 Xingchen Pang1
Xingchen Pang1 Feng Guan
Feng Guan Xiang Li
Xiang Li- 1School of Biotechnology, Jiangnan University, Wuxi, China
- 2College of Life Science, Northwest University, Xi'an, China
- 3Wuxi Medical School, Jiangnan University, Wuxi, China
The three types of blood cells (red blood cells for carrying oxygen, white blood cells for immune protection, and platelets for wound clotting) arise from hematopoietic stem/progenitor cells in the adult bone marrow, and function in physiological regulation and communication with local microenvironments to maintain systemic homeostasis. Hematological malignancies are relatively uncommon malignant disorders derived from the two major blood cell lineages: myeloid (leukemia) and lymphoid (lymphoma). Malignant clones lose their regulatory mechanisms, resulting in production of a large number of dysfunctional cells and destruction of normal hematopoiesis. Glycans are one of the four major types of essential biological macromolecules, along with nucleic acids, proteins, and lipids. Major glycan subgroups are N-glycans, O-glycans, glycosaminoglycans, and glycosphingolipids. Aberrant expression of glycan structures, resulting from dysregulation of glycan-related genes, is associated with cancer development and progression in terms of cell signaling and communication, tumor cell dissociation and invasion, cell-matrix interactions, tumor angiogenesis, immune modulation, and metastasis formation. Aberrant glycan expression occurs in most hematological malignancies, notably acute myeloid leukemia, myeloproliferative neoplasms, and multiple myeloma, etc. Here, we review recent research advances regarding aberrant glycans, their related genes, and their roles in hematological malignancies. Our improved understanding of the mechanisms that underlie aberrant patterns of glycosylation will lead to development of novel, more effective therapeutic approaches targeted to hematological malignancies.
Introduction
Hematological malignancies include numerous forms of acute and chronic lymphoproliferative and myeloproliferative diseases, derived respectively from the two major blood cell lineages: lymphoid and myeloid cells. Notably, lymphoma, lymphocytic leukemia, and myeloma are of lymphoid origin, whereas acute myeloid leukemia (AML), myeloproliferative neoplasm (MPN), and myelodysplastic syndrome (MDS) are of myeloid origin. A crucial factor in development of hematological malignancies is the dynamic interaction between transformed cells and the bone marrow and lymphoid tissue microenvironments. There is increasing evidence that microenvironmental changes produced by neoplastic cells progressively favor survival of these cells and determine the clinical course of disease.
Glycans (polysaccharides), one of the four basic components of plant and animal cells, are the most abundant and diverse type of naturally occurring biopolymer. In addition to their well-known roles as energy sources and structural components, glycans often function as signaling effectors and cell recognition markers, typically attached to cellular proteins and lipids. Such attachment, termed glycosylation, includes N-glycosylation, O-glycosylation, glycosaminoglycans, glycosphingolipids, and glycosylphosphatidylinositol (GPI)-linked proteins, according to the conserved core structure (Figure 1). Glycans are constructed in an ordered, sequential manner that depends on distinct substrate specificities of glycosyltransferase and glycosidase enzymes. Glycosyltransferases synthesize glycan chains, whereas glycosidases hydrolyze specific glycan linkages. Glycans bind to lectins (specific carbohydrate-binding proteins) and sterically modulate molecular interactions in cells, thereby helping to control a variety of physiological mechanisms involved in health maintenance or disease development.
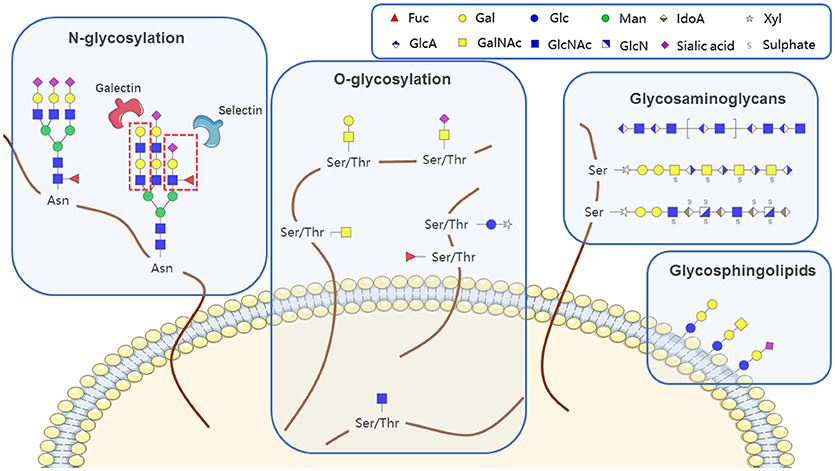
Figure 1. Aberrant features of glycan expression in hematological malignancies.
Aberrant glycan properties, such as hypersialylation and hyperfucosylation, are often associated with interaction of neoplastic cells with the microenvironment (1, 2). Two principal mechanisms that underlie aberrant glycan expression in hematological malignancies are incomplete synthesis and neosynthesis (3), which result mainly from altered transcription of glycosyltransferase and/or glycosidase genes.
Numerous types of glycoconjugates may potentially interfere with neoplastic cell processes or with the microenvironment, leading to malignant progression. We review here recent advances in our knowledge of aberrant glycans, and their related genes, involved in hematological malignancies. Increasingly detailed characterization of the biological functions of glycans and glycan-binding proteins (particularly galectins, selectins, and calreticulin) is progressively clarifying the physiology of hematological malignancies.
N-glycosylation
N-glycans are linked to asparagine (Asn) residues of proteins, specifically a subset residing in the Asn-X-Ser/Thr motif and having a common, conserved five-sugar core structure. Variation in N-glycosylation of proteins, including the well-studied glycoprotein immunoglobulin G (IgG), has physiological significance in hematological malignancies. Mizuochi et al. (4) first reported truncation of N-glycan chain of IgG in a study of myeloma proteins, and many subsequent studies demonstrated effects of N-glycosylation patterns of IgGs on their biological functions. Galactose (Gal) residues are absent from total serum IgG of rheumatoid arthritis patients (5), and this altered glycosylation state activates complement via mannose-binding protein (6). Sialylation of Fc fragment core polysaccharide of IgG enhances its anti-inflammatory properties via distinct Fcγ receptors (7). Addition of fucose (Fuc) to the glycan core interferes with binding of IgG to FcγRIIIA (CD16a), and greatly reduces its capacity for antibody dependent cell-mediated cytotoxicity (ADCC) (8). Lauc et al. (9) used mass spectrometry in combination with genome sequencing to quantify N-linked IgG glycans, and identified nine genetic loci associated with IgG glycosylation, most of which showed strong associations with autoimmune disorders, inflammatory conditions, and/or hematological malignancies. Four of these were glycosyltransferase genes: ST6GAL1, B4GALT1, FUT8, and MGAT3. Leukemia cell function has been shown to be affected by aberrant N-glycosylation of other glycoproteins (e.g., CD79a, μ, CD82, CD95, MPL) besides IgG (Table 1); however, the related N-glycan synthesis genes have not been investigated in detail. Future studies utilizing an “omics” approach (combination of genomics, proteomics, and glycomics) will help elucidate glycosylation processes of single glycoproteins.
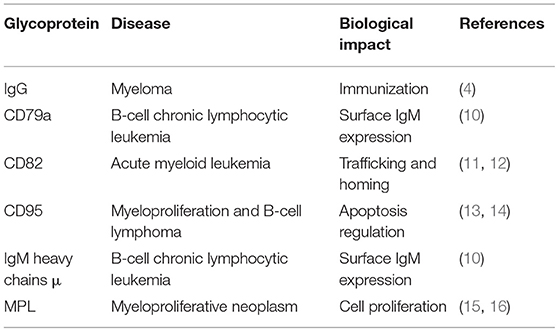
Table 1. Aberrant N-glycosylation of single glycoproteins associated with hematological malignancies.
Biosynthesis of a given N-glycan is a systemic process involving dozens of glycosyltransferases and glycosidases. Aberrant structures typically result from abnormal expression of these enzymes. The three processes most commonly involved in aberrant expression of N-glycan-related enzymes in hematological malignancies are sialylation, fucosylation, and bisecting β D N acetylglucosamine (GlcNAc), as described separately below.
Sialylation
Sialic acids play key roles in cell recognition, adhesion, and signaling. Commonly, sialic acids are attached to galactose or N-acetylgalactosamine residues via a α2,3 or α2,6 linkage and sialic acid can be attached to another sialic acid residue via a α2,8 linkage, forming polysialic acid structure.
