
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 September 2018
Sec. Molecular and Cellular Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00370
This article is part of the Research Topic Cancer Ecosystems View all 19 articles
 Eric Fox1
Eric Fox1 Thomas Oliver1Melissa Rowe1
Thomas Oliver1Melissa Rowe1 Sunil Thomas2
Sunil Thomas2 Yousef Zakharia3
Yousef Zakharia3 Paul B. Gilman1,2
Paul B. Gilman1,2 Alexander J. Muller2,4
Alexander J. Muller2,4 George C. Prendergast2,4*
George C. Prendergast2,4*Exploding interest in immunometabolism as a source of new cancer therapeutics has been driven in large part by studies of tryptophan catabolism mediated by IDO/TDO enzymes. A chief focus in the field is IDO1, a pro-inflammatory modifier that is widely overexpressed in cancers where it blunts immunosurveillance and enables neovascularization and metastasis. The simple racemic compound 1-methyl-D,L-tryptophan (1MT) is an extensively used probe of IDO/TDO pathways that exerts a variety of complex inhibitory effects. The L isomer of 1MT is a weak substrate for IDO1 and is ascribed the weak inhibitory activity of the racemate on the enzyme. In contrast, the D isomer neither binds nor inhibits the purified IDO1 enzyme. However, clinical development focused on D-1MT (now termed indoximod) due to preclinical cues of its greater anticancer activity and its distinct mechanisms of action. In contrast to direct enzymatic inhibitors of IDO1, indoximod acts downstream of IDO1 to stimulate mTORC1, a convergent effector signaling molecule for all IDO/TDO enzymes, thus possibly lowering risks of drug resistance by IDO1 bypass. In this review, we survey the unique biological and mechanistic features of indoximod as an IDO/TDO pathway inhibitor, including recent clinical findings of its ability to safely enhance various types of cancer therapy, including chemotherapy, chemo-radiotherapy, vaccines, and immune checkpoint therapy. We also review the potential advantages indoximod offers compared to selective IDO1-specific blockade, which preclinical studies and the clinical study ECHO-301 suggest may be bypassed readily by tumors. Indoximod lies at a leading edge of broad-spectrum immunometabolic agents that may act to improve responses to many anticancer modalities, in a manner analogous to vaccine adjuvants that act to boost immunity in settings of infectious disease.
Immune therapy has risen to the forefront of cancer therapy in recent years, providing a new approach to cancer therapy, and in some instances has begun to shift the paradigm of cancer care from chemotherapy to immunotherapy. One of the factors crucial to the success of immunotherapy is reversing tumor-mediated immunosuppression (1). The tryptophan catabolic enzyme indoleamine 2,3-dioxygenase-1 (IDO1) has received a great deal of attention as a driver of tumor-mediated suppression (2–4). IDO1 has been shown to be active in many human cancers and its expression has been associated widely with poor prognosis (5, 6). Accordingly, inhibitors of the enzymatic activity and effector functions of IDO1 have been developed as tools to leverage cancer therapy (7).
Elevated tryptophan catabolism as a characteristic of patients with cancer was initially reported over 60 years ago (8). The basis for this observation and later observations in various types of cancer patients was not clear until IDO1 was discovered in the 1960s. An association of elevated tryptophan cata olism with inflammation was established in the 1970s−1980s with demonstrations that IDO1 is induced strongly in the lungs by LPS, viral infection and interferon (9–12). In a seminal line of work in the late 1990s by Munn and Mellor and colleagues, tryptophan catabolism was implicated in immunosuppression during pregnancy, based on the preferential sensitivity of T cells to tryptophan deprivation leading to an impairment of antigen-dependent T cell activation (13–15). In these studies, the key probe in defining this mechanism of immune tolerance was the racemic compound 1-methyl-tryptophan (1MT), a tryptophan mimetic with complex IDO inhibitory effects discussed further below. Indeed, much of the huge amount of subsequent work on IDO and disease pathogenesis has relied on this compound, including most importantly cancer studies.
A causal relationship between IDO1 activity and cancer growth was founded by pivotal studies in the 2000s that have been reviewed in detail elsewhere (7). IDO1 was found to be overexpressed widely in human cancers and 1MT could slow the growth of murine tumors (6, 16, 17). IDO1 overexpression in cancer cells was linked genetically to inactivation of BIN1 (18), a tumor suppressor gene widely attenuated in human cancer (19). Loss of BIN1 empowers IFN/STAT and NFkB mediated IDO1 transcription and later studies also implicated the RAS/MAPK, COX2, and PI3K pathways in driving IDO1 expression (18, 20–22). Interestingly, drugs that target molecules relying on these pathways may act in part by indirectly blocking IDO1 expression, such as the case with imatinib (Gleevec) (23). Pharmacological blockade with 1MT or true catalytic inhibitors of IDO1 enzyme were found to display unimpressive efficacy unless combined with DNA damaging therapies, which led to regression of otherwise unstoppable tumors (18, 24, 25). Preclinical genetic proofs of IDO1 as a valid therapeutic target in cancer were enabled in IDO1-deficient mice, where fundamental connections between IDO1 expression and cancerous inflammatory programs were also established (21, 26, 27).
In the tumor microenvironment or draining lymph nodes, IDO1 activity suppresses the function of T effector cells (Teff) and natural killer (NK) cells and promotes the induction and activation of T regulatory cells (Treg) and the activation, recruitment and expansion of myeloid-derived suppressor cells (MDSC) (Figure 1) [Fallarino(21, 29–36)]. IDO1 effector functions are mediated by the tryptophan catabolite kynurenine (Kyn) and by two stress signals generated by locoregional deprivation of tryptophan (7), as discussed further below. Investigations of IDO1 in immune tolerance have focused heavily on antigen-presenting dendritic cells where IDO1 is upregulated by interferons, TLR ligands and other immune signals (37). Beyond its roles in provoking Treg development, IDO1 also acts in certain dendritic cells to directly suppress effector T cell responses (38, 39).
