 Anna C. Filley
Anna C. Filley Mario Henriquez
Mario Henriquez Mahua Dey
Mahua Dey
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 17 October 2018
Sec. Cancer Immunity and Immunotherapy
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00453
T cell chimeric antigen receptor (CAR) technology has allowed for the introduction of a high degree of tumor selectivity into adoptive cell transfer therapies. Evolution of this technology has produced a robust antitumor immunotherapeutic strategy that has resulted in dramatic outcomes in liquid cancers. CAR-expressing T-cells (CARTs) targeting CD19 and CD20 have been successfully used in the treatment of hematologic malignancies, producing sustained tumor regressions in a majority of treated patients. These encouraging results have led to a historic and unprecedented FDA approval of CTL019, Novartis' CAR T-cell therapy for the treatment of children and young adults with relapsed or refractory B-cell acute lymphoblastic leukemia (ALL). However, the translation of this technology to solid tumors, like malignant gliomas (MG), has thus far been unsuccessful. This review provides a timely analysis of the factors leading to the success of CART immunotherapy in the setting of hematologic malignancies, barriers limiting its success in the treatment of solid tumors, and approaches to overcome these challenges and allow the application of CART immunotherapy as a treatment modality for refractory tumors, like malignant gliomas, that are in desperate need of effective therapies.
The field of oncology has been revolutionized by the emergence of cellular immunotherapies that harness and augment the natural capacity of the immune system to fight cancer. As this ability is often impaired in tumor-bearing patients, one promising approach is to directly bolster deficient endogenous immune responses with adoptive T-cell therapy, which involves the passive infusion of activated, ex vivo expanded autologous lymphocytes that have been activated against tumor-associated antigens (TAAs) (1). These final effectors of the adaptive immune system selectively identify and destroy malignant cells, leaving healthy tissues unharmed. Furthermore, the natural development of memory cells allows for the establishment of long-lasting antitumor immunity and protection from tumor recurrence. However, as the majority of TAAs are poorly immunogenic, it is often difficult to culture a population of lymphocytes whose T-cell receptors (TCRs) have adequate avidity to exert sufficient cytotoxicity to produce lasting tumor eradication (2). This barrier can be overcome with the introduction of engineered surface receptors that have enhanced avidity and affinity for a given TAA. These chimeric antigen receptors (CARs) are comprised of an antibody-derived antigen recognition domain joined to an internal T-cell signaling domain and recognize their antigen targets through a mechanism distinct from classical TCRs (3). In addition to endowing T-cells with antibody-like specificity, these MHC-unrestricted receptors are compatible with patients of all HLA subtypes and can be used to identify tumor cells that have downregulated antigen processing and presentation functions as an adaptation to evade T-cell-mediated destruction (4). In this highly personalized form of immunotherapy, CAR-expressing T-cells (CARTs) combine the strengths of cellular and humoral immunity to equip a patient's immune system with an army of uniquely tumor-specific effector cells that have been functionally enhanced to have superior cytotoxicity, persistence, and antigen recognition capabilities in the face of tumor-induced immunosuppressive influences (5, 6).
Adoptive T-cell therapy with CAR-expressing T-cells has emerged as one of the most promising cancer immunotherapy modalities, demonstrating remarkable antitumor efficacy, particularly in the treatment of hematologic cancers. CARTs targeting CD19, a ubiquitously expressed B-cell surface antigen, have induced durable, sustained antitumor immune responses in patients with acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia, multiple myeloma, and treatment-refractory diffuse large B-cell lymphoma (DLBCL) (7–13). These encouraging results have prompted the recent, first of its kind, FDA approval of CTL019, Novartis' CAR T-cell therapy for children and young adults with relapsed or refractory B-cell ALL (14).
Inspired by this success in liquid tumors, there has been great interest in expanding the use of CART technology to the treatment of solid tumors like glioblastoma (GBM), a highly aggressive form of primary brain cancer for which there is no known cure (15). Supporting the exploration of T-cell-based therapies in solid tumors is the strong positive correlation between the degree of intratumoral infiltration with antigen-specific cytotoxic T-cells (CTLs) and overall patient survival (16, 17). Given the importance of the delicate balance between host and tumor immune responses on the ultimate course of disease, these patients are likely to benefit from highly sophisticated treatments like CART immunotherapy that can both strengthen antitumor immunity and overcome tumor-induced immunosuppressive influences, to tip the balance toward tumor cell death, Figure 1.
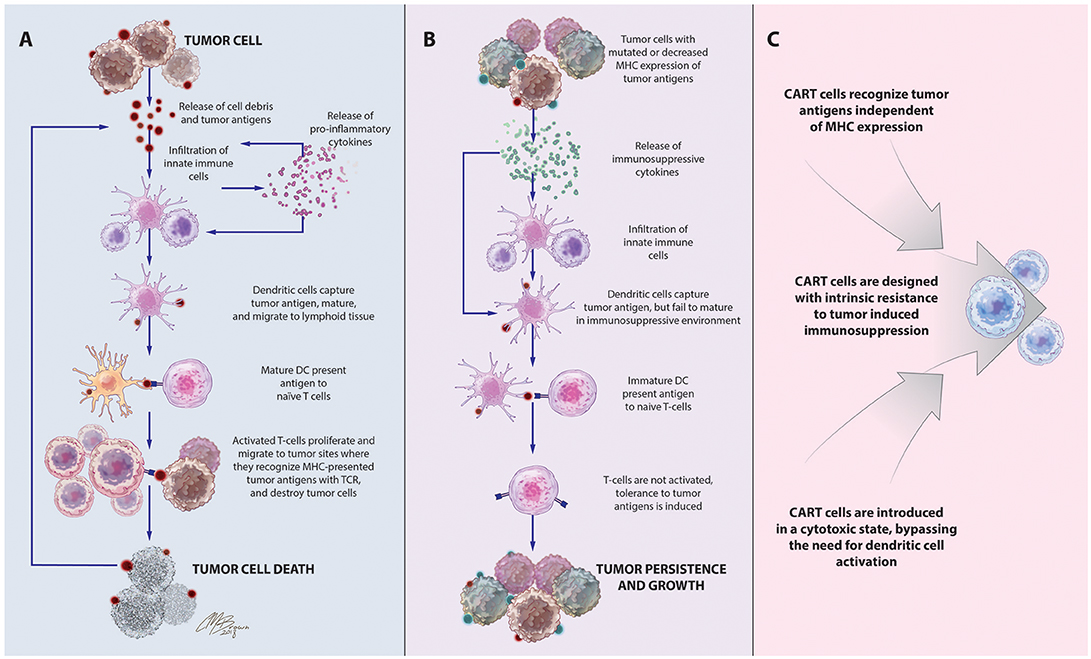
Figure 1. Immune-mediated interactions in solid tumors and rationale for CART immunotherapy. (A) Release of cell debris and tumor antigens from malignant cells activates a cascade of host antitumor immune responses, initiated by innate immune cells that release pro-inflammatory cytokines and contribute to tumor cell destruction. Among these cells are dendritic cells, which capture tumor antigens, mature in response to the pro-inflammatory cytokines in the environment, and travel to lymphoid tissues to stimulate T-cell proliferation and activation of antigen-specific adaptive immune responses leading to tumor death. (B). Tumors often develop adaptations to evade detection and destruction by the host immune system. Through the recruitment of suppressive leukocytes and elaboration of immunosuppressive cytokines, tumors inhibit the function of infiltrating immune cells, including dendritic cells. Incompletely matured DCs are unable to effectively activate naïve T cells, instead inducing T-cell anergy, apoptosis, or tolerance to tumor-associated antigens. Downregulation of antigen-presenting machinery and the development of antigen-loss variants enable tumor cells to escape detection by infiltrating immune cells. (C) CAR T-cells, which recognize antigens via a mechanism distinct from TCR stimulation, bypass the need for DC antigen presentation and are unaffected by MHC downregulation. CAR structure and culture conditions can also be optimized to create CART populations with superior cytotoxicity and resistance to tumor-induced suppressive influences.
CART immunotherapy may also have superior therapeutic efficacy in the treatment of solid cancers that are otherwise poorly accessible to standard therapies, such as malignant gliomas (MG). Nestled within the brain parenchyma, these tumors are particularly dangerous to remove with relatively nonspecific therapies like surgery and radiation due to the risk of damaging surrounding eloquent brain tissue. Furthermore, the highly infiltrative growth pattern of aggressive tumors like GBM makes a complete, curative resection impossible. Intracranial tumors are further isolated from the systemic circulation by the presence of a blood brain barrier, which restricts the passage of most chemotherapeutic agents, preventing their therapeutic accumulation within tumor sites and increasing the risk of systemic toxicity (18). With a known ability to cross the blood brain barrier, activated T-cell-based therapies have potential to overcome these challenges to safely and effectively reach surgically inaccessible malignant cells. A cellular-based approach using functionally enhanced, antigen-specific CTLs, which have cytotoxic functions restricted to antigen-expressing tumor cells, represents a particularly promising treatment strategy for these intracranial tumors (19).