Enhancement of global sialylation, particularly α2,6 and α2,3 linked sialylation resulting from altered expression of the sialyltransferases ST6Gal and ST3Gal, is associated with malignancy (17). Bone marrow hematopoietic stem cells and progenitor cells form surface α2,6-linked sialic acids through the action of circulating ST6Gal1 (originating mostly from the liver), rather than endogenous ST6Gal1 (18). Elevated ST6Gal1 and ST3Gal5 mRNA levels are positively correlated with increased risk of pediatric acute leukemia. Increased expression of ST6Gal1 contributes to multidrug resistance (MDR) of leukemia cells by regulation of P-glycoprotein (P-gp) and MDR-associated protein 1 (MRP1) through phosphoinositide 3-kinase (PI3K)/Akt signaling (2). However, T cell acute lymphoblastic leukemia cell line, CCRF-CEM, decreases ST6Gal expression and, thus, α2,6 sialylation of global membrane glycoprotein to acquire resistance to microtubule targeting drug desoxyepothilone B (19). Elevated ST3Gal4 in chronic myeloid leukemia (CML) cells is associated with imatinib resistance (20). α2,3 linked sialic acids are also often found to modify O-glycans (21). Sialylation of N-glycan and O-glycan on platelets play a part in the regulation of homeostasis and clearance (22, 23). Desialylated platelets can be rapidly cleared by liver macrophages via the recognition of hepatic asialoglycoprotein receptor (also called the Ashwell–Morell receptor) (21, 24, 25).
Polysialic acid (PSA) is a linear homopolymers comprising α2,8-linked sialic acids, typically attached to neural cell adhesion molecule (NCAM). Two polysialyltransferases, ST8SiaII and ST8SiaIV, play dominant roles in PSA synthesis (26). PSA is best known for its proposed role in modulating neuronal development. However, recent researches also suggest a role for PSA in immune regulation. Drake et al. found that human NK cells modulate expression of NCAM and the degree of polymerization of its PSA according to activation state (27). They also found ST8SiaIV−/− mice exhibited a specific defect in T cell development (28). Further research revealed that, Cys-Cys-chemokine receptor 7 (CCR7) and neuropilin-2 (NRP2), which are both important in immune activation, are also targets of polysialylation. After pathogen recognition, dendritic cell (DC) traffic to secondary lymphoid organs to activate naïve T cells through antigen presentation. CCR7 is the central chemokine receptor controlling DC trafficking. Polysialylation of CCR7 is essential for recognition of the CCR7 ligand CCL21. CCL21 adopts an autoinhibited conformation, which is released upon interaction with PSA (29). The interaction of the tail of CCL21 with PSA is needed for efficient ERK1/2 activation in DCs (30). NRP2 is expressed on the surface of DCs. Polysialylation of NRP2 is added to its O-Glycans, and exclusively synthesized by ST8SiaIV (31). Expression of NRP2 is up-regulated during DC maturation, coincident with increased expression of ST8SiaIV and with the appearance of PSA on the cell surface (32). NRP2 enhances the chemotactic migration of DCs toward CCL21 and promotes DC-induced activation and proliferation of T lymphocytes via the enhanced PSA-mediated effect (33, 34).
Fucosylation
Fucosylated glycans are synthesized by a wide variety of fucosyltransferases. Fucosylation, which includes terminal fucosylation and core fucosylation, is a non-extendable modification. There are 11 different, known fucosyltransferases (FUT) that have been cloned to date. FUT1 and FUT2 are involved in the synthesis of α1,2 fucose, while FUT3, 4, 5, 6, 7, and 9 are involved in the synthesis of α1,3/α1,4 fucose, as a terminal modification that is not further elongated. FUT8 exclusively adds α1,6 fucose to the innermost GlcNAc residue of N-glycans, forming core fucosylation. However, the fucosyltransferase activity of either Fut10 or Fut11 has not been confirmed (35). Fucosylation of hematopoietic cells plays an essential functional role in homing to bone marrow, because fucosylation of cell surface molecules is responsible for binding to P- and E-selectins constitutively expressed by microvessels in marrow. Pretreatment with α1,3 fucosyltransferase VI (FucT-VI) enhances interaction of CD34+ stem cells and early progenitor cells with microvessels and thus promotes marrow homing during cord blood transplantation (36). Enhanced expression of sialyl Lewis X (SLex) in adult T cell leukemia cells is dependent on α1,3 fucosyltransferase VII (FucT-VII) activity. Human T-lymphotropic virus 1 (HTLV 1) retrovirus, the etiologic agent of adult T cell leukemia, encodes a transcriptional activator protein (TAX) which regulates the FUT7 gene that encodes FucT-VII, the limiting enzyme controlling SLex synthesis in leukocytes (1). Overexpression of FUT8 and core fucosylation have been demonstrated in several types of cancer (37). FUT8 had an inhibitory effect on hemoglobin production and erythroid differentiation of leukemia cells during hematopoiesis (38, 39). α1,3 and α1,4 fucose does not just modify proteins but also glycosphingolipid. However, so far aberrant α1,3 and α1,4 fucosylation of glycosphingolipid in hematological malignancies has not been documented.
Bisecting GlcNAc
The enzyme β1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase (MGAT3) catalyzes addition of a bisecting GlcNAc residue to glycoproteins, which has a suppressive effect on several types of cancer (40). Elevation of MGAT3 activity was observed in CML patients in blast crisis and in multiple myeloma patients (41). MGAT3 overexpression in K562 leukemia cells enhanced resistance to natural killer cell cytotoxicity and spleen colonization (42), via regulation of cell recognition by bisecting GlcNAc moieties (43).
O-glycosylation
O-glycosylation is a common covalent modification of serine (Ser) and threonine (Thr) residues of glycoproteins. The most common type of protein O-glycosylation involves α linkage via β-D-N-acetylgalactosamine (GalNAc) to the -OH of Ser or Thr by an O-glycosidic bond, resulting in “O-GalNAc glycan”; this process can be extended to various other structures. Other types of O glycans include those attached via GlcNAc, Fuc, or glucose (Glc) (44).
O-GalNAc Glycans
O-GalNAc glycans, also known as mucin-type O glycans, are found mainly in transmembrane and secreted glycoproteins, attached to certain Ser or Thr residues.
Aberrant O-GalNAc glycans, such as the disaccharide Thomsen-Friedenreich antigen (T antigen; CD176), monosaccharide GalNAc (Tn antigen; CD175), and their sialylated forms (ST antigen and STn antigen [CD175s]), result from incomplete synthesis of O-glycans and are associated with malignancy. The first step of mucin O-glycosylation, whereby Tn antigen is formed, is transfer of GalNAc from UDP-GalNAc to Ser or Thr, catalyzed by polypeptide-N-acetyl-galactosaminyltransferase (ppGalNAcT). Tn antigen is strongly expressed in chronic lymphocytic leukemia (CLL) (45), as a result of overexpression of GALNT11, a member of the ppGalNAcT family (46). Mutation or epigenetic silencing of T synthase C1GalT1 specific chaperone 1 (Cosmc; C1GALT1C1) blocked further O glycan elongation and shifted the pathway toward generation of Tn and STn through ST6GalNAc1 action (47, 48). Expression of Cosmc cDNA in Jurkat cells restored C1β3Gal-T activity and T antigen expression (49). Antitumor drugs with T or Tn antigen as therapeutic target have been developed (50, 51).
Mucin1 (MUC1; CD227) is a protein composed of ~1,000 amino acids whose molecular mass can be increased to over 1,000 kDa by addition of hundreds of O-glycans. MUC1 is translated as a single polypeptide and undergoes autoproteolysis to form two subunits: (i) N-terminal subunit (MUC1-N), containing glycosylated tandem repeats, and (ii) C-terminal transmembrane subunit (MUC1-C), which in turn forms a stable non-covalent heterodimer at the cell surface. Aberrant glycoforms of MUC1-N occurring in hematological malignancies may affect oncogenic functions of MUC1-C (52), including cell proliferation (53) and resistance to apoptosis and cytotoxic injury (54–56). MUC1 appears in a variety of hematological malignancies, including T and B cell lymphomas and myelomas (57), and is a potential prognostic marker and therapeutic target for several types of non-Hodgkin lymphoma (58, 59). MUC1 expressed in lymphoma cells is phosphorylated upon T cell receptor ligation, resulting in T cell activation (60).
Some aberrant fucosylation in O-GalNAc glycans were also observed, as referred in part 2.2.
O-GlcNAcylation
O-GlcNAcylation refers to covalent attachment of GlcNAc sugars to Ser or Thr residues of nuclear or cytoplasmic proteins. O-GlcNAcylation, similarly to protein phosphorylation, is an enzymatic modification whose half-life is typically shorter than that of the attached protein. It is catalyzed by a single enzyme (O linked GlcNAc transferase; OGT) that transfers GlcNAc from UDP-GlcNAc to the protein substrate. N-acetyl β-glucosaminidase (OGA; also known as O-GlcNAcase), encoded by MGEA5 gene, rapidly removes the O-GlcNAc modification. These enzymes, acting together, dynamically alter the post-translational state and function of proteins in response to cellular signals. c-Myc is glycosylated by O-linked GlcNAc at Thr-58, a known phosphorylation site and “mutational hot spot” in lymphomas. Hierarchical phosphorylation of Ser-62 and Thr-58 and alternative glycosylation/ phosphorylation of Thr-58, in combination, regulate the many functions of c-Myc in cells (61). On the other hand, O-GlcNAcylation and phosphorylation may act together to trigger increased STAT5 phosphorylation levels and oncogenic transcription in hematopoietic cells (62). In CLL patients, protein O-GlcNAcylation was higher than in normal lymphocytes, and both OGT and OGA protein levels were elevated. Patients whose lymphocytes had the highest levels of O-GlcNAcylated proteins showed better prognosis, as determined by standard prognostic markers (63). The above findings indicate that leukemic lymphocytes have higher O-GlcNAcylation levels than do normal lymphocytes, with consequent blocking of signal pathways essential for rapid leukemia cell proliferation (64). OGT may also act in cooperative fashion with the tumor suppressor Ten-Eleven Translocation-2 (TET2), whose haploinsufficiency initiates myeloid and lymphoid transformation, thereby promoting post-transcriptional modification of histones and facilitating gene expression via double epigenetic modification of both DNA and histones (65, 66). OGT can induce the differentiation of MDS/AML cells in vitro and extend the survival rate of mice carrying leukemic cells expressing mutant ASXL1, which occurs at high frequencies in myeloid malignancies, by conjugating O-GlcNAc to ASXL1-S199 and thereby stabilizing this tumor suppressor protein (67).
