 Pamela Barraza-Flores
Pamela Barraza-Flores Christina R. Bates
Christina R. Bates Dean J. Burkin
Dean J. Burkin- Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, NV, United States
Laminin-α2-related congenital muscular dystrophy (LAMA2-CMD) is a devastating neuromuscular disease caused by mutations in the LAMA2 gene. These mutations result in the complete absence or truncated expression of the laminin-α2 chain. The α2-chain is a major component of the laminin-211 and laminin-221 isoforms, the predominant laminin isoforms in healthy adult skeletal muscle. Mutations in this chain result in progressive skeletal muscle degeneration as early as neonatally. Laminin-211/221 is a ligand for muscle cell receptors integrin-α7β1 and α-dystroglycan. LAMA2 mutations are correlated with integrin-α7β1 disruption in skeletal muscle. In this review, we will summarize laminin-211/221 interactions with integrin-α7β1 in LAMA2-CMD muscle. Additionally, we will summarize recent developments using upregulation of laminin-111 in the sarcolemma of laminin-α2-deficient muscle. We will discuss potential mechanisms of action by which laminin-111 is able to prevent myopathy. These published studies demonstrate that laminin-111 is a disease modifier of LAMA2-CMD through different methods of delivery. Together, these studies show the potential for laminin-111 therapy as a novel paradigm for the treatment of LAMA2-CMD.
Introduction
Laminin-α2-related congenital muscular dystrophy (LAMA2-CMD), also known as merosin deficient congenital muscular dystrophy type 1A (MDC1A), is a genetic disease caused by mutations in the LAMA2 gene encoding the laminin-α2 protein. Severely affected patients present with neonatal hypotonia and delayed motor milestones. Few children achieve independent ambulation, while most develop respiratory insufficiency, proximal joint contractures, and scoliosis. Patients show elevated serum creatine kinase (CK) and inflammatory cell infiltration in muscle biopsies (Konkay et al., 2016). There is currently no cure or effective treatment for LAMA2-CMD.
Laminins are ≈900 kDa heterotrimer glycoproteins composed of α, β, and γ chains. They are expressed in several tissues including skeletal muscle. The main isoforms of laminin expressed in healthy adult skeletal muscle are laminin-211 and 221. Laminin-α2 chain is essential for the assembly of these laminins (review on laminin in skeletal muscle; Holmberg and Durbeej, 2013). The laminin-α2 chain is a 380-kDa protein composed of a 300-kDa N-terminal fragment non-covalently bonded to an 80-kDA C-terminal fragment. The N-terminal domain nucleates the association between other laminin-α2 chains and components of the muscle basal lamina including collagen IV and heparan sulfate proteoglycans (Timpl and Brown, 1996). The basal lamina, along with the fibrillar reticular lamina, is a component of the basal membrane or extracellular matrix (ECM; Sanes, 2003). The C-terminal domain is composed of five laminin G (LG) domains, important for cell receptor binding to, predominantly, integrin-α7β1 and the dystroglycan protein complex (DGC; Timpl and Brown, 1996; Figure 1A). These linkages modulate communication between the basal lamina of the ECM and muscle cell cytoskeleton. They also provide mechanical support and stabilization to the sarcolemma during muscle contraction.
