 Ramon Bartrons1*
Ramon Bartrons1* Helga Simon-Molas1
Helga Simon-Molas1 Ana Rodríguez-García1
Ana Rodríguez-García1 Esther Castaño2
Esther Castaño2 Àurea Navarro-Sabaté1
Àurea Navarro-Sabaté1 Anna Manzano1
Anna Manzano1 Ubaldo E. Martinez-Outschoorn3
Ubaldo E. Martinez-Outschoorn3- 1Unitat de Bioquímica, Departament de Ciències Fisiològiques, Universitat de Barcelona, Institut d'Investigació Biomèdica de Bellvitge (IDIBELL), Catalunya, Spain
- 2Centres Científics i Tecnològics, Universitat de Barcelona, Catalunya, Spain
- 3Department of Medical Oncology, Thomas Jefferson University, Philadelphia, PA, United States
For a long time, pioneers in the field of cancer cell metabolism, such as Otto Warburg, have focused on the idea that tumor cells maintain high glycolytic rates even with adequate oxygen supply, in what is known as aerobic glycolysis or the Warburg effect. Recent studies have reported a more complex situation, where the tumor ecosystem plays a more critical role in cancer progression. Cancer cells display extraordinary plasticity in adapting to changes in their tumor microenvironment, developing strategies to survive and proliferate. The proliferation of cancer cells needs a high rate of energy and metabolic substrates for biosynthesis of biomolecules. These requirements are met by the metabolic reprogramming of cancer cells and others present in the tumor microenvironment, which is essential for tumor survival and spread. Metabolic reprogramming involves a complex interplay between oncogenes, tumor suppressors, growth factors and local factors in the tumor microenvironment. These factors can induce overexpression and increased activity of glycolytic isoenzymes and proteins in stromal and cancer cells which are different from those expressed in normal cells. The fructose-6-phosphate/fructose-1,6-bisphosphate cycle, catalyzed by 6-phosphofructo-1-kinase/fructose 1,6-bisphosphatase (PFK1/FBPase1) isoenzymes, plays a key role in controlling glycolytic rates. PFK1/FBpase1 activities are allosterically regulated by fructose-2,6-bisphosphate, the product of the enzymatic activity of the dual kinase/phosphatase family of enzymes: 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase (PFKFB1-4) and TP53-induced glycolysis and apoptosis regulator (TIGAR), which show increased expression in a significant number of tumor types. In this review, the function of these isoenzymes in the regulation of metabolism, as well as the regulatory factors modulating their expression and activity in the tumor ecosystem are discussed. Targeting these isoenzymes, either directly or by inhibiting their activating factors, could be a promising approach for treating cancers.
Introduction
Otto Warburg, using the Warburg manometer to measure the oxygen consumption in cells, demonstrated that tumor cells showed rapid and intense glycolysis, in which glucose was oxidized into lactate, despite the presence of abundant oxygen (1). This “Warburg effect” is characteristic of proliferating and transformed cells. Warburg postulated that cancer was a result of defects in mitochondrial respiration, which forced the cell to adopt an anaerobic form of energy generation, glycolysis (2). There are different molecular mechanisms that can explain the Warburg effect, including: mitochondrial malfunction (3), oncogenic alterations (4–7), as well as responses to adapt to the tumor microenvironment (TME) (8–10). Even though not all cancer cells have high glycolytic flux (11), the Warburg effect has been observed in most tumor cells and represents an adaptation to support biomass production (10).
At the same time as Otto Warburg, Herbert G. Crabtree studied the heterogeneity of glycolysis in tumors, describing that the magnitude and relationships between respiratory and glycolytic processes were a common feature of uncontrolled proliferation and not specific to malignant tissues (12). He observed considerable variability between respiratory and glycolytic metabolism among different tumors (13). Moreover, he found that glycolytic activity significantly affected the respiration capacity of tumor tissues being respiration and oxidative phosphorylation inhibited by glucose (13). This observation, referred as the “Crabtree effect,” might be advantageous to cancer cells, allowing them to adjust their metabolism to heterogeneous microenvironments in malignant cells, through the glucose-dependent accumulation of essential metabolites, such as serine, phosphoribosyl-pyrophosphate, and glycerol-3-phosphate, which can trigger mitogenic events (11, 14, 15). The Crabtree effect on tumor cells can be eliminated by adding an excess of inorganic phosphate (Pi) (15, 16).
The resurgence of the role of bioenergetics in cancer began in the early 1990s when studies using 2-deoxy-D-glucose (2-DG) in positron emission tomography (PET) showed that most tumors displayed increased glucose uptake in about an order of magnitude higher than that of normal tissue (8). The increased glucose uptake largely depends on the rate of glucose phosphorylation by hexokinases and the upregulation of glucose transporters Glut1 and Glut3 and less often Glut4 (17). More than 90% of primary and metastatic tumors have high glucose uptake, which directly correlates with tumor aggressiveness (8, 18).
For a long time, studies on cancer cell metabolism had focused on investigating a single cell type. However, recent studies have reported a more complex situation in which metabolic heterogeneity within tumors plays a critical role in cancer progression (19–21). Cancer cells display extraordinary plasticity in adapting to changes in their TME, developing strategies to survive and proliferate. Interactions between tumor cells and non-malignant cells of the TME influence cancer initiation and progression as well as patient prognosis (22–24). Local differences in the TME, including acidity and hypoxia, affect cancer cells progression. If located close to blood vessels, cancer cells can proliferate at a higher rate because of the abundant supply of nutrients, growth factors and oxygen. By contrast, if the supply of nutrients and oxygen is reduced, cancer cells rely more on glycolysis, forcing them to develop strategies for survival and proliferation. Thus, it is not surprising that these cells in metabolically deprived environments are usually chemoresistant and have higher malignancy grades (25). Stromal cells, especially cancer-associated fibroblasts (CAFs), influence the homeostasis of the TME. The interactions between TME and cancer cells strongly affect tumor metabolism and growth (26–29). A “two-compartment” model, referred to as the “reversed Warburg effect,” has been proposed as a new perspective of tumor metabolism in which tumor stroma and adjacent host tissues are catabolic, while cancer cells are anabolic (19–21, 28–30). Energy is transferred from the catabolic to the anabolic compartment via the sharing of nutrients, which promotes tumor growth. Although lactate is generally considered a waste product, it has been demonstrated to fuel oxidative metabolism in cancer cells, favoring a symbiosis between glycolytic and oxidative tumor cells (19–21, 28–30). Metabolic reprogramming of cancer and stromal cells involves a complex interplay between oncogenes, tumor suppressors, growth factors and local factors from the TME. These factors can induce the overexpression and increased activity of isoenzymes and other proteins in cancer cells that are different from those found in non-malignant cells (30).
Reprogramming the Glycolytic Phenotype of Cancer Cells
Most tumor cells have a markedly modified energy metabolism in comparison to differentiated cells. Their metabolism, previously based on respiration, changes to another eminently glycolytic, recognized as the glycolytic phenotype (7, 31–34). Several glycolytic genes are usually overexpressed in many tumors and give place to this phenotype (35). This occurs because tumor cells reprogram cellular metabolism increasing the transcription and/or alternative splicing of glycolytic genes induced by the Hypoxia Inducible Factor-1α (HIF-1α), oncogenes and inactivated tumor suppressor genes and distinct growth factors (34, 36–39). The glycolytic phenotype is a distinctive characteristic of tumor cells (7, 31, 33), providing advantages to proliferating cells and allowing them to metabolize the most plentiful nutrient, glucose, to generate energy and anabolic precursors. Even though the yield of ATP per glucose molecule consumed is low, the percentage of cellular ATP generated from glycolysis can surpass that produced from oxidative phosphorylation if the glycolytic flux is high enough (11, 32, 40). Furthermore, glucose metabolism provides intermediates that are needed for biosynthetic pathways, such as ribose sugars for nucleotide synthesis and hexose sugar derivatives, glycerol and citrate for lipid production, non-essential amino acids (serine, glycine, and cysteine) and NADPH. Therefore, the Warburg effect has a positive impact on bioenergetics, biosynthesis and detoxification of reactive oxygen species (ROS) (10).
There are several checkpoints regulating the acquisition of the glycolytic phenotype. The first point of commitment of glucose-6-phosphate to the glycolytic pathway involves the fructose-6-phosphate/fructose-1,6-bisphosphate cycle (Fru-6-P/Fru-1,6-P2) (41) (Figure 1). The following paragraphs describe the specific isoenzymes regulating this substrate cycle, the main properties of which are summarized in Table 1.
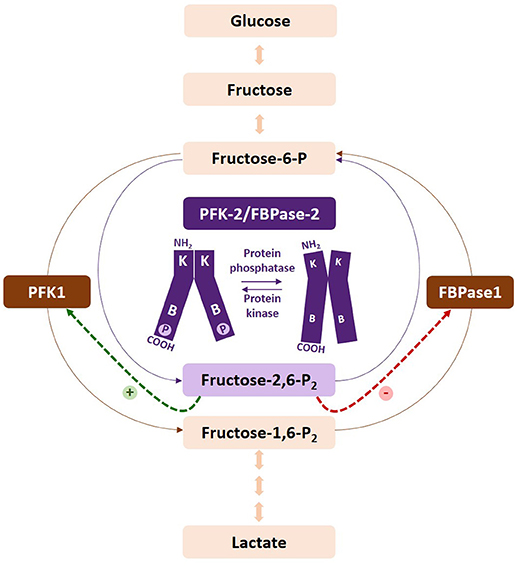
Figure 1. Regulation of the fructose-6-P/fructose-1,6-P2 substrate cycle. Fructose-2,6-P2 is synthesized and degraded by PFK-2/FBPase-2, respectively. Increased levels of the metabolite activate PFK-1 and inhibit FBPase1, increasing the glycolytic flux. Phosphorylation of the PFKFB2 and PFKFB3 isoenzymes increases their kinase activity. The kinases and phosphatases responsible for their regulation vary according to the isoenzyme. PFKFB1 is not represented by this figure, as phosphorylation increases the bisphosphatase activity of the enzyme. At present, PFKFB4 has not been found to be regulated by phosphorylation.
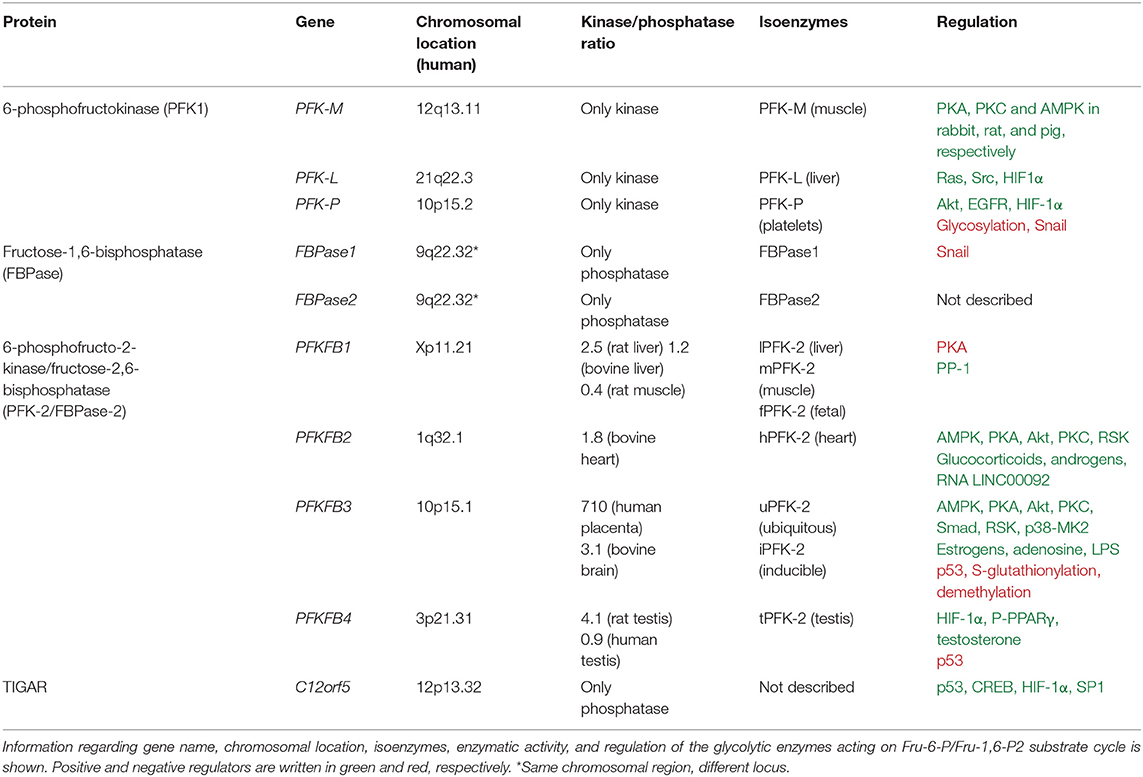
Table 1. Properties of the glycolytic enzymes acting on Fru-6-P/Fru-1,6-P2 substrate cycle.
6-Phosphofructo-1-Kinase (PFK1)
PFK1 is a tetrameric protein, with three genes encoding the PFK-M (muscle), PFK-L (liver), and PFK-P (platelet) human isoforms, with a molecular weight of their subunits of 82.5, 77, and 86.5 kDa, respectively (42). The different isoforms can form homo- or hetero-tetramers depending on the cell type (38, 42). Lactate induces the dissociation of the tetramers into dimers, reducing enzymatic activity and providing negative feedback in the regulation of the glycolytic rate (43). PFK1 isozymes can be phosphorylated by different kinases but this does not produce significant catalytic effects (41, 44, 45), although the covalent modification can affect their association with other proteins (46–48). Localization to the actin filaments of the cytoskeleton has been shown to increase PFK-M activity (49), which can also bind to and modulate protein phosphatase-1 (50). PFK-L, but not PFK-M and PFK-P, assembles into filaments, with fructose-6-P binding essential for their formation (51). These filaments have been observed to localize in the plasma membrane, where they promote the vessel sprouting of endothelial cells (52). PFK-L forms clusters in human cancer cells and colocalizes with other rate-limiting enzymes in both glycolysis and gluconeogenesis, supporting the formation of multienzyme metabolic complexes for glucose metabolism, integrating PFK-L, FBPase1, the pyruvate kinase isoenzyme M2 (PKM2) and phosphoenolpyruvate carboxykinase 1 (PEPCK1), among others, and forming the “glycosome” (53). PFK1 is also regulated by different allosteric effectors, which provides control of the glycolytic flux and coordination of glucose entry into glycolysis. It is a tightly-regulated enzyme and its kinetic and regulatory characteristics depend on the composition of its different subunits. Its regulation involves a series of negative (citrate, ATP, phosphoenolpyruvate, and [H+]) and positive effectors (Fru-2,6-P2, AMP, Fru-1,6-P2, Glu-1,6-P2, NH4+, and Pi) (41, 42), which coordinate its response to the energy status of the cell. PFK1 activity increases in response to proliferation signals alongside elevated glycolysis in proliferating and cancer cells (54), although there are exceptions where its activity does not increase (44, 54). The main isoenzymes expressed in tumor cells are PFK-P and PFK-L (54, 55). In human lymphomas and gliomas, PFK1 is less sensitive to inhibition by ATP and citrate, and more sensitive to activation by Fru-2,6-P2 and AMP (44, 56). PFK1 activity is induced by HIF-1α (57) or the overexpression of the oncogenes RAS and SRC (58). PFK-L and PFK-P can be glycosylated in response to hypoxia, which inhibits PFK1 activity and redirects the glucose flux toward the pentose phosphate pathway (PPP), providing pentose sugars for nucleotide synthesis and NADPH to combat oxidative stress (59). Similarly, the transcription repressor Snail reprograms glucose metabolism by repressing PFK-P, suppressing lactate production and amino acids biosynthesis, while promoting cancer cell survival under metabolic stress (60). Akt can bind to and phosphorylate PFK-P at S386, which inhibits the binding of TRIM21 E3 ligase to PFK-P and its subsequent polyubiquitylation and degradation. This has been shown to increase PFK-P activity, glycolysis, cell proliferation and brain tumor growth (48). Recently, EGFR activation has been reported to elicit lysine acetyl-transferase-5-mediated PFK-P acetylation and subsequent translocation of PFK-P to the plasma membrane, where EGFR phosphorylates PFK-P at Y64. Phosphorylated PFK-P binds to the N-terminal domain of p85α and promotes the activation of PI3K and Akt, leading to increased PFKFB2 activity, Fru-2,6-P2 synthesis and PFK1 activation, which in turn promote cell proliferation and tumorigenesis (61).