Unfortunately, in stark contrast to the success observed with hematologic malignancies, CARTs have exhibited limited efficacy in the treatment of solid tumors. However, this form of immunotherapy is still in its infancy and there have been many exciting advances in recent years with significant potential to revolutionize CART application to solid tumors. In this review, we highlight the landmark discoveries that have led to the success of CART immunotherapy in hematologic malignancies, identify barriers to its application in solid tumors, and propose research avenues for novel approaches to overcome these barriers and allow successful application of CART immunotherapy to treatment-resistant solid tumors, with a special emphasis on the primary malignant brain cancer, GBM.
CARs are synthetic antigen receptors that can be introduced into an immune cell to retarget its cytotoxicity toward a specific tumor antigen with a greater degree of specificity. In contrast to traditional TCRs, which identify intracellularly derived peptide antigens presented by MHC molecules, CARs directly identify antigens expressed on the surface of tumor cells therefore are not restricted by patient HLA subtype and can recognize a variety of antigen structures including proteins, carbohydrates, and glycolipids (20). These membrane-bound fusion proteins couple a high avidity extracellular antigen recognition moiety derived from the single-chain variable fragment of a monoclonal antibody with a modified TCR intracellular signaling domain Figure 2 (3). Originally derived from the CD3ζ chain of the traditional TCR (21), CAR endodomains have undergone generational changes to include one or more costimulatory domains, most commonly CD28 and 41BB, to enhance the persistence and cytotoxicity of CAR-expressing cells (22, 23).
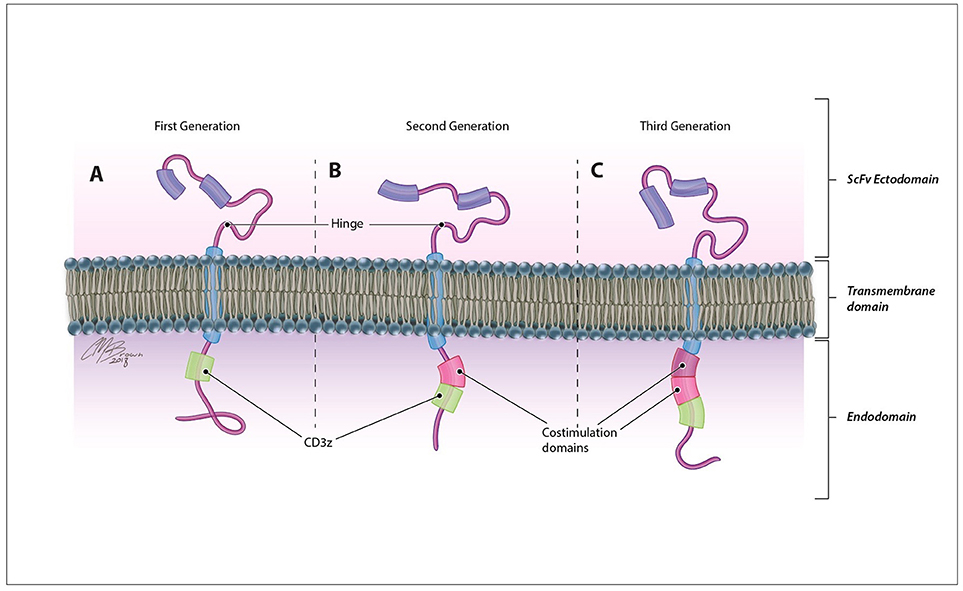
Figure 2. CAR structure. CARs are comprised of an antigen-recognition ectodomain derived from the single-chain variable fragment (scFv) of a monoclonal antibody connected by a flexible hinge and transmembrane segment to an intracellular endodomain. Originally derived from the CD3ζ domain of the classical TCR in (A) first generation CARs, this intracellular signaling component may contain (B) one (second generation) or (C) two (third generation) additional costimulatory domains that enhance the proliferation, persistence, and efficacy of adoptively transferred cells.
Following receptor design, a gene encoding the CAR construct is transfected into the genome of T-cell isolates using gene-therapy viral vectors, and the resulting CAR T-cells are expanded ex vivo and re-infused into the patient. Classical CART therapy is restricted to the use of autologous leukocytes due to the presence of self-identification molecules on the surface of T-cells. However, treatment-related lymphopenia may preclude the ability to isolate sufficient peripheral blood leukocytes for treatment. These patients may benefit from donor cell based therapies that use universal CAR T-cells that have been genetically modified to completely lack endogenous expression of self-identifying molecules like the TCR and HLA class I molecules (24, 25).
Since the first design of chimeric T-cell receptors in 1989, CAR technology and the field of CART immunotherapy have advanced immensely, with several key landmark achievements that have propelled this therapeutic modality to the forefront of cancer treatment, Figure 3 (3). Evaluated in a variety of tumor types, CART immunotherapy has produced particularly successful clinical responses in the treatment of hematologic malignancies. CAR T-cells targeting B-cell surface antigens, particularly CD19, have demonstrated a marked ability to eradicate liquid tumors (27, 28). Upon introduction into human clinical trials, CART immunotherapy induced sustained tumor regression in a majority of treated patients (7–13). This success prompted the recent FDA approval of CTL019, Novartis's CAR T-cell therapy for the treatment of children and young adults with relapsed or refractory B-cell ALL, bringing CART immunotherapy to the front lines of standard cancer treatment (14).
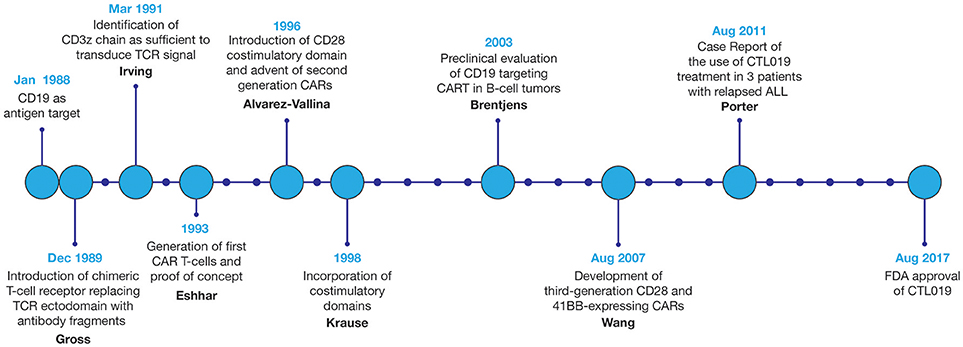
Figure 3. Development of CART immunotherapy. Following development of the first chimeric T-cell receptor in 1989, early preclinical studies of the first CARTs demonstrated the ability to selectively identify and destroy antigen-expressing tumor cells (5, 6). However, upon adoptive transfer into live patients, T-cells expressing these first-generation CARs displayed limited persistence and were often rendered anergic due to the absence of costimulatory signals within the tumor microenvironment (TME) (26). With the introduction of costimulatory domains to provide these necessary activating signals, CART immunotherapy experienced a dramatic improvement in therapeutic efficacy (22). Optimization of CAR structure and ex vivo culture conditions to improve CART persistence, cytotoxicity, and resistance to tumor-induced immunosuppression remains an area of continued research. Evaluated in a variety of tumor types, CART immunotherapy has been markedly successful in the eradication of liquid tumors, culminating in the FDA approval of CART immunotherapy for the treatment of relapsed or refractory B-cell ALL in 2017.
Reflecting on the success of CART immunotherapy in hematologic cancers, tumors of B-cell origin possess several qualities that have provided for this exceptional antitumor efficacy. Primarily, this can be attributed to the virtually ideal properties of CD19 as an antigen target. Ideal antigens for CAR generation are tumor exclusive, expressed by all malignant cells, and have a function crucial to tumor growth and survival. Together, these qualities maximize tumoricidal capacity, prevent immune evasion, and reduce the risk of toxicity stemming from CART destruction of antigen-expressing healthy cells. Uniformly expressed by malignant cells of B-cell cancers CD19 is lineage-restricted B-cell surface marker that is not present on other tissues of the body (29). Although non-cancerous immune cells also express CD19, transient inadvertent destruction of these populations during treatment does not result in therapy-limiting toxicity (30). Additionally, malignant cells of hematologic cancers typically reside in locations of routine T-cell migration (peripheral blood, lymph nodes, and bone marrow), making them readily accessible to systemically delivered CAR T-cells. A relatively disseminated distribution also precludes tumor isolation by physical barriers such as a dense fibrous stroma and the development of a highly immunosuppressive local tumor microenvironment (TME). The combination of these factors has allowed for the rapid, efficient, and uninhibited destruction of CD19-positive malignant cells leading to regression of B-cell cancers.