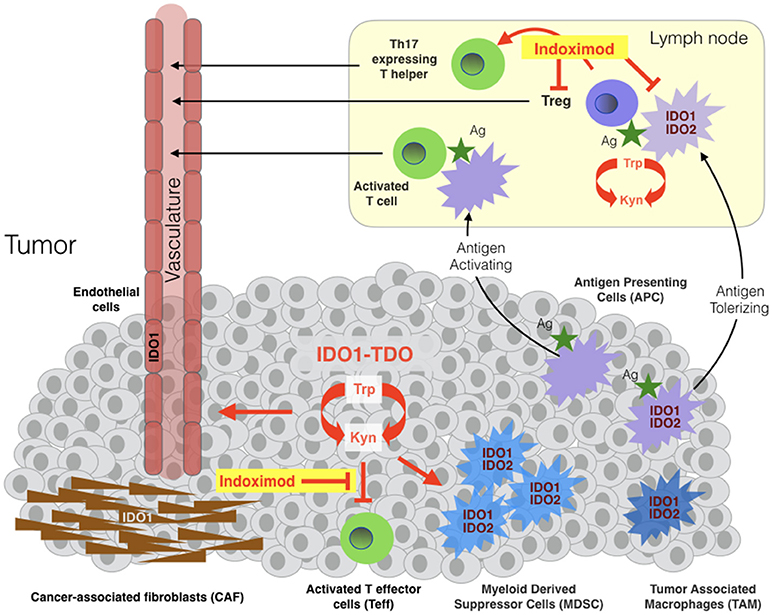
Figure 1. Sites of indoximod action in cancer. IDO1, TDO, and IDO2 are expressed variously in malignant, immune, stromal and vascular cells in the tumor microenvironment and in antigen-presenting cells (APC) of tumor-draining lymph nodes. TDO and IDO2 are relatively more narrowly expressed than IDO1 in human cancers, with TDO overexpressed in some tumors independently or in parallel with IDO1 and IDO2 expressed in antigen-presenting cells including B cells where it may influence IDO1 function (28). Tryptophan catabolism in tumor cells leads to locoregional generation of kynurenine at the cost of tryptophan, enabling suppression of local T effector cells (Teff), licensing and recruitment of myeloid-derived suppressor cells (MDSC) and support of a tumor-enabling vasculature. The tumor microenvironment variously recruits cancer-associated fibroblasts, vascular endothelial and inflammatory myeloid cells that express IDO1 and/or IDO2. Tumor antigens absorbed by IDO1/IDO2-expressing antigen-presenting cells (APC) rove to draining lymph nodes where they promote the formation and activation of T regulatory cells (Tregs). Studies suggest that indoximod enables Teff in tumors and attenuate in draining lymph nodes, in the latter case by acting on dendritic cells leading to suppression and/or reprogramming of Tregs and the formation of Th17-expressing T helper cells.
The racemic compound 1-methyl-D,L-tryptophan (1MT) was first described as a competitive inhibitor of the IDO1 enzyme by Cady and Sono in the early 1990s (40). After the seminal demonstration that 1MT could elicit allogeneic conceptus rejection by ablating T cell tolerance to paternal fetal antigens (13), 1MT was shown to weakly retard the growth of cancer cells in mouse tumor graft or spontaneous transgenic models of cancer (16, 17). While the anticancer effects of 1MT were unremarkable as monotherapy, its striking therapeutic power was revealed in combinations with DNA damaging chemotherapy which elicit regressions of otherwise recalcitrant tumors (18). This discovery was an important advance in providing the first indication of how to use an IDO inhibitor to improve cancer therapy. The regressions achieved by 1MT in combination therapy did not appear to reflect drug-drug interactions that raised the cytotoxicity of the chemotherapies tested, as the efficacy was increased without increasing the known side-effects of the chemotherapies tested (18). Further, T cell depletion in subjects nullified the therapeutic benefits of 1MT administration, establishing that its action was based in provoking T cell attacks in the presence of chemotherapy (18). Overall, these observations challenged the paradigm at the time that active immunotherapy and chemotherapy are fundamentally incompatible by offering one of the first demonstrations of a productive immunochemotherapy regimen based exclusively on small molecule drugs (41).
Careful biochemical studies with purified IDO1 enzyme revealed that only the L racemer of 1MT exerted any catalytic inhibitory activity (42), and it became apparent that L-1MT is actually a weak substrate rather than a true catalytic inhibitor of IDO1 as discussed in detail elsewhere (43). Unexpectedly, the D racemer lacking enzyme inhibitory activity was actually more potent in empowering chemotherapy as well as relieving T cell suppression by IDO1-positive dendritic cells from mouse or human sources (42), although there are conflicting data on T cell suppression (44, 45). Mouse genetic studies were consistent with IDO1 pathway targeting in showing that the anticancer efficacy of D-1MT relied genetically on the presence of a functionally intact IDO1 gene (42), similar to bona fide IDO1 enzyme inhibitors (24, 25). However, subsequent studies of D-1MT make it clear that its antitumor effects in cells and in animals is likely to be complex (7, 43). Indeed, mechanistic studies have made it clear that neither racemer of 1MT is a valid probe of IDO1 enzyme activity, a question ultimately addressed by isolation of several unique structural classes of true enzymatic inhibitors with related antitumor properties, as reviewed elsewhere (7). Cellular mechanisms of action for indoximod have been defined which involve relief of suppression of Teff cells in tumors, limitations on the generation of Tregs, and re-programming of Tregs to Th17 helper cells in draining lymph nodes (Figure 1) (2, 46, 47). The robust preclinical efficacy of D-1MT/indoximod in combination with DNA damaging chemotherapy led to its inclusion on a list of ‘top ten' agents for clinical evaluation by an NCI immunotherapy workshop (48, 49). In 2008, a decision was made to advance D-1MT/indoximod (NLG-8189) to first-in-man trials as a single molecular species through an FDA investigational new drug application by a collaborative team of investigators from the Medical College of Georgia, Lankenau Institute for Medical Research, National Cancer Institute and NewLink Genetics Corporation as corporate sponsor.
Phase 1 studies of indoximod as a monotherapy or in combination with chemotherapy showed it to be well-tolerated in patients with advanced breast cancers or other solid tumors (50, 51). In a first-in-man dose escalation study conducted in advanced breast cancer patients receiving standard of care taxane therapy, the administration of indoximod was well-tolerated to a maximum delivered dose of 1,200 mg twice daily. Four partial responses were observed in the patients studied (n = 22) in the absence of any apparent drug-drug interactions (50). In a larger dose escalation study of advanced cancer patients with various solid tumors, the maximum tolerated dose was not reached until 2,000 mg twice daily (51). Notably, several patients on the indoximod trial who had been treated previously with ipilimumab developed hypophysitis, an autoimmune reaction to the pituitary gland which had been documented in patients treated with ipilimumab. In these patients, stable disease >6 months was observed, encouraging the notion that indoximod can reactivate latent T cell immunity in cancer patients. In the initial trials of indoximod, its relative apparent safety is notable given comparisons to the acute side-effects of immune checkpoint therapy, however, a case of Parkinsonism was reported recently in a patient receiving indoximod treatment (52). While safety studies were not able to identify a maximum tolerated dose (MTD) for indoximod, pharmacokinetic analysis indicated that 1,200 mg twice daily (BID) was the maximum exposure that could be achieved in a patient based on a plateau that occurred in plasma AUC and Cmax beyond this dose. Oral dosing generated a Cmax at 2.9 h with a serum halflife of 10.5 h. Interestingly, there was evidence in indoximod-treated patients of increased levels of both C reactive protein (CRP) and autoantibodies to tumor antigens, consistent with an increased inflammatory response to the chemotherapy onboard (51). Based on these initial studies, multiple Phase 2 studies of indoximod in continuous oral cycles have been conducted at a dose of 1,200 mg twice daily.
Phase 2 data from several trials of indoximod in different types of cancer has been provocative but not uniformly positive in all disease settings examined so far (Table 1). All trials have been conducted in combination with standard of care treatments, including in metastatic cutaneous, mucosal, or uveal melanoma with immune checkpoint therapy; advanced breast cancer (BRCA), acute myeloid leukemia (AML), and pancreatic ductal adenocarcinoma (PDAC) with chemotherapy; and advanced prostate carcinoma (PC) with sipuleucel-T (Provenge®), an approved dendritic cell vaccine. In particular, the melanoma and prostate trials have illustrated significant therapeutic activity of indoximod in empowering anti-PD1 treatment (pembrolizumab) and sipuleucel-T vaccine treatment (Provenge® autologous dendritic cells), respectively.