Fructose 1,6-Bisphosphatase (FBPase1)
FBPase1, a rate-limiting enzyme that catalyzes the opposite reaction to that of PFK1 in the Fru-6-P/Fru-1,6-P2 cycle, exists as two isoenzymes in mammals: FBPase1 and FBPase2. Both isozymes are inhibited by Fru-2,6-P2 synergistically with AMP (41, 62, 63) (Figure 1). FBPase1 can be phosphorylated by different kinases, but this leads to non-significant catalytic effects (41). FBPase1 is ubiquitously expressed and has been reported to be lost in several human cancers (64). FBPase1 overexpression suppresses cancer cell growth (65) and its loss correlates with advanced tumor stage and poor prognosis (66). Snail can repress FBPase1 in breast cancer cells (67), thus tightly controlling glucose flux through the PPP, by suppressing both PFK-P and FBPase1. FBPase1 and PFK-L directly interact forming multienzyme complexes that can modulate their activities (53). By contrast, FBPase2 is restricted to muscle cells and participates in the synthesis of glycogen from carbohydrate precursors (62).
6-Phosphofructo-2-Kinase/Fructose 2,6-Bisphosphatase (PFK-2/FBPase-2) Isoenzymes
Fru-2,6-P2 (Figure 2), a powerful allosteric modulator of the Fru-6-P/Fru-1,6-P2 substrate cycle, was discovered in 1980 when its concentration was observed greatly increased in hepatocytes upon incubation with glucose and disappeared in the presence of glucagon, providing a refined regulatory mechanism between glycolysis and gluconeogenesis (41, 68–71) (Figure 2). Fru-2,6-P2 has a dual function, increasing the affinity of PFK1 for Fru-6-P and releasing the enzyme from ATP-mediated inhibition. It also synergistically increases the affinity of PFK1 for AMP, a positive allosteric effector of the enzyme. By contrast, both Fru-2,6-P2 and AMP inhibit FBPase1 (41, 68–71). Furthermore, Fru-2,6-P2 stabilizes PFK1 (68) and promotes its association into tetramers and higher oligomers with enhanced activity (72). Therefore, changes in the concentration of this metabolite regulate the activities of PFK1 and FBPase1, thereby conferring a key role to Fru-2,6-P2 in the regulation of the Fru-6-P/Fru-1,6-P2 substrate cycle and the intensity and direction of glycolysis and gluconeogenesis (Figure 1). Since Fru-2,6-P2 does not take part as an intermediary in any metabolic interconversion, and given its lability in acid extracts used to measure phosphoric acid esters in tissues, this metabolite managed to escape discovery until 1980 (41, 70). Fru-2,6-P2 has been shown to carry out a leading function in regulating glycolysis in other eukaryotic cells (41, 68, 69).
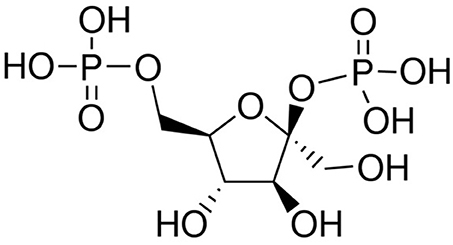
Figure 2. Structure of β-Fructose 2,6-bisphosphate.
Fru-2,6-P2 concentration is significantly higher in tumor cells than in normal cells (56, 73, 74) and is regulated by different bifunctional isoenzymes called 6-phosphofructo-2-kinases/fructose 2,6-bisphosphatases (PFK-2/FBPase-2), which catalyze the synthesis and degradation of this metabolite (68–71, 75). The balance between the activity of 6-phosphofructo-2-kinase (PFK-2), which synthesizes Fru-2,6-P2 from Fru-6-P and ATP, and that of fructose 2,6-bisphosphatase (FBPase-2), which hydrolyzes Fru-2,6-P2 into Fru-6-P and inorganic phosphate, ultimately determines the concentration of this metabolite (Figure 1). PFK-2/FBPase-2 is one of the few homodimeric bifunctional enzymes, composed of two 55-kDa subunits. Each monomer presents both kinase and bisphosphatase domains in the same polypeptide chain, with the kinase domain at the N-terminal end of the protein and the bisphosphatase domain at the C-terminal (68–71, 75, 76) (Figure 3). The amino acids located near the N- and C-terminal ends of the PFK-2/FBPase2 isoenzymes protein are responsible for its post-translational regulation as they can be phosphorylated by different protein kinases. The protein is derived from the fusion of two genes that express a kinase domain, evolutionarily related to the family of proteins that link mononucleotides, and a bisphosphatase domain, which is related to the family of phosphoglycerate phosphatases and acid phosphatases (75–77). The regulatory function of Fru-2,6-P2 in carbohydrate metabolism implies that the modulation of Fru-2,6-P2 synthesis and degradation must be very well compensated to adapt Fru-2,6-P2 concentration to the needs of the cell. The degree of complexity involved in regulating Fru-2,6-P2 levels in each tissue and physiological condition is reflected by the existence of different PFK-2/FBPase-2 isoenzymes that are capable of adapting to different conditions (75, 76, 78). These isoenzymes, encoded by four genes (PFKFB1-PFKFB4), display variances in their kinetic properties and distribution, as well as in their responses to allosteric, hormonal, and growth factors (75, 76). The PFKFB1 gene encodes the isoforms that were originally identified in the liver, muscle and fetal tissue, while the PFKFB2 gene encodes the isoenzyme occurring in the heart and kidney and in some cancer cells. The PFKFB3 gene encodes the isoforms present in the brain, placenta and adipose tissue, and is the most expressed PFKFB gene in proliferating and cancer cells. Finally, the PFKFB4 gene encodes the isoenzyme occurring in the testis, although it has also been found in several types of tumor cells (Table 1).
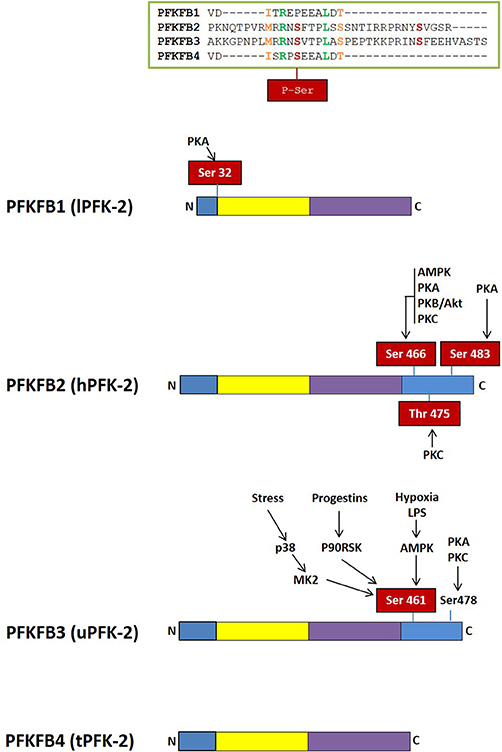
Figure 3. Domain organization of PFK-2/FBPase-2 isoenzymes. The N-terminal PFK-2 domain is shown in yellow and the C-terminal FBPase-2 domain is in purple. Regulatory regions with residues susceptible of phosphorylation by different protein kinases are shown in blue.
PFKFB1
The PFKFB1 gene was cloned from rat and human liver (71, 79, 80) and is composed of 60,944 bp. It contains 17 exons under the control of different promoters and gives rise to three different transcripts, mRNAs L (liver), M (muscle), and FL (fetal) (68, 70, 75, 81). The muscle and fetal transcripts have the same sequence as that of the liver, except for the exon encoding the N-terminal end containing the S32 residue, that can be phosphorylated by the cAMP-dependent protein kinase (PKA) in response to glucagon and dephosphorylated by protein phosphatase 2A (PP2A), activating the bisphosphatase and inhibiting the kinase activities of the liver isoenzyme, respectively (68, 70, 82) (Figure 3). The FL mRNA variant, present in several rat-derived cell lines and proliferating tissues, contains two non-coding exons (1aF and 1bF) (83). Although the liver, muscle and fetal isoenzymes come from the same gene, they are regulated differently, since glucagon induces glucose synthesis in the liver but not in other tissues. PFKFB1 has not been found to be overexpressed in cancer cells.
PFKFB2
The human PFKFB2 gene was cloned from human heart and it contains 15 exons spanning 27,961 bp. This gene generates nine transcripts, four of which encode full-length proteins, two encode truncated proteins and the other three contain an open reading frame without producing any protein (84). PFKFB2 is a homodimeric protein, with isoform A being a 58-kDa protein containing 505 amino acids and isoform B a 54-kDa protein containing 471 amino acids. The sequence of the catalytic site is preserved, but those of the N- and C-terminal regions exhibit more variances (75, 76, 84, 85). PFKFB2 is mainly expressed in the heart, being also located in other tissues, but in lesser proportion (76, 86). Moreover, it is expressed in cancer cells from different origins (76, 86, 87).
PFKFB2 can undergo multisite phosphorylation, integrating signals from many pathways (Figure 3). The C-terminal domain residues S29, S466, T475 and S483 can be phosphorylated by protein kinases such as 3-phosphoinositide-dependent kinase-1 (PDPK-1), AMP-activated protein kinase (AMPK), PKA, protein kinase B (PKB; also known as Akt), mitogen-activated protein kinase 1 (MAPK-1), and p70 ribosomal S6 kinase (S6K1). PFKFB2 phosphorylation at three conserved residues (S466, T475, and S483) results in the activation of the enzyme, decreasing its Km for Fru-6-P and increasing the Vmax of PFK-2 activity (75). PFK-2 activity, however, varies depending on the kinase that activates it (75, 88). Moreover, it has been proposed that the 14-3-3 proteins, which promote cell survival (89), bind to PFKFB2 when it is phosphorylated at S483 by Akt in response to insulin and IGF-1 or when transfected with active forms of Akt, mediating growth factors-induced glycolysis (90). Oncogenic BRAF V600E has also been found to activate p90 ribosomal S6 kinase (RSK), which phosphorylates and activates PFKFB2, that then binds to 14-3-3 to promote glycolysis and melanoma cell growth (91). Furthermore, amino acids increase Fru-2,6-P2 synthesis and glycolysis in cardiomyocytes and cancer cell lines by PI3K and Akt-mediated phosphorylation of PFKFB2 at S483 (92). Moreover, EGFR activation induces PFK-P phosphorylation (Y64), which binds to the N-terminal SH2 domain of p85α and promotes Akt-dependent PFKFB2 phosphorylation (S483), glycolysis, cell proliferation and brain tumorigenesis (61). Adrenalin promotes PFKFB2 phosphorylation by PKA at S466 and S483, while AMPK activation during ischemia or hypoxia induces PFKFB2 phosphorylation at S466, which increases Fru-2,6-P2 levels and stimulates glycolysis (75, 88). PFKFB2 is also a substrate of PKC, which phosphorylates S84, S466, and T475 residues (75, 93) (Figure 3). Several studies have reported that HIF-1α can regulate PFKFB2 expression in vivo but this appears to be cell-specific (86). Citrate, whose concentration is increased by the oxidation of fatty acids and ketone bodies, competitively blocks Fru-6-P binding, down-regulating PFKFB2 expression and glycolysis through the “glucose-sparing effect” (85).
PFKFB2 is one of the genes increased in lymphoblasts from glucocorticoid (GC)-treated children suffering from acute lymphoblastic leukemia (94). Surprisingly, overexpression of the two PFKFB2 splice variants seems to have little effect on lactate and ATP production, two metabolites that are reduced after GC treatment, and cell survival, suggesting that this gene is not an essential regulator of the anti-leukemic effects of GC (95). The androgen receptor (AR) is a key regulator of prostate growth, promoting glycolysis and anabolic metabolism, and the principal drug target for the treatment of prostate cancer (96). One of the mechanisms behind this phenotype is the transcriptional upregulation of PFKFB2, possibly under the control of the AR-CAMKII-AMPK signaling pathway (96). Androgens have been shown to stimulate glycolysis for de novo lipid synthesis. Androgens promote transcriptional up-regulation of de novo, mediated by binding of ligand-activated AR to its promoter, and phosphorylation of PFKFB2 generated by the PI3K/Akt signaling pathway. Moreover, blocking PFKFB2 expression with siRNA or inhibiting PFK-2 activity with LY294002 (PI3K inhibitor) has been observed to reduce glucose uptake and lipogenesis, suggesting that the induction of de novo lipid synthesis by androgens requires the transcriptional up-regulation of PFKFB2 (97). PFKFB2 expression is also enhanced in human gastric malignant tumors, being associated with increased levels of HIF-1α dependent genes, vascular endothelial growth factor (VEGF) and Glut1, indicating that HIF-1α could be responsible for the induction of PFKFB2 expression (87). In hepatocellular carcinoma, high expression of metastasis-associated in colon cancer protein 1 (MACC1), a key regulator of the hepatocyte growth factor (HGF)/c-Met pathway, has been noted to correlate with the high expression of PFKFB2, this correlation being associated to TNM stage (classification of malignant tumors), overall survival and Edmondson-Steier classification (98). Furthermore, MAPK-activated RSK, which directly phosphorylates PFKFB2, is required to maintain glycolytic metabolism in BRAF-mutated melanoma cells. RSK inhibition reduces PFKFB2 activity and glycolytic flux, suggesting an important role for RSK in BRAF-mediated metabolic rewiring (91).
Another observation highlighting the importance of PFKFB2 in metabolic reprogramming is its contribution to osteosarcoma development. Slit guidance ligand 2 (SLIT2) binds to round about guidance receptor 1 (ROBO1) and plays important roles in various physiological and pathological conditions, such as axon guidance, organ development, and angiogenesis (99). The SLIT2/ROBO1 axis promotes proliferation, inhibits apoptosis and contributes to the Warburg effect in osteosarcoma cells via activation of the SRC/ERK/c-MYC/PFKFB2 pathway (99).
PFKFB2 expression has also been linked to the regulation of non-coding RNAs. In ovarian cancer, the long non-coding RNA LINC00092 has been identified as a nodal driver of CAF-mediated metastasis. The pro-metastatic properties of CAFs have been linked to the elevated expression of both the chemokine CXCL14 and PFKFB2, correlating with poor prognosis. Mechanistic studies have demonstrated that LINC00092 binds PFKFB2, thereby promoting metastasis by inducing a glycolytic phenotype in these tumors and sustaining the local supportive function of CAFs (100). Similarly, the long non-coding RNA UCA1/miR-182 has been observed to be a nodal driver of metastasis in glioma that is mediated by glioblastoma-associated stromal cells (GASCs) and the GASC-secreted chemokine CXCL14. In clinical specimens, CXCL14 upregulation in GASCs cells was seen to correlate with poor prognosis. Interestingly, GASCs expressing high levels of CXCL14 have been shown to upregulate lncRNA UCA1 and downregulate miR-182, with miR-182 directly binding to PFKFB2 to modulate CXCL14 secretion, glycolysis, and the invasion of glioma cells (101).