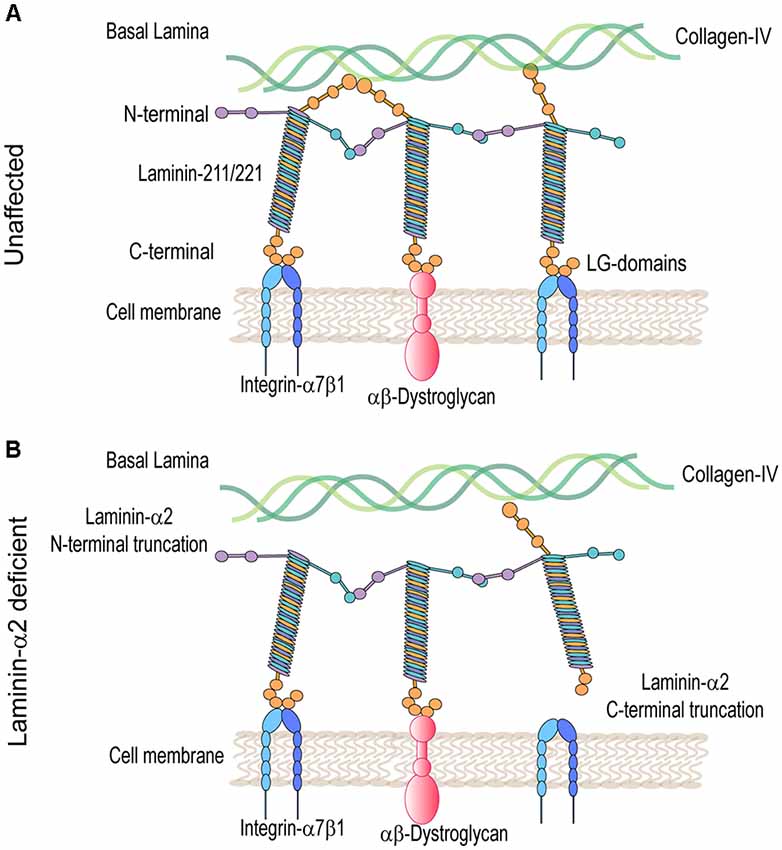
Figure 1. Molecular model in unaffected and laminin-α2-deficient patient muscle. (A) Laminin 211/221 heterotrimeric proteins bind through their N-terminal domain to the collagen-IV-rich basal lamina and through their C-terminal domain to muscle cell receptors heterodimers integrin-α7β1 and αβ-dystroglycan protein complexes. (B) In LAMA2-CMD, laminins are unable to bind to collagen-IV and/or integrin-α7β1 and α-dystroglycan protein complexes disrupting communication between the basal and muscle cell membrane.
Mutations in the LAMA2 gene result in various truncations of the laminin-α2 protein that can result in differential disease presentation and progression. Loss-of-function mutations in the LAMA2 gene are the most common cause of severe LAMA2-related congenital muscular dystrophy (LAMA2-CMD). These are mutations affecting the C-terminal domain, the N-terminal domain, or causing complete ablation of the laminin-α2 protein. Alternatively, variant missense mutations, in-frame deletions, and splice site mutations in the LAMA2 gene often result in a milder, limb-girdle-like, late-onset muscular dystrophy (Geranmayeh et al., 2010; Mohassel et al., 2018). Laminin-α2 deficiency causes disruption of the basal lamina, leading to increased susceptibility to mechanical stress and damage of the myofibers within the muscle. Loss of the laminin-211-rich microenvironment negatively impacts satellite cells (SC) and results in defects in muscle regeneration. Muscle damage results in an inflammatory cell infiltrate leading to replacement of functional skeletal muscle with fibrotic tissue, which exacerbates disease progression (Pegoraro et al., 1996; Nguyen et al., 2019).
Laminin-α2-deficient muscles in both mice and patients exhibit major transcriptome and proteome dysregulation. The major upregulated proteins are components of the ECM and proteins related to muscle regeneration (Taniguchi et al., 2006; van Lunteren et al., 2006; Yanay et al., 2019; Kölbel et al., 2019). The main laminin-α2 cell receptors, α-dystroglycan and integrin-α7β1, are also dysregulated in LAMA2-CMD (Figure 1B). Integrin-α7β1 plays an important role during SC activation, myoblast adhesion, and survival. Consequently, this integrin is important during embryonic development, regeneration, and repair of adult skeletal muscle. Integrin-α7β1 is found to be disrupted in laminin-α2-deficient muscle of multiple mouse models and human biopsies, and therefore, it was studied in LAMA2-CMD. Therapeutic approaches that aim to restore the basement membrane in LAMA2-CMD include LAMA2 gene replacement, engineered linker proteins mini-agrin and αLNND (Reinhard et al., 2017), and laminin-111 treatment. In this review, we will explore laminin-α2 and its receptor integrin-α7 in the LAMA2-CMD disease context, as well as laminin-111 as a disease modifier and therapeutic target for the treatment of LAMA2-CMD.