CARs have also been developed against a variety of solid tumor surface antigens including mesothelin (31), carcinoembryonic antigen (CEA) (32), disialoganglioside (GD2) (33), interleukin-13 receptor α2 (IL-13Rα2) (34), mucin-1 (MUC1) (35, 36), ephrin type-A receptor 2 (EphA2), (37), human epidermal growth factor receptor 2 (HER2) (38) and other growth factor receptors. Currently there are several ongoing clinical trials assessing the safety and efficacy of CART immunotherapy in various solid malignancies (Table 1).
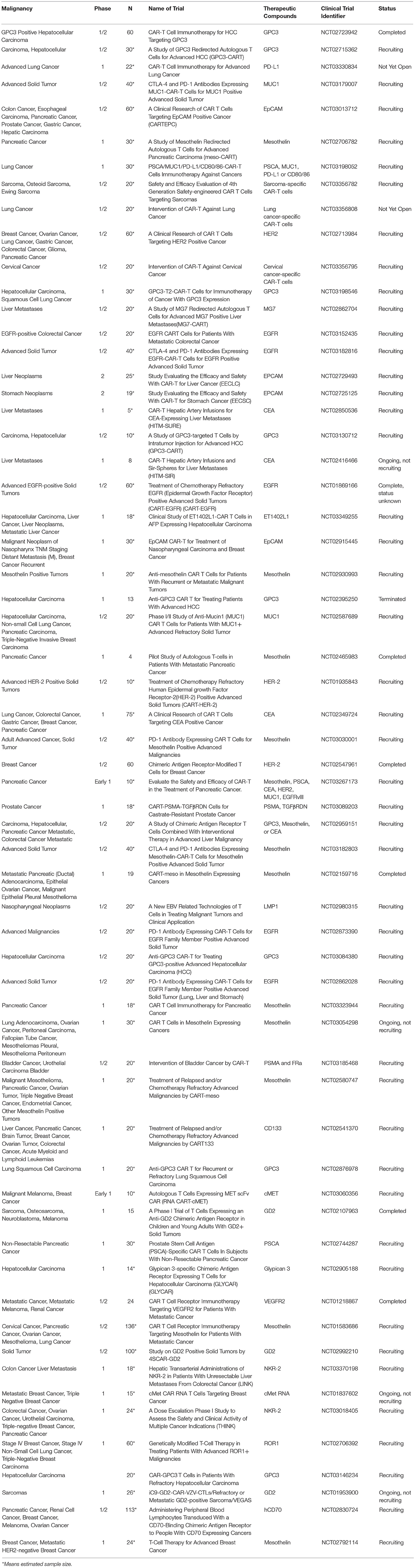
Table 1. List of CART immunotherapy clinical trials in solid tumors (except gliomas).
With respect to GBM, much of the current CAR development is focused on the following antigen targets: EGFRvIII, IL-13Rα2, and HER2. EGFRvIII is a mutated form of the epidermal growth factor receptor (EGFR), resulting from a tumor-specific in-frame deletion creating a constitutively active surface receptor protein. Present in approximately 30% of GBMs, this mutant receptor enhances glioma cell proliferation, angiogenesis, and invasiveness (39) and is independently associated with a poor prognosis (40). Preclinical studies have established the ability of T-cells targeting this unique, tumor-specific epitope to proliferate and release cytokines in response to stimulation with the mutant EGFRvIII antigen, but not wild-type EGFR (41). In preclinical studies, EGFRvIII-targeting CARTs effectively traffic to tumor sites and suppress the growth of glioma xenografts in murine models (42). In human clinical trials, preliminary reports from a phase I study in 10 patients with recurrent GBM established the safety and feasibility of EGFRvIII-targeting CART immunotherapy. Adoptively transferred cells were shown to proliferate within the peripheral blood and traffic to intracranial tumor sites, exerting antitumor effects without any evidence of cross-toxicity with wild-type EGFR. Interestingly, analysis of pre- and post-treatment tumor samples revealed post-treatment decreases in antigen expression and an increased presence of inhibitory immune checkpoint molecules and regulatory T-cell infiltrates, indicative of evasive tumor responses. The median overall survival was approximately 8 months, with one patient experiencing residual stable disease at 18 months (43). Clinical trials are currently ongoing specifically to assess efficacy.
Another encouraging target for patients with GBM is interleukin-13 receptor alpha-2 (IL-13Rα2). Not expressed by healthy tissues, IL-13Rα2 is overexpressed in nearly all GBM tumors (44). IL-13Rα2-targeting CARTs have been shown to selectively target and kill IL-13Rα2-positive tumor cells in vitro and in vivo, producing regression of xenograft glioma tumors in a murine model (34). Based on this success, several human clinical trials were initiated with IL-13Rα2-targeting CARTs. Transient anti-glioma responses, without significant associated toxicities, were observed in a subset of treated patients (45, 46).
Human epidermal growth factor receptor 2 (HER2) is a transmembrane tyrosine kinase receptor expressed on a variety of healthy tissues. Overexpressed in many solid tumors, including approximately 15% of GBM tumors, HER2 has been identified as an independent negative prognostic indicator for GBM patient survival (47). In a recent phase I dose-escalation study of HER2-specific CAR-modified virus-specific T-cells for the treatment of progressive HER2-positive glioblastoma, therapy was well tolerated without any dose-limiting toxicities. Median overall survival was 11.1 months from the time of first infusion, and 24.5 months from initial diagnosis (48).
CARTs targeting these and other TAAs have shown significant antitumor activity in in vitro and in vivo preclinical studies (Table 2), and many of these therapies have reached the stage of human clinical trials for patients with GBM (Table 3). Across all solid tumors, one of the most promising studies has occurred in the treatment of neuroblastoma with GD2-targeting CARTs, where complete remission was achieved in 3 of 11 treated patients (57). However, in stark contrast to the efficacy in hematologic malignancies, no CAR T-cell therapy has been shown to induce consistent, lasting regression of solid tumor in human patients.
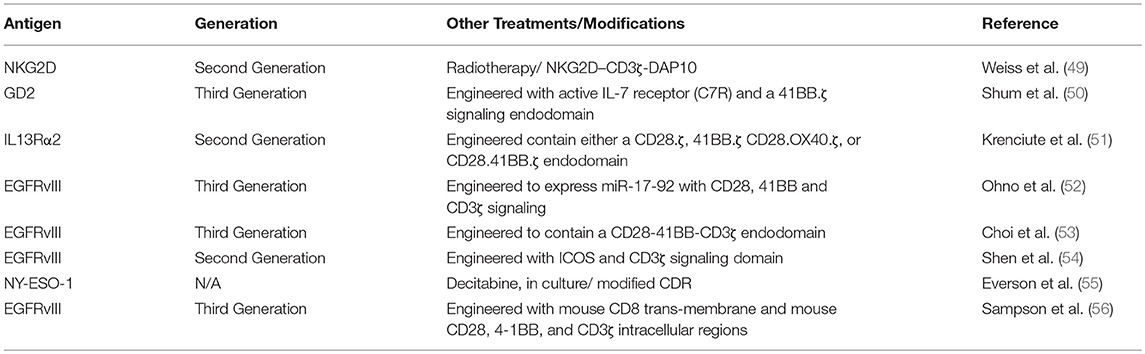
Table 2. List of antigens targeted by CART in pre-clinical glioma model.