PFKFB2 has also important roles in the physiology of human CD3+ T cells. Treatment of activated human CD3+ T cells with the proinflammatory chemokine CCL5 induces the activation of AMPK and PFKFB2 phosphorylation and activation, promoting glycolytic flux and suggesting that both glycolysis and AMPK signaling are required for efficient T cell activation in response to CCL5 (102).
PFKFB3
The PFKFB3 gene was cloned from a cDNA library of fetal brain and has been found expressed in all the tissues studied (103–107). It spans 109,770 bp and is composed of 19 exons. The variable C-terminal domain can undergo alternative splicing to produce six different isoforms. The two main isoforms are generated by alternative splicing of exon 15 and differ in their C-terminal sequence, the 4,553 bp mRNA variant initially named as ubiquitous PFK-2 (uPFK-2) (107) and the 4,226 bp mRNA inducible PFK-2 (iPFK-2) variant (106). Four additional splice variants have also been described (108). As many proto-oncogenes and pro-inflammatory cytokines, PFKFB3 has multiple copies of the AUUUA sequence in the 3′UTR of its mRNA, which confer instability and enhanced translational activity (106). It has been found that miR-26b and miR-206 interact with the 3′UTR of the PFKFB3 mRNA, decreasing glycolysis in osteosarcoma and breast cancer cells, respectively (109, 110).
PFKFB3 gene expression is induced by different stimuli, such as hypoxia (31, 111, 112), progestin (104, 113), estrogens (114) and stress stimuli (115), through the interactions of HIF-1α, the progesterone receptor (PR), estrogen receptor (ER), and the serum response factor (SRF). These factors bind to specific sequences in PFKFB3 promoter which are the consensus hypoxia response element (HRE), the progesterone response element (PRE), estrogen response element (ERE), and the serum response element (SRE), respectively. PFKFB3 expression can also be stimulated by growth factors such as insulin (73), pro-inflammatory molecules (106) such as interleukin-6 (IL-6) (116, 117), lipopolysaccharide (LPS) and adenosine (118), mitogenic lectins such as concanavalin A (ConA) (119) and phytohemagglutinin (PHA) (120), and the transforming growth factor beta 1 (TGF-β1) (121).
PFKFB3 gene expresses an isoenzyme that has high kinase and low bisphosphatase activity (K/B = 710), favoring the net synthesis of Fru-2,6-P2 and eliciting high concentrations of this metabolite in proliferating and tumor cells (122). The presence of a serine instead of an arginine at position 302 in the bisphosphatase active site gives place to the low bisphosphatase activity (123).
Different protein kinases, such as AMPK (124, 125), RSK (113), MK2 (115), PKA, PKB (119), and PKC (125) regulate the PFKFB3 isoenzyme by covalent modification of its C-terminal domain (Figure 3). PI3K/Akt also controls the PFKFB3 isoenzyme downstream of growth factors signaling (119, 126, 127). Phosphorylated PFKFB3 has increased Vmax of its kinase activity and decreased Km for fructose-6-P (119, 124). ROS-mediated S-glutathionylation (128) or demethylation (129) also regulate PFKFB3 in cancer cells, decreasing its catalytic activity and redirecting the glycolytic flux to the PPP, increasing NADPH and decreasing ROS levels. Similarly, cell damage-mediated induction of p53 stimulates nucleotide biosynthesis by inhibiting PFKFB3 expression and enhancing the flux of glucose through the PPP to increase nucleotide production, which promotes DNA repair and cell survival (130) (Figure 4).
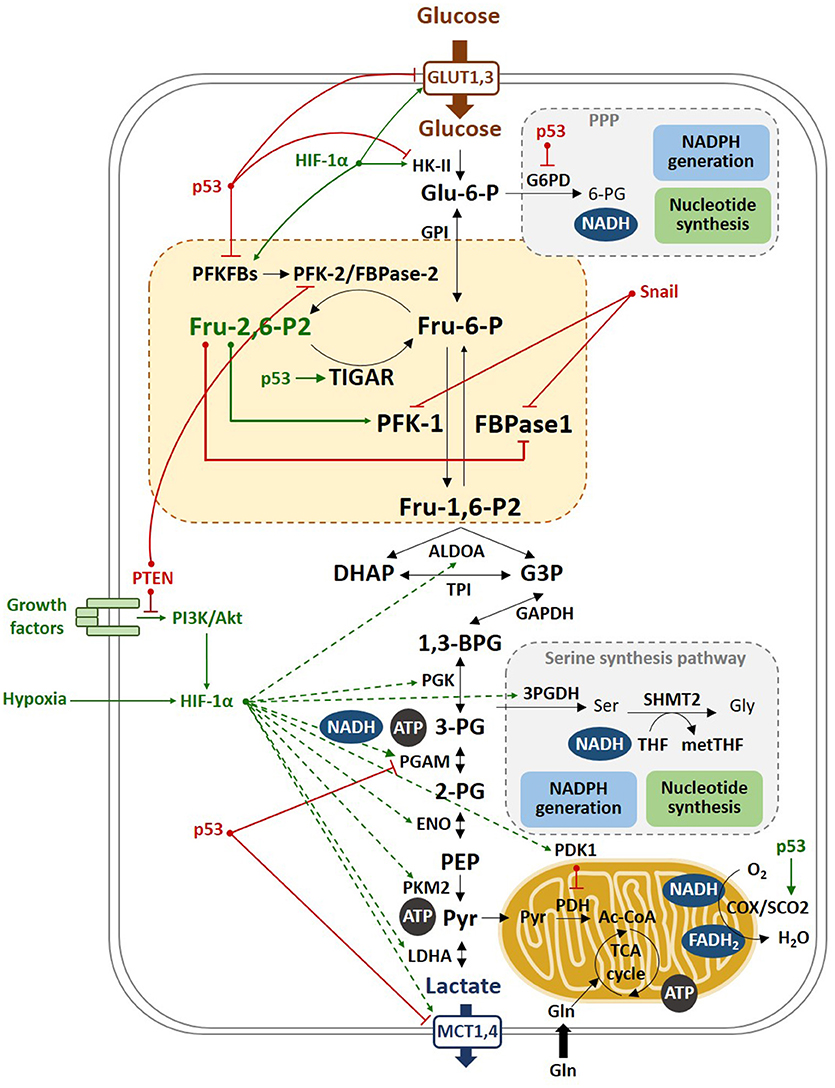
Figure 4. Molecular mechanisms of the Warburg effect. Hypoxia stabilizes HIF-1α, which transactivates most of the glycolytic genes, such as HK-II and PFKFBs, as well as PDK1, which in turn inhibits PDH, that catalyzes the conversion of pyruvate to Ac-CoA. PDH inhibition blocks the entry of pyruvate into the TCA, promoting the oxidation of pyruvate to lactate. Lactate is then excreted through the MCT transporters, which expression is also increased by HIF-1α. p53 inhibits the glycolytic genes PFKFB3, PFKFB4, and PGAM and the lactate transporter MCT1, and induces TIGAR, reducing the glycolytic flux. This results in increased flux of the pentose phosphate shunt to produce NADPH and ribose-5P. Moreover, p53 stimulates respiration by inducing SCO2, a component of cytochrome c oxidase (COX). In tumors, limited access to oxygen and mutations in the p53 gene drive to increased glycolysis, a phenomenon named as the Warburg effect.
The PFKFB3 isoenzyme can also be regulated through the ubiquitin-proteasome system (131), as it contains a KEN box that can be ubiquitinated by the E3 ubiquitin ligase of the anaphase-promoting complex (APC/C), which is activated by Cdh1, in a similar way to that of other proteins in the cell cycle. The proliferative response depends on the reduced activity of APC/C-Cdh1 to activate proliferation and glycolysis (132). PFKFB3 silencing prevents cell cycle progression, illustrating that this isoenzyme is essential for cell division (133). The tumor suppressor PTEN promotes APC/C-Cdh1 activity (127, 134) and cells from mice that overexpress PTEN show APC/C-Cdh1-mediated degradation of PFKFB3 and glutaminase, resulting in a decrease of glycolysis and proliferation, and an increase in resistance to oncogenic transformation (127). PFKFB3 isoenzyme levels have also been shown to increase in proliferating cells through the activation of cyclin D and E2F1, two downstream effectors of the PI3K-Akt-mTOR pathway (126), localizing in the nucleus and regulating proliferation through cyclin-dependent kinases (135). The nuclear targeting of PFKFB3 has been shown to increase cyclin-dependent kinase 1 (CDK1) expression among other cell cycle proteins. In particular, Fru-2,6-P2 stimulates the phosphorylation of the Cip/Kip protein p27 mediated by CDK1, which in turn elicits p27 ubiquitination and proteasomal degradation (136). PFKFB3 also interacts with CDK4, inhibiting its degradation via the ubiquitin proteasome pathway to promote cell cycle progression (137).
PFKFB3 expression is induced by endotoxin in human macrophages (106, 124). Macrophage Toll-4 receptor agonists cooperate with adenosine to increase glycolysis by heightening PFKFB3 gene expression and Fru-2,6-P2 synthesis, which increases glycolysis and favors ATP synthesis, developing the long-term defensive and reparative functions of macrophages (118). Macrophage glycolysis and pro-inflammatory activation mainly depend on HIF-1α and its effects on glucose uptake and the expression of hexokinase-II (HK-2) and PFKFB3 (138) (Figure 4). These findings indicate that hypoxia enhances glycolytic flux in macrophages through HIF-1α and PFKFB3 proportionally to the upregulation of pro-inflammatory activities in these cells (138). These in vivo observations suggest that HIF-1α antagonists or PFKFB3 inhibitors could be used to alleviate inflammatory diseases. Furthermore, cytosolic viral recognition by secondary interferon signaling has been demonstrated to upregulate glycolysis preferentially in macrophages through PFKFB3 induction, promoting the extrinsic antiviral capacity of macrophages and being a crucial component of innate antiviral immunity (139).
PFKFB3 is constitutively overexpressed in different cancer cell lines and in several human leukemias and solid tumors (140, 141), including ovarian and thyroid carcinomas (142), colon adenocarcinoma, breast cancer, gastric tumors and pancreatic cancer (73, 87, 111, 113, 143), and has been associated with lymph node metastasis and the TNM stage (143). PFKFB3 can also represent a biomarker and an anti-neoplastic target in gastric cancer (144). Furthermore, PFKFB3 expression is required for cell growth and increased metabolic activity in myeloproliferative neoplasms expressing the oncogenic JAK2V617F kinase, which is a very common mutation in these malignancies. JAK2V617F and active STAT5 overexpress PFKFB3 and PFKFB3 silencing reduces cell growth under normoxic and hypoxic conditions and prevents tumor formation (145). In chronic myeloid leukemia (CML), PFKFB3 has been found to be strongly associated with resistance to the BCR-ABL tyrosine kinase inhibitors. PFKFB3 silencing or pharmacological inhibition of its kinase activity enhances the sensitivity of CML cells to these inhibitors (146). In acute myeloid leukemia (AML), mTOR-mediated up-regulation of PFKFB3 is essential for cell survival, as mTORC1 up-regulates PFKFB3 in a HIF1α-dependent manner, and PFKFB3 silencing suppresses glycolysis and cell proliferation and activates apoptosis (147).
In malignant human colon tumors, PFKFB3 overexpression and phosphorylation of its S461 residue (P-PFKFB3 S461) has been observed (148). The cytokine IL-6 increases glycolysis by inducing PFKFB3 expression through STAT3 activation (116), suggesting a functional role of PFKFB3 in chronic inflammation and in the development of colorectal cancer (117). Similar results have been reported in TGF-β1-induced lung fibrosis, in which PFKFB3 inhibition attenuates prefibrotic phenotypes and blocks the differentiation of lung fibroblasts (149). Furthermore, we have shown that TGF-β1 overexpresses PFKFB3 mRNA and protein in glioblastoma cells through the activation of the Smad, p38 MAPK and PI3K/Akt signaling pathways. Inhibiting PFKFB3 expression or activity significantly suppressed the ability of T98G cells to form colonies, which is one of the hallmarks of cell transformation (121).
High-grade astrocytomas also contain increased PFKFB3 protein levels (150). The expression of the PFKFB splice variant UBI2K4 prevents tumor cell growth, acting as a tumor suppressor in astrocytic tumors (151). Besides, loss of heterozygosity in 10p14-p15, which leads to the allelic deletion of UBI2K4, has been detected in 55% of glioblastomas and is associated with poor prognosis (152).
PFKFB3 has also a key role in the interaction between cancer cells and other cells in the TME. Endothelial cells (ECs) depend on glycolysis more than on oxidative phosphorylation for ATP synthesis and loss of PFKFB3 in ECs impairs vessel formation (52). Inhibition of glycolysis by silencing PFKFB3 expression or pharmacologically blocking its kinase activity has been observed to inhibit pathological angiogenesis, such as ocular and inflammatory disease, without causing systemic effects (153). Recently, an article reported that targeting PFKFB3 in ECs significantly impeded metastasis by normalizing tumor vessels and improved the delivery and efficacy of chemotherapy (154). Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) regulates endothelial glycolysis and proliferation through the transcriptional regulation of PFKFB3, VEGFA, FOXO1, and MYC (155), with a positive correlation occurring between Nrf2, HIF-1α, and PFKFB3 expression in breast cancer cells, and cancer patients with high PFKFB3 expression showing poorer overall survival (156).
PFKFB3 expression is also linked to hepatocellular carcinoma (HCC) growth. PFKFB3 overexpression has been associated with a large tumor size and poor survival in patients, while PFKFB3 knockdown inhibits HCC growth by reducing glucose consumption and impeding DNA repair, which leads to cell cycle arrest at the G2/M phase and apoptosis. Silencing PFKFB3 expression decreases Akt phosphorylation and reduces the expression of ERCC1, a protein involved in DNA repair (157). The combination of aspirin and sorafenib has been shown to perform a synergistic effect against liver cancer. PFKFB3 overexpression, associated with high glycolytic flux, is frequently observed with sorafenib resistance, which can be overcome by aspirin. By inhibiting PFKFB3, sorafenib plus aspirin induce apoptosis in tumors without eliciting weight loss, hepatotoxicity and inflammation, suggesting that their combination may be an effective treatment for HCC (158).
A large number of studies have reported that increased PFKFB3 expression promotes proliferation and carcinogenesis, indicating that its inhibition could be crucial for treating inflammation and cancer. Indeed, siRNA suppression of PFKFB3 has been reported to reduce cancer cell viability (159, 160) while small molecule inhibitors of the PFKFB3 isoenzyme have been developed (161).
PFKFB4
PFKFB4, firstly cloned from rat (162) and human testis (163), is a 44,332 bp gene composed of 14 exons. Several splice variants have been reported, with the PFK-2 core domain being conserved among all of them (162, 164). The PFKFB4 gene encodes an isoenzyme that is expressed in the testis under the regulation of testosterone (165, 166). Moreover, it has been demonstrated that PFKFB4 mRNA and protein levels are regulated by hypoxia and glucose levels in different cancer cell lines from the prostate, liver, colon, bladder, stomach and pancreas (86, 87, 167–171). PFKFB4 is a prognostic marker in invasive bladder cancer (172), where its expression is activated by HIF-1α (171) (Figure 4). In hepatic cancer cell lines, sulforaphane-induced apoptosis was shown to decrease PFKFB4 protein expression and glucose consumption whereas HIF-1α induced PFKFB4 expression under hypoxic conditions (173). PFKFB4 expression is also controlled by heme-oxygenase-2 in HepG2 cancer cells (174). In HCC, upregulated Peroxisome proliferator-activating receptor γ (PPARγ) induces PFKFB4 expression through the transcriptional activity of its promoter, regulating glycolysis and cell proliferation (175). High PFKFB4 mRNA and protein expression have been described in three different glioblastoma stem-like cell lines, with shRNA-mediated knockdown of PFKFB4 promoting apoptosis (176) and no phenotypic effect occurring in PFKFB4-silenced normal neural stem cells. Furthermore, HIF1α-induced PFKFB4 mRNA expression correlates with glioma tumor grade (176).