Integrin and Laminin Interaction in Laminin-α2-Deficient Skeletal Muscle
Integrins are heterodimeric transmembrane glycoprotein receptors made of non-covalently bound α and β subunits (Figure 2). They are essential for cell attachment to the ECM, cell migration, and regulate cellular signal transduction. Integrin clustering and ligand binding to the ECM, including binding to fibronectin, laminins, and collagen, induce conformational changes in integrin, regulating cell adhesion, proliferation, migration, and differentiation (Boppart and Mahmassani, 2019).
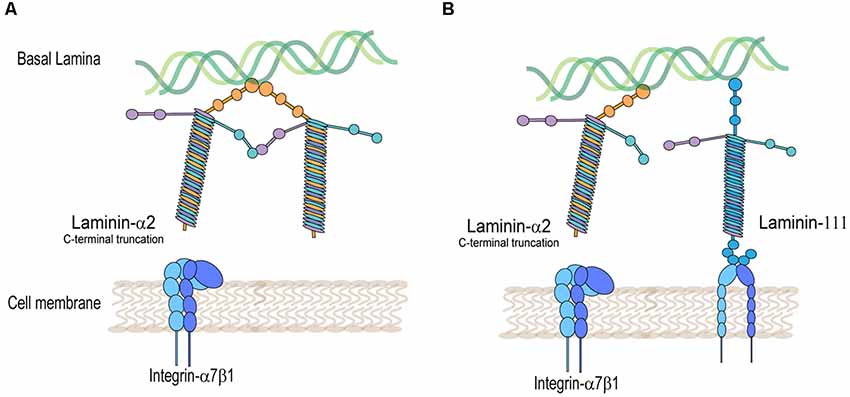
Figure 2. Integrin-α7β1 protein complex is disrupted in laminin-α2-deficient muscle. (A) Decreased detection of integrin-α7β1 in the muscle sarcolemma in laminin-α2-truncated muscle. (B) Rescued expression of integrin-α7β1 in the muscle sarcolemma by expression of laminin-111 in laminin-α2-deficient muscle.
Integrin-α7β1 plays a crucial role during embryonic development and in adult skeletal muscle repair by facilitating myoblast adhesion to laminin-111, -211, and -221 (Crawley et al., 1997). Integrin-α7 is encoded by the ITGA7 gene with multiple isoforms produced by developmentally regulated RNA splicing. Alternative RNA splicing of ITGA7 results in integrin-α7A and integrin-α7B cytoplasmic isoforms, as well as integrin-α7X1 and integrin-α7X2 extracellular domain isoforms. The cytoplasmicα7A domain is produced during terminal differentiation of myoblasts and is found in myofibers at the sarcolemma. The cytoplasmic domain α7B-integrin was originally identified during myoblast proliferation (Ziober et al., 1993). The extracellular domain isoform integrin-α7X1 binds strongly to laminin-411, laminin-511, and laminin-521, predominant during muscle embryonic development, while integrin α7X2 binds to laminin-111, expressed during early muscle development. Both integrin α7X1 and α7X2 isoforms bind to laminin-211. This suggests that integrin-α7X1 is involved during early embryogenesis, while integrin-α7X2 is involved at later stages of skeletal muscle development (von der Mark et al., 2002).
The main alternative isoforms for integrin-β1 include integrin β1A and β1D. Whereas integrins β1B and β1C are considered minor isoforms found only in humans, the integrin β1D subunit is expressed in skeletal muscle sarcolemma and associates with integrin-α7 subunit (Belkin et al., 1997).
Laminin-α2 is the main ligand for integrin-α7β1 in skeletal muscle. Integrin-α7 is localized at the neuromuscular and myotendinous junctions and throughout the muscle sarcolemma (Burkin and Kaufman, 1999). Targeted deletion of the integrin-α7 gene in mice results in a mild form of congenital muscular dystrophy and vascular defects (Mayer, 2003; Flintoff-Dye et al., 2005; Rooney et al., 2006). Histopathological studies of this mouse show variation in fiber size, a high percentage of centrally nucleated fibers, elevated CK, mononuclear cell infiltration, and little to no degeneration/regeneration cycles. Expression of laminin-α2 in this mouse, as well as expression of integrin-β1, is unaffected (Mayer et al., 1997). Consistent with this mouse model, patients identified as integrin-α7-negative and laminin-α2-positive present with mild myopathy from birth with no evidence of constituent muscle regeneration (Hayashi et al., 1998). This indicates that other integrin subunits that pair with integrin β1 can partially compensate for the loss of integrin-α7 in muscle.