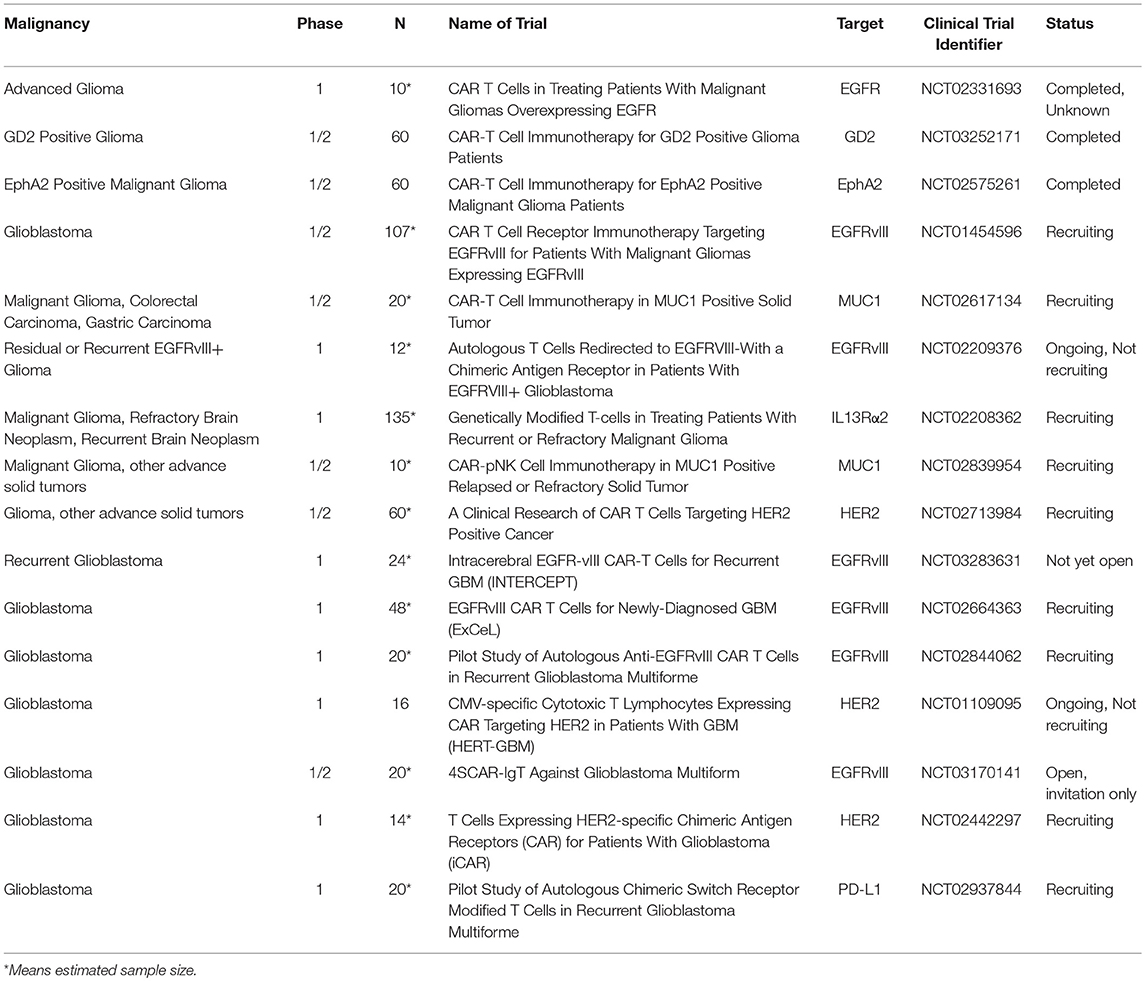
Table 3. List of CART immunotherapy clinical trials in malignant gliomas.
For CAR T-cells to effectively eradicate solid tumors in human patients, they must be able to migrate to and infiltrate tumor tissues, proliferate and persist long enough to exert therapeutic effects, and identify and destroy only antigen-expressing cells within the TME. However, the ability for CART cells to these goals has thus far been hindered by a variety of factors unique to solid tumors, Figure 4.
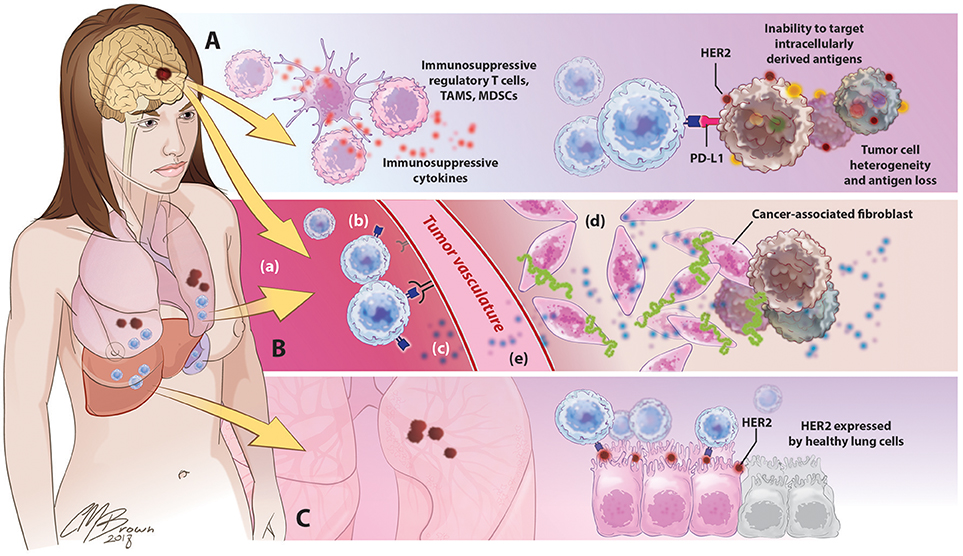
Figure 4. Barriers to successful CART immunotherapy in solid tumors. (A) Factors within the tumor microenvironment: Solid tumors contain an abundance of immunosuppressive leukocytes, immune checkpoint molecules, and suppressive cytokines. The tumor cells themselves are highly heterogeneous, preventing the identification of uniformly expressed targets for CAR design; antigen selection is further limited by an inability of CARTs to target intracellularly derived antigens. (B) Barriers to CART migration and entry into tumor sites: In contrast to the disseminated nature of hematologic cancers, solid tumors are often found in isolated locations that are difficult to access, like the brain. (a) following adoptive cancer, CARTs have been shown to preferentially accumulate in organs such as the lungs, liver, and spleen, with limited natural trafficking to tumor sites. (b) downregulated expression of ICAMs and other adhesion molecules on tumor vasculature limits lymphocyte extravasation, (c) reduced release of lymphocyte-attracting chemokines such as CXCL9-11 precludes CART homing to tumor sites, (d) in addition to supporting the growth and persistence of malignant cells, tumor-associated stroma provides both a physical and immunologic barrier to CART immunotherapy, (e) release of angiogenic factors such as VEGF promotes the develop of abnormal, tortuous, high-pressure vasculature that impedes lymphocyte entry.(C) Toxicity secondary to off-target effects: the use of overexpressed self-antigens as CAR targets introduces the risk of significant toxicity associated with CART identification and destruction of normal, healthy cells expressing these antigens.
Perhaps the greatest challenge facing the successful application of CART technology to solid tumors in human patients is the selection of acceptable antigen targets. Solid tumors develop from the accumulation of a variety different mutations that promote unchecked cellular growth and proliferation, some of which result in the creation of tumor-exclusive epitopes that can be used to selectively identify malignant cells. However, rather than occurring linearly with the progression of a single homogenous line of cells expressing all accumulated mutations, tumorigenesis transpires through the simultaneous evolution and adaptation of individual malignant cells. As a result, solid tumors are comprised of highly molecularly heterogeneous subpopulations expressing a diverse, overlapping profile of unique TAAs. As a result, the current pool of potential antigens for CAR development in solid tumors is comprised of suboptimal targets that either lack uniformity of expression or tumor exclusivity, introducing additional concerns for efficacy and safety. In addition to precluding the identification of commonly and uniformly expressed targets for CAR development, this naturally-derived molecular heterogeneity both between patients and within individual tumors is difficult to reproduce in the human cell line-derived relatively homogeneous tumors utilized in preclinical experiments, limiting the ability of these studies to truly approximate human disease and predict patient responses to treatment (58).
To prevent inadvertent destruction of healthy tissues, CARTs are ideally designed to target antigens exclusively expressed by malignant cells. However, the vast majority of mutations producing novel epitopes in solid tumors occur through the processing of intracellular proteins, which are presented in the context of MHC molecules and therefore not accessible to traditional CARs. Due to a lack of known feasible tumor-exclusive antigens, many TAAs under current evaluation for CART immunotherapy are derived from overexpressed endogenous molecules, particularly those that promote tumor proliferation and persistence (i.e., growth factor receptors). Their disproportionately high degree of upregulation within tumors allows for preferential targeting of malignant cells. However, there are several important challenges and health risks associated with targeting overexpressed self-antigens that must be considered. Due to the natural development of tolerance to endogenous peptides, the majority of these overexpressed TAAs are poorly immunogenic. However, enhancing CART cytotoxicity against epitopes not restricted to malignant cells is limited by the danger of simultaneously promoting CAR recognition of target antigen expressed by healthy tissues. Toxicities secondary to unintentional destruction of non-cancerous cells has been observed to varying degrees following CART therapy targeting overexpressed self-antigens like CEA, a tumor-associated antigen that is also expressed in normal gastrointestinal epithelium. In a study of CEA-targeting CART immunotherapy for metastatic colorectal cancer, tumor regression was accompanied by severe inflammatory colitis in all treated patients, due to the destruction of healthy epithelial cells (59). Depending on the location of off-target antigen expression, the resulting toxicities may have life-threatening consequences, as occurred in a patient receiving CART therapy for refractory HER2-overexpressing metastatic colorectal cancer. This patient experienced fatal pulmonary toxicity shortly after CART infusion secondary to CAR recognition of low levels of HER2 on lung epithelium (60).