PFKFB4 is required to balance glycolytic activity and antioxidant production to maintain the cellular redox balance in prostate cancer cells (169). PFKFB4 mRNA expression has been found to be greater in metastatic prostate cancer cells than in primary tumors. PFKFB4 silencing selectively increases Fru-2,6-P2 concentration in prostate cancer cells, suggesting that it mainly functions as a fructose-2,6-bisphosphatase in these particular cells. The increase in Fru-2,6-P2 levels should direct glucose 6-phosphate toward the glycolytic pathway, thereby reducing the activity of the PPP. This would explain why prostate cancer cells show lower NADPH and glutathione levels after PFKFB4 silencing, which results in enhanced oxidative stress and cell death (169). Furthermore, p53 decreases PFKFB4 gene expression by binding to its promoter to mediate transcriptional repression via histone deacetylases. PFKFB4 depletion also attenuates biosynthetic activity and induced ROS accumulation and cell death in the absence of p53 (177).
In a study investigating the differences in glucose metabolism between two forms of prostate cancer, small cell neuroendocrine carcinoma (SCNC) was found to be more glycolytic than adenocarcinoma, CD44 being a key regulator of glucose metabolism. PFKFB4 expression in benign prostate tissue was lower than that in the adenocarcinoma, and significantly higher in SCNC. CD44 ablation in SCNC cells reduced both mRNA and protein levels of PFKFB4 (178). Thus, CD44 can modulate the aggressive phenotype of prostate cancer cells by increasing PFKFB4 expression (179).
PFKFB4 can also regulate autophagy by influencing the redox balance. In PC3 prostate cancer cells, PFKFB4 inhibition was observed to cause p62 accumulation, which is usually associated with the inhibition of autophagy. However, the autophagic flux was increased in these cells. The combination of antioxidants and PFKFB4 inhibition prevented p62 accumulation, which was instead mediated by Nrf2, thus avoiding autophagy. Hence, PFKFB4 expression is required for appropriate ROS detoxification in these cells (180). It was recently found that epithelial and endothelial tyrosine kinase interacts with PFKFB4 modulating chemoresistance of small-cell lung cancer by regulating autophagy (181).
Solid malignant tumors of the breast present a higher PFKFB4 expression compared to non-malignant tissue. In several breast cancer cell lines, PFKFB4 expression increased upon exposure to hypoxia (87). PFKFB4 was recently shown to act as a protein kinase phosphorylating the oncogenic steroid receptor coactivator-3 (SRC-3) and enhancing its transcriptional activity to drive breast cancer (182). PFKFB4 suppression or ectopic expression of a phosphorylation-deficient S857A mutant of SRC-3 abolished SRC-3-mediated transcription. Mechanistically, SRC-3 phosphorylation increases its binding with the ATF4 transcription factor by stabilizing the recruitment of SRC-3 and ATF4 to target gene promoters. Functionally, PFKFB4-induced SRC-3 activation directs the glucose flux toward the PPP and the synthesis of purines by transcriptionally upregulating the expression of transketolase. PFKFB4 or SRC-3 silencing inhibits breast tumor growth and metastasis (182).
Apart from the importance of PFKFB4 in regulating cancer cell glycolysis, its expression also determines the metabolic adaptation of non-tumor cells. In mitogen-stimulated rat thymocytes, ConA was shown to induce the expression of PFKFB3 and PFKFB4 as well as increase glycolysis, cell proliferation and protein synthesis. This supports a role for these two proteins in coupling glycolysis to cell proliferation in lymphoid tissues (119).
TIGAR
The c12orf5 gene was discovered during a computer-based analysis of microarray data trying to find novel p53-regulated genes that are activated in response to ionizing radiation (183). This gene was cloned and characterized, and named as TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) (184) (Figure 4). TIGAR is a target of p53 that becomes rapidly activated by low levels of stress. The human TIGAR gene consists of six exons spanning about 38,835 bp, coding for a unique mRNA transcript variant of 8.2 kb with a 813 bp coding sequence. TIGAR promoter contains two p53 binding sites, one upstream of the first exon and the other within the first intron, the latter being the most efficient (184). TIGAR can be induced by Nutlin-3, an antagonist of Mdm2 that increases p53 levels (185), radiotherapy (183, 186), glutamine (29), chemotherapy (187), UV light (187), TNFα, and radiotherapy mimetics (188) or by the Akt signaling pathway in response to the metabolic stress caused by PFKFB3 knockdown (189). TIGAR expression can also be regulated in a p53 independent manner (186, 189) by linking the CRE-binding protein (CREB) to the TIGAR promoter (190). Another transcription factor, the specificity protein 1 (SP1), can bind to TIGAR promoter and is considered important for its basal activity (191). TIGAR can be induced in response to hypoxia in myocytes (192) and some studies have identified HIF-1α as a regulator of cytochrome C-oxidase-2 (SCO2) and TIGAR gene expression in response to hypoxia (193). SCO2, a metallochaperone that is involved in the biogenesis of cytochrome C oxidase subunit II, participates in the mitochondrial chain, it is also induced by p53 and its blockage leads to the glycolytic phenotype (194).
The human TIGAR protein is composed of 270 amino acids and has a molecular weight of 30 kDa. It contains a bisphosphatase active center in which two histidine residues, H11 and H198, and one glutamic acid, E102, are essential for its activity (184). TIGAR contains a catalytic domain similar to the histidine phosphatase superfamily of proteins with a histidine forming a transient phosphoenzyme during catalysis (195). This domain shares similarity with those of the phosphoglycerate mutase (PGAM) family of enzymes and with the bisphosphatase domain of PFK-2/FBPase-2 isoenzymes (196). TIGAR bisphosphatase activity hydrolyzes Fru-2,6-P2 into Fru-6-P, which can then enter in the PPP to synthesize NADPH and ribose-5-phosphate, thus reducing ROS and producing nucleotide precursors that are essential for biosynthesis, DNA repair and cell proliferation (184) (Figure 4). As TIGAR has no kinase domain, it behaves as a kinase-deficient PFKFB isoform. Thus, cells overexpressing FBPase-2 show similar enhanced PPP flux and resistance to oxidative stress (197). The FBPase catalytic activity of TIGAR is several orders of magnitude lower than that of the FBPase-2 component of PFK-2/FBPase-2 isoenzymes, pointing out that Fru-2,6-P2 could not be its main physiological substrate (198).
TIGAR mRNA is expressed in all the tissues in which it has been analyzed to date and is overexpressed in several cancer cells. It localizes mainly in the cytoplasm, but has been observed to relocalize to the outer mitochondrial membrane under hypoxic conditions to form complexes with HK-2, limiting ROS production (199).
TIGAR has also been linked to autophagy. For example, TIGAR overexpression and reduced ROS levels have been observed alongside suppressed autophagy in cells exposed to stress conditions, and TIGAR suppression induced autophagy that subsequently mediates apoptosis by restraining ROS levels (200). The relationship between autophagy and apoptosis is regulated distinctively according to the stimulus and cell type. Thus, treatment of neuroblastoma cells with D-galactose induces necroptosis and autophagy, as reflected in the upregulation of BMF, BNIP3, ATG5, and TIGAR, without affecting the expression of the genes associated with apoptosis (201). Decreased mRNA levels of TIGAR and reduced levels of the damage-regulated autophagy modulator (DRAM) have been reported in HepG2 cells exposed to high oxidative stress or nutrient starvation (202). Upon disruption of the homeostasis balance of the cell, TIGAR is activated and provides protection through its antioxidant properties rather than by inhibiting autophagy, while other transcriptionally activated targets, such as DRAM, enhance autophagy (203). Some studies have proposed that p53 regulates stress-induced autophagy by balancing TIGAR and DRAM, which have opposite effects (204, 205).
Like the PFKFB isoenzymes, TIGAR can also play a role in cancer. The function of TIGAR in a specific cell type depends on the metabolic state of the cell and PFKFBs activities that determine Fru-2,6-P2 concentration and glycolytic flux. TIGAR activity could limit glycolysis and produce antioxidant molecules and precursors for nucleotide synthesis, thereby limiting cancer development. However, TIGAR overexpression can also promote the growth of tumor cells with high ROS levels. TIGAR has been reported not be necessary for normal growth and development in mice, but plays an important function in intestinal regeneration. The lack of TIGAR causes growth defects which are recovered by ROS scavengers and nucleosides (206). Besides, TIGAR deficiency has been reported to reduce tumor growth and improve survival in a mouse intestinal adenoma model, while elevated TIGAR expression supported cancer progression (206). TIGAR expression is increased in human breast, gastric and lung cancer, inversely correlating with p53 expression levels (207–209). Furthermore, TIGAR downregulation inhibits growth in several cancer cell lines (184). In a model of nasopharyngeal cancer, 1-(3-C-ethynyl-beta-d-ribo-pentofuranosyl)cytosine (ECyd), an RNA-nucleoside anti-metabolite with potent anticancer activity, was shown to downregulate TIGAR and deplete NADPH. TIGAR overexpression was able to recover the growth inhibition induced by ECyd (210). In the same model, c-Met protein kinase maintained TIGAR expression, whereas c-Met silencing significantly decreased TIGAR expression and subsequently depleted intracellular NADPH, which lead to cell death (211). TIGAR silencing induces also apoptosis and autophagy in HepG2 cells (212), while RNAi-mediated knockdown of citrate synthase in human cervical carcinoma cells accelerates cancer cell metastasis and proliferation deregulating the p53/TIGAR pathway (213). In HeLa cervical carcinoma cells, TIGAR can be induced in an Akt-dependent manner in response to the inhibition of glycolysis. PFKFB3 depletion by RNAi increases ROS levels and decreases cell viability, this effect being highly exacerbated when TIGAR is also inhibited. However, TIGAR inhibition alone does not have an impact on HeLa cell survival (189). Furthermore, some studies have reported that TIGAR regulates the cell cycle by de-phosphorylating the retinoblastoma protein (RB) and stabilizing RB-E2F1 complex, thus delaying entry into the S phase (187, 214).
In multiple myeloma cells, inhibition of MUC1-C oncoprotein increases ROS levels and downregulates TIGAR expression, resulting in decreased NADPH and glutathione levels and promoting ROS-mediated apoptosis/necrosis (215). Sensitivity to fludarabine and p53-mediated TIGAR induction has been described in chronic lymphocytic leukemia. The sensitivity to fludarabine varied despite all patients presented wild-type p53 (216). Glioblastoma cells overexpress TIGAR which reduces cell death induced by restricting glucose and oxygen (217). These results indicate the potential therapeutic use of TIGAR as an antitumoral target (186). Similarly, TIGAR expression was found decreased with a sonodynamic therapy tested in a neuroblastoma cell model which decreases cell proliferation, possibly through increased ROS levels (218). In glioblastoma-derived cell lines, TIGAR abrogation increased radiation-induced cell destruction, providing a new therapeutic strategy that could be used to increase cell death in glial tumors, thus allowing the use of lower doses of radiotherapy. Gliomas are resistant to radiotherapy and to TNFα-induced killing. Radiation-induced TNFα increases radioresistance through nuclear factor κB (NFκB). Thus, the existence of an ATM-NFκB axis regulating TIGAR indicates its involvement in the inflammation and resistance to radiomimetics (188). It was recently reported that TIGAR regulates NF-κB activation by suppressing phosphorylation and activation of the upstream IKKβ, which occurs through a direct binding competition between NEMO and TIGAR for the linear ubiquitination assembly complex (LUBAC), preventing the linear ubiquitination of NEMO required for the activation of IKKβ and other downstream targets. Furthermore, a TIGAR mutant with impaired phosphatase activity was equally effective as wild-type TIGAR in inhibiting the linear ubiquitination of NEMO, IKKβ phosphorylation/activation and NF-κB signaling, indicating that the effect of TIGAR on NF-κB signaling is due to a non-enzymatic molecular function, that directly inhibits the E3 ligase activity of LUBAC (219).
In a co-culture system, oxidative stress-induced autophagy correlated with caveolin-1 (CAV1) downregulation in CAFs and TIGAR overexpression in adjacent breast cancer cells (Figure 5). Reduced CAV1 expression in fibroblasts reduces mitochondrial function and induces glycolysis through HIF-1α and NF-kB signaling (30). Consequently, autophagic CAFs supply recycled substrates for the cancer cell metabolism and, also, avoid cancer cell death by overexpressing TIGAR, thereby conferring resistance to apoptosis and autophagy (220). Other studies by the same group have shown that the metabolic coupling between cancer cells and fibroblasts contribute to tamoxifen resistance, as CAFs enhanced TIGAR activity in cancer cells, that protected against tamoxifen-induced apoptosis (221). In another study, glutamine was described to increase TIGAR and be needed for CAV1 downregulation in CAFs, decreasing mediators and markers of autophagy in cancer cells. In this model, glutamine from autophagic fibroblasts may serve to fuel cancer cell mitochondrial activity. Thus, a cycle of nutrients between catabolic stromal cells and anabolic tumor cells has been suggested to account for the relationship between cells in the TME (19–21, 28–30, 222). In addition, TIGAR overexpression has been shown to reprogram carcinoma and stromal cells in breast cancer (29), as well as to increase oxygen consumption rates and ATP levels in the presence of glutamine and lactate, leading to enhanced ATP synthesis. Moreover, when carcinoma cells overexpress TIGAR in co-cultures with fibroblasts, a glycolytic phenotype is induced in the fibroblasts, inducing HIF-1α expression as well as increasing glucose uptake and the expression of PFKFB3 and lactate dehydrogenase-A. TIGAR overexpression in carcinoma cells increases tumor growth and proliferation rates in vivo (29, 30). All these data support a two-compartment model of tumor metabolism (Figure 5). The mechanisms by which TIGAR overexpression increases mitochondrial activity remain unknown to date (29, 217).
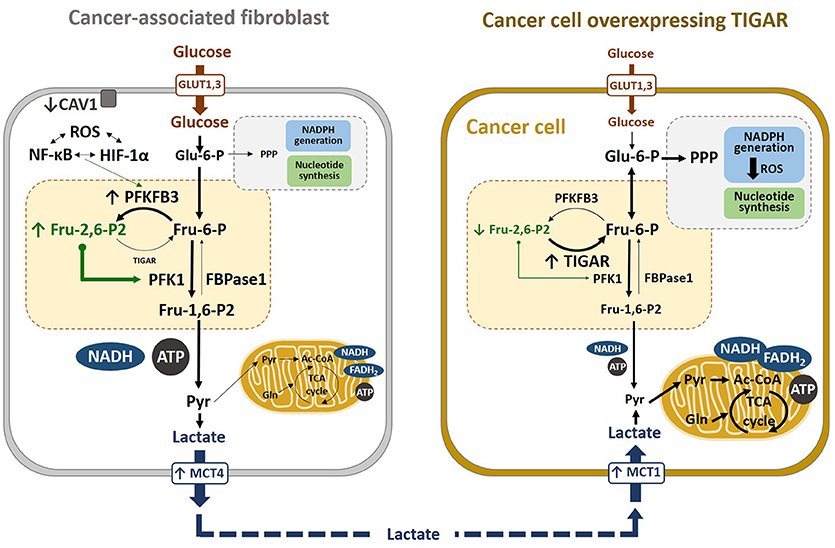
Figure 5. Metabolic reprogramming in cancer cells. Lactate transport is crucial in the crossed-regulation of metabolism between cancer-associated fibroblasts (CAFs) and cancer cells. In the fibroblasts close to carcinoma cells, ROS, HIF-1α, and NF-κB induce glycolysis, downregulating CAV1 and upregulating MCT4, which results in increased lactate secretion in CAFs. Conversely, TIGAR overexpression in carcinoma cells alters their metabolic state, increasing the pentose phosphate pathway (PPP). Lactate released from CAFs is taken by cancer cells via MCT1 and converted to pyruvate, which can enter the TCA cycle. These cancer cells have high mitochondrial OXPHOS and low glycolysis, which is associated with high proliferation and low apoptosis rates, resulting in increased tumor growth.