On the other hand, mutations in the laminin-α2 protein disrupt the expression of integrin-α7β1 in skeletal muscle (Figure 2A). This was shown in several laminin-α2-deficient mouse models and human biopsies of LAMA2-CMD. Using immunofluorescence, studies typically show a decreased detection of integrin-α7B and integrin-β1 subunits in the sarcolemma of laminin-α2-deficient muscle. The dy/dy mouse model of LAMA2-CMD, which has low expression of full-length laminin-α2, exhibits reduced levels of integrin-α7A, integrin-β1D, and irregular integrin-α7B expression (Vachon et al., 1997). Similarly, the dy2J/dy2J mouse model of LAMA2-CMD, which has a mutation in the N-terminal domain, also shows a reduction in integrin-α7A and patchy expression in integrin-α7B (Hodges et al., 1997; Vachon et al., 1997; Cohn et al., 1999). Durbeej (2010) summarizes in a comprehensive review the different mouse models of LAMA2-CMD (Durbeej, 2010).
A case study using biopsies from congenital muscular dystrophy patients with laminin-α2 chain deficiency showed reduced integrin-α7B expression in the sarcolemma of six LAMA2-CMD patients. However, the level of laminin-α2 expression did not correlate with the level of integrin-α7β1 reduction. Restoration of laminin-α2 rescued expression of integrin-α7β1 in the sarcolemma (Vachon et al., 1997). Moreover, the integrin-α7 knockout mouse show no change in laminin-α2 levels (Mayer et al., 1997). Gawlik and Durbeej (2015) reported that loss of integrin-α7 and laminin-α2 in the dy3k/dy3k mouse model, did not result in more severe myopathy. This double knockout mouse presented similar levels of fibrosis, apoptosis, lifespan, and body weight. This same group had previously showed that loss of β-sarcoglycan or dystroglycan in laminin-α2-deficient dy3k/dy3k mice resulted in a dramatically worse muscle disease (Gawlik et al., 2014). This may indicate that the integrin-α7β1 complex has intersecting roles with laminin-α2, while the DGC complex, which includes β-sarcoglycan and dystrophin, may have non-redundant complementary roles. These observations indicate a strong dependence in the expression of integrin-α7β1 receptor to laminin-α2 ligand. This may also indicate that laminin-α2 ligand can compensate for its interaction with secondary integrin complexes.
Doe et al. (2011) showed improvements in myopathy in the dyW mouse model of LAMA2-CMD by transgenically overexpressing integrin-α7. The dyW model presents downregulated and N-terminally truncated levels of laminin-α2 along with severe, early myopathy (Guo et al., 2003). This study showed a threefold increase in lifespan, reduced CLN percent, and reduced macrophage infiltration (Doe et al., 2011). A previous study showed that overexpressing rat integrin-α7 in C2C12 myoblasts promoted adhesion to laminin and overall cell survival, while it did not affect differentiation (Liu et al., 2008). Increased myoblast adhesion to other laminin isoforms may explain the improvements in integrin-α7 overexpressing dyW mouse; however, this remains to be fully explored in vivo.
Other integrin dysregulation may drive disease progression in laminin-α2-deficient muscle. Integrin-αV, integrin-α5, integrin-β1, and integrin-β3 were shown to be upregulated in the dyW model of LAMA2-CMD (Accorsi et al., 2015). Integrin-αV was proposed to have a role in TGF-β-mediated fibrosis. More research investigating aberrant disruption of integrin receptors in laminin-α2-deficient skeletal muscle is needed to further our understanding of their roles in LAMA2-CMD disease progression.