Identification of a tumor-exclusive antigen target feasible for CART immunotherapy is further complicated by the often inconsistent and incomplete degree of expression of unique tumor epitopes throughout the tissue. As a result, there likely exists a subpopulation of malignant cells lacking expression of any given target antigen that will therefore evade destruction by a corresponding CAR T-cell. In addition to reducing overall therapeutic efficacy this creates a selective pressure for the accumulation of cells with absent or mutated target antigen expression, introducing the potential for tumor recurrence through expansion of antigen-negative cell populations. This is particularly applicable when targeting epitopes derived from mutations that are not crucial to oncogenesis and tumor survival, as these structures can be readily modified or lost without compromising vital tumor functions. Indeed, therapy resistance coinciding with the emergence of antigen-loss variants is commonly observed in human patients with GBM and other solid tumors following treatment with CART immunotherapy (43, 45, 46, 61, 62).
In contrast to the simplicity and ease of encountering of malignant cells in hematologic cancers, CARTs for solid tumors face the additional challenge of migrating to and infiltrating tumor sites. In humans and mice, CART persistence and intratumoral accumulation following systemic adoptive transfer is characteristically poor (63), with some studies showing initial trafficking to organs such as the lung, spleen, and liver, without any preferential accumulation in tumor sites (64). This limited trafficking may be multifactorial and impacted by both lymphocyte and tumor related influences, including settings of mismatch between tumor-expressed molecules and corresponding lymphocyte receptors (65) and the development of structural barriers surrounding tumor sites (66). With a reduced capacity to proliferate and relatively short overall persistence, CART immunotherapy in solid tumors becomes a “race against the clock,” as adoptively transferred cells have a limited amount of time to reach and destroy malignant cells, including those at sites distant to the bulk tumor.
Normal leukocyte migration and extravasation from the bloodstream is a multistep process that can be separated into four characteristic stages: (1) tethering/rolling, (2) activation, (3) adhesion/arrest, and (4) transmigration. Transient interactions between leukocyte carbohydrates and endothelial cell selectin receptors slows leukocyte velocity, causing them to roll along the endothelial wall in the direction of blood flow. These tethering interactions bring leukocytes in closer proximity to chemokines secreted by vascular endothelium, which bind leukocyte G-protein coupled receptors that activate the expression and adhesiveness of surface integrins, including lymphocyte function antigen-1 (LFA-1) and very late antigen (VLA)-4. Binding of these integrins to their ligands, like VCAM-1 and ICAM-1, present on tumor vasculature leads to a firm arrest of leukocyte motion. This stronger binding provides for transmigration into tumor sites (67).
In the setting of cancer, preferential migration of immune cells to tumor sites is mediated by tumor-secreted chemoattractants such as CCL2, CXCL9, CXCL10, and CXCL11 (65). An expression of corresponding receptors allows activated T-cells to respond to these factors, inducing migration in the direction of increasing chemokine concentration. This relationship between tumor-expressed chemoattractants and the profile of corresponding T-cell homing receptors is crucial in dictating a leukocyte's capacity to traffic to tumor sites (68). In a study of CTL trafficking in murine brain tumors, Okada et al. observed that CNS homing capacity was restricted to CTLs polarized toward a type 1, as compared to type 2, cytokine profile, demonstrating the importance of tumor tropism. These type 1 CTLs exhibited comparatively higher levels of endogenous VLA-4 and CXCR3, receptors that are crucial for CNS tropism (2). GBM and other highly vascular tumors characteristically overproduce vascular endothelial growth factor (VEGF), which, in addition to promoting angiogenesis, inhibits the secretion of lymphocyte-attracting chemokines like CXCL10 and CXCL11 (69) and downregulates tumor endothelial cell expression of adhesion molecules like ICAM-1 and ICAM-2, VCAM-1 and CD34 (70). Also abundant within the TME is the immunosuppressive cytokine TGF-β, which also downregulates endothelial cell expression of cellular adhesion molecules, thereby inhibiting T-cell transmigration (17).
Solid tumors also induce the development of structural modifications to the adjacent tissue that can prevent intratumoral lymphocyte accumulation. Many solid tumors are surrounded by a dense, peritumoral fibrous stroma that insulates nests of tumor cells from the surrounding environment (66). Forming the structural framework of this stroma are cancer-associated fibroblasts (CAFs), which play an important role in regulating tumor metabolism, growth, and persistence and mediate tumor invasion and metastasis through remodeling of the extracellular matrix (70, 71). Endogenous T-cells express heparanase, an ECM-degrading enzyme, which enhances their ability to penetrate this stroma; however, heparanase expression is often lost during in vitro culture, preventing CART from entering tumor sites (71). Abundantly secreted by tumor and stromal cells are CXCL-12, VEGF, and PDGF, which recruit bone marrow-derived endothelial cells and promote the development of abnormal, tortuous vessels leading to the development of abnormal, leaky vascular networks and elevated intratumoral interstitial pressures that resist lymphocyte infiltration (66).
A major barrier to the efficacy of CART immunotherapy in solid tumors is a characteristic state of profound tumor-induced suppression of host antitumor immunity (72–74). The complex network of signals orchestrated by malignant and non-malignant cells of a tumor induces the development of an environment that is hostile to immune cell function and survival and favors tumor persistence. Through the elaboration of immunosuppressive cytokines and other soluble mediators, recruitment of inhibitory leukocytes, and activation of immune checkpoints, tumors create a microenvironment that inhibits effector cell activity and resists immune-mediated destruction. This multitude of immunosuppressive influences is responsible for the rapid loss of effector function observed upon CART entry into tumor sites (75).
Through surface expression of immunosuppressive molecules, solid tumors exploit endogenous regulatory pathways to directly inhibit T-cell effector functions. Among these immune checkpoints are the programmed cell death-1 (PD-1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4) pathways. Activation of CTLA-4 receptors expressed by naïve T cells prevents their initial activation and stimulation of PD-1 on activated T-cells induces anergy, apoptosis, or development of immunosuppressive regulatory T-cells (Tregs). By upregulating PD-L1 and enhancing T-cell CTLA-4 and PD-1 expression, tumor cells are able to suppress the activity of incoming immune cells (76).
Present in high concentrations within the glioma microenvironment are immunoregulatory cytokines like transforming growth factor-β (TGF-β) and interleukin 10 (IL-10), which support the development of a type-2 polarized environment. In addition to suppressing MHC II molecule expression and antigen presenting cell maturation and function, these cytokines are potent inhibitors of T-cell differentiation, proliferation, and cytotoxicity (77, 78). Accumulating within gliomas and other solid tumors are inhibitory leukocytes like regulatory T-cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), which are potent inhibitors of antitumor immunity (66). MDSCs constitute a diverse pool of immature myeloid cells that accumulate in settings of inflammation and malignancy. Through cell-cell contacts and the release of soluble mediators, MDSCs suppress the proliferation and function of antigen-specific CTLs and induce the development of Treg cells (79).
Although the application of CART immunotherapy to solid tumors continues to be faced with significant challenges, this therapeutic modality still holds the potential to make a significant impact in the field of solid tumors as it did for liquid malignancies.
Moving forward, the identification of commonly expressed antigen targets remains a constant challenge plaguing CART development for solid tumors. However, given the high degree of solid tumor heterogeneity existing across different patients, identification of such a broadly applicable antigen has been problematic. Despite the challenges facing antigen selection in solid tumors, CARs developed against a variety of TAAs have demonstrated encouraging antitumor efficacy and safety profiles in preliminary studies. One potential solution involves the study of individual intratumoral mutation profiles, which has revealed an exciting new category of possible antigen targets that may allow for the development of highly specific and effective personalized therapies (80, 81). Arising through somatic mutations within an individual tumor, these “neo-antigens” are inherently both tumor-specific and immunogenic. Endogenous T-cells naturally activated against these unique, patient-specific epitopes have been recovered from the tumors and peripheral blood of patients with a variety of solid cancers (82–84). Interestingly, patients with higher numbers of these immunogenic neo-antigens have been shown to possess a greater expansion of antigen-reactive CD8+ T-cells and elevated immune checkpoint expression, which may suggest synergy between CART immunotherapy and immune checkpoint blockade (85). Furthermore, the generation of these novel epitopes can be enhanced with other forms of immunotherapy known to trigger epitope spreading and activate antitumor immune responses, like dendritic cell-based vaccination or oncolytic virotherapy (86, 87). Aiding in the development of neo-antigen-based therapies, tumor exome and proteome analysis allows for a characterization of the full profile of neo-antigens within a single tumor and identification of epitopes that may be particularly immunogenic (88–90). Using this information, Castle et al. demonstrated the ability to induce T-cell immune responses against patient-specific neo-antigens via peptide vaccination against immunogenic epitopes (91). Given that the majority of these novel epitopes created within solid tumors are peptide-based antigens, CARTs have been developed in which the CAR ectodomain consists of a TCR-like construct that recognizes MHC-presented intracellular antigens (92). Indeed, transfection of T-cells with neo-antigen-specific receptors has been shown to be a viable option in preclinical studies (93, 94).