Fru-2,6-P2 Metabolism and the Regulation of Glycolysis and the Pentose Phosphate Pathway in Cancer Cells
The different PFKFB isoenzymes and TIGAR have important roles in the TME. The genes encoding these isoenzymes are commonly overexpressed in tumors and can be induced in response to cellular stress. These isoenzymes, regulating Fru-2,6-P2 concentration, have contrary effects on cell metabolism and different studies have dealt with the cross-regulation of these enzymes (Figure 4). The contribution of TIGAR to the regulation of Fru-2,6-P2 levels is expected to vary depending on the expression levels of the PFKFB1–PFKFB4 isoenzymes. TIGAR overexpression is associated with decreased survival of patients with acute myeloid leukemia (AML) (223) and other tumors (217, 224). Overexpression of PFKFB2-4 has similar outcomes (97, 98, 113, 161, 176). However, crosstalk between these genes must exist in cancer cells since it has been observed that when TIGAR is eliminated in leukemia cells expressing high levels of TIGAR, PFKFB3 expression increase, and when TIGAR is overexpressed in cells with low levels of TIGAR, PFKFB3 expression decrease (223). Moreover, we have reported that PFKFB3 silencing in HeLa cells elevates ROS levels and overexpresses TIGAR through Akt signaling, preserving against DNA damage and apoptosis (189). These results indicate that cancer cells sustain high glycolytic flux in order to fuel biosynthetic pathways under basal conditions. Nevertheless, if glycolysis is impeded, for example by ROS-induced S-glutathionylation or demethylation of PFKFB3 (128, 129), PFKFB3 interference, or PFKFB3 inhibitors (189), cancer cells divert the flow toward the PPP. Cancer cells can also inhibit glycolysis to decrease ROS in response to DNA damage. In this respect, following DNA damage, the function of p53 reducing the mRNA and protein levels of PFKFB3 (130) and PFKFB4 (177) as well as the glycolytic flux, while increasing TIGAR expression and the levels of NADPH and nucleotides through the PPP (184, 186), enhances DNA repair and cell survival. In this sense, in p53-deficient cells exposed to UV radiation, glycolysis is not impeded supporting that p53 is required for this regulatory role (130, 177).
The FBPase catalytic efficiency of TIGAR has been reported to be several orders of magnitude lower than that of the FBPase-2 activity of PFKFBs (195, 198), thus challenging the concept that TIGAR acts primarily on Fru-2,6-P2. TIGAR could also exert its effects by directly increasing flux through the terminal part of glycolysis, given that it was found to act as the phosphoglycolate-independent 2,3-bisphosphoglycerate phosphatase (225), with 2,3-bisphosphoglycerate (2,3-BPG), 2-phosphoglycerate (2-PG) and phosphoenolpyruvate (PEP) being better substrates than Fru-2,6-P2 (198). These effects could also be enhanced by the presence of PKM2 in cancer cells, which has low affinity for PEP and provides a large amount of metabolic precursors for biosynthesis (14, 226). The concentration of 2,3-BPG in cells is three orders of magnitude lower than that in erythrocytes (227) and very little is known about its function, apart from the fact that it is an essential cofactor for the phosphoglycerate mutase. This adds a novel layer of complexity to the function of TIGAR that should be taken into account in future studies.
PFKFB4 expression is essential for prostate and p53-null colon cancer cell survival, maintaining the balance between the use of glucose for energy generation and the synthesis of antioxidants (177). PFKFB4 silencing increases Fru-2,6-P2 levels in prostate cancer cells, suggesting that it mainly functions as a fructose-2,6-bisphosphatase in these particular cells, diverting glucose 6-phosphate toward the PPP (169). By contrast, PFKFB4 that is expressed in other types of transformed cells and tumors synthesizes Fru-2,6-P2 and is required for the glycolytic reprogramming of cancer cells (170). Specific analyses of the enzymatic activity and regulation of PFKFB4 are needed to characterize its potential role as a FBPase-2 proposed in some studies (169, 177).
PFKFB4 has been shown to act as a protein kinase of SRC-3 resulting in the upregulation of transketolase (182). This finding could explain the effect of PFKFB4 overexpression in some cancer cells, such as the transcription of a key non-oxidative enzyme of the PPP and the redirection of glycolytic intermediates to the non-oxidative arm of PPP.
Other inhibitory glycolytic effects that contribute to cancer development, such as the glycosylation of PFK1 in response to hypoxia (59) and the Snail repression of PFK-P (60), redirect glucose flux through the PPP, thereby conferring a selective growth advantage to cancer cells. Furthermore, FBPase1 overexpression suppresses cancer cell growth (65), its loss correlating with advanced tumor stage and poor prognosis (66). Snail can also repress FBPase1 in breast cancer cells (67), regulating glucose flux toward glycolysis or the PPP by suppressing either FBPase1 or PFK-P, respectively.
Finally, FBPase1 and PFK-L are part of the “glycosome,” a complex that modulates the activities of these enzymes and integrates them with others such as PKM2 and PEPCK1. Quantitative high-content imaging assays indicate that the direction of glucose flux between glycolysis, the PPP and serine biosynthesis seems to be spatially regulated by these multienzyme complexes in a cluster size-dependent manner, providing new mechanistic insight into how a cell regulates the direction of glucose flux between energy metabolism and anabolic biosynthesis (53).
Targeting the Tumor Metabolic Ecosystem
The ability to selectively modulate the metabolism of cancer cells could have high therapeutic potential. Tumor cells expressing active oncogenes and/or defects in their tumor suppressors enter apoptosis when glucose oxidation is limited. Peculiarly, these same cells often show resistance to other forms of apoptotic stimuli (radiation and chemotherapy) and the use of glycolytic inhibitors sensitizes cells to these stimuli and promotes their death (21, 228, 229).
The dependence of cancer cells on glucose consumption led to the development of different therapeutic approaches. Pharmacological inhibition of glycolysis has emerged as a novel strategy since high glycolytic activity is considered a metabolic hallmark of cancer (6, 21, 31, 34). HIF-1α overexpression and the induction of glycolytic isoenzymes, present in many tumors, have been shown to generate resistance to chemotherapy and radiation (39, 230, 231). Therefore, inhibiting HIF-1α could be an important component of cancer therapy (232, 233).
One of the most studied inhibitors has been 2-DG, a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen. 2-DG can be phosphorylated by hexokinase, but it cannot be metabolized by phosphohexose-isomerase. Therefore, its intracellular accumulation produces a competitive inhibition of hexokinases. 2-DG has cytotoxic effects on different types of cancer cells, especially those overexpressing HIF-1α and with mitochondrial defects (229, 234). Accordingly, 2-DG significantly increases the response to treatment with adriamycin and paclitaxel in human osteosarcoma-bearing mice and of small cell lung cancer (234). However, the high doses needed to compete with glucose can induce toxicity (21, 235). Inhibition of other glycolytic enzymes has been shown to successfully suppress tumor cell growth, although systemic toxicity and lack of therapeutic benefit has precluded further development in numerous preclinical studies (21).
The fact that the PFKFB2-4 genes are overexpressed in different tumors and are activated by hypoxia and/or oncogenes indicates that their role is necessary in the development of the glycolytic phenotype, facilitating the adaptation and survival of tumor cells in hypoxic micro-environments. Thus, small molecule inhibitors of PFKFBs could be used to improve the efficiency and specificity of cancer treatment (161, 236).
The expression of more than one PFKFB isoenzyme in some cells suggests the use of less specific PFKFB kinase inhibitors to effectively reduce Fru-2,6-P2 concentrations. In this sense, targeting of both PFKFB3 and PFKFB4 isoenzymes has been proposed to be advantageous due to their high expression in some cancer cells (173). PFKFB isoenzyme inhibitors could also be used in combination with agents that mimic hypoxic conditions, increasing cellular dependence on the upregulation of glycolysis and PFKFBs. The use of chemotherapeutic drugs together with PFKFB3 inhibitors may improve response rates as well as progression-free survival in cancer patients. This is corroborated by recent data demonstrating that sorafenib resistance in HCC can be overcome by aspirin, through PFKFB3 inhibition (158). This type of anti-glycolytic approach substantially differs from previous cancer treatments that attempted to block glycolysis entirely and in a permanent way, causing significant adverse effects. Given that Fru-2,6-P2 is not part of a main metabolic pathway, and is not a biosynthetic precursor or intermediate in energy production, its concentration can be independently controlled, making PFKFB isoenzymes more specific targets.
TIGAR has important functions in the regulation of cell processes such as apoptosis, autophagy, DNA repair, and the control of oxidative stress. The elevated levels of TIGAR expression in some types of tumors (207, 217) and the action of different therapeutic agents associated with decreased TIGAR expression, highlight the importance of TIGAR in tumor cell survival. TIGAR can support tumorigenesis by reducing ROS production and generating precursors for biosynthesis. These data indicate that the inhibition of TIGAR might confer advantages in cancer treatments (29, 186, 206, 221). Moreover, TIGAR silencing has been shown to increase sensitivity of glioblastoma cells to radiotherapy (186, 237) and increase cell death mediated by PFKFB3 inhibition (189).
The current results show that the metabolic phenotype of tumor cells is heterogeneous and that metabolic coupling occurs between different cell populations of the TME with complementary metabolic profiles. The metabolic differences between tumor and non-tumor cells can potentially be exploited therapeutically. There are currently no glycolytic inhibitors that have been approved as anticancer agents (21) and little is known about the degree of glycolysis inhibition in tumor vs. non-tumor tissues that can be achieved with glycolytic inhibitors, but the preclinical results obtained with these molecules look promising.
The targeting of mitochondrial oxidative metabolism and antioxidant effectors also hold promise as anticancer strategies. Arsenic trioxide, an inhibitor of the mitochondrial oxidative metabolism, has been approved for the treatment of acute promyelocytic leukemia (238). Metformin, an inhibitor of complex I of oxidative phosphorylation (239), has been shown to increase lactate levels and induce apoptosis in a clinical trial in head and neck squamous cell cancer (240). In the same clinical trial, metformin was shown to induce CAV1 expression in CAFs, preventing the metabolic coupling between stromal and cancer cells (240, 241). Another example of effective anti-metabolic cancer therapies is the use of N-acetyl cysteine (NAC), whose antioxidant potential reduced both the proliferation of cancer cells and the expression of the metabolic coupling monocarboxylate transporter 4 (MCT4) in stromal cells in a clinical trial in breast cancer (242). In summary, inhibiting oxidative metabolism or altering the redox state of tumors appear as promising approaches for the treatment of cancer.
Conclusions
In this review, we summarize current knowledge on the enzymes regulating the Fru-6-P/Fru-1,6-P2 cycle and their role in cancer and TME cells. PFKFB and TIGAR enzymes control this cycle and are overexpressed in cancer cells, acting as prognostic markers. Small molecule inhibitors of PFKFB2-4 in combination with other drugs could increase the efficiency of cancer treatment. Further preclinical data on PFKFBs inhibitors are required to confirm their potential clinical use.
In summary, glycolysis in tumor cells is a complex phenomenon in which this and other metabolic pathways are reprogrammed to increase energy production and biomolecular synthesis required for cell proliferation. Understanding the regulation of genes and glycolytic isoenzymes in cancer cells and other cells of the TME will have implications for cancer diagnosis and prognosis and for the development of more selective therapies.
Author Contributions
All authors jointly developed the structure and arguments of the paper, prepared the manuscript, reviewed it and approved the final version. RB supervised each of the tasks.
Funding
The authors are supported by the Instituto de Salud Carlos III–Fondo de Investigaciones Sanitarias (grants PI13/0096 and PI17/00412) and the Fondo Europeo de Desarrollo Regional (FEDER).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank E. Adanero for her helpful assistance and T. Evans for English correction. AR-G and HS-M were recipients of a fellowship from the University of Barcelona and the Generalitat de Catalunya, respectively.
Abbreviations
2,3-BPG, 2,3-bisphosphoglycerate; 2-DG, 2-deoxy-D-glucose; AML, acute myeloid leukemia; AMPK, AMP-activated protein kinase; APC/C, anaphase-promoting complex; AR, androgen receptor; CAF, cancer-associated fibroblast; CAV1, caveolin-1; CDK1, cyclin-dependent kinase 1; CML, chronic myeloid leukemia; ConA, concanavalin A; CREB, CRE-binding protein; DRAM, damage-regulated autophagy modulator; EC, endothelial cell; ECyd, 1-(3-C-ethynyl-beta-d-ribo-pentofuranosyl)cytosine; ER, estrogen receptor; ERE, estrogen response element; FBPase1, fructose 1,6-bisphosphatase; FBPase-2, fructose 2,6-bisphosphatase; Fru-1, 6-P2, fructose-1,6-bisphosphate; Fru-2, 6-P2, fructose 2,6-bisphosphate; Fru-6-P, fructose-6-phosphate; GASC, glioblastoma-associated stromal cell; GC, glucocorticoid; Glut1, glucose transporter 1; HCC, hepatocellular carcinoma; HIF-1α, hypoxia inducible factor-1α; HK-2, hexokinase-II; IL-6, interleukin-6; LPS, lipopolysaccharide; LUBAC, linear ubiquitination assembly complex; MACC1, metastasis-associated in colon cancer protein 1; MAPK-1, mitogen-activated protein kinase 1; MCT4, metabolic coupling monocarboxylate transporter 4; NFκB, nuclear factor κB; PDK1, pyruvate dehydrogenase kinase; PEP, phosphoenolpyruvate; PEPCK1, phosphoenolpyruvate carboxykinase 1; PET, positron emission tomography; PFK1, 6-phosphofructo-1-kinase; PFK1/FBPase1, 6-phosphofructo-1-kinase/fructose 1,6-bisphosphatase; PFK-2, 6-phosphofructo-2-kinase; PFK-2/FBPase-2, 6-fosfofructo-2-kinase/fructose 2,6-bisphosphatase; PGAM, phosphoglycerate mutase; PKA, cAMP-dependent protein kinase; PKB, protein kinase B; PKM2, pyruvate kinase isoenzyme M2; PP-1, protein phosphatase 1; PPARγ, peroxisome proliferator-activating receptor γ; PPP, pentose phosphate pathway; PR, progesterone receptor; PRE, progesterone response element; RB, retinoblastoma; ROBO1, round about guidance receptor 1; ROS, reactive oxygen species; RSK, p90 ribosomal S6 kinase; SCNC, small cell neuroendocrine carcinoma; SCO2, cytochrome C-oxidase-2; SLIT2, slit guidance ligand 2; SP1, specificity protein 1; SRC-3, steroid receptor coactivator-3; TGF-β1, transforming growth factor beta 1; TIGAR, TP53-induced glycolysis and apoptosis regulator; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
References
1. Otto Warburg B, Wind F, Negelein N. The metabolism of tumors in the body. J Gen Physiol. (1926) 309:397–519.
2. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
3. Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol. (2005) 70:363–74. doi: 10.1101/sqb.2005.70.035
4. Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. (1999) 24:68–72. doi: 10.1016/S0968-0004(98)01344-9
5. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
7. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
8. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi: 10.1038/nrc1478
9. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer (2010) 10:267–77. doi: 10.1038/nrc2817
10. Liberti M V, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
11. Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem. (1993) 212:95–9. doi: 10.1111/j.1432-1033.1993.tb17637.x
12. Crabtree HG. The carbohydrate metabolism of certain pathological overgrowths. Biochem J. (1928) 22:1289–98. doi: 10.1042/bj0221289
13. Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. (1929) 23:536–45. doi: 10.1042/bj0230536
14. Eigenbrodt E, Gerbracht U, Mazurek S, Presek P, Friis R. Carbohydrate metabolism and neoplasia: new perspectives for diagnosis and therapy. In: Biochemical and Molecular Aspects of Selected Cancers, Vol. 2. Boston, MA: Academic Press (1994) p. 311–85.
15. Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: on the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta Bioenerg. (2011) 1807:568–76. doi: 10.1016/j.bbabio.2010.08.010
16. Koobs DH. Phosphate mediation of the Crabtree and Pasteur effects. Science (1972) 178:127–33. doi: 10.1126/science.178.4057.127
17. Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta Rev Cancer (2013) 1835:164–9. doi: 10.1016/j.bbcan.2012.12.004
18. Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol. (2007) 244:703–13. doi: 10.1016/j.jtbi.2006.09.010
19. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. (2008) 118:3930–42. doi: 10.1172/JCI36843
20. Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. (2014) 41:195–216. doi: 10.1053/j.seminoncol.2014.03.002
21. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:113. doi: 10.1038/nrclinonc.2017.1
22. Hanahan D, Coussens LM, Bissell MJ, Lindblom P, Betsholtz C, Gerhardt H, et al. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
23. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125:5591–6. doi: 10.1242/jcs.116392
24. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
25. Icard P, Kafara P, Steyaert J-M, Schwartz L, Lincet H. The metabolic cooperation between cells in solid cancer tumors. Biochim Biophys Acta (2014) 1846:216–25. doi: 10.1016/j.bbcan.2014.06.002
26. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121:335–48. doi: 10.1016/j.cell.2005.02.034
27. Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol Mech Dis. (2012) 7:423–67. doi: 10.1146/annurev-pathol-011811-120856
28. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
29. Ko Y-H, Domingo-Vidal M, Roche M, Lin Z, Whitaker-Menezes D, Seifert E, et al. TP53-inducible glycolysis and apoptosis regulator (TIGAR) metabolically reprograms carcinoma and stromal cells in breast cancer. J Biol Chem. (2016) 291:26291–303. doi: 10.1074/jbc.M116.740209
30. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic coupling and the Reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. (2017) 44:198–203. doi: 10.1053/j.seminoncol.2017.10.004
31. Bartrons R, Caro J. Hypoxia, glucose metabolism and the Warburg's effect. J Bioenerg Biomembr. (2007) 39:223–9. doi: 10.1007/s10863-007-9080-3
32. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
33. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi: 10.1038/nrc3038
34. Pavlova NN, Thompson CB. The Emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
35. Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, Kim H, et al. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. (2002) 62:6674–81.
36. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
37. Flöter J, Kaymak I, Schulze A. Regulation of metabolic activity by p53. Metabolites (2017) 7:21. doi: 10.3390/metabo7020021
38. Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. (2009) 9:1084–101. doi: 10.2174/138955709788922610
39. Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. (2017) 36:252–9. doi: 10.15252/embj.201695204
40. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science (2001) 292:504–7. doi: 10.1126/science.1058079
41. Hers HG, Van Schaftingen E. Fructose 2,6-bisphosphate 2 years after its discovery. Biochem J. (1982) 206:1–12. doi: 10.1042/bj2060001
42. Dunaway GA, Kasten TP, Sebo T, Trapp R. Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem J. (1988) 251:677–83. doi: 10.1042/bj2510677
43. Costa Leite T, Da Silva D, Guimarães Coelho R, Zancan P, Sola-Penna M. Lactate favours the dissociation of skeletal muscle 6-phosphofructo-1-kinase tetramers down-regulating the enzyme and muscle glycolysis. Biochem J. (2007) 408:123–30. doi: 10.1042/BJ20070687
44. Staal GE, Kalff A, Heesbeen EC, van Veelen CW, Rijksen G. Subunit composition, regulatory properties, and phosphorylation of phosphofructokinase from human gliomas. Cancer Res. (1987) 47:5047–51.
45. Foe LG, Kemp RG. Isozyme composition and phosphorylation of brain phosphofructokinase. Arch Biochem Biophys. (1984) 228:503–11. doi: 10.1016/0003-9861(84)90016-X
46. Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci USA. (2005) 102:2402–7. doi: 10.1073/pnas.0409741102
47. Deng H, Yu F, Chen J, Zhao Y, Xiang J, Lin A. Phosphorylation of bad at Thr-201 by JNK1 promotes glycolysis through activation of phosphofructokinase. J Biol Chem. (2008) 283:20754–60. doi: 10.1074/jbc.M800024200
48. Lee J-H, Liu R, Li J, Zhang C, Wang Y, Cai Q, et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Commun. (2017) 8:949. doi: 10.1038/s41467-017-00906-9
49. Real-Hohn A, Zancan P, Da Silva D, Martins ER, Salgado LT, Mermelstein CS, et al. Filamentous actin and its associated binding proteins are the stimulatory site for 6-phosphofructo-1-kinase association within the membrane of human erythrocytes. Biochimie (2010) 92:538–44. doi: 10.1016/j.biochi.2010.01.023
50. Zhao S, Lee EYC. Targeting of the catalytic subunit of protein phosphatase-1 to the glycolytic enzyme phosphofructokinase. Biochemistry (1997) 36:8318–24. doi: 10.1021/bi962814r
51. Webb BA, Dosey AM, Wittmann T, Kollman JM, Barber DL. The glycolytic enzyme phosphofructokinase-1 assembles into filaments. J Cell Biol. (2017) 216:2305–13. doi: 10.1083/jcb.201701084
52. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BWW, Cantelmo ARR, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell (2013) 154:651–63. doi: 10.1016/j.cell.2013.06.037
53. Kohnhorst CL, Kyoung M, Jeon M, Schmitt DL, Kennedy EL, Ramirez J, et al. Identification of a multienzyme complex for glucose metabolism in living cells. J Biol Chem. (2017) 292:9191–203. doi: 10.1074/jbc.M117.783050
54. Vora S, Halper JP, Knowles DM. Alterations in the activity and isozymic profile of human phosphofructokinase during malignant transformation in vivo and in vitro: transformation- and progression-linked discriminants of malignancy. Cancer Res. (1985) 45:2993–3001.
55. Moon J-S, Kim HE, Koh E, Park SH, Jin W-J, Park B-W, et al. Krüppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J Biol Chem. (2011) 286:23808–16. doi: 10.1074/jbc.M111.236737
56. Colomer D, Vives-Corrons JL, Pujades A, Bartrons R. Control of phosphofructokinase by fructose 2,6-bisphosphate in B-lymphocytes and B-chronic lymphocytic leukemia cells. Cancer Res. (1987) 47:1859–62.
57. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. (1994) 269:23757–63.
58. Kole HK, Resnick RJ, Van Doren M, Racker E. Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch Biochem Biophys. (1991) 286:586–90. doi: 10.1016/0003-9861(91)90084-V
59. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337:975–80. doi: 10.1126/science.1222278
60. Kim NH, Cha YH, Lee J, Lee S-H, Yang JH, Yun JS, et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun. (2017) 8:14374. doi: 10.1038/ncomms14374
61. Lee J-H, Liu R, Li J, Wang Y, Tan L, Li X-J, et al. EGFR-phosphorylated platelet isoform of phosphofructokinase 1 promotes PI3K activation. Mol Cell (2018) 70:197–210.e7. doi: 10.1016/j.molcel.2018.03.018
62. Dzugaj A. Localization and regulation of muscle fructose-1,6-bisphosphatase, the key enzyme of glyconeogenesis. Adv Enzyme Regul. (2006) 46:51–71. doi: 10.1016/j.advenzreg.2006.01.021
63. Gizak A, Maciaszczyk E, Dzugaj A, Eschrich K, Rakus D. Evolutionary conserved N-terminal region of human muscle fructose 1,6-bisphosphatase regulates its activity and the interaction with aldolase. Proteins Struct Funct Bioinforma. (2008) 72:209–16. doi: 10.1002/prot.21909
64. Dai J, Ji Y, Wang W, Kim D, Fai LY, Wang L, et al. Loss of fructose-1,6-bisphosphatase induces glycolysis and promotes apoptosis resistance of cancer stem-like cells: an important role in hexavalent chromium-induced carcinogenesis. Toxicol Appl Pharmacol. (2017) 331:164–73. doi: 10.1016/j.taap.2017.06.014
65. Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD, Mathew LK, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature (2014) 513:251–5. doi: 10.1038/nature13557
66. Zhang J, Wang J, Xing H, Li Q, Zhao Q, Li J. Down-regulation of FBP1 by ZEB1-mediated repression confers to growth and invasion in lung cancer cells. Mol Cell Biochem. (2016) 411:331–40. doi: 10.1007/s11010-015-2595-8
67. Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23:316–31. doi: 10.1016/j.ccr.2013.01.022
68. Van Schaftingen E. Fructose 2,6-bisphosphate. Adv Enzymol Relat Areas Mol Biol. (1987) 59:315–95. doi: 10.1002/9780470123058.ch7
69. Hue L, Rider MH. Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem J. (1987) 245:313–24. doi: 10.1042/bj2450313
70. Rider MH, Bartrons R. Fructose 2,6-bisphosphate: the last milestone of the 20th century in metabolic control? Biochem J. (2010) 32:1–5. doi: 10.1042/BJ20091921
71. Pilkis SJ, Claus TH, Kurland IJ, Lange AJ. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a metabolic signaling enzyme. Annu Rev Biochem. (1995) 64:799–835. doi: 10.1146/annurev.bi.64.070195.004055
72. Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life (2010) 62:791–6. doi: 10.1002/iub.393
73. Riera L, Manzano A, Navarro-Sabaté A, Perales JC, Bartrons R. Insulin induces PFKFB3 gene expression in HT29 human colon adenocarcinoma cells. Biochim Biophys Acta (2002) 1589:89–92. doi: 10.1016/S0167-4889(02)00169-6
74. Rousseau GG, Hue L. Mammalian 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a bifunctional enzyme that controls glycolysis. Prog Nucleic Acid Res Mol Biol. (1993) 45:99–127. doi: 10.1016/S0079-6603(08)60868-5
75. Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J. (2004) 381:561–79. doi: 10.1042/BJ20040752
76. Okar DA, Manzano A, Navarro-Sabatè A, Riera L, Bartrons R, Lange AJ. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci. (2001) 26:30–5. doi: 10.1016/S0968-0004(00)01699-6
77. Jedrzejas MJ. Structure, function, and evolution of phosphoglycerate mutases: comparison with fructose-2,6-bisphosphatase, acid phosphatase, and alkaline phosphatase. Prog Biophys Mol Biol. (2000) 73:263–87. doi: 10.1016/S0079-6107(00)00007-9
78. Goren N, Manzano A, Riera L, Ambrosio S, Ventura F, Bartrons R. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase expression in rat brain during development. Brain Res Mol Brain Res. (2000) 75:138–42. doi: 10.1016/S0169-328X(99)00319-8
79. Darville MI, Crepin KM, Vandekerckhove J, Van Damme J, Octave JN, Rider MH, et al. Complete nucleotide sequence coding for rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase derived from a cDNA clone. FEBS Lett. (1987) 224:317–21. doi: 10.1016/0014-5793(87)80476-3
80. Algaier J, Uyeda K. Molecular cloning, sequence analysis, and expression of a human liver cDNA coding for fructose-6-P,2-kinase:fructose-2,6-bisphosphatase. Biochem Biophys Res Commun. (1988) 153:328–33. doi: 10.1016/S0006-291X(88)81226-9
81. Lee Y-H, Li Y, Uyeda K, Hasemann CA. Tissue-specific structure/function differentiation of the liver isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. J Biol Chem. (2003) 278:523–30. doi: 10.1074/jbc.M209105200
82. Bartrons R, Hue L, Van Schaftingen E, Hers HG. Hormonal control of fructose 2,6-bisphosphate concentration in isolated rat hepatocytes. Biochem J. (1983) 214:829–37. doi: 10.1042/bj2140829
83. Cosin-Roger J, Vernia S, Alvarez MS, Cucarella C, Boscá L, Martin-Sanz P, et al. Identification of a novel Pfkfb1 mRNA variant in rat fetal liver. Biochem Biophys Res Commun. (2013) 431:36–40. doi: 10.1016/j.bbrc.2012.12.109
84. Heine-Suñer D, Díaz-Guillén MA, Lange AJ, Rodríguez de Córdoba S. Sequence and structure of the human 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase heart isoform gene (PFKFB2). Eur J Biochem. (1998) 254:103–10.
85. Crochet RB, Kim J-D, Lee H, Yim Y-S, Kim S-G, Neau D, et al. Crystal structure of heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and the inhibitory influence of citrate on substrate binding. Proteins (2017) 85:117–24. doi: 10.1002/prot.25204
86. Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. (2003) 554:264–70. doi: 10.1016/S0014-5793(03)01179-7
87. Bobarykina AY, Minchenko DO, Opentanova IL, Moenner M, Caro J, Esumi H, et al. Hypoxic regulation of PFKFB-3 and PFKFB-4 gene expression in gastric and pancreatic cancer cell lines and expression of PFKFB genes in gastric cancers. Acta Biochim Pol. (2006) 53:789–99.
88. Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. (2000) 10:1247–55. doi: 10.1016/S0960-9822(00)00742-9
89. Masters SC, Subramanian RR, Truong A, Yang H, Fujii K, Zhang H, et al. Survival-promoting functions of 14-3-3 proteins. Biochem Soc Trans. (2002) 30:360–5. doi: 10.1042/bst0300360
90. Pozuelo Rubio M, Peggie M, Wong BHC, Morrice N, MacKintosh C. 14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase. EMBO J. (2003) 22:3514–23. doi: 10.1093/emboj/cdg363
91. Houles T, Gravel S-P, Lavoie G, Shin S, Savall M, Méant A, et al. RSK regulates PFK-2 activity to promote metabolic rewiring in melanoma. Cancer Res. (2018) 78:2191–204. doi: 10.1158/0008-5472.CAN-17-2215
92. Novellasdemunt L, Tato I, Navarro-Sabate A, Ruiz-Meana M, Méndez-Lucas A, Perales JC, et al. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J Biol Chem. (2013) 288:10640–51. doi: 10.1074/jbc.M113.455998
93. Kitamura K, Kangawa K, Matsuo H, Uyeda K. Phosphorylation of myocardial fructose-6-phosphate,2-kinase: fructose-2,6-bisphosphatase by cAMP-dependent protein kinase and protein kinase C. Activation by phosphorylation and amino acid sequences of the phosphorylation sites. J Biol Chem. (1988) 263:16796–801.