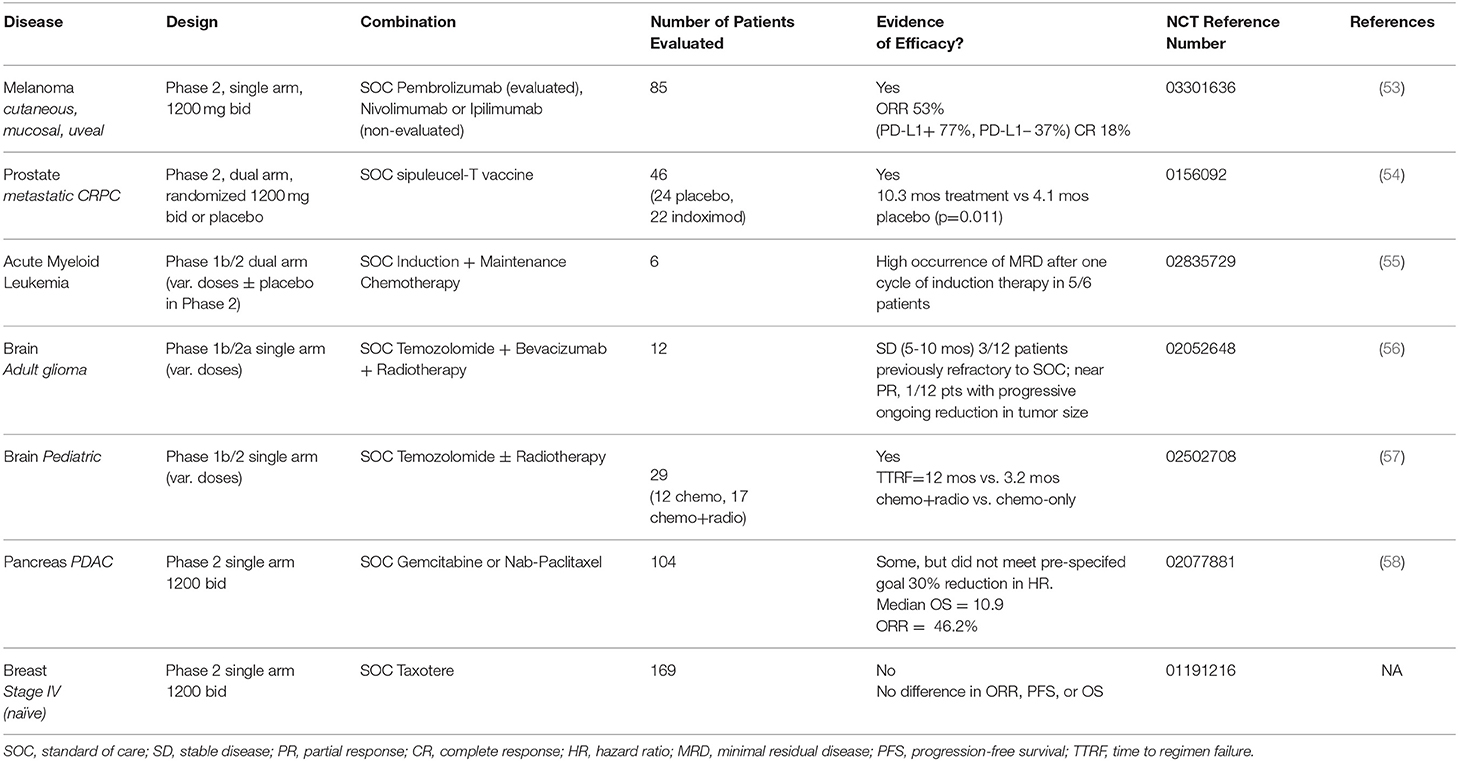
Table 1. Overview of indoximod clinical data (phase 1b/2, phase 2 trials).
The initial phase 1b study in melanoma illustrated the safety of indoximod in combination with the anti-CTLA4 antibody ipilimumab, the standard of care treatment for metastatic melanoma at the time of testing. Nine patients with unresectable stage 3 or 4 melanoma patients were treated with escalating doses of indoximod (600 mg BID, then 1,200 mg BID). Unlike an IDO1 enzyme inhibitor (epacadostat) which yields dose-limiting toxicity (DLT) in combination with ipilimumab, no DLT was encountered with indoximod. Thus, the pre-specified highest dose of indoximod (1,200 mg BID) was deemed tolerable and used as the recommended phase 2 dose (RP2D) in combination with checkpoint inhibitors (59).
The phase 2 melanoma study enrolled over 100 patients in a single-arm trial of indoximod plus provider choice of immune checkpoint antibodies (ipilimumab or the anti-PD1 antibodies nivolimumab or pembrolizumab) (NCT03301636). A preclinical treatment rationale was provided by a study showing that indoximod could improve the response of B16 murine melanoma tumors to immune checkpoint therapy (60). In this single-arm trial (53), 85 patients were treated with pembrolizumab plus indoximod with on-treatment imaging to meet a pre-specified definition of evaluable for efficacy. Overall response rate (ORR) was 53% with a rate of complete response (CR) of 18% and disease control rate (DCR) of 73%. Median progression-free survival (PFS) was 12.4 months (95% confidence interval: 7.1, 24.9). Notably, these efficacy data paralleled those achieved by the approved combination of nivolumab and ipilimumab, but without the elevated rate of severe autoimmune side-effects experienced by patients treated with these agents (61). Stratifying the data by PD-L1 expression status, the ORR in PD-L1 positive (+) patients was 77% vs. 37% in PD-L1 negative (-) patients. Some responses seen in uveal melanomas were encouraging given its extremely aggressive nature and complete lack of response to immune checkpoint therapy (62). Overall, these data suggest the ability of indoximod to safely augment anti-PD1 antibody responses, strongly encouraging a randomized Phase 3 trial in this disease setting. These data are striking in light of the failure of epacadastat, a direct IDO1 enzyme inhibitor, to show any benefit to melanoma patients in the phase 3 ECHO-301 study when administered in combination with pembrolizumab. Given the different mechanism of action of indoximod, its independent evaluation must not be dismissed out of hand.
Further significant evidence of the efficacy of indoximod as an immunometabolic adjuvant has been documented in advanced prostate cancer. In a randomized study of metastatic castrate-resistant disease (NCT01560923), 46 patients treated with the dendritic cell vaccine sipuleucel-T (Provenge®) received placebo (n = 24) or indoximod (n = 22) with the latter cohort displaying a >2-fold increase in overall survival (OS) (54). Indoximod was administered for 10 weeks with 3 additional months in cases where an absence of radiographic or clinical progression was documented. Immune monitoring of patients was the same as performed for the IMPACT study which led to approval of sipuleucel-T (63). Indoximod was well tolerated with no significant difference in adverse events between the two study arms. Median OS had not yet been achieved at the time of report, but median radiographic PFS was 10.3 months in the treatment arm vs. 4.1 months in placebo arm (p = 0.011). Notably, the PFS on the placebo arm was identical to that reported in the pivotal IMPACT study for sipuleucel-T. These positive data align with recent evidence that epithelial-mesenchyme transition (EMT) drives IDO1 expression as part of this key step in metastatic progression of prostate cancer to its deadly castrate-resistant form (64). Overall, the findings of this randomized phase 2 trial with a placebo control arm strongly encourages further study of indoximod as an immunometabolic adjuvant for prostate cancer treatment.