Targeting multiple TAAs has potential to enhance overall specificity for tumor cells, lessen the risk of off-target effects, and reduce the emergence of antigen loss variants and therapy-resistant tumors. After studying single-cell coexpression patterns of the TAAs HER2, IL-13Rα2, and EphA2 in primary GBM samples, Hegde et al. developed a mathematical model of antigen expression to predict the odds of complete tumor elimination with CART therapy. This model determined that a maximal expansion of CART therapeutic potential in all tumors can be achieved by co-targeting HER2 and IL-13Rα2, without any added advantage to targeting a third antigen. They subsequently developed bi-specific HER2 and IL-13Rα2-targeting CAR T-cells that exhibited enhanced antitumor activity and an ability to offset antigen escape in in vitro immunoassays and an orthotopic xenogeneic murine model (95). Tumor cell selectivity of bi-specific CAR T-cells can be further refined to only target cells expressing a specific antigen combination. This is achieved by physically separating CAR signaling domains between two distinct CARs specific for two different antigens, such that binding of both CARs to their individual target antigens is required to transmit an activating signal to induce T-cell cytotoxicity (96). This technique may be particularly effective against tumors that overexpress two antigens that are individually not exclusively expressed by malignant cells, but when coupled together, result in acceptable tumor specificity due to a lack of overlapping expression on non-target tissues.
Considering tumor-associated stroma is also important for therapy development. For example, in tumors with a high degree of antigen expression, CART immunotherapy has been shown to indirectly eradicate antigen loss variants, a process dependent on tumor stromal cell cross-presentation of TAAs (97). Further studies demonstrated that in settings of low antigen expression, local irradiation or chemotherapy was shown to cause sufficient release of antigen to sensitize stromal cells for T-cell mediated destruction, supporting introduction of CART immunotherapy into standard treatment regiments including radio and chemotherapy (98). Directly targeting antigens present on tumor-supporting structures such as tumor vasculature or associated stroma is also a promising option. CARTs directed against antigens preferentially expressed by tumor vasculature have been shown to directly disrupt these vascular networks leading to tumor regression in murine models of ovarian cancer and other vascular tumors (99). CARTs targeting fibroblast activation protein-α (FAP), a tumor stromal-associated antigen expressed by CAFs, delivered in combination with an EphA2-targeting CAR population was shown to control the tumor growth in an A549 lung cancer model more effectively than either agent alone (37).
A variety of methods have been successfully employed to enhance CART trafficking to tumor sites including augmenting CART responsiveness to tumor-secreted cytokines, targeting tumor-associated stroma and the TME, and exploration of regional routes of delivery.
Fundamentally, T-cell migration and intratumoral accumulation will be facilitated by the utilization of T-cell populations expressing a profile of homing receptors allowing them to respond to the unique factors secreted by an individual tumor. To overcome tumor chemokine and lymphocyte receptor mismatches, CAR T-cells can be transduced with specific chemokine receptors known to dictate the appropriate tissue tropism (100, 101). For example, transfection of CARTs with a gene for CCRb2, the chemokine receptor for the T-cell chemoattractant CCL2, has been shown to enhance the migration and intratumoral accumulation of CARTs in xenograft models of multiple solid tumor types (33, 102). Brown et al. reported that glioma-derived CCL2/MCP-1 is ultimately responsible for the in vivo chemotaxis of adoptively transferred T-cells to glioma xenograft tumor sites (103), introducing promise for similar studies in glioma. CAR T-cells have been engineered to express heparanase, a modification that enhances intratumoral accumulation and overall antitumor activity (71). Combining multiple immunotherapeutic strategies to improve trafficking, one study evaluated CARTs delivered together with an oncolytic adenovirus engineered to express both a chemokine and T-cell growth factor. The combination of RANTES and IL-15 was shown to improve T-cell trafficking to tumor sites as well as create a favorable environment within the TME to enhance immune cell persistence (104).
In theory, any therapy that favorably modulates the TME or enhances the elaboration of relevant chemokines will enhance the migration of CARTs to tumor sites. In addition to mollifying the tumor-induced suppression of T-cell activity, immune checkpoint blockade with anti-PD-1 therapy augments lymphocyte trafficking in tumor-bearing mice by promoting the elaboration of the lymphocyte-attracting cytokines interferon-γ (IFN-γ) and CXCL10 (105). Similar effects are achieved with radiotherapy, which leads to IFN-γ-enhanced expression of adhesion molecules on tumor-associated vasculature (106). Anti-angiogenic therapy also promotes lymphocyte infiltration into tumor sites, and administration in combination with CART immunotherapy has been shown to be more therapeutically effective than treatment with either modality alone (107, 108). In addition to normalization of tumor vasculature, inhibition of VEGF signaling in a B16 melanoma model resulted in an 18-fold increase in intratumoral T-cell infiltration, which was associated with increased expression of CXCL10 and CXCL11 (69).
Proposed to circumnavigate many of the barriers limiting the trafficking of systemically delivered CARTs, regional CART administration may be a superior delivery strategy for CARTs in solid tumors. In contrast to the need for systemically delivered cells to traffic to tumor sites, intratumorally delivered cells have been shown largely to remain at the site of inoculation with minimal systemic absorption, which suggests that regionally delivered CARTs may carry a lower risk of off-target toxicities (63, 109). Several additional studies evaluating the efficacy of systemic and regionally delivered CART therapy have found regional delivery methods to result in superior T-cell persistence and overall therapeutic efficacy (32, 110). In one such comparison, regional intra-pleural delivery of mesothelin-targeting CAR T-cells resulted in earlier intratumoral accumulation, increased CD8+ T-cell proliferation and persistence, enhanced cytokine secretion, and improved overall therapeutic efficacy in an orthotopic model of human pleural malignancy. Adoptively transferred cells exhibited long-term persistence and protection against tumor rechallenge after 200 days (110). The application of such a regional delivery method may be especially advantageous in the setting of poorly accessible intracranial tumors like GBM that, nestled within a strongly immunosuppressive microenvironment, are significantly isolated from the systemic circulation. With respect to intracranial tumors, an in vivo study of breast cancer xenograft tumors with intracranial metastases was conducted to compare two regional CART delivery methods: intraventricular and a more localized, intracranial method. With both methods, functionally active, antigen-specific CD4+ and CD8+ T-cells were detected at tumor sites 1 week following administration. In this study, CARTs were rarely detectable by two weeks following administration Although equivalent antitumor activity was observed, some mice receiving the less localized intraventricular therapy exhibited more delayed therapeutic responses, demonstrating the need for adoptively transferred cells to traffic from the site of delivery (ventricle) to tumor sites (111). This delayed onset of action associated with T-cell trafficking makes the persistence of adoptively transferred cells of utmost importance. Specific to glioma model, intracerebral delivery of EGFRvIII-targeting CARTs has been shown to induce tumor regression in murine models of glioma (53). Several human clinical trials have employed regional CART delivery methods for the treatment of patients with progressive GBM. In one such phase I human clinical trial, following surgical tumor debulking, IL13Rα2-targeted CARTs were infused into the resection cavity via a Rickham catheter. Although this study did not assess the duration of CART persistence within the target tissue or at other locations, it proved that this route of administration was well tolerated and established feasibility for future evaluations (45). More recently, Brown et al. reported observations regarding two different regional CART delivery routes employed in the treatment of one patient with multifocal GBM. Transient anti-glioma responses were seen following initial intracranial CART delivery. After tumor recurrence, the patient was treated with intraventricular CART infusions and experienced robust tumor regression over the course of 7.5 months, though eventually succumbing to multifocal relapsed disease. Investigators evaluated CART persistence and cytokine levels in relation to intraventricular infusions. CAR T-cells were detected within the CSF for up to 1 week following infusion, with peak concentrations occurring after 2 days. Despite this lack of robust persistence, there was a significant induction of inflammatory cytokines, including IFN-γ, TNF-α, IL-12, IL-5, IL-6, IL-8, IL-10, CXCL9, CXCL10, CCR2, and IL-1Rα (46). Further studies will be needed to characterize and contrast the T-cell persistence and overall therapeutic efficacy associated with regional and systemically delivered CART therapies in the setting of GBM.
The persistence of adoptively transferred T-cells is directly correlated with patient clinical responses and the degree of tumor regression (112). Although factors impacting CART persistence remain poorly understood, there have been several key advances in this field that have shown promise for optimizing persistence and associated therapeutic efficacy.