94. Schmidt S, Rainer J, Riml S, Ploner C, Jesacher S, Achmüller C, et al. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood. (2006) 107:2061–9. doi: 10.1182/blood-2005-07-2853
95. Carlet M, Janjetovic K, Rainer J, Schmidt S, Panzer-Grümayer R, Mann G, et al. Expression, regulation and function of phosphofructo-kinase/fructose-biphosphatases (PFKFBs) in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia cells. BMC Cancer (2010) 10:638. doi: 10.1186/1471-2407-10-638
96. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. (2011) 30:2719–33. doi: 10.1038/emboj.2011.158
97. Moon J-S, Jin W-J, Kwak J-H, Kim H-J, Yun M-J, Kim J-W, et al. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J. (2011) 433:225–33. doi: 10.1042/BJ20101104
98. Ji D, Lu Z-T, Li Y-Q, Liang Z-Y, Zhang P-F, Li C, et al. MACC1 expression correlates with PFKFB2 and survival in hepatocellular carcinoma. Asian Pac J Cancer Prev. (2014) 15:999–1003. doi: 10.7314/APJCP.2014.15.2.999
99. Zhao S-J, Shen Y-F, Li Q, He Y-J, Zhang Y-K, Hu L-P, et al. SLIT2/ROBO1 axis contributes to the Warburg effect in osteosarcoma through activation of SRC/ERK/c-MYC/PFKFB2 pathway. Cell Death Dis. (2018) 9:390. doi: 10.1038/s41419-018-0419-y
100. Zhao L, Ji G, Le X, Wang C, Xu L, Feng M, et al. Long noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. (2017) 77:1369–82. doi: 10.1158/0008-5472.CAN-16-1615
101. He Z, You C, Zhao D. Long non-coding RNA UCA1/miR-182/PFKFB2 axis modulates glioblastoma-associated stromal cells-mediated glycolysis and invasion of glioma cells. Biochem Biophys Res Commun. (2018) 500:569–76. doi: 10.1016/j.bbrc.2018.04.091
102. Chan O, Burke JD, Gao DF, Fish EN. The chemokine CCL5 regulates glucose uptake and AMP kinase signaling in activated T cells to facilitate chemotaxis. J Biol Chem. (2012) 287:29406–16. doi: 10.1074/jbc.M112.348946
103. Ventura F, Ambrosio S, Bartrons R, El-Maghrabi MR, Lange AJ, Pilkis SJ. Cloning and expression of a catalytic core bovine brain 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Biochem Biophys Res Commun. (1995) 209:1140–8. doi: 10.1006/bbrc.1995.1616
104. Hamilton JA, Callaghan MJ, Sutherland RL, Watts CK. Identification of PRG1, a novel progestin-responsive gene with sequence homology to 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Mol Endocrinol. (1997) 11:490–502. doi: 10.1210/mend.11.4.9909
105. Manzano A, Rosa JL, Ventura F, Pérez JX, Nadal M, Estivill X, et al. Molecular cloning, expression, and chromosomal localization of a ubiquitously expressed human 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase gene (PFKFB3). Cytogenet Cell Genet. (1998) 83(3–4):214–7.
106. Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, et al. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci USA. (1999) 96:3047–52. doi: 10.1073/pnas.96.6.3047
107. Navarro-Sabaté A, Manzano A, Riera L, Rosa JL, Ventura F, Bartrons R. The human ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene (PFKFB3): promoter characterization and genomic structure. Gene (2001) 264:131–8. doi: 10.1016/S0378-1119(00)00591-6
108. Kessler R, Eschrich K. Splice isoforms of ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in human brain. Brain Res Mol Brain Res. (2001) 87:190–5. doi: 10.1016/S0169-328X(01)00014-6
109. Ge X, Lyu P, Cao Z, Li J, Guo G, Xia W, et al. Overexpression of miR-206 suppresses glycolysis, proliferation and migration in breast cancer cells via PFKFB3 targeting. Biochem Biophys Res Commun. (2015) 463:1115–21. doi: 10.1016/j.bbrc.2015.06.068
110. Du J-Y, Wang L-F, Wang Q, Yu L-D. miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol Rep. (2015) 33:1890–8. doi: 10.3892/or.2015.3797
111. Obach M, Navarro-Sabaté A, Caro J, Kong X, Duran J, Gómez M, et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem. (2004) 279:53562–70. doi: 10.1074/jbc.M406096200
112. Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, et al. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J Biol Chem. (2002) 277:6183–7. doi: 10.1074/jbc.M110978200
113. Novellasdemunt L, Obach M, Millán-Ariño L, Manzano A, Ventura F, Rosa JL, et al. Progestins activate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in breast cancer cells. Biochem J. (2012) 442:345–56. doi: 10.1042/BJ20111418
114. Imbert-Fernandez Y, Clem BF, O'Neal J, Kerr DA, Spaulding R, Lanceta L, et al. Estradiol stimulates glucose metabolism via 6-phosphofructo-2-kinase (PFKFB3). J Biol Chem. (2014) 289:9440–8. doi: 10.1074/jbc.M113.529990
115. Novellasdemunt L, Bultot L, Manzano A, Ventura F, Rosa JLL, Vertommen D, et al. PFKFB3 activation in cancer cells by the p38/MK2 pathway in response to stress stimuli. Biochem J. (2013) 452:531–43. doi: 10.1042/BJ20121886
116. Ando M, Uehara I, Kogure K, Asano Y, Nakajima W, Abe Y, et al. Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J Nippon Med Sch. (2010) 77:97–105. doi: 10.1272/jnms.77.97
117. Han J, Meng Q, Xi Q, Zhang Y, Zhuang Q, Han Y, et al. Interleukin-6 stimulates aerobic glycolysis by regulating PFKFB3 at early stage of colorectal cancer. Int J Oncol. (2015) 48:215–24. doi: 10.3892/ijo.2015.3225
118. Ruiz-Garcia A, Monsalve E, Novellasdemunt L, Navarro-Sabate A, Manzano A, Rivero S, et al. Cooperation of adenosine with macrophage toll-4 receptor agonists leads to increased glycolytic flux through the enhanced expression of PFKFB3 gene. J Biol Chem. (2011) 286:19247–58. doi: 10.1074/jbc.M110.190298
119. Houddane A, Bultot L, Novellasdemunt L, Johanns M, Gueuning M-A, Vertommen D, et al. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell Signal (2017) 34:23–37. doi: 10.1016/j.cellsig.2017.02.019
120. Simon-Molas H, Arnedo-Pac C, Fontova P, Vidal-Alabró A, Castaño E, Rodríguez-García A, et al. PI3K-Akt signaling controls PFKFB3 expression during human T-lymphocyte activation. Mol Cell Biochem. (2018) [Epub ahead of print]. doi: 10.1007/s11010-018-3325-9
121. Rodríguez-García A, Samsó P, Fontova P, Simon-Molas H, Manzano A, Castaño E, et al. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. (2017) 284:3437–54. doi: 10.1111/febs.14201
122. Sakakibara R, Uemura M, Hirata T, Okamura N, Kato M. Human placental fructose-6-phosphate,2-kinase/fructose-2,6-bisphosphatase: its isozymic form, expression and characterization. Biosci Biotechnol Biochem. (1997) 61:1949–52. doi: 10.1271/bbb.61.1949
123. Cavalier MC, Kim S-G, Neau D, Lee Y-H. Molecular basis of the fructose-2,6-bisphosphatase reaction of PFKFB3: transition state and the C-terminal function. Proteins (2012) 80:1143–53. doi: 10.1002/prot.24015
124. Marsin A-S, Bouzin C, Bertrand L, Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem. (2002) 277:30778–83. doi: 10.1074/jbc.M205213200
125. Okamura N, Sakakibara R. A common phosphorylation site for cyclic AMP-dependent protein kinase and protein kinase C in human placental 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Biosci Biotechnol Biochem. (1998) 62:2039–42. doi: 10.1271/bbb.62.2039
126. Duran J, Obach M, Navarro-Sabate A, Manzano A, Gómez M, Rosa JL, et al. Pfkfb3 is transcriptionally upregulated in diabetic mouse liver through proliferative signals. FEBS J. (2009) 276:4555–68. doi: 10.1111/j.1742-4658.2009.07161.x
127. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VCJ, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149:49–62. doi: 10.1016/j.cell.2012.02.030
128. Seo M, Lee Y-H. PFKFB3 regulates oxidative stress homeostasis via its S-glutathionylation in cancer. J Mol Biol. (2014) 426:830–42. doi: 10.1016/j.jmb.2013.11.021
129. Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. (2014) 5:3480. doi: 10.1038/ncomms4480
130. Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, et al. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. (2016) 6:38067. doi: 10.1038/srep38067
131. Riera L, Obach M, Navarro-Sabaté A, Duran J, Perales JC, Viñals F, et al. Regulation of ubiquitous 6-phosphofructo-2-kinase by the ubiquitin-proteasome proteolytic pathway during myogenic C2C12 cell differentiation. FEBS Lett. (2003) 550:23–9. doi: 10.1016/S0014-5793(03)00808-1
132. Almeida A, Bolaños JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci USA. (2010) 107:738–41. doi: 10.1073/pnas.0913668107
133. Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH, Moncada S. Molecular architecture of the DNA replication origin activation checkpoint. EMBO J. (2011) 108:3381–94. doi: 10.1073/pnas.1102247108
134. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. (2011) 144:187–99. doi: 10.1016/j.cell.2010.12.020
135. Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol. (2009) 86:174–9. doi: 10.1016/j.yexmp.2009.01.003
136. Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O'Neal J, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. (2014) 5:e1337. doi: 10.1038/cddis.2014.292
137. Jia W, Zhao X, Zhao L, Yan H, Li J, Yang H, et al. Non-canonical roles of PFKFB3 in regulation of cell cycle through binding to CDK4. Oncogene (2018) 37:1685–98. doi: 10.1038/s41388-017-0072-4
138. Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. (2015) 35:1463–71. doi: 10.1161/ATVBAHA.115.305551
139. Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi M, et al. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br J Pharmacol. (2017) 174:893–908. doi: 10.1111/bph.13762
140. Chesney J. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care. (2006) 9:535–9. doi: 10.1097/01.mco.0000241661.15514.fb
141. Novellasdemunt L, Navarro-Sabaté À, Manzano A, Rodríguez-García A, Bartrons R. PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3). Atlas Genet Cytogenet Oncol Haematol. (2013) 17:609–22.
142. Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. (2002) 62:5881–7.
143. Marotta LLC, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest. (2011) 121:2723–35. doi: 10.1172/JCI44745
144. Han J, Meng Q, Xi Q, Wang H, Wu G. PFKFB3 was overexpressed in gastric cancer patients and promoted the proliferation and migration of gastric cancer cells. Cancer Biomarkers (2017) 18:249–56. doi: 10.3233/CBM-160143
145. Reddy MM, Fernandes MS, Deshpande A, Weisberg E, Inguilizian HV, Abdel-Wahab O, et al. The JAK2V617F oncogene requires expression of inducible phosphofructokinase/fructose-bisphosphatase 3 for cell growth and increased metabolic activity. Leukemia. (2012) 26:481–9. doi: 10.1038/leu.2011.225
146. Zhu Y, Lu L, Qiao C, Shan Y, Li H, Qian S, et al. Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor. Oncogene (2018) 37:2837–49. doi: 10.1038/s41388-018-0157-8
147. Feng Y, Wu L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem Biophys Res Commun. (2017) 483:897–903. doi: 10.1016/j.bbrc.2017.01.031
148. Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. (2005) 11:5784–92. doi: 10.1158/1078-0432.CCR-05-0149
149. Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu R-M, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. (2015) 192:1462–74. doi: 10.1164/rccm.201504-0780OC
150. Kessler R, Bleichert F, Warnke J-P, Eschrich K. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is up-regulated in high-grade astrocytomas. J Neurooncol. (2008) 86:257–64. doi: 10.1007/s11060-007-9471-7
151. Zscharnack K, Kessler R, Bleichert F, Warnke JP, Eschrich K. The PFKFB3 splice variant UBI2K4 is downregulated in high-grade astrocytomas and impedes the growth of U87 glioblastoma cells. Neuropathol Appl Neurobiol. (2009) 35:566–78. doi: 10.1111/j.1365-2990.2009.01027.x
152. Fleischer M, Kessler R, Klammer A, Warnke J-P, Eschrich K. LOH on 10p14-p15 targets the PFKFB3 gene locus in human glioblastomas. Genes Chromosom Cancer (2011) 50:1010–20. doi: 10.1002/gcc.20914
153. Schoors S, De Bock K, Cantelmo ARR, Georgiadou M, Ghesquière B, Cauwenberghs S, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. (2014) 19:37–48. doi: 10.1016/j.cmet.2013.11.008
154. Cantelmo AR, Conradi L-C, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell (2016) 30:968–85. doi: 10.1016/j.ccell.2016.10.006
155. Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkänen H-K, et al. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. (2018) 46:1124–38. doi: 10.1093/nar/gkx1155
156. Zhang H-S, Du G-Y, Zhang Z-G, Zhou Z, Sun H-L, Yu X-Y, et al. NRF2 facilitates breast cancer cell growth via HIF1α-mediated metabolic reprogramming. Int J Biochem Cell Biol. (2018) 95:85–92. doi: 10.1016/j.biocel.2017.12.016
157. Shi W-K, Zhu X-D, Wang C-H, Zhang Y-Y, Cai H, Li X-L, et al. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis. (2018) 9:428. doi: 10.1038/s41419-018-0435-y
158. Li S, Dai W, Mo W, Li J, Feng J, Wu L, et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int J Cancer (2017) 141:2571–84. doi: 10.1002/ijc.31022
159. Calvo MN, Bartrons R, Castaño E, Perales JC, Navarro-Sabaté A, Manzano A. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett. (2006) 580:3308–14. doi: 10.1016/j.febslet.2006.04.093
160. Telang S, Yalcin A, Clem AL, Bucala R, Lane AN, Eaton JW, et al. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene (2006) 25:7225–34. doi: 10.1038/sj.onc.1209709
161. Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. (2013) 12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097
162. Sakata J, Abe Y, Uyeda K. Molecular cloning of the DNA and expression and characterization of rat testes fructose-6-phosphate,2-kinase:fructose-2,6-bisphosphatase. J Biol Chem. (1991) 266:15764–70.
163. Manzano A, Pérez JX, Nadal M, Estivill X, Lange A, Bartrons R. Cloning, expression and chromosomal localization of a human testis 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene. Gene (1999) 229:83–9.
164. Minchenko DO, Mykhalchenko VG, Tsuchihara K, Kanehara S, Yavorovsky OP, Zavgorodny IV, et al. Alternative splice variants of rat 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase-4 mRNA. Ukr Biokhim Zh (1999) (2008) 80:66–73.