In a Phase 1b trial that includes a randomized Phase 2a component to treat AML, patients with newly diagnosed disease received remission-induction chemotherapy (cytarabine plus idarubicin) plus consolidation chemotherapy (high dose cytarabine), a standard of care regimen, with the addition of indoximod or placebo as maintenance therapy (55) (NCT02835729). The dose escalation was a standard 3+3 design for the phase 1 portion aimed at gauging toxicities in combination with the chemotherapy regimen [400, 600, 1,000, 1,200 mg indoximod]. A different schedule was used in this trial, with indoximod provided every 8 h starting on day 8 of induction therapy, avoiding administration on days that patients received consolidation chemotherapy, and then stopping it 4 weeks prior to hematopoietic stem cell allo-transplanation. At the time of the report, the evidence presented indicated that indoximod did not add significant toxicity to standard of care treatment, and early response data suggested a high occurrence of minimal residual disease after one cycle of induction chemotherapy.
Phase 1b/2 single-arm trials in adult and pediatric brain cancers are being conducted in which indoximod is combined with chemotherapy or chemo-radiotherapy, with some early but intriguing efficacy data being reported. A preclinical treatment rationale was established in a robust orthotopic model of malignant brain cancer (glioblastoma), where the synergistic effects of indoximod were demonstrated in combination with temozolomide (TMZ) and radiation as a cooperative DNA damaging modality (65). In the latest report from the adult trial (NCT02052648) (56), 12 patients who had progressed on standard of care therapy with TMZ were enrolled in a traditional 3+3 dose escalation study of indoximod (600, 1,000, or 1,200 mg twice daily). No dose-limiting toxicity was encountered nor did indoximod cause a delay or reduction in TMZ dosing in any patient. The best responses documented were 1 patient with partial response per Response Assessment in Neuro-Oncology (RANO) criteria at 15 months and 4 patients with stable disease lasting between 4 and 11 months (66). A phase 2 expansion of the study is ongoing at the 1,200 mg twice daily dose in combination with TMZ, bevacizumab and ateriotactic radiation (SRS) (NCT02052648).
In the pediatric brain cancer trial (NCT02502708) (57), the first trial to evaluate indoximod both in children and in the context of radiotherapy, 17 patients from an original cohort of 29 heavily pretreated patients in the dose escalation phase 1b study who were eligible to receive further treatment were administered indoximod and radiotherapy followed by standard of care cycles of TMZ with indoximod as maintenance therapy. The other 12 patients received only indoximod and TMZ. Both treatments were well tolerated with minimal toxicity attributed to indoximod. Overall, at the time of the report, 29 patients in the dose-escalation phase of the study exhibited a median PFS of 6.2 months and median time to regimen failure (TTRF) of 11.7 months, which compares favorably with historical controls. Notably, patients receiving radiotherapy appeared to benefit significantly when indoximod was added, with a median TTRF of 12 months observed vs. 3.2 months without radiotherapy (p = 0.04). These data suggested a dose-sparing effect of indoximod on conventional chemo-radiotherapy, potentially extending efficacious responses. The notion that targeting the IDO pathway may improve chemo-radiotherapy is supported a recent study in lung cancer (67). Encouraged by these response data, the same regimen is now being tested in patients with diffuse intrinsic pontine glioma (DIPG), a dismal disease with no effective treatment option. Thus far, 3/6 patients enrolled are reported to have achieved good symptomatic and radiographic response.
In contrast to the trials above, two phase 2 studies of >100 patients in pancreatic or breast cancer have shown little to no evidence of efficacy. In a single-arm study of metastatic PDAC (NCT02077881), 104 of 135 patients enrolled to receive a standard of care regimen of gemcidabine or nab-paclitaxel plus indoximod were judged evaluable for efficacy by a pre-specified definition (58). Patients were enrolled with treatment-naïve disease or first line therapy following earlier resection and adjuvant therapy. Treatment was administered until disease progression or toxicity occurred. Median OS was 10.9 months with an ORR of 46.2%. Notably, responding patients exhibited an increased density of intratumoral CD8+ T cells. This study did not meet its pre-specified goal of a hazard ratio (HR) = 0.70, but the increased ORR that was observed correlated with a positive immunological response. In contrast, a study of metastatic BRCA patients failed to produce any evidence of efficacy. In this study of 169 newly diagnosed patients treated with taxotere and indoximod (NCT01191216), no statistically significant difference in PFS, OS, or ORR was observed. While these two types of aggressive cancer set a high bar for improvements in efficacy, the selection of subjects who were not heavily pre-treated opened a window of opportunity for indoximod. Taken together, clinical findings clearly encourage further study of indoximod as an immunometabolic adjuvant for immunotherapy in treatment of melanoma and prostate cancer, and possibly for DNA damaging modalities in treatment brain cancer and AML, a diverse set of diseases and combinations that illustrate the potentially broad uses indoximod may realize in the clinical setting.
The molecular mechanisms of action of indoximod as an inhibitor of the IDO pathway are a subject of continued study. However, only one mechanism of action has been described that is consistent with pharmacokinetic analyses of the blood serum levels of indoximod that are actually achieved in human subjects (68). Specifically, in cells subjected to IDO/TDO-mediated tryptophan depletion, indoximod has been shown to relieve suppression of the master metabolic kinase mTORC1 that occurs in tryptophan-depleted cells, with an IC50 (~70 nM) that is more potent than L-tryptophan itself (68). mTORC1 controls protein synthesis, coordinating nutrient levels to different cellular physiological responses of autophagy vs. growth. In T cells, mTORC1 is pivotal in determining autophagy/tolerance vs. growth/activation. mTORC1 is downregulated by depletion of essential amino acids like tryptophan, to which it responds by activating autophagy as an attempt to access tryptophan from intracellular stores. Accordingly, depletion of tryptophan by IDO/TDO activation downregulates mTORC1 and promotes autophagy which indoximod reverses as a tryptophan mimetic (Figure 2). Although the precise connections between IDO/TDO-mediated downregulation of mTORC1 in T cells are not well understood, there is evidence of an intermediate role for the amino acid sensing kinase GLK1 which acts upstream to regulate not only mTORC1 but also PKC-θ, a T cell receptor regulatory kinase (69). Thus, GLK1 may be a linchpin between tryptophan catabolism by IDO/TDO enzymes and mTORC1 downregulation in T cells (7).