O-Fuc and O-Glc
Epidermal growth factor (EGF)-like repeats are small protein motifs (~40 amino acids) defined by six conserved cysteine residues which form three disulfide bonds. They are found in hundreds of cell surface and secreted animal proteins, and some have unusual O-linked Fuc or Glc residues. The enzymes O-fucosyltransferase 1 (POFUT1) and O-glucosyltransferase 1 (POGLUT1; hCLP46), are responsible for addition of O-Fuc and O-Glc, respectively. O-Fuc and O-Glc modifications are essential for proper Notch function. POGLUT1 is overexpressed in primary AML, T cell acute lymphoblastic leukemia (ALL), and other leukemia cell lines (68, 69). POGLUT1 overexpression enhances Notch activation and regulates cell proliferation in a cell type-dependent manner (70, 71). POFUT1 regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions (72).
Glycosaminoglycans
Proteoglycans consist of a core protein to which one or more glycosaminoglycan (GAG) chains are covalently attached to Ser or Thr. GAGs are unbranched, often long, polysaccharides with a repeating disaccharide structure; they include heparan sulfate, chondroitin sulfate, dermatan sulfate, and keratin sulfate. The location is determined by the core protein. GAG chains, the essential functional parts, are produced by various biosynthetic pathways and are often highly sulfated, with resulting capability to bind cytokines, chemokines, or growth factors. Through such binding, they can modulate cell growth and differentiation, and thus help control embryogenesis, angiogenesis, and homeostasis. In multiple myeloma cells, the secretory-vesicle proteoglycan serglycin is the major proteoglycan expressed and constitutively secreted. High serglycin levels are present in bone marrow aspirates of >30% of newly diagnosed multiple myeloma patients, and are required for adhesion, in vivo growth, and vascularization of multiple myeloma cell (73, 74). Serglycin level is correlated with drug resistance in hematological malignancy cell lines (75). Serglycin is a selective marker for immature myeloid cells, and can distinguish AML from Philadelphia chromosome negative (Ph-) MPN (76). Serglycin attaches to CS4 and CS6 moieties, but not to heparin or heparan sulfate, and interacts with CD44 in a variety of hematopoietic cells (77). Cell surface proteoglycan syndecan-1 (CD138) is highly expressed in multiple myeloma cells (78) and function as a coreceptor for HGF that promotes HGF/Met signaling in MM cells (79). Heparanase is an endo-ß-d-glucuronidase that trims the heparan sulfate chains of proteoglycans, releasing biologically active fragments of heparan sulfate. Heparanase enhances shedding of syndecan-1 and high levels of heparanase and shed syndecan-1 in the tumor microenvironment are associated with elevated angiogenesis and poor prognosis in myeloma, by activating integrin and VEGF receptors on adjacent endothelial cells thereby stimulating tumor angiogenesis (80, 81). Heparan sulfate is a complex molecule because of modification by sulfation and epimerization. Heparan sulfate of syndecan-1 has more sulfated motifs than in normal plasma cells. These highly sulfated motifs bind various angiogenic and growth factors and present them to their respective receptors, and therefore play crucial roles in multiple myeloma cell survival, proliferation, and metastasis (82). A large number of transferases and modifying enzymes are involved in regulating fine structural properties of GAGs. The synthetic mechanisms responsible for generating cellular GAG structures remain poorly understood.
Hyaluronan (HA) is a unique GAG, not sulfated and not attached to a protein or lipid, that is secreted into extracellular compartments. It can interact non-covalently with various matrix components, and can be bound at the cell surface by receptors such as the adhesion molecule CD44 (83). CD44/HA interaction in monocytic cells plays crucial roles in cell migration, inflammation, and immune responses. CD44 binding ability is regulated by sialidase induced in response to cytokine stimulation (84). CD44 and HA cooperate with stromal cell-derived factor 1 (SDF-1) in trafficking of human CD34+ stem/ progenitor cells to bone marrow (85). A CD44 glycoform expressed on primitive CD34+ human hematopoietic progenitor cells is the ligand of both E-selectin (86) and L-selectin (87), through sialylated, fucosylated binding determinants on N-glycans. Ex vivo glycan engineering of mesenchymal stromal cell CD44, derived from adipose tissue or marrow (88), confering the native CD44 glycoform tropism to bone, which is essential for stem cell-based tissue engineering and other adoptive cellular therapies (89).
Glycosphingolipids (GSLs)
GSLs, composed of a glycan linked to a lipid ceramide (Cer), include a series of neutral “core” structures and gangliosides, which carry one or several sialic acids. Lipid rafts on cell membranes, which contain GSLs and protein receptors, play key roles in hematological malignancies through regulation of retention, quiescence, mobilization, and homing of hematopoietic stem/progenitor cells (90). Several GSLs are markers of hematological malignancies; e.g., Gg3 in Hodgkin lymphoma (91) and Gb3 in Burkitt lymphoma (92), which result from precursor accumulation during incomplete GSL synthesis. Cer, a central molecule in sphingolipid metabolism, functions effectively as a tumor-suppressing lipid, inducing antiproliferative and apoptotic responses in a variety of cancer cells (93). Cer can be phosphorylated to form Cer-1-P, or glycosylated to form glucosylceramide (GlcCer). These derivatives have functions antagonistic to those of Cer, including anti-apoptotic effects (94). The normal balance between Cer and its derivatives is disrupted in hematological malignancies, particularly those displaying chemotherapeutic resistance (95). GlcCer was shown to be a marker for MDR cancers; it is present consistently in MDR cell lines and absent (or present at very low levels) in corresponding drug-sensitive cells (96). GlcCer synthase, the enzyme that attaches the Glc moiety to Cer, is co-overexpressed with P-glycoprotein in MDR leukemia cells (97, 98), and blocks drug-induced cell cycle arrest (99).
Gangliosides are often expressed aberrantly in malignant cells. They are important in cancer biology as both cell surface structures and molecules shed by malignant cells (100). Pediatric ALL patients showed enhanced levels of ST3GAL5 (GM3 synthase), which catalyzes α2-3-sialylation of lactosylceramide (LacCer), and resulting ganglioside GM3 (101). On the other hand, membrane type- and ganglioside-specific sialidase NEU3 was downregulated in these patients, and overexpression of NEU3 in ALL cells led to a significant increase of Cer, and induction of apoptosis in lymphoblasts (101). GM3 was found to induce monocytic differentiation in leukemia cells (102). In contrast, NEU3 inhibited megakaryocytic differentiation by degrading membrane sialic acids, maintain low level of GM3, and downregulating the PKC/ERKs/p38 MAPK pathway (103, 104).
Lectins
Lectins are glycan-binding proteins that are typically highly selective for specific glycan structures. They include selectins, galectins, and the molecular chaperones calreticulin. Aberrant glycosylation alters the abundance of ligands for endogenous lectins, and thereby affects multiple cellular mechanisms involving the corresponding glycans.
Selectins
SLex and SLea determinants are sialylated antigens associated with malignancy and also well-studied ligands for selectins, which are vascular cell adhesion molecules (VCAMs) belonging to the C type lectin family. There are three types of selectin: E-selectin in endothelial cells, L-selectin in lymphocytes, and P-selectin in platelets and endothelial cells. High expression of ST3 β-galactoside α2,3-sialyltransferase 6 (ST3GAL6) promotes transendothelial migration to bone marrow and survival of multiple myeloma cells, through generation of functional SLex determinants, the ligands of E-selectins on endothelial cells (105). Leukemia cells initiate interactions between Lex determinants and E-selectins by directly activating resting endothelial cells. The activated endothelial cells then induce E-selectin-mediated adhesion of a subset of leukemia cells. The adherent leukemia cells are sequestered in a quiescent state and are unaffected by chemotherapy (106). Defective homing of umbilical cord blood cells to bone marrow, resulting in delayed engraftment of cord blood transplantation, is related to low fucosylation levels of cell surface molecules responsible for binding to P- and E-selectins constitutively expressed by marrow microvessels (36). L-selectin is the key factor controlling binding of B-cell CLL cells to high endothelial venule (HEV) walls of lymph nodes in vivo, which is related to cell growth and drug resistance of CLL (107). P-selectin glycoprotein ligand 1 (PSGL-1) is the primary ligand for L- and P-selectin, and can bind E-selectin if appropriately glycosylated (108). PSGL-1 plays a crucial role in hematogenous metastasis of lymphoid cancer cells, and led to liver and spleen colonization in in vivo experiments (109). The number of circulating PSGL-1-positive microparticles released from activated or apoptotic cells is a prognostic indicator for leukemia and lymphoma patients following allogeneic stem cell transplantation (110). PSGL-1 can also be a therapeutic target; immunotherapy with anti-PSGL-1 mAbs was used in combination with mCRP blockage-induced, complement-mediated lysis of multiple myeloma cells in vivo (111).