Cell-Based Laminin-111 Treatment of Skeletal Muscle
Skeletal muscle has the capacity to regenerate due to the presence of resident stem cells also known as SCs. These cells are located between the sarcolemma and the basement membrane of muscle fibers (Mauro, 1961). SCs are myogenic progenitors activated in response to muscle injuries and trauma. Upon activation, they proliferate, migrate, and differentiate into myotubes fusing into damaged muscle fibers or generating de novo fibers (Morgan and Partridge, 2003; Lepper et al., 2011; Murphy et al., 2011; Sambasivan et al., 2011). There are two types of SC division: apical and symmetrical. Apical or asymmetric division gives rise to two daughter cells of which one is located basally with respect to the other cell, while undergoing differentiation. Symmetrical division, on the other hand, will give rise to two daughter cells that undergo quiescence and maintain the SC pool (Yin et al., 2013).
The ECM, while essential for maintaining muscle fiber integrity (Grounds et al., 2005), also has a crucial function in supporting SC proliferation, migration, and differentiation (Öcalan et al., 1988; Sanes, 2003; Silva Garcia et al., 2019). Disruption of the ECM surrounding SCs, also known as the SC niche, has negative impact on SCs that can lead to failed myogenesis and impaired muscle regeneration (Thomas et al., 2015). The major protein components of the ECM in this niche include collagen (Gillies and Lieber, 2011) and laminin (Yurchenco and Patton, 2009). Laminin-211 and laminin-221, previously known as merosin and merosin-S, are the most abundant laminin isoforms in the adult skeletal muscle (Nissinen et al., 1991; Sasaki et al., 2002). Disrupted levels of laminin-α2 in LAMA2-CMD compromises the integrity of the SC niche, affecting the overall regenerative capacity of the skeletal muscle (Mohassel et al., 2018; Yurchenco et al., 2018). An in vitro study by Summers and Parsons (1981) showed that SCs isolated from the dy/dy mouse model of LAMA2-CMD, presented an 80% reduction in muscle colony formation compared to the wild-type controls at 5 months of age. This defect was not shown neonatally or at 1 week of age.
Another in vitro study using LAMA2-deficient embryonic stem cells showed that these cells had no defect in differentiating into cardiomyocytes, smooth muscle, and myotubes compared to the wild type. However, the myotubes formed were unstable, detached, collapsed, and degenerated (Kuang et al., 1998a). They found no proliferation or differentiation defects but potential ineffective/abortive development of myotubes (Summers and Parsons, 1981). This points at the role for laminin-α2 in the maintenance of fully differentiated and mature myofibers, essential for myotube stability and survival. An in vivo study in mice showed that homozygous LacZ insertion in the LAMA2 gene, effectively disrupting LAMA2 expression, results in incomplete muscle repair and presence of immature myofibers. This study also showed that the early expression of laminin-α2 is important in early embryonic and adult myogenesis (Kuang et al., 1999). Treatment with laminin-α2 protein rescued the instability of LAMA2-CMD myotubes isolated from patient samples (Vachon et al., 1996). An in vivo study found that laminin-α2 expression increased levels of SCs in dystrophic gastrocnemius and triceps of dy/dy mice. This provided evidence on the importance of laminin-α2 in the process of skeletal muscle early myogenesis and regeneration of adult muscle. LAMA2 gene replacement in dyW/dyW and dy2J/dy2J mice was capable of restoring the overall health and lifespan of these mouse models of LAMA2-CMD and could serve as a potential therapeutic target (Kuang et al., 1998b). However, immune response against exogenous and now foreign laminin-α2 protein in LAMA2-CMD could impair the benefits of this therapy and exacerbate disease severity.
To address this issue, other laminin isoforms are the target of interest for the treatment of LAMA2-CMD. Laminin-α4 and laminin-α5 are present and upregulated in laminin-α2-deficient muscle (Patton et al., 1997; Kölbel et al., 2019). These laminin isoforms are involved in myogenesis. However, their increased expression in laminin-α2-deficient muscle does not fully rescue its regenerative capacity. Moreover, upon SC activation, an increase in laminin-α4 and laminin-α5 expression is detected, followed by a transient deposition of laminin-α1 in the SC niche (Ishii et al., 2018; Rayagiri et al., 2018). Loss of laminin-α1 impairs SC proliferation and self-renewal leading to defective muscle regeneration indicating its role in SC cycle. Laminin-α1 effects on SC are mediated through integrin-α6 cell receptor and promote SC apical cell division (Rayagiri et al., 2018).