The selection of different costimulatory motifs can confer unique properties in terms of cellular metabolism, cytokine secretion, cytotoxicity, proliferation, persistence, and memory cell generation. Costimulation with 4-1BB derived moieties promotes the generation of long-lived central memory cells with enhanced mitochondrial biogenesis and fatty acid oxidation. Signaling through a different pathway, CD28 leads to the differentiation of shorter-lived effector memory cells and metabolic changes leading to increased reliance on aerobic glycolysis for proliferative energy (113). A diverse combination of different subpopulations of CARTs possessing the unique strengths and weaknesses of differential costimulation may provide the most effective overall antitumor effect.
In addition to structural CAR modifications, CART persistence is classically enhanced by pretreatment lymphodepletion with cyclophosphamide or fludarabine (114). Lymphodepletion is thought to support T-cell engraftment by depleting populations of suppressive host lymphocytes, like Tregs, and eliminating competition for cytokines like IL-7, IL-15, and IL-21, which promote T-cell activation and expansion (115). This effect can be further augmented with the administration of supportive cytokines (116). The most widely studied is IL-2, however in addition to supporting CTL persistence and function, IL-2 also promotes the expansion of immunosuppressive regulatory T-cells (117). In contrast, IL-15 has been shown to preferentially promote cytotoxic T-cell persistence and function even in the presence of Tregs (118). Persistence and cytotoxicity are also influenced by the composition of the transferred CART lymphocyte population (119), with infusions containing higher numbers of CD4+ and memory T-cells exhibiting longer overall longevity (57).
The clinical success of CART immunotherapy depends upon the ultimate ability of tumor-infiltrating CTLs to retain their ability to identify and destroy malignant cells within this suppressive microenvironment. The immunosuppressive effects of the TME can be addressed through a variety of strategies that enhance the effector functions of adoptively transferred cells and/or directly counteract mechanisms of tumor-induced immunosuppression. In addition to exogenous administration of adjunctive therapies, CAR T-cells themselves can be genetically engineered to secrete immune-modulating compounds or to reduce their susceptibility to tumor-induced immunosuppression.
The success of CART immunotherapy can also be improved by strategies that focus on altering the anti-inflammatory and immunosuppressive nature of the TME by armoring them with immune modulating cytokines. CARTs engineered to express IL-12 upon antigen binding release this pro-inflammatory cytokine into the surrounding milieu, which functions to promote an environment favoring antitumor immunity. By counteracting the immunosuppressive nature of the TME, IL-12 lessens the recruitment and suppressive influences of immature dendritic cells, TAMs, Tregs, and MDSCs, and recruits additional innate immune cells that may be able to destroy tumor cells that are not visible to CARs (120). IL-12-expressing CARTs also exhibit improved antitumor efficacy and have been shown to be intrinsically resistant to Treg-mediated immunosuppression (121). Overall, delivery of pro-inflammatory cytokines like IL-12 has been shown to counter the immunosuppressive nature of the local microenvironment by promoting the polarization T-cell responses toward the T helper-1 phenotype, enhancing IFN-γ release, augmenting granzyme and perforin production, suppressing angiogenesis, and reducing the population of infiltrating MDSCs (122, 123). Antibody-mediated therapy to deplete suppressor cells, including MDSCs and Tregs, has also been shown to further reduce tumor burden (32). Alternatively, CARTs can be modified to resist immunosuppressive influences within the TME. Present in high amounts within tumor sites, TGF-β has a profound inhibitory influence on the activity of immune effector cells. CTLs transfected with a dominant negative TGF-β receptor are rendered insensitive to TGF-β-mediated suppression and were shown to have enhanced antitumor activity as compared to unmodified CTLs (124). Engineering CAR T-cells to be resistant to the multitude of immunosuppressive influences within the TME is a constant goal in the development of more sophisticated therapies.
Given the complexity of immune interactions within solid tumors and their importance in disease outcome, there inherently exists significant synergistic potential in combination treatment with CARTs and other forms of immunotherapy, such as dendritic cell-based vaccination, oncolytic virotherapy, and immune checkpoint blockade (87, 125). Supporting this assertion, simultaneous PD pathway blockade has been shown to improve the cytotoxic activity of adoptively transferred CAR T-cells, reduce the presence of MDSCs, and enhance tumor growth inhibition in murine models (126). Overall, in addition to attracting CARTs to tumor sites, the resulting enhanced secretion of IFN-γ and CXCL10 creates a pro-inflammatory TME favoring the development of antitumor immunity (105). Immune checkpoint blockade with anti-PD pathway antibodies can be achieved either through the exogenous coadministration or via CART secretion. Anti-PD-L1 antibodies secreted by CARTs have been shown to induce an efficient, localized PD blockade that promoted T-cell cytotoxicity, NK cell recruitment, and tumor growth control in a murine model of colorectal carcinoma (127). An alternative, potentially more efficacious, strategy to blocking immune checkpoint signaling is exchanging the inhibitory signal transmitted by checkpoint activation for a stimulatory one. T-cells can also be engineered to express decoy receptors containing an extracellular domain similar to CTLA-4 or PD-1 that transmits an activating signal. CARTs expressing a PD-1-CD28 switch receptor were shown to be more resistant to tumor-induced immunosuppression, and in a solid tumor mouse model resulted in enhanced TIL infiltration and tumor regression as compared to treatment with CART therapy or PD-1 blockade alone (128).
In conclusion, CART therapy has significant unrealized potential to revolutionize the treatment of solid tumors, as it has already done for hematologic cancers. A better understanding of factors leading to the success of CART immunotherapy in these liquid cancers and an analysis of those limiting the applicability of CART therapy to solid tumors will be necessary to move this field forward. Strategies that address the unique aspects of solid tumor biology like molecular heterogeneity, isolated tumor location, and associated immunosuppression must be thoroughly studied to extend the use of CART immunotherapy to these tumors. Further studies involving optimization of ex vivo culture conditions, genetic manipulation of CAR and CART structure, and investigation of combination therapies are necessary to provide a lasting solution.
AF and MH contributed to literature review and writing. MD contributed to article conception, writing, and overall supervision.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the NIH K08NS092895 grant (MD). Authors would like to thank Christopher Brown MS for his help with the figure illustrations.
1. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
2. Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, Ueda R, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol. (2009) 29:1–42. doi: 10.1615/CritRevImmunol.v29.i1.10
3. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
4. Rabinovich GA, Gabrilovich D, and Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
5. Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, and Eshhar Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. (1993) 151:6577–82.
6. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
7. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
8. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
9. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
10. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
11. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
12. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. (2011) 3:95ra73. doi: 10.1126/scitranslmed.3002842
13. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR t cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
14. Novartis. Novartis Receives First Ever FDA Approval for a CAR-T Cell Therapy, Kymriah(TM) (CTL019), for Children and Young Adults with B-Cell ALL That is Refractory or Has Relapsed at Least Twice. (2017)
15. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
16. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. (2013) 264:71–83. doi: 10.1016/j.jneuroim.2013.08.013
17. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. (2011) 17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557
18. Miura Y, Takenaka T, Toh K, Wu S, Nishihara H, Kano MR, et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano. (2013) 7:8583–92. doi: 10.1021/nn402662d
19. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, and Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. (2007) 204:2023–30. doi: 10.1084/jem.20070064
20. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. (2014) 257:107–26. doi: 10.1111/imr.12131
21. Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell (1991) 64:891–901. doi: 10.1016/0092-8674(91)90314-O
22. Alvarez-Vallina L, Hawkins RE. Antigen-specific targeting of CD28-mediated T cell co-stimulation using chimeric single-chain antibody variable fragment-CD28 receptors. Eur J Immunol. (1996) 26:2304–9. doi: 10.1002/eji.1830261006
23. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. (2004) 172:104–13. doi: 10.4049/jimmunol.172.1.104
24. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119:5697–705. doi: 10.1182/blood-2012-01-405365
25. MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther. (2017) 25:949–61. doi: 10.1016/j.ymthe.2017.02.005
26. Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res. (2007) 13(18 Pt 1): 5271–9. doi: 10.1158/1078-0432.CCR-07-1030
27. Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. (2003) 9:279–86. doi: 10.1038/nm827
28. Milone MC, Fish JD, Carpenito C, Carroll RG, binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
29. Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood (1988) 71:13–29.