Galectins
Galectins are a family of lectin molecules that display intracellular and extracellular effects making them useful agents in treatment of inflammation and tumor progression. They are classified as (i) “prototype” galectins (e.g., galectin-1 and−7) that have one carbohydrate recognition domain (CRD) and can undergo dimerization; (ii) “tandem-repeat” galectins (e.g., galectin-9) that contain two homologous CRDs in tandem in a single polypeptide chain; and (iii) galectin-3, which contains a CRD connected to a non-lectin N-terminal region responsible for oligomerization. Poly-LacNAc structure (elongated β1,4 GalT glycan chain) is a ligand for galectins. During hematopoiesis, galectin-glycan interactions maintain formation of microenvironmental niches, modulate acute and chronic inflammatory responses, and provide “on-and-off” signals that help control the balance between immune cell responsiveness and tolerance (112, 113).
The roles of galectin-1 and−3 in hematological malignancies have been well-studied. Overexpression of galectin-1 is correlated with activation of AP-1 pathway in malignant cells of Hodgkin lymphoma. Galectin-1 can serve as a predictive biomarker for relapsed or refractory Hodgkin lymphoma (114), and its serum levels reflect tumor burden and adverse clinical features (115). In clinical studies, neutralization of galectin-1 was an effective therapeutic strategy (114). In CLL, increased level of galectin-1 derived from myeloid cells was required for full stimulation of malignant cells through reduced threshold of B-cell receptor signaling (116). In multiple myeloma, galectin-1 displayed dual functions depending on CD45RA expression of malignant cells: it promoted viability and proliferation of CD45RA− cells through aggregation of β1-integrin, and induced growth arrest of CD45RA+ cells through inhibition of ERK phosphorylation (117). Aberrant expression of galectin-1 and/or−3 in patients with MPNs is also discovered to correlate with JAK2 mutation in these diseases and state of cell differentiation (118).
Galectin-3 is overexpressed in diffuse large B-cell lymphoma, the most common type of non-Hodgkin lymphoma in adults. Cell-surface galectin-3 binds a subset of highly glycosylated CD45, the major receptor tyrosine phosphatase in B-cells, and enhances its phosphatase activity, with consequent increase of anti-apoptotic activity (119). Bone marrow stromal cells also participate in drug resistance of AML and ALL by secreting galectin-3. Soluble galectin-3 is internalized by leukemia cells and transported to the nucleus, stimulates transcription of endogenous LGALS3 mRNA and thus activates the Wnt/β-catenin signaling pathway in leukemia cells, which is critical in cytotoxic drug resistance (120, 121). In AML, high galectin-3 expression in bone marrow is an independent unfavorable prognostic factor for overall patient survival (both M3 and non-M3) (122, 123). In CML, malignant cells in chronic phase showed galectin-3 levels much higher than those in bone marrow hematopoietic cells from control subjects or acute leukemia patients. Co-culture of five CML cell lines with bone marrow stromal cells induced galectin-3 expression and increased proliferative potential and resistance to genotoxic agents (124). In multiple myeloma, the supporting effect of galectin-3 was reduced, and malignant cells were sensitized to chemotherapeutic drugs, by treatment with two agents: galectin-3 antagonist GCS-100 and an N-terminally truncated form of galectin-3 (124).
Galectin-9, in cooperation with its highly expressed ligand TIM-3, leads to T cell exhaustion and increased survival of AML cells (125). In multiple myeloma, a recombinant protease-resistant mutant form of human galectin-9 had a strong antiproliferative effect on malignant cells, dependent in part on MAPK pathway activation (126).
Calreticulin (CALR)
Ph- MPNs are a group of diseases involving excessive bone marrow cell production, including essential thrombocytosis (ET), polycythemia vera (PV), and primary myelofibrosis (PMF). Thrombopoietin (TPO) receptor (MPL/TpoR) is the key cytokine receptor in MPN development, and activates MPL-JAK-STAT signaling in MPN stem cells (127). Incomplete MPL glycosylation was a common feature of Ph- MPNs, which was first discovered in PV patients (15). N-glycosylation on the MPL exert essential regulatory roles on MPL cell surface localization and function (128).
Calreticulin (CALR) and Calnexin(CNX), a pair of homologous C-type (calcium-dependent) lectins found in endoplasmic reticulum, act together as molecular chaperones to ensure proper folding and function of newly synthesized N-glycoproteins (129). CALR mutations are the second most frequent mutation after JAK2 in ET and PMF, nevertheless, no CALR mutations were found in patients with PV, which is specifically associated with JAK2 mutations. Moreover, CALR mutations are mutually exclusive with mutations in both JAK2 and MPL, indicating it may be responsible for the initiation of this disease (130, 131). The most frequent CALR mutations, type 1 (52-bp deletion; c.1092_1143del) and type 2 (5-bp insertion; c.1154_1155insTTGTC) accounted for 53.0% and 31.7% of all CALR mutations, respectively (130). All the mutations of CALR are insertion or deletion mutations in the last exon (exon 9) encoding the C-terminal amino acids of the protein, resulting in a novel C-terminal positively charged polypeptide tail and absence of last four amino acids (KDEL) contain the endoplasmic reticulum–retention signal (130).
CALR mutations prevent proper maturation of certain proteins (132), notably MPL in the case of Ph- MPNs. CALR mutants show alterations in MPL stability, maturation, trafficking (133), and N-glycan pattern. The majority of MPL in these mutants remains in an immature, high high-mannose form that is EndoH-sensitive (16). Pathogenic CALR mutants interacted with extracellular N-glycosylated residues of MPL via a glycan-binding site on the C-terminal tail (134, 135). Such interaction led to activation of MPL and downstream JAK2, and subsequent promotion of TPO-independent growth, accompanied by STAT5 phosphorylation. In a mouse model, engraftment of CALR mutant bone marrow cells resulted in thrombocytosis, but did not show same effect in MPL knockout mice, further confirming the MPL-dependence of calreticulin mutants (136). Moreover, CALR mutants are secreted proteins, which may be able to activate other cells, especially monocytes, to secrete inflammatory cytokines (137).
MPN patients carrying CALR mutations presented with higher platelet counts and lower hemoglobin levels than patients with mutated JAK2 (138), thus have a lower risk of thrombosis and have longer overall survival than those with a JAK2 mutation (130). CALR-positive MPNs have a more benign clinical course than the corresponding disorders associated with JAK2 or MPL mutations.
Ruxolitinib is a JAK inhibitor that ameliorates splenomegaly and constitutional symptoms associated with myelofibrosis (both PMF and post-ET/PV) (139), and is also superior to standard therapy in controlling the hematocrit, reducing the spleen volume, and improving symptoms associated with PV (140). And ruxolitinib ameliorated the thrombocytosis in CALR mutant mice and attenuated the increase in number of BM megakaryocytes and HSCs, revealing a vital role for MPL and STAT5 activation in CALR mutation-induced MPN (141).
The WHO added CALR mutations to the 2016 revision of MPN diagnostic criteria (142), while accurater and simpler prognostic models are needed to be validated for routine clinical practice (143).
Conclusion and Perspectives
Glycoconjugates are major components of animal cells and play essential roles in many physiological processes. Progress in glycobiology has led to the discovery of increasing numbers of aberrant glycans, and elucidation of the functions of certain glycans and related genes. Several serological markers currently used in the clinic are based on the detection of circulating glycoproteins or glycoconjugates with altered glycosylation. Most discoveries of aberrant glycosylation in hematological malignancy are based on genomics analysis using patient samples, mostly focusing on the abnormal expression of genes for glycosyltransferases and glycosidases. In recent years, the development of mass spectrometry-based proteomic and glycomic techniques make it possible to discover the abnormal glycoproteins and glycolipids, and cancer-related and glycosite-specific glycan structures, with the limited samples (144). Tracing back the abnormal glycan structures to the corresponding genes may sparkle this field in hematological malignancy study.
Recent studies revealed that aberrant glycosylation of hematopoietic microenvironment impact the malignant process by interacting with neoplastic cells, such as high serglycin level in multiple myeloma (74). The microenvironment of bone marrow or lymphoid tissue can be active participants to help neoplastic cells to resist drug therapy and avoid immune response. Future works are needed to understand the mechanisms of those aberrant glycan patterns not only in the aspect of cellular self-regulation, but also in the aspect of cell-cell communication within their microenvironment. The glycosylation of microenvironment could be one promising direction of hematological malignancy research in future.