Laminin-α1 contains the highest amino acid homology to laminin-α2 compared to all other laminin chains, and therefore, it is a therapeutic target for LAMA2-CMD. The Laminin-111 heterotrimer differs only in homologous α1 subunit compared to laminin-211. Laminin-111 can be purified from the Engelbreth–Holm–Swarm (EHS) murine sarcoma cell line, where it was first discovered. Several groups reported that in vitro treatment with EHS laminin-111 promotes myoblast survival while promoting proliferation and migration (Silva-Barbosa et al., 2008; Goudenege et al., 2010). In a biomaterial study, it was shown that increasing concentrations of laminin-111 integrated into fibrin hydrogels presented a dose-dependent myogenic marker effect. C2C12 myoblasts seeded onto fibrin hydrogel conjugated with a low concentration of laminin-111 showed an increase in myogenin transcription factor, promoting differentiation. At a ninefold increased concentration of laminin-111–fibrin hydrogel, C2C12s showed an increase in MyoD expression, promoting proliferation. This same study reported a dose-dependent increase in VEGF and decrease in IL-6 cytokine secretions effect after 4 days post-seeding onto laminin-111-supported fibrin gels (Marcinczyk et al., 2017). Another study developed hydrogels composed of poly-ethyl glycol conjugated with diacrylate (PEGDA) with and without 5% and 10% laminin-111 (PEGLM). The 5% PEGLM hydrogel showed a more porous structure compared to the other gels, indicating a more ECM-like structure. C2C12s were also seeded in each condition showing flat and spread out morphology in the 5% PEGLM ideal for myofiber formation, compared to rounded, multicellular clusters found in the other hydrogel conditions. A 5% PEGLM C2C12s showed a significant increase in EGF and IL-6 secretion as well as myogenin transcription factor (Ziemkiewicz et al., 2018).
Laminin-111–hydrogel studies show the potential for this biological to affect ECM morphology, alter myoblast differentiation and myokine secretion. Consistent with these studies, SCs expanded in vitro on laminin-111–hydrogel were later engrafted onto mdx mouse model of Duchenne muscular dystrophy (DMD) and more efficiently regenerated into the dystrophic muscle compared to cells expanded without laminin-111 (Ross et al., 2012). Further studies are necessary to characterize the SC niche and its role in normal muscle regeneration and in the context of LAMA2-CMD. These studies indicate the potential for laminin-111 to structurally support myogenic activity, activate cellular signaling pathways to promote survival, enhance SC engraftment, and promote regeneration in laminin-α2-deficient skeletal muscle.
Laminin-111 Protein Therapeutics in LAMA2-CMD Mouse Models
Several groups tested the hypothesis that upregulation of endogenous laminin-α1 or exogenous treatment with laminin-111 protein can alleviate disease progression in mouse models of DMD and LAMA2-CMD (Table 1). A study by Rooney et al. (2009) treated the mdx mouse model of DMD with weekly intraperitoneal doses of EHS laminin-111. Their results showed an increase in protein levels of integrin-α7 in mice and human DMD myoblasts. They also showed reduced CK, Evans Blue dye-positive fibers, and centrally located nuclei fibers, indicating an increase in sarcolemma stability in dystrophin-deficient muscle (Rooney et al., 2009). These studies were followed by intramuscular treatments with EHS laminin-111 in the golden retriever muscular dystrophy (GRMD) dog model of DMD. The GRMD dog model more closely recapitulates disease progression in DMD compared to the mdx mouse model (Kornegay, 2017). These studies showed an increase in muscle regeneration and repair and in vivo force measurements in the dog’s hindlimbs (Barraza-Flores et al., 2019).