30. Porter DL, Levine BL, Kalos M, Bagg A, and June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
31. Morello A, Sadelain M, and Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. (2016) 6:133-46. doi: 10.1158/2159-8290.CD-15-0583
32. Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. (2016) 23:142–8. doi: 10.1038/cgt.2016.14
33. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
34. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. (2004) 64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454
35. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. (2008) 180:4901–9. doi: 10.4049/jimmunol.180.7.4901
36. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
37. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
38. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. (2010) 16:474–85. doi: 10.1158/1078-0432.CCR-09-1322
39. Yang J, Yan J, Liu B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett. (2017) 403:224–30. doi: 10.1016/j.canlet.2017.06.024
40. Feldkamp MM, Lala P, Lau N, Roncari L, and Guha A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery (1999) 45:1442–53. doi: 10.1097/00006123-199912000-00034
41. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra22. doi: 10.1126/scitranslmed.aaa4963
42. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE (2014) 9:e94281. doi: 10.1371/journal.pone.0094281
43. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
44. Debinski W, Gibo DM, Hulet SW, Connor JR, and Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. (1999) 5:985–90.
45. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-Redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
46. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
47. Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, koch M, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol. (2003) 26:332–5. doi: 10.1097/01.COC.0000020922.66984.E7
48. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
49. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR-T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. (2017) 78:1031–43. doi: 10.1158/0008-5472.CAN-17-1788
50. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Disc. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
51. Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
52. Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer (2013) 1:21. doi: 10.1186/2051-1426-1-21
53. Choi BD, Suryadevara CM, Hedeon PC, Herndon JE II, Sanchez-Perez L, Bigner DD, et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci. (2014) 21:189–90. doi: 10.1016/j.jocn.2013.03.012
54. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. (2013) 6:33. doi: 10.1186/1756-8722-6-33
55. Everson RG, Antonios JP, Lisiero DN, Soto H, Scharnweber R, Garrett MC, et al. Efficacy of systemic adoptive transfer immunotherapy targeting NY-ESO-1 for glioblastoma. Neuro Oncol. (2016) 18:368–78. doi: 10.1093/neuonc/nov153
56. Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. (2014) 20:972–84. doi: 10.1158/1078-0432.CCR-13-0709
57. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
58. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. (2011) 71:4055–60. doi: 10.1158/0008-5472.CAN-11-0153
59. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
60. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
61. Krenciute G, Krebs S, Torres D, Wu MF, Liu H, Dotti G, et al. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Ralpha2-positive glioma. Mol Ther. (2016) 24:354–63. doi: 10.1038/mt.2015.199
62. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. (2002) 99:16168–73. doi: 10.1073/pnas.242600099
63. Parente-Pereira AC, burnet J, Ellison D, Foster J, Davies DM, van der Stegen S, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. (2011) 31:710–8. doi: 10.1007/s10875-011-9532-8
64. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
65. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
66. Kakarla S, Song XT, Gottschalk S. Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy (2012) 4:1129–38. doi: 10.2217/imt.12.112
67. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell (1994) 76:301–14. doi: 10.1016/0092-8674(94)90337-9
68. Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity (2005) 22:175–84. doi: 10.1016/j.immuni.2004.12.008
69. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. (2015) 29:227–38. doi: 10.1096/fj.14-250985
70. Slaney CY, Kershaw MH, and Darcy PK. Trafficking of T cells into tumors. Cancer Res. (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
71. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
72. Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. (1999) 100:216–32. doi: 10.1016/S0165-5728(99)00203-9
73. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
74. Zou JP, Morford LA, Chougnet C, Dix AR, Brooks AG, Torres N, et al. Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J Immunol. (1999) 162:4882–92.
75. Moon EK, Wang LC, Dolfi DV, Wilcon CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
76. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. (2013) 94:25–39. doi: 10.1189/jlb.1212621
77. Kaminska B, Wesolowska A, Danilkiewicz M. TGF beta signalling and its role in tumour pathogenesis. Acta Biochim Pol. (2005) 52:329–37.
78. Hishii M, Nitta T, Ishida H, Ebato M, Kurosu A, Yagita H, et al. Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery (1995) 37:1160–6; discussion 1166–7. doi: 10.1227/00006123-199512000-00016
79. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
80. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature (2015) 520:692–6. doi: 10.1038/nature14426
81. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
82. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
83. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
84. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. (2015) 125:3981–91. doi: 10.1172/JCI82416
85. Brown SD, Warren RL, Gobbs EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. (2014) 24:743–50. doi: 10.1101/gr.165985.113
86. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. a dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348:803–8. doi: 10.1126/science.aaa3828
87. Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. (2017) 7:106. doi: 10.3389/fonc.2017.00106
88. Kalaora S, Barnea E, Merhavi-Shoham E, Qutob N, Teer JK, Shimony N, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget (2016) 7:5110–7. doi: 10.18632/oncotarget.6960
89. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Erratum: Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature (2015) 523:370. doi: 10.1038/nature14567
90. van Rooij N, van Buuren MM, Phillips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:e439–42. doi: 10.1200/JCO.2012.47.7521
91. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. (2012) 72:1081–91. doi: 10.1158/0008-5472.CAN-11-3722
92. Walseng E, Koksal H, Sektioglu IM, Fane A, Skorstad G, Kvalheim G, et al. A TCR-based chimeric antigen receptor. Sci Rep. (2017) 7:10713. doi: 10.1038/s41598-017-11126-y
93. Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, et al. Stable, nonviral expression of mutated tumor neoantigen-specific t-cell receptors using the sleeping beauty transposon/transposase system. Mol Ther. (2016) 24:1078–89. doi: 10.1038/mt.2016.51
94. Hofflin S, Prommersberger S, Uslu U, Schuler G, Schmidt CW, Lennerz V, et al. Generation of CD8(+) T cells expressing two additional T-cell receptors (TETARs) for personalised melanoma therapy. Cancer Biol Ther. (2015) 16:1323–31. doi: 10.1080/15384047.2015.1070981
95. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
96. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. (2013) 1:43–53. doi: 10.1158/2326-6066.CIR-13-0008
97. Spiotto MT, Rowley DA, and Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. (2004) 10:294–8. doi: 10.1038/nm999
98. Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. (2007) 204:49–55. doi: 10.1084/jem.20062056
99. Santoro SP, Kim S, Motz GT, Alatzoglou D, Li C, Irving M, et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res. (2015) 3:68–84. doi: 10.1158/2326-6066.CIR-14-0192
100. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
101. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res. (2010) 16:5458–68. doi: 10.1158/1078-0432.CCR-10-0712
102. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
103. Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. (2007) 179:3332–41. doi: 10.4049/jimmunol.179.5.3332
104. Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology (2015) 4:e988098. doi: 10.4161/21505594.2014.988098
105. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. (2012) 72:5209–18. doi: 10.1158/0008-5472.CAN-12-1187
106. Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. (2008) 180:3132–9. doi: 10.4049/jimmunol.180.5.3132
107. Chinnasamy D, Tran E, Yu Z, Morgan RA, Roestifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. (2013) 73:3371–80. doi: 10.1158/0008-5472.CAN-12-3913
108. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, and Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
109. van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity?. J Immunol. (2013) 191:4589–98. doi: 10.4049/jimmunol.1301523
110. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
111. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res. (2017) 24:95–105. doi: 10.1158/1078-0432.CCR-17-2041
112. Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. (2004) 173:7125–30. doi: 10.4049/jimmunol.173.12.7125
113. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
114. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
115. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. (2005) 26:111–7. doi: 10.1016/j.it.2004.12.003
116. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
117. Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, et al. Monoclonal antibody blockade of IL-2 receptor alpha during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood (2011) 118:3003–12. doi: 10.1182/blood-2011-02-334565
118. Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res. (2013) 19:106–17. doi: 10.1158/1078-0432.CCR-12-2143
119. Moeller M, Haynes NM, Kershaw MH, Jackson JT, Teng MW, Street SE, et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood (2005) 106:2995–3003. doi: 10.1182/blood-2004-12-4906
120. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. (2014) 257:83–90. doi: 10.1111/imr.12125
121. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
122. Chinnasamy D, Yu Z, Kerhar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. (2012) 18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050
123. Chmielewski M, Abken H. CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. (2012) 61:1269–77. doi: 10.1007/s00262-012-1202-z
124. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. (2008) 31:500–5. doi: 10.1097/CJI.0b013e318177092b
125. Filley AC, Dey M. Dendritic cell based vaccination strategy: an evolving paradigm. J Neurooncol. (2017) 133:223–35. doi: 10.1007/s11060-017-2446-4
126. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
127. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
Keywords: glioblastoma, malignant glioma, CAR T-cells, chimeric antigen receptor, immunotherapy
Citation: Filley AC, Henriquez M and Dey M (2018) CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Front. Oncol. 8:453. doi: 10.3389/fonc.2018.00453
Received: 26 July 2018; Accepted: 26 September 2018;
Published: 17 October 2018.
Edited by:
Arian Dominic John Laurence, Newcastle University, United KingdomReviewed by:
Georgina Cornish, King's College London, United KingdomCopyright © 2018 Filley, Henriquez and Dey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahua Dey, bWRleUBpdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.