The discovery of aberrant glycans and exploration of underling their mechanisms would expand their applications as diagnosis marker or therapeutic target for hematological malignancies.
For example, Roneparstat (SST0001), a 100% N-acetylated and glycol split heparin, inhibited myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis (145). As an heparanase inhibitor used in phase I clinical trial in hematological malignancies, Roneparstat presented an excellent safety profile, without clinically relevant systemic reactions, and an excellent tolerability profile (146). In other hands, glyco-engineered antibodies, such as anti-CD20 monoclonal antibody (Obinutuzumab [GA101]) may enhance the effective treatment for CD20+ B-cell non-Hodgkin's lymphoma (NHL), CD20+ follicular NHL, and CLL (147). In vitro glyco-engineering could be a powerful approach to develop monoclonal antibodies with homogenous humanized glycosylation in clinical trials of hematological malignancies.
The integrated reseach of glycomics and glycobiology with hematological malignancy will further boost the scientists to oversee the merging of biological disciplines and molecular mechanisms into physiology and disease.
Author Contributions
FG and XL conceived and designed the article frame. XP, HL, XL, and FG wrote the paper.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81672537, 81770123, and 81470294), and the Natural Science Foundation of Shaanxi Province (No. 2018JM3014), Hundred-Talent Program of Shaanxi Province. The authors are grateful to Dr. S. Anderson for English editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FL and the handling Editor declared their shared affiliation.
References
1. Hiraiwa N, Yabuta T, Yoritomi K, Hiraiwa M, Tanaka Y, Suzuki T, et al. Transactivation of the fucosyltransferase VII gene by human T-cell leukemia virus type 1 Tax through a variant cAMP-responsive element. Blood (2003) 101:3615–21. doi: 10.1182/blood-2002-07-2301
2. Ma H, Cheng L, Hao K, Li Y, Song X, Zhou H, et al. Reversal effect of ST6GAL 1 on multidrug resistance in human leukemia by regulating the PI3K/Akt pathway and the expression of P-gp and MRP1. PLoS ONE (2014) 9:e85113. doi: 10.1371/journal.pone.0085113
3. Hakomori S, Kannagi R. Glycosphingolipids as tumor-associated and differentiation markers. J Natl Cancer Inst. (1983) 71:231–51.
4. Mizuochi T, Taniguchi T, Shimizu A, Kobata A. Structural and numerical variations of the carbohydrate moiety of immunoglobulin G. J Immunol. (1982) 129:2016–20.
5. Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature (1985) 316:452–7. doi: 10.1038/316452a0
6. Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med. (1995) 1:237–43. doi: 10.1038/nm0395-237
7. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (2006) 313:670–3. doi: 10.1126/science.1129594
8. Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. (2002) 277:26733–40. doi: 10.1074/jbc.M202069200
9. Lauc G, Huffman JE, Pucic M, Zgaga L, Adamczyk B, Muzinic A, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. (2013) 9:e1003225. doi: 10.1371/journal.pgen.1003225
10. Vuillier F, Dumas G, Magnac C, Prevost MC, Lalanne AI, Oppezzo P, et al. Lower levels of surface B-cell-receptor expression in chronic lymphocytic leukemia are associated with glycosylation and folding defects of the mu and CD79a chains. Blood (2005) 105:2933–40. doi: 10.1182/blood-2004-09-3643
11. Wang H, Zhang W, Zhao J, Zhang L, Liu M, Yan G, et al. N-Glycosylation pattern of recombinant human CD82 (KAI1), a tumor-associated membrane protein. J Proteomics (2012) 75:1375–85. doi: 10.1016/j.jprot.2011.11.013
12. Marjon KD, Termini CM, Karlen KL, Saito-Reis C, Soria CE, Lidke KA, et al. Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene (2016) 35:4132–40. doi: 10.1038/onc.2015.449
13. Charlier E, Conde C, Zhang J, Deneubourg L, Di Valentin E, Rahmouni S, et al. SHIP-1 inhibits CD95/APO-1/Fas-induced apoptosis in primary T lymphocytes and T leukemic cells by promoting CD95 glycosylation independently of its phosphatase activity. Leukemia (2010) 24:821–32. doi: 10.1038/leu.2010.9
14. Shatnyeva OM, Kubarenko AV, Weber CE, Pappa A, Schwartz-Albiez R, Weber AN, et al. Modulation of the CD95-induced apoptosis: the role of CD95 N-glycosylation. PLoS ONE (2011) 6:e19927. doi: 10.1371/journal.pone.0019927
15. Moliterno AR, Spivak JL. Posttranslational processing of the thrombopoietin receptor is impaired in polycythemia vera. Blood (1999) 94:2555–61.
16. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127:1325–35. doi: 10.1182/blood-2015-11-681932
17. Yogeeswaran G, Salk PL. Metastatic potential is positively correlated with cell surface sialylation of cultured murine tumor cell lines. Science (1981) 212:1514–6. doi: 10.1126/science.7233237
18. Nasirikenari M, Veillon L, Collins CC, Azadi P, Lau JT. Remodeling of marrow hematopoietic stem and progenitor cells by non-self ST6Gal-1 sialyltransferase. J Biol Chem. (2014) 289:7178–89. doi: 10.1074/jbc.M113.508457
19. Nakano M, Saldanha R, Gobel A, Kavallaris M, Packer NH. Identification of glycan structure alterations on cell membrane proteins in desoxyepothilone B resistant leukemia cells. Mol Cell Proteomics (2011) 10:M111.009001. doi: 10.1074/mcp.M111.009001
20. Zhou H, Li Y, Liu B, Shan Y, Li Y, Zhao L, et al. Downregulation of miR-224 and let-7i contribute to cell survival and chemoresistance in chronic myeloid leukemia cells by regulating ST3GAL IV expression. Gene (2017) 626:106–18. doi: 10.1016/j.gene.2017.05.030
21. Li Y, Fu J, Ling Y, Yago T, McDaniel JM, Song J, et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc Natl Acad Sci USA. (2017) 114:8360–5. doi: 10.1073/pnas.1707662114
22. Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, et al. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc Natl Acad Sci USA. (2002) 99:10042–7. doi: 10.1073/pnas.142005099
23. Sorensen AL, Rumjantseva V, Nayeb-Hashemi S, Clausen H, Hartwig JH, Wandall HH, et al. Role of sialic acid for platelet life span: exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood (2009) 114:1645–54. doi: 10.1182/blood-2009-01-199414
24. Jansen AJ, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIbalpha metalloproteinase-mediated cleavage in mice. Blood (2012) 119:1263–73. doi: 10.1182/blood-2011-05-355628
25. Li J, Van Der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nature Commun. (2015) 6:7737. doi: 10.1038/ncomms8737
26. Angata K, Suzuki M, Fukuda M. ST8Sia II and ST8Sia IV polysialyltransferases exhibit marked differences in utilizing various acceptors containing oligosialic acid and short polysialic acid the basis for cooperative polysialylation by two enzymes. J Biol Chem. (2002) 277:36808–17. doi: 10.1074/jbc.M204632200
27. Drake PM, Nathan JK, Stock CM, Chang PV, Muench MO, Nakata D, et al. Polysialic acid, a glycan with highly restricted expression, is found on human and murine leukocytes and modulates immune responses. J Immunol. (2008) 181:6850–8. doi: 10.4049/jimmunol.181.10.6850