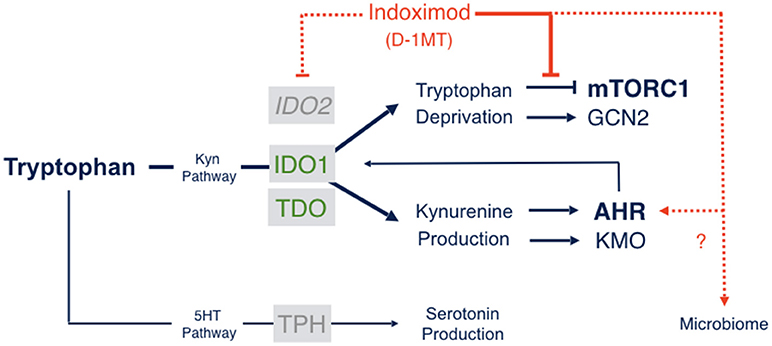
Figure 2. Indoximod mechanisms of action. Tryptophan catabolism proceeds through pathways leading to serotonin or NAD production, the latter through the kynurenine pathway which handles ~95% of Trp catabolism in mammals. Indoximod is a Trp mimetic which mTORC1 interprets as L-Trp under conditions of high Trp catabolism and autophagy. Thus, the drug acts to block the suppressive signal on mTORC1 function generated by IDO/TDO activity. Other suggested mechanisms of action include an indirect suppression of IDO2 activity; a modulation of kynurenine-regulated AHR function, which may also influence feedback on IDO1 expression and activity; and an influence on gut microbial physiology influencing systemic immunity (see the text).
By restoring mTORC1 activity, indoximod acts to reverse mTORC1-activated autophagy triggered by tryptophan depletion (68). Since indoximod is a D-tryptophan analog, it cannot support protein translation, but nevertheless it is interpreted by the mTORC1 kinase as a high-potency L-tryptophan mimetic. Why this is the case is unclear, but a mammalian capability to recognize (if not use) D-amino acids might reflect immune crosstalk with the microbiome given their use in bacteria (70). In any case, mTORC1 has a critical role in human Teff cell activity and indoximod acts directly in human T cells where it exerts a direct effect, unlike IDO1 enzyme inhibitors (71).
There are at least three implications of this mechanism of action. First, by targeting a downstream effector molecule, indoximod differs from IDO1 enzyme inhibitors in being agnostic to the IDO/TDO enzyme(s) contributing to cancer pathogenesis. Thus, indoximod is rationalized to treat tumor cells overexpressing IDO1, IDO2 or TDO (or any combination thereof), which is not the case for an enzyme-selective inhibitor. This is a useful feature in heterogenous plastic tumors which represent the norm in advanced cancer patients. Second, by targeting a convergent effector mechanism used by all IDO/TDO enzymes, indoximod may prove less sensitive to inherent or acquired resistance that may arise in patients due to IDO1 mutation, IDO1 overexpression or other target bypass mechanisms that heterogeneous cancers evolve. On this point, preclinical genetic studies illustrate clearly how tumoral bypass of an IDO1-specific blockade is associated with IDO1-independent elevation of regional kynurenine levels (21), suggesting the availability of resistance pathways via TDO2 or IDO2 activation. The critical question of inherent and acquired resistance to IDO1 selective blockade is discussed in greater depth in a separate review of the failed ECHO-301 phase 3 clinical trial in melanoma patients of pembrolizumab with epacadastat, a direct IDO1 enzyme inhibitor that added no benefit to the immune checkpoint therapy under the conditions of study (72). Lastly, mTORC1 is implicated in tumor cell growth and proliferation as well as in T cell activation. Thus, if indoximod also provokes mTORC1 activation in tumor cells, the drug may also empower tumor cell killing when combined with chemotherapeutic drugs, which generally exhibit greater cytotoxicity against growing cells.
Overall, the evidence that indoximod may broaden the efficacy of pembrolizumab (53) suggests that restoring mTORC1 in effector T cells might be sufficient to improve therapeutic responses with reduced risks of resistance due to IDO1 bypass. On this point, it is known that mTORC1 drives expression of ICOS, a positive-acting T cell co-regulatory receptor, and that elevated expression of ICOS in melanoma patients receiving immune checkpoint therapy correlates with the most favorable outcomes (73). In efforts to further leverage its features as an IDO/TDO effector pathway inhibitor, novel salts of indoximod and a pro-drug form of the drug (NLG-802) with superior pharmacokinetic properties have recently been described which have entered clinical testing (71).
Indoximod clearly has complex immunomodulatory properties, as illustrated, for example, by its ability to act on B cells to relieve inflammation in a murine model of autoimmune rheumatoid arthritis (74, 75). Thus, other mechanisms of action that have been described for indoximod are likely to illuminate its therapeutic properties.
The catalytic activity of IDO2 has been shown to be inhibited indirectly by indoximod in human kidney cells where the IDO2 gene is expressed normally (76). There is conflicting data in dendritic cells, which express IDO2 as well as IDO1, on the ability of indoximod in this setting to block T cell suppression (42, 77, 78). However, mouse genetic studies support a link between indoximod action and IDO2 function, for example, in demonstrating that the therapeutic benefits of indoximod administration in a model of rheumatoid arthritis that relies on the presence of the Ido2 gene (74), which interacts genetically with IDO1 in IDO1-mediated activation of Treg cells in the mouse (28). Here we note that the ability of indoximod to limit rheumatoid arthritis is highly relevant to combination treatments with immune checkpoint antibodies, which often cause autoimmune side-effect in patients. In this sense, indoximod co-administration with immune checkpoint antibodies may widen the therapeutic window at both ends, by extending efficacy and reducing side-effects, unlike IDO1-selective enzyme inhibitors.
At high concentrations in cell culture (1 mM), evidence has been presented that D-1MT/indoximod can elevate transcription of IDO1 leading to increased production of kynurenine in cancer cells (79), but the concentrations used in this study, which exceed by ~100-fold the serum levels of indoximod achieved in patients in clinical trials (50), cast doubt on the physiological relevance of this observation. However, a very recent report offers additional support for the related idea that indoximod may somehow affect IDO1 expression in cell-specific ways via AHR (80), a transcription factor that binds and is activated by kynurenine (81) as a convergent effector pathway downstream of all IDO/TDO enzymes (7). Indeed, other evidence has been presented for an autocrine feedback pathway involving IDO1, AHR, and IL-6 that controls IDO1 expression in cancer cells (82).
The AHR connection for indoximod is complex. There are binding sites for AHR in the IDO1 gene and other genes that influence the differentiation of dendritic cells, T helper cells and Tregs and the proliferation of Teffs and Tregs where AHR has influence (83). In a recent study reported at the 2018 AACR conference (80), indoximod was reported to modulate AHR-dependent transcriptional activity in human liver and primary T cells, in the latter case altering the transcription of genes associated with T helper and Treg phenotypes. These effects were reversed by an AHR inhibitor, suggesting that indoximod acts upstream of AHR (80). In plasmacytoid dendritic cells in vitro and in vivo (in tumor-draining lymph nodes), indoximod was found to downregulate IDO1 expression and function, decrease kynurenine production and increase T cell proliferation, while promoting a phenotypic shift in T cells from Treg to Th17-producing T helper cells (80). Thus, in addition to resuscitating Teff cells in tumors, indoximod may also act in draining lymph nodes to reprogram the AHR effector pathway to shift Tregs to Th17 cells.
Immune homeostasis involves a dynamic balance between tolerance of commensals and suitable immune responses to eradicate or otherwise control pathogens (84, 85). Tolerance is important to avoid tissue injury but at the potential costs of chronic infections and inflammation which in the long term become factors in metabolic diseases, autoimmunity, and, in certain settings, cancer (85). Regarding indoximod mechanisms this is an important area to survey given evidence that the therapeutic impact of anti-PD1 therapy is determined by microbiome character, in both preclinical models (86, 87) and clinical settings (88–90).