165. Gómez M, Manzano A, Navarro-Sabaté A, Duran J, Obach M, Perales JC, et al. Specific expression of pfkfb4 gene in spermatogonia germ cells and analysis of its 5′-flanking region. FEBS Lett. (2005) 579:357–62. doi: 10.1016/j.febslet.2004.11.096
166. Gómez M, Manzano A, Figueras A, Viñals F, Ventura F, Rosa JL, et al. Sertoli-secreted FGF-2 induces PFKFB4 isozyme expression in mouse spermatogenic cells by activation of the MEK/ERK/CREB pathway. Am J Physiol Endocrinol Metab. (2012) 303:E695–707. doi: 10.1152/ajpendo.00381.2011
167. Minchenko O, Opentanova I, Minchenko D, Ogura T, Esumi H. Hypoxia induces transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1α activation. FEBS Lett. (2004) 576:14–20. doi: 10.1016/j.febslet.2004.08.053
168. Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. (2014) 20:13705. doi: 10.3748/wjg.v20.i38.13705
169. Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. (2012) 2:328–43. doi: 10.1158/2159-8290.CD-11-0234
170. Chesney J, Clark J, Klarer AC, Imbert-Fernandez Y, Lane AN, Telang S. Fructose-2,6-bisphosphate synthesis by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) is required for the glycolytic response to hypoxia and tumor growth. Oncotarget (2014) 5:6670–86. doi: 10.18632/oncotarget.2213
171. Zhang H, Lu C, Fang M, Yan W, Chen M, Ji Y, et al. HIF-1α activates hypoxia-induced PFKFB4 expression in human bladder cancer cells. Biochem Biophys Res Commun. (2016) 476:146–52. doi: 10.1016/j.bbrc.2016.05.026
172. Yun SJ, Jo S-W, Ha Y-S, Lee O-J, Kim WT, Kim Y-J, et al. PFKFB4 as a prognostic marker in non-muscle-invasive bladder cancer. Urol Oncol. (2012) 30:893–9. doi: 10.1016/j.urolonc.2010.08.018
173. Jeon YK, Yoo DR, Jang YH, Jang SY, Nam MJ. Sulforaphane induces apoptosis in human hepatic cancer cells through inhibition of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase4, mediated by hypoxia inducible factor-1-dependent pathway. Biochim Biophys Acta (2011) 1814:1340–8. doi: 10.1016/j.bbapap.2011.05.015
174. Li B, Takeda K, Ishikawa K, Yoshizawa M, Sato M, Shibahara S, et al. Coordinated expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 and heme oxygenase 2: evidence for a regulatory link between glycolysis and heme catabolism. Tohoku J Exp Med. (2012) 228:27–41. doi: 10.1620/tjem.228.27
175. Shu Y, Lu Y, Pang X, Zheng W, Huang Y, Li J, et al. Phosphorylation of PPARγ at Ser84 promotes glycolysis and cell proliferation in hepatocellular carcinoma by targeting PFKFB4. Oncotarget (2016) 7:76984–94. doi: 10.18632/oncotarget.12764
176. Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S, et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene (2012) 31:3235–43. doi: 10.1038/onc.2011.490
177. Ros S, Flöter J, Kaymak I, Da Costa C, Houddane A, Dubuis S, et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene (2017) 36:3287–99. doi: 10.1038/onc.2016.477
178. Li W, Cohen A, Sun Y, Squires J, Braas D, Graeber TG, et al. The role of CD44 in glucose metabolism in prostatic small cell neuroendocrine carcinoma. Mol Cancer Res. (2016) 14:344–53. doi: 10.1158/1541-7786.MCR-15-0466
179. Li W, Qian L, Lin J, Huang G, Hao N, Wei X, et al. CD44 regulates prostate cancer proliferation, invasion and migration via PDK1 and PFKFB4. Oncotarget (2017) 8:65143–51. doi: 10.18632/oncotarget.17821
180. Strohecker AM, Joshi S, Possemato R, Abraham RT, Sabatini DM, White E. Identification of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase as a novel autophagy regulator by high content shRNA screening. Oncogene (2015) 34:5662–76. doi: 10.1038/onc.2015.23
181. Wang Q, Zeng F, Sun Y, Qiu Q, Zhang J, Huang W, et al. Etk interaction with PFKFB4 modulates chemoresistance of small-cell lung cancer by regulating autophagy. Clin Cancer Res. (2018) 24:950–62. doi: 10.1158/1078-0432.CCR-17-1475
182. Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature (2018) 556:249–54. doi: 10.1038/s41586-018-0018-1
183. Jen K-Y, Cheung VG. Identification of novel p53 target genes in ionizing radiation response. Cancer Res. (2005) 65:7666–73. doi: 10.1158/0008-5472.CAN-05-1039
184. Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
185. Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S, et al. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia (2009) 23:2090–101. doi: 10.1038/leu.2009.171
186. Peña-Rico MA, Calvo-Vidal MN, Villalonga-Planells R, Martínez-Soler F, Giménez-Bonafé P, Navarro-Sabaté À, et al. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol. (2011) 101:132–9. doi: 10.1016/j.radonc.2011.07.002
187. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget (2011) 2:948–57. doi: 10.18632/oncotarget.389
188. Sinha S, Ghildiyal R, Mehta VS, Sen E. ATM-NFκB axis-driven TIGAR regulates sensitivity of glioma cells to radiomimetics in the presence of TNFα. Cell Death Dis. (2013) 4:e615. doi: 10.1038/cddis.2013.128
189. Simon-Molas H, Calvo-Vidal MN, Castaño E, Rodríguez-García A, Navarro-Sabaté À, Bartrons R, et al. Akt mediates TIGAR induction in HeLa cells following PFKFB3 inhibition. FEBS Lett. (2016) 590:2915–26. doi: 10.1002/1873-3468.12338
190. Zou S, Wang X, Deng L, Wang Y, Huang B, Zhang N, et al. CREB, another culprit for TIGAR promoter activity and expression. Biochem Biophys Res Commun. (2013) 439:481–6. doi: 10.1016/j.bbrc.2013.08.098
191. Zou S, Gu Z, Ni P, Liu X, Wang J, Fan Q. SP1 plays a pivotal role for basal activity of TIGAR promoter in liver cancer cell lines. Mol Cell Biochem. (2012) 359:17–23. doi: 10.1007/s11010-011-0993-0
192. Kimata M, Matoba S, Iwai-Kanai E, Nakamura H, Hoshino A, Nakaoka M, et al. p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. Am J Physiol Heart Circ Physiol. (2010) 299:H1908–16. doi: 10.1152/ajpheart.00250.2010
193. Rajendran R, Garva R, Ashour H, Leung T, Stratford I, Krstic-Demonacos M, et al. Acetylation mediated by the p300/CBP-associated factor determines cellular energy metabolic pathways in cancer. Int J Oncol. (2013) 42:1961–72. doi: 10.3892/ijo.2013.1907
194. Matoba S, Kang J-G, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 Regulates mitochondrial respiration. Science (2006) 312:1650–3. doi: 10.1126/science.1126863
195. Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). J Biol Chem. (2009) 284:1748–54. doi: 10.1074/jbc.M807821200
196. Rigden DJ. The histidine phosphatase superfamily: structure and function. Biochem J. (2008) 409:333–48. doi: 10.1042/BJ20071097
197. Boada J, Roig T, Perez X, Gamez A, Bartrons R, Cascante M, et al. Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. (2000) 480:261–4. doi: 10.1016/S0014-5793(00)01950-5
198. Gerin I, Noël G, Bolsée J, Haumont O, Van Schaftingen E, Bommer GT. Identification of TP53-induced glycolysis and apoptosis regulator (TIGAR) as the phosphoglycolate-independent 2,3-bisphosphoglycerate phosphatase. Biochem J. (2014) 458:439–48. doi: 10.1042/BJ20130841
199. Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci USA. (2012) 109:20491–6. doi: 10.1073/pnas.1206530109
200. Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. (2009) 28:3015–26. doi: 10.1038/emboj.2009.242
201. Li N, He Y, Wang L, Mo C, Zhang J, Zhang W, et al. D-galactose induces necroptotic cell death in neuroblastoma cell lines. J Cell Biochem. (2011) 112:3834–44. doi: 10.1002/jcb.23314
202. Kim S-J, Jung H-J, Lim C-J. Reactive oxygen species-dependent down-regulation of tumor suppressor genes PTEN, USP28, DRAM, TIGAR, and CYLD under oxidative stress. Biochem Genet. (2013) 51:901–15. doi: 10.1007/s10528-013-9616-7
203. Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. (2013) 23:310–22. doi: 10.1016/j.semcancer.2013.05.008
204. Zhang X, Qin Z, Wang J. The role of p53 in cell metabolism. Acta Pharmacol Sin. (2010) 31:1208–12. doi: 10.1038/aps.2010.151
205. Cui L, Song Z, Liang B, Jia L, Ma S, Liu X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep. (2016) 35:3639–47. doi: 10.3892/or.2016.4752
206. Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev Cell. (2013) 25:463–77. doi: 10.1016/j.devcel.2013.05.001
207. Won KY, Lim S-J, Kim GY, Kim YW, Han S-A, Song JY, et al. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum Pathol. (2012) 43:221–8. doi: 10.1016/j.humpath.2011.04.021
208. Liu J, Lu F, Gong Y, Zhao C, Pan Q, Ballantyne S, et al. High expression of synthesis of cytochrome c oxidase 2 and TP53-induced glycolysis and apoptosis regulator can predict poor prognosis in human lung adenocarcinoma. Hum Pathol. (2018) 77:54–62. doi: 10.1016/j.humpath.2017.12.029
209. Kim SH, Choi SI, Won KY, Lim S-J. Distinctive interrelation of p53 with SCO2, COX, and TIGAR in human gastric cancer. Pathol Res Pract. (2016) 212:904–10. doi: 10.1016/j.prp.2016.07.014
210. Lui VWY, Lau CPY, Cheung CSF, Ho K, Ng MHL, Cheng SH, et al. An RNA-directed nucleoside anti-metabolite, 1-(3-C-ethynyl-beta-d-ribo-pentofuranosyl)cytosine (ECyd), elicits antitumor effect via TP53-induced glycolysis and apoptosis regulator (TIGAR) downregulation. Biochem Pharmacol. (2010) 79:1772–80. doi: 10.1016/j.bcp.2010.02.012
211. Lui VWY, Wong EYL, Ho K, Ng PKS, Lau CPY, Tsui SKW, et al. Inhibition of c-Met downregulates TIGAR expression and reduces NADPH production leading to cell death. Oncogene (2011) 30:1127–34. doi: 10.1038/onc.2010.490
212. Ye L, Zhao X, Lu J, Qian G, Zheng JC, Ge S. Knockdown of TIGAR by RNA interference induces apoptosis and autophagy in HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun. (2013) 437:300–6. doi: 10.1016/j.bbrc.2013.06.072
213. Lin C-C, Cheng T-L, Tsai W-H, Tsai H-J, Hu K-H, Chang H-C, et al. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. (2012) 2:785. doi: 10.1038/srep00785
214. Madan E, Gogna R, Kuppusamy P, Bhatt M, Pati U, Mahdi AA. TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex. Br J Cancer (2012) 107:516–26. doi: 10.1038/bjc.2012.260
215. Yin L, Kosugi M, Kufe D. Inhibition of the MUC1-C oncoprotein induces multiple myeloma cell death by down-regulating TIGAR expression and depleting NADPH. Blood (2012) 119:810–6. doi: 10.1182/blood-2011-07-369686
216. López-Guerra M, Trigueros-Motos L, Molina-Arcas M, Villamor N, Casado FJ, Montserrat E, et al. Identification of TIGAR in the equilibrative nucleoside transporter 2-mediated response to fludarabine in chronic lymphocytic leukemia cells. Haematologica (2008) 93:1843–51. doi: 10.3324/haematol.13186
217. Wanka C, Steinbach JP, Rieger J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem. (2012) 287:33436–46. doi: 10.1074/jbc.M112.384578
218. Canaparo R, Varchi G, Ballestri M, Foglietta F, Sotgiu G, Guerrini A, et al. Polymeric nanoparticles enhance the sonodynamic activity of meso-tetrakis (4-sulfonatophenyl) porphyrin in an in vitro neuroblastoma model. Int J Nanomed. (2013) 8:4247–63. doi: 10.2147/IJN.S51070
219. Tang Y, Kwon H, Neel BA, Kasher-Meron M, Pessin J, Yamada E, et al. The fructose-2,6-bisphosphatase TIGAR suppresses NF-κB signaling by directly inhibiting the linear ubiquitin assembly complex LUBAC. J Biol Chem. (2018) 293:7578–91. doi: 10.1074/jbc.RA118.002727
220. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle (2010) 9:3515–33. doi: 10.4161/cc.9.17.12928
221. Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. (2011) 43:1045–51. doi: 10.1016/j.biocel.2011.01.023
222. Ko Y-H, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells. Cancer Biol Ther. (2011) 12:1085–97. doi: 10.4161/cbt.12.12.18671
223. Qian S, Li J, Hong M, Zhu Y, Zhao H, Xie Y, et al. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J Hematol Oncol. (2016) 9:128. doi: 10.1186/s13045-016-0360-4
224. Zhou X, Xie W, Li Q, Zhang Y, Zhang J, Zhao X, et al. TIGAR is correlated with maximal standardized uptake value on FDG-PET and survival in non-small cell lung cancer. PLoS ONE (2013) 8:e80576. doi: 10.1371/journal.pone.0080576
225. Pons G, Carreras J. Functional characterization of the enzymes with 2,3-bisphosphoglycerate phosphatase activity from pig skeletal muscle. Comp Biochem Physiol B (1986) 85:879–85. doi: 10.1016/0305-0491(86)90191-4
226. Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. (2011) 43:969–80. doi: 10.1016/j.biocel.2010.02.005
227. Tauler A, Gil J, Bartrons R, Carreras J. Levels of glycerate 2,3-P2, 2,3-bisphosphoglycerate synthase and 2,3-bisphosphoglycerate phosphatase activities in rat tissues. A method to quantify blood contamination of tissue extracts. Comp Biochem Physiol B (1987) 86:11–3. doi: 10.1016/0305-0491(87)90167-2
228. Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs (2008) 17:1533–45. doi: 10.1517/13543784.17.10.1533
229. El Mjiyad N, Caro-Maldonado A, Ramírez-Peinado S, Muñoz-Pinedo C. Sugar-free approaches to cancer cell killing. Oncogene (2011) 30:253–64. doi: 10.1038/onc.2010.466
230. Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. (2002) 64:993–8. doi: 10.1016/S0006-2952(02)01168-1
231. Semenza GL. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med. (2012) 18:534–43. doi: 10.1016/j.molmed.2012.08.001
232. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
233. Hu Y, Liu J, Huang H. Recent agents targeting HIF-1α for cancer therapy. J Cell Biochem. (2013) 114:498–509. doi: 10.1002/jcb.24390
234. Pelicano H, Martin DS, Xu R-H, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene (2006) 25:4633–46. doi: 10.1038/sj.onc.1209597
235. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, DiPaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2013) 71:523–30. doi: 10.1007/s00280-012-2045-1
236. Chesney J, Clark J, Lanceta L, Trent JO, Telang S. Targeting the sugar metabolism of tumors with a first-in-class 6-phosphofructo-2-kinase (PFKFB4) inhibitor. Oncotarget. (2015) 6:18001–11. doi: 10.18632/oncotarget.4534
237. Zhang Y, Chen F, Tai G, Wang J, Shang J, Zhang B, et al. TIGAR knockdown radiosensitizes TrxR1-overexpressing glioma in vitro and in vivo via inhibiting Trx1 nuclear transport. Sci Rep. (2017) 7:42928. doi: 10.1038/srep42928
238. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al., Study Alliance Leukemia. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. (2013) 369:111–21. doi: 10.1056/NEJMoa1300874
239. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
240. Curry J, Johnson J, Tassone P, Vidal MD, Menezes DW, Sprandio J, et al. Metformin effects on head and neck squamous carcinoma microenvironment: window of opportunity trial. Laryngoscope (2017) 127:1808–15. doi: 10.1002/lary.26489
241. Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Caveolae and signalling in cancer. Nat Rev Cancer (2015) 15:225–37. doi: 10.1038/nrc3915
Keywords: fructose 2, 6-bisphosphate, cancer metabolism, glycolysis, PFKFB isoenzymes, TIGAR, tumor microenvironment
Citation: Bartrons R, Simon-Molas H, Rodríguez-García A, Castaño E, Navarro-Sabaté À, Manzano A and Martinez-Outschoorn UE (2018) Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front. Oncol. 8:331. doi: 10.3389/fonc.2018.00331
Received: 07 June 2018; Accepted: 01 August 2018;
Published: 04 September 2018.
Edited by:
Michael Breitenbach, University of Salzburg, AustriaReviewed by:
Markus Schosserer, Universität für Bodenkultur Wien, AustriaJohannes A. Mayr, Paracelsus Medical University Salzburg, Austria
Copyright © 2018 Bartrons, Simon-Molas, Rodríguez-García, Castaño, Navarro-Sabaté, Manzano and Martinez-Outschoorn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramon Bartrons, rbartrons@ub.edu