28. Drake PM, Stock CM, Nathan JK, Gip P, Golden KP, Weinhold B, et al. Polysialic acid governs T-cell development by regulating progenitor access to the thymus. Proc Natl Acad Sci USA. (2009) 106:11995–2000. doi: 10.1073/pnas.0905188106
29. Kiermaier E, Moussion C, Veldkamp CT, Gerardy-Schahn R, de Vries I, Williams LG, et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science (2016) 351:186–90. doi: 10.1126/science.aad0512
30. Hjortø GM, Larsen O, Steen A, Daugvilaite V, Berg C, Fares S, et al. Corrigendum: differential CCr7 targeting in dendritic cells by three naturally occurring CC-chemokines. Front Immunol. (2017) 8:89. doi: 10.3389/fimmu.2017.00089
31. Rollenhagen M, Buettner FF, Reismann M, Jirmo AC, Grove M, Behrens GM, et al. Polysialic acid on neuropilin-2 is exclusively synthesized by the polysialyltransferase ST8SiaIV and attached to mucin-type o-glycans located between the b2 and c domain. J Biol Chem. (2013) 288:22880–92. doi: 10.1074/jbc.M113.463927
32. Curreli S, Arany Z, Gerardy-Schahn R, Mann D, Stamatos NM. Polysialylated neuropilin-2 is expressed on the surface of human dendritic cells and modulates dendritic cell-T lymphocyte interactions. J Biol Chem. (2007) 282:30346–56. doi: 10.1074/jbc.M702965200
33. Rey-Gallardo A, Escribano C, Delgado-Martín C, Rodriguez-Fernández JL, Gerardy-Schahn R, Rutishauser U, et al. Polysialylated neuropilin-2 enhances human dendritic cell migration through the basic C-terminal region of CCL21. Glycobiology (2010) 20:1139–46. doi: 10.1093/glycob/cwq078
34. Rey-Gallardo A, Delgado-Martín C, Gerardy-Schahn R, Rodríguez-Fernández JL, Vega MA. Polysialic acid is required for neuropilin-2a/b-mediated control of CCL21-driven chemotaxis of mature dendritic cells and for their migration in vivo. Glycobiology (2011) 21:655–62. doi: 10.1093/glycob/cwq216
35. Miyoshi E, Moriwaki K, Nakagawa T. Biological function of fucosylation in cancer biology. J Biochem. (2008) 143:725–9. doi: 10.1093/jb/mvn011
36. Popat U, Mehta RS, Rezvani K, Fox P, Kondo K, Marin D, et al. Enforced fucosylation of cord blood hematopoietic cells accelerates neutrophil and platelet engraftment after transplantation. Blood (2015) 125:2885–92. doi: 10.1182/blood-2015-01-607366
37. Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, et al. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc Natl Acad Sci USA. (2013) 110:630–5. doi: 10.1073/pnas.1220425110
38. Sasaki H, Toda T, Furukawa T, Mawatari Y, Takaesu R, Shimizu M, et al. alpha-1,6-Fucosyltransferase (FUT8) inhibits hemoglobin production during differentiation of murine and K562 human erythroleukemia cells. J Biol Chem. (2013) 288:16839–47. doi: 10.1074/jbc.M113.459594
39. Kuila N, Nayak KB, Halder A, Agatheeswaran S, Biswas G, Biswas S, et al. Ecotropic viral integration site I regulates alpha1, 6-fucosyl transferase expression and blocks erythropoiesis in chronic myeloid leukemia. Leuk Lymphoma (2017) 58:1941–7. doi: 10.1080/10428194.2016.1266622
40. Taniguchi N, Miyoshi E, Ko JH, Ikeda Y, Ihara Y. Implication of N-acetylglucosaminyltransferases III and V in cancer: gene regulation and signaling mechanism. Biochim Biophys Acta (1999) 1455:287–300. doi: 10.1016/S0925-4439(99)00066-6
41. Yoshimura M, Nishikawa A, Ihara Y, Nishiura T, Nakao H, Kanayama Y, et al. High expression of UDP-N-acetylglucosamine: beta-D mannoside beta-1,4-N-acetylglucosaminyltransferase III (GnT-III) in chronic myelogenous leukemia in blast crisis. Int J Cancer (1995) 60:443–9. doi: 10.1002/ijc.2910600404
42. Yoshimura M, Ihara Y, Ohnishi A, Ijuhin N, Nishiura T, Kanakura Y, et al. Bisecting N-acetylglucosamine on K562 cells suppresses natural killer cytotoxicity and promotes spleen colonization. Cancer Res (1996) 56:412–8.
43. Andre S, Kozar T, Schuberth R, Unverzagt C, Kojima S, Gabius HJ. Substitutions in the N-glycan core as regulators of biorecognition: the case of core-fucose and bisecting GlcNAc moieties. Biochemistry (2007) 46:6984–95. doi: 10.1021/bi7000467
44. Brockhausen I, Schachter H, Stanley P. O-GalNAc Glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME editors. Essentials of Glycobiology, 2nd ed. New York, NY: Cold Spring Harbor (2009).
45. Aller CT, Kucuk O, Springer GF, Gilman-Sachs A. Flow cytometric analysis of T and Tn epitopes on chronic lymphocytic leukemia cells. Am J Hematol. (1996) 52:29–38. doi: 10.1002/(SICI)1096-8652(199605)52:1<29::AID-AJH5>3.0.CO;2-8
46. Libisch MG, Casas M, Chiribao M, Moreno P, Cayota A, Osinaga E, et al. GALNT11 as a new molecular marker in chronic lymphocytic leukemia. Gene (2014) 533:270–9. doi: 10.1016/j.gene.2013.09.052
47. Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, Stowell SR, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. (2008) 68:1636–46. doi: 10.1158/0008-5472.CAN-07-2345
48. Mi R, Song L, Wang Y, Ding X, Zeng J, Lehoux S, et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing tn antigen. J Biol Chem. (2012) 287:41523–33. doi: 10.1074/jbc.M112.371989
49. Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci USA. (2002) 99:16613–8. doi: 10.1073/pnas.262438199
50. Yi B, Zhang Z, Zhang M, Schwartz-Albiez R, Cao Y. CD176 antiserum treatment leads to a therapeutic response in a murine model of leukemia. Oncol Rep. (2013) 30:1841–7. doi: 10.3892/or.2013.2639
51. Sedlik C, Heitzmann A, Viel S, Ait Sarkouh R, Batisse C, Schmidt F, et al. Effective antitumor therapy based on a novel antibody-drug conjugate targeting the Tn carbohydrate antigen. Oncoimmunology (2016) 5:e1171434. doi: 10.1080/2162402X.2016.1171434
52. Cloosen S, Gratama J, van Leeuwen EB, Senden-Gijsbers BL, Oving EB, von Mensdorff-Pouilly S, et al. Cancer specific Mucin-1 glycoforms are expressed on multiple myeloma. Br J Haematol. (2006) 135:513–6. doi: 10.1111/j.1365-2141.2006.06331.x
53. Kawano T, Ahmad R, Nogi H, Agata N, Anderson K, Kufe D. MUC1 oncoprotein promotes growth and survival of human multiple myeloma cells. Int J Oncol. (2008) 33:153–9. doi: 10.3892/ijo.33.1.153
54. Liu S, Yin L, Stroopinsky D, Rajabi H, Puissant A, Stegmaier K, et al. MUC1-C oncoprotein promotes FLT3 receptor activation in acute myeloid leukemia cells. Blood (2014) 123:734–42. doi: 10.1182/blood-2013-04-493858
55. Bar-Natan M, Stroopinsky D, Luptakova K, Coll MD, Apel A, Rajabi H, et al. Bone marrow stroma protects myeloma cells from cytotoxic damage via induction of the oncoprotein MUC1. Br J Haematol. (2017) 176:929–38. doi: 10.1111/bjh.14493
56. Yin L, Tagde A, Gali R, Tai YT, Hideshima T, Anderson K, et al. MUC1-C is a target in lenalidomide resistant multiple myeloma. Br J Haematol. (2017) 178:914–26. doi: 10.1111/bjh.14801
57. Brossart P, Schneider A, Dill P, Schammann T, Grunebach F, Wirths S, et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-Lymphocytes. Cancer Res. (2001) 61:6846–50.
58. Rassidakis GZ, Goy A, Medeiros LJ, Jiang Y, Thomaides A, Remache Y, et al. Prognostic significance of MUC-1 expression in systemic anaplastic large cell lymphoma. Clin Cancer Res. (2003) 9:2213–20.