Cross-regulatory circuitry between IDO1 and AHR is a key factor in mediating disease tolerance (91). For example, exposure to bacterial lipopolysaccharide will program a state of refractoriness to further LPS challenge (endotoxin tolerance), a phenomenon reflecting the engagment of AHR in long-term control of systemic inflammation only when IDO1 is active, which responds late upon initial stimulation but earlier upon subsequent challenge. Mechanistic studies have revealed a feedback control cycle, with SRC kinase as an intermediate between kynurenine-activated AHR and IDO1 expression in regulating tolerance to bacterial endotoxins, a state that protects against immunopathology in Gram-negative and Gram-positive infections. In this fundamental way, IDO1 and especially AHR contribute to immunologic host fitness (91).
IDO1 and AHR are highly expressed in the small and large intestine (92). IDO1 expression increases further during aging, a key factor in the likelihood of a positive therapeutic response to anti-PD1 treatment (93). In the intestine of adult germ-free mice, IDO1 levels are reduced suggesting that commensal microorganisms mediate the age-dependent increase in IDO1. Supporting the likelihood that it modulates mucosal immunity to intestinal microbiota, IDO1-deficient mice exhibit resistance to enteric pathogens, for example, to Citrobacter rodentium (94). Tryptophan catabolites produced by microbiota such as gut Lactobacillus can also act as AHR ligands, confounding a clear interpretation of the link between IDO1 and cancer that may involve microbiota-mediated tryptophan catabolism (85).
In melanoma studies of anti-PD-1/PD-L1 it appears that gut commensals of Bifidobacteria can enhance therapeutic efficacy (86, 88–90). Given evidence that indoximod can heighten the benefit of anti-PD1 therapy, it will be important to evaluate Bifidobacteria as a potential mediator in this effects, which raises the possibility of conceptualizing indoximod as a prebiotic substance. In one clue that this may be the case, indoximod was able to reverse the effects of IDO1 activity in models of colitis that are quieted by Bifidobacteria (95). While still in their infancy, studies of the effects on indoximod and the IDO/TDO/AHR pathways on gut microbial physiology and cancer immunity is a rich area for exploration.
Recent studies suggest that indoximod may exert a variety of benefits as an immunometabolic adjuvant on the quality of life of cancer patients and survivors. The conditions that are improved are not critical to overall survival, but are of major importance to affected individuals and their oncologists and caregivers. As noted above, one interesting feature of indoximod is its ability to limit autoimmune arthritis in preclinical models, possibly by limiting IDO2 function implicated in this condition (96). Autoimmune joint inflammation is a common short and long term side effect of immune checkpoint therapy in cancer patients which indoximod may limit. This potential may be confirmed through long-term follow up of melanoma patients receiving combinations of indoximod and pembrolizumab in the phase 2 trial discussed above. Other beneficial effects of indoximod that have been described are behavioral, as evaluated in preclinical models of depression, anhedonia, anxiety or pain (97–100), one or more of which occur commonly in cancer patients and survivors. Given its relative safety in trials to date, it may be possible to consider uses in these settings, not only during cancer therapy but as a palliative adjunctive therapy. In summary, indoximod is a unique immunometabolic adjuvant with a wide potential range of uses to improve cancer therapy in adults and children, not only safely but with possible collateral benefits to quality of life.
All authors contributed to composing the text. GP composed the figures. YZ, PG, AM, and GP made final edits to the text and figures.
GP and AM disclose equity ownership in NewLink Genetics reflecting inventorship of licensed IDO intellectual property including indoximod and its uses in cancer treatment from the Lankenau Institute of Medical Research, as described in U.S. Patents Nos. 7705022, 7714139, 8008281, 8058416, 8383613, 8389568, 8436151, 8476454, and 8586636. GP additionally discloses equity ownership in Incyte and Merck; former and present advisory board roles for NewLink Genetics and Kyn Therapeutics, respectively; and a board director role for Meditope Biosciences. AM additionally discloses roles as an advisory board member and grant recipient for I-O Biotech AG, which is developing IDO vaccines for cancer treatment. YZ discloses research and travel support from NewLink Genetics and advisory board roles for Amgen, Roche Diagnostics, Novartis, Eisai, Castle Bioscience and Exelixis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our many colleagues and collaborators and apologize for omitting citations to many primary research reports cited through reviews due to space limitations. GP and AM acknowledge grant support from the NCI (R01 CA191119) and The W.W. Smith Trust, with additional support from the Lankenau Medical Center Foundation and the Main Line Health System. GP holds The Havens Chair for Biomedical Research at the Lankenau Institute for Medical Research.
1. Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn't talk then but need to now. Cancer Res. (2007) 67:3500–4. doi: 10.1158/0008-5472.CAN-06-4626
2. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
3. Amobi A, Qian F, Lugade AA, Odunsi K. Tryptophan catabolism and cancer immunotherapy targeting IDO mediated immune suppression. Adv Exp Med Biol. (2017) 1036:129–44. doi: 10.1007/978-3-319-67577-0_9
4. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol. (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
5. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. (2011) 17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331
6. Theate I, Van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. (2015) 3:161–72. doi: 10.1158/2326-6066.CIR-14-0137
7. Prendergast GC, Malachowski WP, Duhadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. (2017) 77:6795–811. doi: 10.1158/0008-5472.CAN-17-2285
8. Boyland E, Williams DC. The metabolism of tryptophan. 2. the metabolism of tryptophan in patients suffering from cancer of the bladder. Biochem J. (1956) 64:578–82. doi: 10.1042/bj0640578
9. Yoshida R, Hayaishi O. Induction of pulmonary indoleamine 2,3-dioxygenase by intraperitoneal injection of bacterial lipopolysaccharide. Proc Natl Acad Sci USA. (1978) 75:3998–4000. doi: 10.1073/pnas.75.8.3998
10. Yoshida R, Urade Y, Tokuda M, Hayaishi O. Induction of indoleamine 2,3-dioxygenase in mouse lung during virus infection. Proc Natl Acad Sci USA. (1979) 76:4084–6. doi: 10.1073/pnas.76.8.4084
11. Yoshida R, Imanishi J, Oku T, Kishida T, Hayaishi O. Induction of pulmonary indoleamine 2,3-dioxygenase by interferon. Proc Natl Acad Sci USA. (1981) 78:129–32. doi: 10.1073/pnas.78.1.129
12. Yasui H, Takai K, Yoshida R, Hayaishi O. Interferon enhances tryptophan metabolism by inducing pulmonary indoleamine 2,3-dioxygenase: its possible occurence in cancer patients. Proc Natl Acad Sci USA. (1986) 83:6622–6. doi: 10.1073/pnas.83.17.6622
13. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
14. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
15. Mellor AL, Sivakumar J, Chandler PKS, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat. Immunol. (2001) 2:64–8. doi: 10.1038/83183
16. Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer (2002) 101:151–5. doi: 10.1002/ijc.10645
17. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
18. Muller AJ, Duhadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene Bin1, potentiates cancer chemotherapy. Nat Med. (2005) 11:312–9. doi: 10.1038/nm1196
19. Prendergast GC, Muller AJ, Ramalingam A, Chang MY. BAR the door: cancer suppression by amphiphysin-like genes. Biochim Biophys Acta (2009) 1795:25–36. doi: 10.1016/j.bbcan.2008.09.001
20. Basu GD, Tinder TL, Bradley JM, Tu T, Hattrup CL, Pockaj BA, et al. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: role of IDO. J Immunol. (2006) 177:2391–402. doi: 10.4049/jimmunol.177.4.2391
21. Smith C, Chang MY, Parker KH, Beury DW, Duhadaway JB, Flick HE, et al. IDO Is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. (2012) 2:722–35. doi: 10.1158/2159-8290.CD-12-0014
22. Hennequart M, Pilotte L, Cane S, Hoffmann D, Stroobant V, De Plaen E, et al. Constitutive IDO1 expression in human tumors is driven by Cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol Res. (2017). 5:695–709. doi: 10.1158/2326-6066.CIR-16-0400
23. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. (2011) 17:1094–100. doi: 10.1038/nm.2438
24. Banerjee T, Duhadaway JB, Gaspari P, Sutanto-Ward E, Munn DH, Mellor AL, et al. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene (2008) 27:2851–7. doi: 10.1038/sj.onc.1210939
25. Kumar S, Jaller D, Patel B, Lalonde JM, Duhadaway JB, Malachowski WP, et al. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J Med Chem. (2008) 51:4968–77. doi: 10.1021/jm800512z
26. Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA.III, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci USA. (2008) 105:17073–78. doi: 10.1073/pnas.0806173105
27. Muller AJ, Duhadaway JB, Chang MY, Ramalingam A, Sutanto-Ward E, Boulden J, et al. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol Immunother. (2010) 59:1655–63. doi: 10.1007/s00262-010-0891-4
28. Metz R, Smith C, Duhadaway JB, Chandler P, Baban B, Merlo LM, et al. IDO2 is critical for IDO1-mediated T cell regulation and exerts a non-redundant function in inflammation. Int Immunol. (2014) 26:357–67. doi: 10.1093/intimm/dxt073
29. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Diff. (2002) 9:1069–77. doi: 10.1038/sj.cdd.4401073
30. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. (2002) 196:459–68. doi: 10.1084/jem.20020121
31. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science (2002) 297:1867–70. doi: 10.1126/science.1073514
32. Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. (2002) 196:447–57. doi: 10.1084/jem.20020052
33. Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, et al. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. (2004) 16:1391–401. doi: 10.1093/intimm/dxh140
34. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
35. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108:4118–25. doi: 10.1182/blood-2006-03-006700
36. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP., et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
37. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. (2008) 8:74–80. doi: 10.1038/nri2233
38. Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. (2003) 24:242–8. doi: 10.1016/S1471-4906(03)00072-3
39. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
40. Cady SG, Sono M. 1-methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch Biochem Biophys. (1991) 291:326–33. doi: 10.1016/0003-9861(91)90142-6
41. Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. (2005) 65:8065–8. doi: 10.1158/0008-5472.CAN-05-2213
42. Hou DY, Muller AJ, Sharma MD, Duhadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. (2007) 67:792–801. doi: 10.1158/0008-5472.CAN-06-2925
43. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
44. Lob S, Konigsrainer A, Schafer R, Rammensee HG, Opelz G, Terness P. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood (2008) 111:2152–4. doi: 10.1182/blood-2007-10-116111
45. Qian F, Villella J, Wallace PK, Mhawech-Fauceglia P, Tario JD Jr, Andrews C, et al. Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2,3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. (2009) 69:5498–504. doi: 10.1158/0008-5472.CAN-08-2106
46. Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. (2016) 39:1–6. doi: 10.1016/j.coi.2015.10.009
47. Munn DH, Sharma MD, Johnson TS, Rodriguez P. IDO, PTEN-expressing tregs and control of antigen-presentation in the murine tumor microenvironment. Cancer Immunol Immunother. (2017) 66:1049–58. doi: 10.1007/s00262-017-2010-2
48. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. (2008) 222:357–68. doi: 10.1111/j.1600-065X.2008.00604.x
49. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. (2009) 15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737
50. Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget (2014) 5:8136–46. doi: 10.18632/oncotarget.2357
51. Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget (2016) 7:22928–38. doi: 10.18632/oncotarget.8216
52. Floyd M, Osta BE, Tang SC. First report of parkinsonism associated with indoximod, an immune-modulating agent. J Global Oncol. (2018) 4:1–2. doi: 10.1200/JGO.2016.007492
53. Zakharia Y, Rixe O, Ward JH, Drabick JJ, Shaheen MF, Milhem MM, et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J Clin Oncol. (2018) 36:9512.
54. Jha GG, Gupta S, Tagawa ST, Koopmeiners JS, Vivek S, Dudek AZ, et al. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol. (2017) 35:3066. doi: 10.1200/JCO.2017.35.15
55. Emadi A, Holtzman NG, Imran M, El-Chaer F, Koka M, Singh Z, et al. Indoximod in Combination with Idarubicin and Cytarabine for Upfront Treatment of Patients with Newly Diagnosed Acute Myeloid Leukemia (AML): Phase 1 Report. II Congress of the European Hematology Association. (2017).
56. Colman H, Mott F, Spira AI, Johnson TS, Zakharia Y, Vahanian NN, et al. A phase 1b/2 study of the combination of the IDO pathway inhibitor indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors: Safety analysis and preliminary efficacy of the phase 1b component. J Clin Oncol. (2015) 33:2070. doi: 10.1200/jco.2015.33.15
57. Johnson TS. Radio-chemo-immunotherapy using the IDO-inhibitor indoximod for childhood brain cancer. In: 2017 International Pediatric Neuro-oncology Conference. (Houston TX, Texas Children's Hospital) (2017).
58. Bahary N, Wang-Gillam A, Haraldsdottir S, Somer BG, Lee JS, O'rourke MA, et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. J Clin Oncol. (2018) 36:4015. doi: 10.1200/JCO.2018.36.15
59. Zakharia Y, Drabick JJ, Khleif SN, Munn DH, Link CJ, Vahanian NN, et al. Results of a Phase 1b trial of the indoleamine 2,3-dioxygenase (IDO) pathway inhibitor indoximod plus ipilimumab for the treatment of unresectable stage 3 or 4 melanoma. In: European Cancer Congress 2015 (18th ECCO/40th ESMO), Vienna. abstract #514 (2015).
60. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. (2013) 210:1389–402. doi: 10.1084/jem.20130066
61. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, Mcdermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
62. Zakharia Y, Mcwilliams R, Shaheen M, Grossman K, Drabick J, Milhem M, et al. Interim analysis of the Phase 2 clinical trial of the IDO pathway inhibitor indoximod in combination with pembrolizumab for patients with advanced melanoma. Cancer Res. (2017) 77:CT117. doi: 10.1158/1538-7445.AM2017-CT117
63. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
64. Kolijn K, Verhoef EI, Smid M, Bottcher R, Jenster GW, Debets R, et al. Epithelial-mesenchymal transition in human prostate cancer demonstrates enhanced immune evasion marked by IDO1 expression. Cancer Res. (2018) 78:4671–9. doi: 10.1158/0008-5472.CAN-17-3752
65. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca S-B, Bolduc AK, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer (2014) 2:21. doi: 10.1186/2051-1426-2-21
66. Zakharia Y, Colman H, Mott F, Lukas R, Vahanian N, Link C, et al. IMCT-21 updates on phase 1b/2 combination study of the IDO pathway inhibitor indoximod with temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors. Neuro-Oncology (2015) 17:v112. doi: 10.1093/neuonc/nov218.21
67. Wang W, Huang L, Jin JY, Jolly S, Zang Y, Wu H, et al. IDO immune status after chemoradiation may predict survival in lung cancer patients. Cancer Res. (2018) 78:809–16. doi: 10.1158/0008-5472.CAN-17-2995
68. Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology (2012) 1:1460–8. doi: 10.4161/onci.21716
69. Chuang HC, Lan JL, Chen DY, Yang CY, Chen YM, Li JP, et al. The kinase GLK controls autoimmunity and NF-kappaB signaling by activating the kinase PKC-theta in T cells. Nat Immunol. (2011) 12:1113–8. doi: 10.1038/ni.2121
70. Thomas S, Izard J, Walsh E, Batich K, Chongsathidkiet P, Clarke G, et al. The host microbiome regulates and maintains human health: a primer and perspective for non-microbiologists. Cancer Res. (2017) 77:1783–812. doi: 10.1158/0008-5472.CAN-16-2929
71. Mautino MR, Kumar S, Zhuang H, Waldo J, Jaipuri F, Potturi H, et al. a novel prodrug of indoximod with enhanced pharmacokinetic properties. Cancer Res. (2017) 77:4076. doi: 10.1158/1538-7445.AM2017-4076
72. Muller A, Manfredi M, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. (in press).
73. Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. (2011) 71:5445–54. doi: 10.1158/0008-5472.CAN-11-1138
74. Merlo LM, Pigott E, Duhadaway JB, Grabler S, Metz R, Prendergast GC, et al. IDO2 Is a critical mediator of autoantibody production and inflammatory pathogenesis in a mouse model of autoimmune arthritis. J Immunol. (2014) 92:2082–90. doi: 10.4049/jimmunol.1303012
75. Merlo LM, Duhadaway JB, Grabler S, Prendergast GC, Muller AJ, Mandik-Nayak L. IDO2 modulates T Cell-dependent autoimmune responses through a B Cell-intrinsic mechanism. J Immunol. (2016) 196:4487–97. doi: 10.4049/jimmunol.1600141
76. Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. (2007) 67:7082–7. doi: 10.1158/0008-5472.CAN-07-1872
77. Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. (2009) 58:153–7. doi: 10.1007/s00262-008-0513-6
78. Qian F, Liao J, Villella J, Edwards R, Kalinski P, Lele S, et al. Effects of 1-methyltryptophan stereoisomers on IDO2 enzyme activity and IDO2-mediated arrest of human T cell proliferation. Cancer Immunol Immunother. (2012) 61:2013–20. doi: 10.1007/s00262-012-1265-x
79. Opitz CA, Litzenburger UM, Opitz U, Sahm F, Ochs K, Lutz C, et al. The indoleamine-2,3-dioxygenase (IDO) inhibitor 1-methyl-D-tryptophan upregulates IDO1 in human cancer cells. PLoS ONE (2011) 6:e19823. doi: 10.1371/journal.pone.0019823
80. Brincks EL, Adams J, Essmann M, Turner BA, Wang L, Ke J, et al. Indoximod modulates AhR-driven transcription of genes that control immune function. Cancer Res. (2018) 78. doi: 10.1158/1538-7445.AM2018-3753
81. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature (2011) 478:197–203. doi: 10.1038/nature10491
82. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget (2014) 5:1038–51. doi: 10.18632/oncotarget.1637
83. Zhou L. AHR Function in Lymphocytes: emerging Concepts. Trends Immunol. (2016) 37:17–31. doi: 10.1016/j.it.2015.11.007
84. Raberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science (2007) 318:812–4. doi: 10.1126/science.1148526
85. Zelante T, Iannitti RG, Fallarino F, Gargaro M, De Luca A, Moretti S, et al. Tryptophan Feeding of the IDO1-AhR Axis in Host-Microbial Symbiosis. Front Immunol. (2014) 5:640. doi: 10.3389/fimmu.2014.00640
86. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350:1084–9. doi: 10.1126/science.aac4255
87. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350:1079–84. doi: 10.1126/science.aad1329
88. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359:97–103. doi: 10.1126/science.aan4236
89. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (2018) 359:104–8. doi: 10.1126/science.aao3290
90. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359:91–7. doi: 10.1126/science.aan3706
91. Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature (2014) 511:184–90. doi: 10.1038/nature13323
92. Rhee SJ, Walker WA, Cherayil BJ. Developmentally regulated intestinal expression of IFN-gamma and its target genes and the age-specific response to enteric Salmonella infection. J Immunol. (2005) 175:1127–36. doi: 10.4049/jimmunol.175.2.1127
93. Kugel CH III, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age correlates with response to Anti-PD1, reflecting age-related differences in intratumoral effector and Regulatory T-Cell populations. Clin Cancer Res. (in press). doi: 10.1158/1078-0432.CCR-18-1116
94. Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Shi HN, et al. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect Immun. (2008) 76:3045–53. doi: 10.1128/IAI.00193-08
95. Zhao L, Suolang Y, Zhou D, Tang Y, Zhang Y. Bifidobacteria alleviate experimentally induced colitis by upregulating indoleamine 2, 3-dioxygenase expression. Microbiol Immunol. (2018) 62:71–9. doi: 10.1111/1348-0421.12562
96. Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in immunomodulation and autoimmune disease. Front Immunol. (2014) 5:585. doi: 10.3389/fimmu.2014.00585
97. Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O'connor JC. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav. (2012) 62:202–9. doi: 10.1016/j.yhbeh.2012.03.010
98. Liu YN, Peng YL, Liu L, Wu TY, Zhang Y, Lian YJ, et al. TNFalpha mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw. (2015) 26:15–25. doi: 10.1684/ecn.2015.0362
99. Huang L, Ou R, Rabelo De Souza G, Cunha TM, Lemos H, Mohamed E, et al. Virus Infections Incite Pain Hypersensitivity by Inducing Indoleamine 2,3 Dioxygenase. PLoS Pathog. (2016) 12:e1005615. doi: 10.1371/journal.ppat.1005615
Keywords: immunometabolism, immune adjuvant, Immunotherapy, immuno-chemotherapy, immuno-radiotherapy
Citation: Fox E, Oliver T, Rowe M, Thomas S, Zakharia Y, Gilman PB, Muller AJ and Prendergast GC (2018) Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 8:370. doi: 10.3389/fonc.2018.00370
Received: 11 July 2018; Accepted: 21 August 2018;
Published: 11 September 2018.
Edited by:
Ubaldo Emilio Martinez-Outschoorn, Thomas Jefferson University, United StatesReviewed by:
Paolo Puccetti, University of Perugia, ItalyCopyright © 2018 Fox, Oliver, Rowe, Thomas, Zakharia, Gilman, Muller and Prendergast. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George C. Prendergast, cHJlbmRlcmdhc3RAbGltci5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.