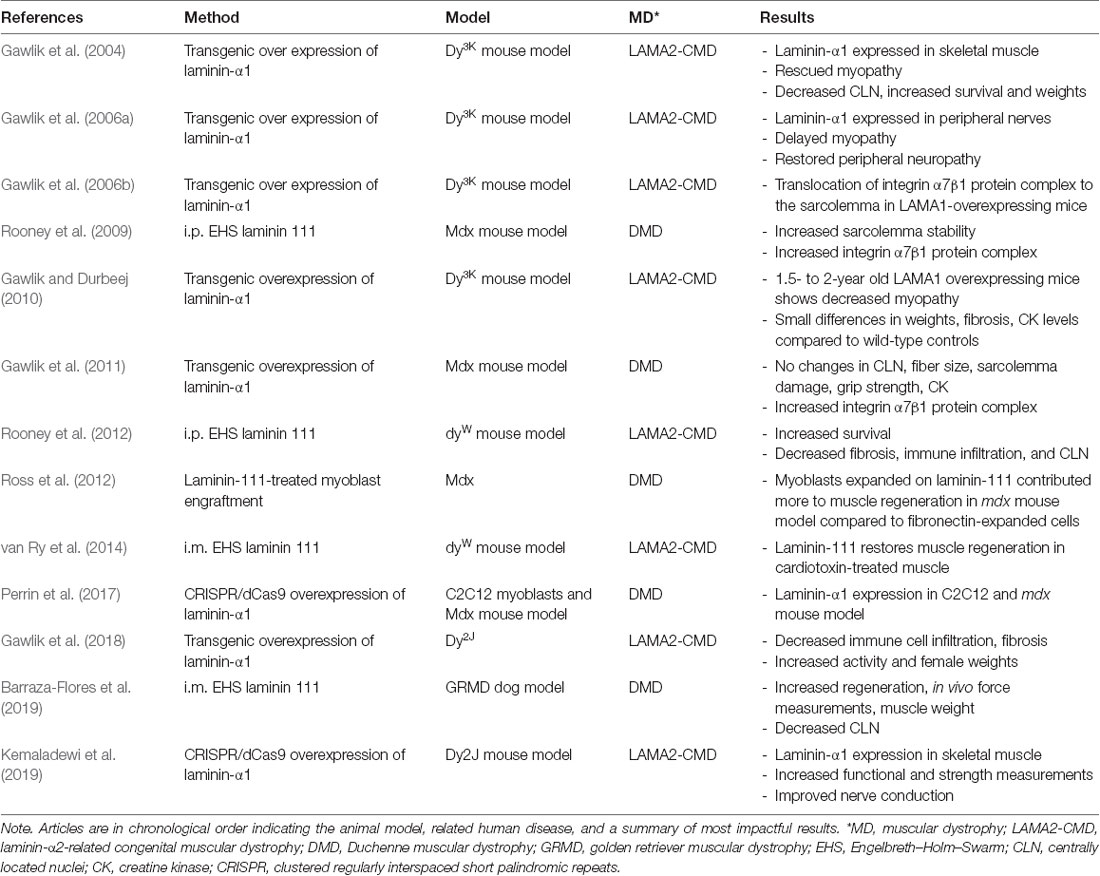
Table 1. Laminin-111 therapy articles in muscular dystrophy animal models.
Similarly, Rooney et al. (2012) treated the dyW mouse model of LAMA2-CMD, with weekly intraperitoneal injections of EHS laminin-111. These studies reported an increase in life-span and a decrease in centrally located nuclei fibers, fibrosis, and immune infiltration. In a subsequent study in dyW mouse, intramuscular injections with EHS laminin-111 showed improvements in muscle repair post cardiotoxin treatment of hindlimbs. Treated muscles showed increased integrin-α7β1 complex, increased myofiber size, and number (van Ry et al., 2014).