59. Jain S, Stroopinsky D, Yin L, Rosenblatt J, Alam M, Bhargava P, et al. Mucin 1 is a potential therapeutic target in cutaneous T-cell lymphoma. Blood (2015) 126:354–62. doi: 10.1182/blood-2015-02-628149
60. Mukherjee P, Tinder TL, Basu GD, Gendler SJ. MUC1 (CD227) interacts with lck tyrosine kinase in Jurkat lymphoma cells and normal T cells. J Leukoc Biol. (2005) 77:90–9. doi: 10.1189/jlb.0604333
61. Kamemura K, Hayes BK, Comer FI, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins - Alternative glycosylation/phosphorylation of Thr-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J Biol Chem. (2002) 277:19229–35. doi: 10.1074/jbc.M201729200
62. Freund P, Kerenyi MA, Hager M, Wagner T, Wingelhofer B, Pham HT, et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia (2017) 31:2132–42. doi: 10.1038/leu.2017.4
63. Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia (2010) 24:1588–98. doi: 10.1038/leu.2010.152
64. Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem. (2005) 280:32944–56. doi: 10.1074/jbc.M503396200
65. Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature (2013b) 493:561–4. doi: 10.1038/nature11742
66. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia (2014) 28:485–96. doi: 10.1038/leu.2013.337
67. Inoue D, Fujino T, Sheridan P, Zhang YZ, Nagase R, Horikawa S, et al. A novel ASXL1–OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia (2018) 32:1327–37. doi: 10.1038/s41375-018-0083-3
68. Teng Y, Liu QH, Ma H, Liu F, Han ZG, Wang YX, et al. Cloning, expression and characterization of a novel human CAP10-like gene hCLP46 ftom CD34+stem/progenitor cells. Gene (2006) 371:7–15. doi: 10.1016/j.gene.2005.08.027
69. Wang Y, Chang N, Zhang T, Liu H, Ma W, Chu Q, et al. Overexpression of human CAP10-like protein 46 KD in T-acute lymphoblastic leukemia and acute myelogenous leukemia. Genet Test Mol Biomark. (2010) 14:127–33. doi: 10.1089/gtmb.2009.0145
70. Ma W, Du J, Chu Q, Wang Y, Liu L, Song M, et al. hCLP46 regulates U937 cell proliferation via Notch signaling pathway. Biochem Biophys Res Commun. (2011) 408:84–8. doi: 10.1016/j.bbrc.2011.03.124
71. Chu Q, Liu L, Wang W. Overexpression of hCLP46 enhances Notch activation and regulates cell proliferation in a cell type-dependent manner. Cell Prolif. (2013) 46:254–62. doi: 10.1111/cpr.12037
72. Yao D, Huang YS, Huang XR, Wang WH, Yan QJ, Wei LB, et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood (2011) 117:5652–62. doi: 10.1182/blood-2010-12-326074
73. Skliris A, Labropoulou VT, Papachristou DJ, Aletras A, Karamanos NK, Theocharis AD. Cell-surface serglycin promotes adhesion of myeloma cells to collagen type I and affects the expression of matrix metalloproteinases. FEBS J. (2013) 280:2342–52. doi: 10.1111/febs.12179
74. Purushothaman A, Toole BP. Serglycin proteoglycan is required for multiple myeloma cell adhesion, in vivo growth, and vascularization. J Biol Chem. (2014) 289:5499–509. doi: 10.1074/jbc.M113.532143
75. Beyer-Sehlmeyer G, Hiddemann W, Wormann B, Bertram J. Suppressive subtractive hybridisation reveals differential expression of serglycin, sorcin, bone marrow proteoglycan and prostate-tumour-inducing gene I (PTI-1) in drug-resistant and sensitive tumour cell lines of haematopoetic origin. Eur J Cancer (1999) 35:1735–42. doi: 10.1016/S0959-8049(99)00202-6
76. Niemann CU, Kjeldsen L, Ralfkiaer E, Jensen MK, Borregaard N. Serglycin proteoglycan in hematologic malignancies: a marker of acute myeloid leukemia. Leukemia (2007) 21:2406–10. doi: 10.1038/sj.leu.2404975
77. Toyama-Sorimachi N, Kitamura F, Habuchi H, Tobita Y, Kimata K, Miyasaka M. Widespread expression of chondroitin sulfate-type serglycins with CD44 binding ability in hematopoietic cells. J Biol Chem. (1997) 272:26714–9. doi: 10.1074/jbc.272.42.26714
78. Dhodapkar MV, Abe E, Theus A, Lacy M, Langford JK, Barlogie B, et al. Syndecan-1 is a multifunctional regulator of myeloma pathobiology: control of tumor cell survival, growth, and bone cell differentiation. Blood (1998) 91:2679–88.
79. Derksen PW, Keehnen RM, Evers LM, Van Oers MH, Spaargaren M, Pals ST. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood (2002) 99:1405–10. doi: 10.1182/blood.V99.4.1405
80. Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, et al. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. (2003) 63:8749–56.
81. Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, et al. Heparanase enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood (2010) 115:2449–57. doi: 10.1182/blood-2009-07-234757
82. Delcommenne M, Klingemann HG. Detection and characterization of syndecan-1-associated heparan sulfate 6-O-sulfated motifs overexpressed in multiple myeloma cells using single chain antibody variable fragments. Hum Antibodies (2012) 21:29–40. doi: 10.3233/HAB-2012-0259
83. Banerji S, Wright AJ, Noble M, Mahoney DJ, Campbell ID, Day AJ, et al. Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat Struct Mol Biol. (2007) 14:234–9. doi: 10.1038/nsmb1201
84. Gee K, Kozlowski M, Kumar A. Tumor necrosis factor-alpha induces functionally active hyaluronan-adhesive CD44 by activating sialidase through p38 mitogen-activated protein kinase in lipopolysaccharide-stimulated human monocytic cells. J Biol Chem. (2003) 278:37275–87. doi: 10.1074/jbc.M302309200
85. Avigdor A, Goichberg P, Shivtiel S, Dar A, Peled A, Samira S, et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood (2004) 103:2981–9. doi: 10.1182/blood-2003-10-3611
86. Dimitroff CJ, Lee JY, Rafii S, Fuhlbrigge RC, Sackstein R. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J Cell Biol. (2001) 153:1277–86. doi: 10.1083/jcb.153.6.1277
87. Dimitroff CJ, Lee JY, Fuhlbrigge RC, Sackstein R. A distinct glycoform of CD44 is an L-selectin ligand on human hematopoietic cells. Proc Natl Acad Sci USA. (2000) 97:13841–6. doi: 10.1073/pnas.250484797
88. Pachon-Pena G, Donnelly C, Ruiz-Canada C, Katz A, Fernandez-Veledo S, Vendrell J, et al. A glycovariant of human CD44 Is characteristically expressed on human mesenchymal stem cells. Stem Cells (2017) 35:1080–92. doi: 10.1002/stem.2549
89. Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. (2008) 14:181–7. doi: 10.1038/nm1703
90. Ratajczak MZ, Adamiak M. Membrane lipid rafts, master regulators of hematopoietic stem cell retention in bone marrow and their trafficking. Leukemia (2015) 29:1452–7. doi: 10.1038/leu.2015.66
91. Kniep B, Monner DA, Burrichter H, Diehl V, Muhlradt PF. Gangliotriaosylceramide (asialo GM2), a glycosphingolipid marker for cell lines derived from patients with Hodgkin's disease. J Immunol. (1983) 131:1591–4.
92. Mangeney M, Lingwood CA, Taga S, Caillou B, Tursz T, Wiels J. Apoptosis induced in Burkitt's lymphoma cells via Gb3/CD77, a glycolipid antigen. Cancer Res. (1993) 53:5314–9.
93. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer (2004) 4:604–16. doi: 10.1038/nrc1411
94. Ekiz HA, Baran Y. Therapeutic applications of bioactive sphingolipids in hematological malignancies. Int J Cancer (2010) 127:1497–506. doi: 10.1002/ijc.25478
95. Itoh M, Kitano T, Watanabe M, Kondo T, Yabu T, Taguchi Y, et al. Possible role of ceramide as an indicator of chemoresistance: decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin Cancer Res. (2003) 9:415–23.
96. Lucci A, Cho WI, Han TY, Giuliano AE, Morton DL, Cabot MC. Glucosylceramide: a marker for multiple-drug resistant cancers. Anticancer Res. (1998) 18:475–80.
97. Turzanski J, Grundy M, Shang SL, Russell N, Pallis M. P-glycoprotein is implicated in the inhibition of ceramide-induced apoptosis in TF-1 acute myeloid leukemia cells by modulation of the glucosylceramide synthase pathway. Exp Hematol. (2005) 33:62–72. doi: 10.1016/j.exphem.2004.10.005
98. Xie P, Shen YF, Shi YP, Ge SM, Gu ZH, Wang J, et al. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk Res. (2008) 32:475–80. doi: 10.1016/j.leukres.2007.07.006
99. Rani CS, Abe A, Chang Y, Rosenzweig N, Saltiel AR, Radin NS, et al. Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem. (1995) 270:2859–67. doi: 10.1074/jbc.270.6.2859
100. Hakomori S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res. (1989) 52:257–331. doi: 10.1016/S0065-230X(08)60215-8
101. Mondal S, Chandra S, Mandal C. Elevated mRNA level of hST6Gal I and hST3Gal V positively correlates with the high risk of pediatric acute leukemia. Leuk Res. (2010) 34:463–70. doi: 10.1016/j.leukres.2009.07.042
102. Nakamura M, Ogino H, Nojiri H, Kitagawa S, Saito M. Characteristic incorporation of ganglioside GM3, which induces monocytic differentiation in human myelogenous leukemia HL-60 cells. Biochem Biophys Res Commun. (1989) 161:782–9. doi: 10.1016/0006-291X(89)92668-5
103. Jin UH, Ha KT, Kim KW, Chang YC, Lee YC, Ko JH, et al. Membrane type sialidase inhibits the megakaryocytic differentiation of human leukemia K562 cells. Biochim Biophys Acta (2008) 1780:757–63. doi: 10.1016/j.bbagen.2008.01.019
