebinger MR, Giangreco A, Groot KR, Prichard L, Allen K, Simpson C, et al. Squamous cell cancers contain a side population of stem-like cells that are made chemosensitive by ABC transporter blockade. Br J Cancer (2008) 98:380–7. doi: 10.1038/sj.bjc.6604185
168. Luo Y, Ellis LZ, Dallaglio K, Takeda M, Robinson WA, Robinson SE, et al. Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J Invest Dermatol. (2012) 132:2440–50. doi: 10.1038/jid.2012.161
169. Wu C, Wei Q, Utomo V, Nadesan P, Whetstone H, Kandel R, et al. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. (2007) 67:8216–22. doi: 10.1158/0008-5472.CAN-07-0999
170. Bonnefoix T, Bonnefoix P, Verdiel P, Sotto JJ. Fitting limiting dilution experiments with generalized linear models results in a test of the single-hit Poisson assumption. J Immunol Methods (1996) 194:113–9. doi: 10.1016/0022-1759(96)00077-4
171. Dick JE, Bhatia M, Gan O, Kapp U, Wang JC. Assay of human stem cells by repopulation of NOD/SCID mice. Stem Cells (1997) 15 (Suppl. 1):199–203. discussion 4–7. doi: 10.1002/stem.5530150826
172. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods (2009) 347:70–8. doi: 10.1016/j.jim.2009.06.008
173. Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. (2008) 68:4674–82. doi: 10.1158/0008-5472.CAN-07-6353
174. Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature (2010) 465:704–12. doi: 10.1038/nature09229
175. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
176. Hochedlinger K, Blelloch R, Brennan C, Yamada Y, Kim M, Chin L, et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. (2004) 18:1875–85. doi: 10.1101/gad.1213504
177. Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu DT, Chen DT, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA (2008) 14:2115–24. doi: 10.1261/rna.1162708
178. Koga C, Kobayashi S, Nagano H, Tomimaru Y, Hama N, Wada H, et al. Reprogramming using microRNA-302 improves drug sensitivity in hepatocellular carcinoma cells. Ann Surg Oncol. (2014) 21 (Suppl. 4):S591–600. doi: 10.1245/s10434-014-3705-7
179. Debeb BG, Lacerda L, Xu W, Larson R, Solley T, Atkinson R, et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells (2012) 30:2366–77. doi: 10.1002/stem.1219
180. Chang G, Miao YL, Zhang Y, Liu S, Kou Z, Ding J, et al. Linking incomplete reprogramming to the improved pluripotency of murine embryonal carcinoma cell-derived pluripotent stem cells. PLoS ONE (2010) 5:e10320. doi: 10.1371/journal.pone.0010320
181. Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, et al. Generation of iPSCs from cultured human malignant cells. Blood (2010) 115:4039–42. doi: 10.1182/blood-2009-07-231845
182. Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA. (2010) 107:40–5. doi: 10.1073/pnas.0912407107
183. Kim J, Zaret KS. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. (2015) 34:739–47. doi: 10.15252/embj.201490736
184. Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature (1998) 396:370–3. doi: 10.1038/24620
185. Loebel DA, Watson CM, De Young RA, Tam PP. Lineage choice and differentiation in mouse embryos and embryonic stem cells. Dev Biol. (2003) 264:1–14. doi: 10.1016/S0012-1606(03)00390-7
186. Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development (2011) 138:3593–612. doi: 10.1242/dev.063610
187. Thesleff I, Sharpe P. Signalling networks regulating dental development. Mech Dev. (1997) 67:111–23. doi: 10.1016/S0925-4773(97)00115-9
188. Ulloa F, Briscoe J. Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle (2007) 6:2640–9. doi: 10.4161/cc.6.21.4822
189. Zeller R, Lopez-Rios J, Zuniga A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat Rev Genet. (2009) 10:845–58. doi: 10.1038/nrg2681
190. Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE (2010) 5:e12445. doi: 10.1371/journal.pone.0012445
191. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci USA. (2011) 108:9951–6. doi: 10.1073/pnas.1016912108
192. Suva ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell (2014) 157:580–94. doi: 10.1016/j.cell.2014.02.030
193. Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell (2011) 145:835–50. doi: 10.1016/j.cell.2011.05.019
194. Young RA. Control of the embryonic stem cell state. Cell (2011) 144:940–54. doi: 10.1016/j.cell.2011.01.032
195. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell (2010) 143:313–24. doi: 10.1016/j.cell.2010.09.010
196. Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. (2010) 70:10433–44. doi: 10.1158/0008-5472.CAN-10-2638
197. Eberle I, Pless B, Braun M, Dingermann T, Marschalek R. Transcriptional properties of human NANOG1 and NANOG2 in acute leukemic cells. Nucleic Acids Res. (2010) 38:5384–95. doi: 10.1093/nar/gkq307
198. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. (2008) 22:2454–72. doi: 10.1101/gad.1693608
199. Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers (Basel) (2015) 7:1554–85. doi: 10.3390/cancers7030851
200. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell (1996) 85:841–51. doi: 10.1016/S0092-8674(00)81268-4
201. Satheesha S, Manzella G, Bovay A, Casanova EA, Bode PK, Belle R, et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene (2016) 35:2020–30. doi: 10.1038/onc.2015.267
202. Roma J, Almazan-Moga A, Sanchez de Toledo J, Gallego S. Notch, wnt, and hedgehog pathways in rhabdomyosarcoma: from single pathways to an integrated network. Sarcoma (2012) 2012:695603. doi: 10.1155/2012/695603
203. Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. (2014) 15:642–56. doi: 10.15252/embr.201438638
204. Zibat A, Missiaglia E, Rosenberger A, Pritchard-Jones K, Shipley J, Hahn H, et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene (2010) 29:6323–30. doi: 10.1038/onc.2010.368
205. Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell (2014) 26:273–87. doi: 10.1016/j.ccr.2014.05.029
206. Eisinger-Mathason TS, Mucaj V, Biju KM, Nakazawa MS, Gohil M, Cash TP, et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc Natl Acad Sci USA. (2015) 112:E3402–11. doi: 10.1073/pnas.1420005112
207. Fullenkamp CA, Hall SL, Jaber OI, Pakalniskis BL, Savage EC, Savage JM, et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget (2016) 7:30094–108. doi: 10.18632/oncotarget.8979
208. Deel MD, Li JJ, Crose LE, Linardic CM. A review: molecular aberrations within hippo signaling in bone and soft-tissue sarcomas. Front Oncol. (2015) 5:190. doi: 10.3389/fonc.2015.00190
209. Park JH, Shin JE, Park HW. The role of hippo pathway in cancer stem cell biology. Mol Cells (2018) 41:83–92. doi: 10.14348/molcells.2018.2242
210. Slemmons KK, Crose LES, Riedel S, Sushnitha M, Belyea B, Linardic CM. A novel notch-yap circuit drives stemness and tumorigenesis in embryonal rhabdomyosarcoma. Mol Cancer Res. (2017) 15:1777–91. doi: 10.1158/1541-7786.MCR-17-0004
211. Deel MD, Slemmons KK, Hinson AR, Genadry KC, Burgess BA, Crose LES, et al. The transcriptional coactivator TAZ is a potent mediator of alveolar rhabdomyosarcoma tumorigenesis. Clin Cancer Res. (2018) 24:2616–30. doi: 10.1158/1078-0432.CCR-17-1207
212. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284:770–6. doi: 10.1126/science.284.5415.770
213. Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature (2006) 442:823–6. doi: 10.1038/nature04940
214. Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. (2010) 70:709–18. doi: 10.1158/0008-5472.CAN-09-1681
215. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. (2017) 36:41. doi: 10.1186/s13046-017-0507-3
216. Roma J, Masia A, Reventos J, Sanchez de Toledo J, Gallego S. Notch pathway inhibition significantly reduces rhabdomyosarcoma invasiveness and mobility in vitro. Clin Cancer Res. (2011) 17:505–13. doi: 10.1158/1078-0432.CCR-10-0166
217. Ignatius MS, Hayes MN, Lobbardi R, Chen EY, McCarthy KM, Sreenivas P, et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. (2017) 19:2304–18. doi: 10.1016/j.celrep.2017.05.061
218. Francis P, Namlos HM, Muller C, Eden P, Fernebro J, Berner JM, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics (2007) 8:73. doi: 10.1186/1471-2164-8-73
219. Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene (2009) 433:1–7. doi: 10.1016/j.gene.2008.12.008
220. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. (2017) 51:1357–69. doi: 10.3892/ijo.2017.4129
221. Chien AJ, Moore EC, Lonsdorf AS, Kulikauskas RM, Rothberg BG, Berger AJ, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA. (2009) 106:1193–8. doi: 10.1073/pnas.0811902106
222. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer (2013) 13:11–26. doi: 10.1038/nrc3419
223. Singh S, Vinson C, Gurley CM, Nolen GT, Beggs ML, Nagarajan R, et al. Impaired Wnt signaling in embryonal rhabdomyosarcoma cells from p53/c-fos double mutant mice. Am J Pathol. (2010) 177:2055–66. doi: 10.2353/ajpath.2010.091195
224. Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM, et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/beta-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA. (2014) 111:5349–54. doi: 10.1073/pnas.1317731111
225. Kephart JJ, Tiller RG, Crose LE, Slemmons KK, Chen PH, Hinson AR, et al. Secreted frizzled-related protein 3 (SFRP3) is required for tumorigenesis of PAX3-FOXO1-positive alveolar rhabdomyosarcoma. Clin Cancer Res. (2015) 21:4868–80. doi: 10.1158/1078-0432.CCR-14-1797
226. Barham W, Frump AL, Sherrill TP, Garcia CB, Saito-Diaz K, VanSaun MN, et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. (2013) 3:1286–301. doi: 10.1158/2159-8290.CD-13-0138
227. Nagayama S, Fukukawa C, Katagiri T, Okamoto T, Aoyama T, Oyaizu N, et al. Therapeutic potential of antibodies against FZD 10, a cell-surface protein, for synovial sarcomas. Oncogene (2005) 24:6201–12. doi: 10.1038/sj.onc.1208780
228. Hanaoka H, Katagiri T, Fukukawa C, Yoshioka H, Yamamoto S, Iida Y, et al. Radioimmunotherapy of solid tumors targeting a cell-surface protein, FZD10: therapeutic efficacy largely depends on radiosensitivity. Ann Nucl Med. (2009) 23:479–85. doi: 10.1007/s12149-009-0265-1
229. Watson AL, Rahrmann EP, Moriarity BS, Choi K, Conboy CB, Greeley AD, et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. (2013) 3:674–89. doi: 10.1158/2159-8290.CD-13-0081
230. Mo W, Chen J, Patel A, Zhang L, Chau V, Li Y, et al. CXCR4/CXCL12 mediate autocrine cell-cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell (2013) 152:1077–90. doi: 10.1016/j.cell.2013.01.053
231. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell (2005) 122:947–56. doi: 10.1016/j.cell.2005.08.020
232. Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. (2006) 38:431–40. doi: 10.1038/ng1760
233. Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. (2006) 8:188–94. doi: 10.1038/ncb1353
234. Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. (2004) 18:1263–71. doi: 10.1101/gad.1198204
235. Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. (2006) 8:532–8. doi: 10.1038/ncb1403
236. Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. (2013) 27:1318–38. doi: 10.1101/gad.219626.113
237. Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, et al. H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell (2015) 60:584–96. doi: 10.1016/j.molcel.2015.10.025
238. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature (2007) 448:553–60. doi: 10.1038/nature06008
239. Margaritis T, Holstege FC. Poised RNA polymerase II gives pause for thought. Cell (2008) 133:581–4. doi: 10.1016/j.cell.2008.04.027
240. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell (2006) 125:315–26. doi: 10.1016/j.cell.2006.02.041
241. Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, et al. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell (2007) 1:299–312. doi: 10.1016/j.stem.2007.08.003
242. Amador-Arjona A, Cimadamore F, Huang CT, Wright R, Lewis S, Gage FH, et al. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc Natl Acad Sci USA. (2015) 112:E1936–45. doi: 10.1073/pnas.1421480112
243. Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. (2007) 39:237–42. doi: 10.1038/ng1972
244. Rodriguez J, Munoz M, Vives L, Frangou CG, Groudine M, Peinado MA. Bivalent domains enforce transcriptional memory of DNA methylated genes in cancer cells. Proc Natl Acad Sci USA. (2008) 105:19809–14. doi: 10.1073/pnas.0810133105
245. Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. (2012) 26:344–9. doi: 10.1101/gad.184341.111
246. Mazzu YZ, Hu Y, Soni RK, Mojica KM, Qin LX, Agius P, et al. miR-193b-regulated signaling networks serve as tumor suppressors in liposarcoma and promote adipogenesis in adipose-derived stem cells. Cancer Res. (2017) 77:5728–40. doi: 10.1158/0008-5472.CAN-16-2253
247. Megiorni F, Camero S, Ceccarelli S, McDowell HP, Mannarino O, Marampon F, et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget (2016) 7:79342–56. doi: 10.18632/oncotarget.12688
248. Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. (2012) 6:620–36. doi: 10.1016/j.molonc.2012.10.006
249. Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, et al. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle (2009) 8:1991–6. doi: 10.4161/cc.8.13.8929
250. Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. (2009) 69:9211–8. doi: 10.1158/0008-5472.CAN-09-1622
251. Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, et al. Ovarian cancer stem cell-like side populations are enriched following chemotherapy and overexpress EZH2. Mol Cancer Ther. (2011) 10:325–35. doi: 10.1158/1535-7163.MCT-10-0788
252. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell (2011) 19:86–100. doi: 10.1016/j.ccr.2010.10.035
253. Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beausejour M, Beaulieu JF, et al. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res. (2013) 319:1463–70. doi: 10.1016/j.yexcr.2013.04.006
254. Yang J, Chai L, Liu F, Fink LM, Lin P, Silberstein LE, et al. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci USA. (2007) 104:10494–9. doi: 10.1073/pnas.0704001104
255. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. (2009) 29:8884–96. doi: 10.1523/JNEUROSCI.0968-09.2009
256. Changchien YC, Tatrai P, Papp G, Sapi J, Fonyad L, Szendroi M, et al. Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J Transl Med. (2012) 10:216. doi: 10.1186/1479-5876-10-216
257. Ciarapica R, Carcarino E, Adesso L, De Salvo M, Bracaglia G, Leoncini PP, et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer (2014) 14:139. doi: 10.1186/1471-2407-14-139
258. Kawano S, Grassian AR, Tsuda M, Knutson SK, Warholic NM, Kuznetsov G, et al. Preclinical evidence of anti-tumor activity induced by EZH2 inhibition in human models of synovial sarcoma. PLoS ONE (2016) 11:e0158888. doi: 10.1371/journal.pone.0158888
259. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. (2014) 166C:350–66. doi: 10.1002/ajmg.c.31410
260. Kohashi K, Yamamoto H, Yamada Y, Kinoshita I, Taguchi T, Iwamoto Y, et al. SWI/SNF Chromatin-remodeling complex status in SMARCB1/INI1-preserved epithelioid sarcoma. Am J Surg Pathol. (2018) 42:312–8. doi: 10.1097/PAS.0000000000001011
261. McBride MJ, Kadoch C. Disruption of mammalian SWI/SNF and polycomb complexes in human sarcomas: mechanisms and therapeutic opportunities. J Pathol. (2018) 244:638–49. doi: 10.1002/path.5042
262. Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell (2013) 153:71–85. doi: 10.1016/j.cell.2013.02.036
263. Li ZY, Yang J, Gao X, Lu JY, Zhang Y, Wang K, et al. Sequential recruitment of PCAF and BRG1 contributes to myogenin activation in 12-O-tetradecanoylphorbol-13-acetate-induced early differentiation of rhabdomyosarcoma-derived cells. J Biol Chem. (2007) 282:18872–8. doi: 10.1074/jbc.M609448200
264. Taulli R, Foglizzo V, Morena D, Coda DM, Ala U, Bersani F, et al. Failure to downregulate the BAF53a subunit of the SWI/SNF chromatin remodeling complex contributes to the differentiation block in rhabdomyosarcoma. Oncogene (2014) 33:2354–62. doi: 10.1038/onc.2013.188
265. Bohm M, Wachtel M, Marques JG, Streiff N, Laubscher D, Nanni P, et al. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J Clin Invest. (2016) 126:4237–49. doi: 10.1172/JCI85057
266. Grunewald TG, Herbst SM, Heinze J, Burdach S. Understanding tumor heterogeneity as functional compartments–superorganisms revisited. J Transl Med. (2011) 9:79. doi: 10.1186/1479-5876-9-79
267. Pande V. Understanding the complexity of epigenetic target space. J Med Chem. (2016) 59:1299–307. doi: 10.1021/acs.jmedchem.5b01507
268. Rosenberg SA. Decade in review-cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol. (2014) 11:630–2. doi: 10.1038/nrclinonc.2014.174
Keywords: stemness, cancer stem cells, sarcoma, epigenetic plasticity, developmental pathways, soft tissue sarcoma
Citation: Genadry KC, Pietrobono S, Rota R and Linardic CM (2018) Soft Tissue Sarcoma Cancer Stem Cells: An Overview. Front. Oncol. 8:475. doi: 10.3389/fonc.2018.00475
Received: 18 March 2018; Accepted: 05 October 2018;
Published: 26 October 2018.
Edited by:
Thomas G. P. Grünewald, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Dominique Heymann, University of Nantes, FranceBarak Rotblat, Ben-Gurion University of the Negev, Israel
Copyright © 2018 Genadry, Pietrobono, Rota and Linardic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corinne M. Linardic, linar001@mc.duke.edu
†These authors have contributed equally to this work
‡These authors have contributed equally to this work