104. Tringali C, Lupo B, Cirillo F, Papini N, Anastasia L, Lamorte G, et al. Silencing of membrane-associated sialidase Neu3 diminishes apoptosis resistance and triggers megakaryocytic differentiation of chronic myeloid leukemic cells K562 through the increase of ganglioside GM3. Cell Death Differ. (2009) 16:164–74. doi: 10.1038/cdd.2008.141
105. Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood (2014) 124:1765–76. doi: 10.1182/blood-2014-03-560862
106. Pezeshkian B, Donnelly C, Tamburo K, Geddes T, Madlambayan GJ. Leukemia mediated endothelial cell activation modulates leukemia cell susceptibility to chemotherapy through a positive feedback loop mechanism. PLoS ONE (2013) 8:e60823. doi: 10.1371/journal.pone.0060823
107. Lafouresse F, Bellard E, Laurent C, Moussion C, Fournie JJ, Ysebaert L, et al. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood (2015) 126:1336–45. doi: 10.1182/blood-2015-02-626291
108. Moore KL. Structure and function of P-selectin glycoprotein ligand-1. Leuk Lymphoma (1998) 29:1–15. doi: 10.3109/10428199809058377
109. Raes G, Ghassabeh GH, Brys L, Mpofu N, Verschueren H, Vanhecke D, et al. The metastatic T-cell hybridoma antigen/P-selectin glycoprotein ligand 1 is required for hematogenous metastasis of lymphomas. Int J Cancer (2007) 121:2646–52. doi: 10.1002/ijc.23067
110. Trummer A, De Rop C, Stadler M, Ganser A, Buchholz S. P-selectin glycoprotein ligand-1 positive microparticles in allogeneic stem cell transplantation of hematologic malignancies. Exp Hematol. (2011) 39:1047–55. doi: 10.1016/j.exphem.2011.08.007
111. Tripodo C, Florena AM, Macor P, Di Bernardo A, Porcasi R, Guarnotta C, et al. P-selectin glycoprotein ligand-1 as a potential target for humoral immunotherapy of multiple myeloma. Curr Cancer Drug Tar. (2009) 9:617–25. doi: 10.2174/156800909789056971
112. Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. Functions of cell surface galectin-glycoprotein lattices. Curr Opin Struct Biol. (2007) 17:513–20. doi: 10.1016/j.sbi.2007.09.002
113. Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann N Y Acad Sci. (2010) 1183:158–82. doi: 10.1111/j.1749-6632.2009.05131.x
114. Kamper P, Ludvigsen M, Bendix K, Hamilton-Dutoit S, Rabinovich GA, Moller MB, et al. Proteomic analysis identifies galectin-1 as a predictive biomarker for relapsed/refractory disease in classical Hodgkin lymphoma. Blood (2011) 117:6638–49. doi: 10.1182/blood-2010-12-327346
115. Ouyang J, Plutschow A, Pogge von Strandmann E, Reiners KS, Ponader S, Rabinovich GA, et al. Galectin-1 serum levels reflect tumor burden and adverse clinical features in classical Hodgkin lymphoma. Blood (2013) 121:3431–3. doi: 10.1182/blood-2012-12-474569
116. Croci DO, Morande PE, Dergan-Dylon S, Borge M, Toscano MA, Stupirski JC, et al. Nurse-like cells control the activity of chronic lymphocytic leukemia B cells via galectin-1. Leukemia (2013) 27:1413–6. doi: 10.1038/leu.2012.315
117. Abroun S, Otsuyama K, Shamsasenjan K, Islam A, Amin J, Iqbal MS, et al. Galectin-1 supports the survival of CD45RA(-) primary myeloma cells in vitro. Br J Haematol. (2008) 142:754–65. doi: 10.1111/j.1365-2141.2008.07252.x
118. Moura LG, Tognon R, Nunes NS, Rodrigues LC, Ferreira AF, Kashima S, et al. Different expression patterns of LGALS1 and LGALS3 in polycythemia vera, essential thrombocythemia and primary myelofibrosis. J Clin Pathol. (2016) 69:926–9. doi: 10.1136/jclinpath-2016-203948
119. Clark MC, Pang M, Hsu DK, Liu FT, De Vos S, Gascoyne RD, et al. Galectin-3 binds to CD45 on diffuse large B cell lymphoma cells to regulate susceptibility to cell death. Blood (2012) 120:4635–44. doi: 10.1182/blood-2012-06-438234
120. Hu K, Gu Y, Lou L, Liu L, Hu Y, Wang B, et al. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/β-catenin signaling pathway. J Hematol Oncol. (2015) 8:1. doi: 10.1186/s13045-014-0099-8
121. Fei F, Joo EJ, Tarighat SS, Schiffer I, Paz H, Fabbri M, et al. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget (2015) 6:11378. doi: 10.18632/oncotarget.3409
122. Cheng CL, Hou HA, Lee MC, Liu CY, Jhuang JY, Lai YJ, et al. Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood (2015) 121:3172–80. doi: 10.1182/blood-2012-07-443762
123. Gao N, Wang XX, Sun JR, Yu WZ, Li XZ. Clinical impact of galectin-3 in newly diagnosed t (15; 17)(q22; q21)/PML-RARa acute promyelocytic leukemia treated with all-trans retinoic acid and arsenic trioxide-based regimens. Ann Hematol. (2017) 96:711–8. doi: 10.1007/s00277-017-2948-3
124. Yamamoto-Sugitani M, Kuroda J, Ashihara E, Nagoshi H, Kobayashi T, Matsumoto Y, et al. Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc Natl Acad Sci USA. (2011) 108:17468–73. doi: 10.1073/pnas.1111138108
125. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
126. Kobayashi T, Kuroda J, Ashihara E, Oomizu S, Terui Y, Taniyama A, et al. Galectin-9 exhibits anti-myeloma activity through JNK and p38 MAP kinase pathways. Leukemia (2010) 24:843–50. doi: 10.1038/leu.2010.25
127. Mead AJ, Mullally A. Myeloproliferative neoplasm stem cells. Blood (2017) 129:1607–16. doi: 10.1182/blood-2016-10-696005
128. Albu RI, Constantinescu SN. Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels. Front Endocrinol. (2011) 2:71. doi: 10.3389/fendo.2011.00071
129. Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. (2008) 283:10221–5. doi: 10.1074/jbc.R700048200
130. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. (2013) 369:2379–90. doi: 10.1056/NEJMoa1311347
131. Vainchenker W, Constantinescu SN, Plo I. Recent advances in understanding myelofibrosis and essential thrombocythemia. F1000Research (2016) 5:700. doi: 10.12688/f1000research.8081.1
132. Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, et al. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell (2004) 13:125–35. doi: 10.1016/S1097-2765(03)00494-5
133. Cleyrat C, Darehshouri A, Steinkamp MP, Vilaine M, Boassa D, Ellisman MH, et al. Mpl traffics to the cell surface through conventional and unconventional routes. Traffic (2014) 15:961–82. doi: 10.1111/tra.12185
134. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood (2016) 127:1307–16. doi: 10.1182/blood-2015-09-671172
135. Elf S, Abdelfattah NS, Chen E, Perales-Paton J, Rosen EA, Ko A, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. (2016) 6:368–81. doi: 10.1158/2159-8290.CD-15-1434
136. Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood (2016) 127:1317–24. doi: 10.1182/blood-2015-11-679571
137. Garbati MR, Welgan CA, Landefeld SH, Newell LF, Agarwal A, Dunlap JB, et al. Mutant calreticulin-expressing cells induce monocyte hyperreactivity through a paracrine mechanism. Am J Hematol. (2016) 91:211–9. doi: 10.1002/ajh.24245
138. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
139. Guglielmelli P, Rotunno G, Bogani C, Mannarelli C, Giunti L, Provenzano A, et al. Ruxolitinib is an effective treatment for CALR-positive patients with myelofibrosis. Br J Haematol. (2016) 173:938–40. doi: 10.1111/bjh.13644
140. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. New Eng J Med. (2015) 372:426–35. doi: 10.1056/NEJMoa1409002
141. Harrison C, Kiladjian JJ, Al-Ali, HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. x JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. New Eng J Med. (2015) 366, 787–98. doi: 10.1056/NEJMoa1110556
142. Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi AM, Tefferi A. The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev. (2016) 30:453–9. doi: 10.1016/j.blre.2016.06.001
143. Rozovski U, Verstovsek S, Manshouri T, Dembitz V, Bozinovic K, Newberry K, et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematol. (2016) 102:79–84. doi: 10.3324/haematol.2016.149765
144. Chen JY, Huang HH, Yu SY, Wu SJ, Kannagi R, Khoo KH. Concerted mass spectrometry-based glycomic approach for precision mapping of sulfo sialylated N-glycans on human peripheral blood mononuclear cells and lymphocytes. Glycobiology (2017) 28:9–20. doi: 10.1093/glycob/cwx091
145. Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. (2011) 17:1382–93. doi: 10.1158/1078-0432.CCR-10-2476
146. Galli M, Chatterjee M, Grasso M, Specchia G, Magen H., Einsele H, et al. Phase I study of the heparanase inhibitor Roneparstat: an innovative approach for multiple myeloma therapy. Haematologica (2018) doi: 10.3324/haematol.2017.182865. [Epub ahead of print].
Keywords: glycan, hematological malignancies, N-glycosylation, O-glycosylation, glycosaminoglycan, glycosphingolipid, lectin
Citation: Pang X, Li H, Guan F and Li X (2018) Multiple Roles of Glycans in Hematological Malignancies. Front. Oncol. 8:364. doi: 10.3389/fonc.2018.00364
Received: 17 July 2018; Accepted: 17 August 2018;
Published: 06 September 2018.
Edited by:
Alessandro Isidori, Ematologia e Centro Trapianti, Ospedali Riuniti Marche Nord, ItalyReviewed by:
Sara Galimberti, Università degli Studi di Pisa, ItalyFederica Loscocco, Ematologia e Centro Trapianti, Ospedali Riuniti Marche Nord, Italy
Copyright © 2018 Pang, Li, Guan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Guan, Z3VhbmZlbmdAbnd1LmVkdS5jbg==
Xiang Li, eGlhbmdsaUBud3UuZWR1LmNu