Other groups explored the transgenic overexpression of laminin-α1 in several mouse models of LAMA2-CMD and DMD. Overexpression of laminin-α1 results in proper assembly of laminin-111 and translocation to the sarcolemma as demonstrated by several transgenic mice using immunofluorescence. Gawlik et al. (2011) showed that the transgenic overexpression of laminin-α1 in the mdx mouse model of DMD, while increasing levels of integrin-α7β1, had little to no effect in myopathy. This could mean that the presence of endogenous laminin-211/221 in the mdx mice may reduce the efficacy of laminin-α1 transgenic expression. These studies may also indicate that in dystrophin-deficient muscle, exogenous treatment can repair sarcolemmal integrity, perhaps through signaling stimulated exclusively through an external laminin-111 receptor binding.
Transgenic overexpression of laminin-α1 was also achieved in two different models of LAMA2-CMD. Transgenic expression of laminin-α1 in dy3K mice showed a drastic increase in survival, weights, decreased centrally located nuclei, and alleviated myopathy (Gawlik et al., 2004). This model was also used to investigate the role integrin-α7β1 plays in laminin-α1 treatment of LAMA2-CMD. They found that the non-transgenic dy3K mouse already shows significantly increased synthesis of integrin-α7β1 (via transcript and protein levels) compared to the wild type. However, the integrin-α7 subunit increase was not detected in the skeletal muscle sarcolemma, indicating that it was not translocated from the cytoplasm to the sarcolemma. Interestingly, when laminin-α1 was transgenically overexpressed, integrin-α7 was detected in the sarcolemma of the muscle (Gawlik et al., 2006b) perhaps due to available ligand-binding receptors (Figure 2B). This provides a potential mechanism of action in which overexpression of integrin-α7 is able to prevent muscle disease in LAMA2-CMD. Additionally, aged dy3K mice overexpressing laminin-α1 had a lifespan of up to 1.5–2 years of age, and all non-transgenic dy3K mice died. These older laminin-α1 transgenic dy3K mice had improved survival; however, they had significantly lower weights, muscle strength, and exhibited muscle fibrosis compared to the wild type (Gawlik and Durbeej, 2010).
A new transgenic mouse able to express laminin-α1 in the peripheral nervous system of the dy3K mouse was reported. Results showed rescued peripheral neuropathy such as improved Schwann cell basement membrane and nerve conduction (Gawlik et al., 2006a). In order to show that laminin-111 is a disease modifier of LAMA2-CMD, laminin-α1 was overexpressed in a model with a different LAMA2 mutation, the dy2J. These studies provided a comparison between the most severe, laminin-α2 negative, and the less severe, laminin-α2-reduced mouse models. Transgenic dy2J mouse also showed laminin-α1 expression in the heart, skeletal muscle, and sciatic nerve. Functional assessments showed increased grip strength and activity, while histopathology revealed reduced central nucleation and fibrosis (Gawlik et al., 2018). Together, these studies indicate that laminin-111 can modify muscle disease progression in LAMA2-CMD independent of LAMA2 mutation and is a therapeutic target for LAMA2-CMD.
Ubiquitous expression of laminin-α1 in dystrophic skeletal muscle could be achieved through gene replacement using adeno-associated virus (AAV) delivery. However, this approach is currently limited by the ~10-kb size of the LAMA1 and LAMA2 cDNAs. Production of micro-laminin-α2 retains the activity if the full protein is a possibility, but challenges include producing constructs that successfully associate with laminin-β1, laminin-β2, and laminin-γ1 chains and are functional.
Recent advancements in CRISPR/Cas9 technology showed the capacity for catalytically inactive dCas9 (or dead Cas9) nuclease to upregulate genes of interest by activating to gene promoters. A recent study in the dy2J mouse model used the Streptococcus pyogenes dead Cas9 (sadCas9) and fused it to eight copies of the transcriptional activator VP16. Five guide RNAs (gRNAs) were designed to target upstream LAMA1 promoter. CRISPR and VP64–sadCas9 along with gRNAs were packaged into AAVs and delivered in vitro. A combination of gRNAs was found optimal for the upregulation of LAMA1 and tested in vivo. Laminin-α1 was successfully upregulated in the skeletal muscle and sciatic nerve of dy2J mice via LAMA1 promoter activation. Histopathology and functional studies showed reduced fibrosis, increased fiber diameters, and myelination of sciatic nerves, as well as improvements in activity, force measurements, and nerve conduction after a single injection of AAV-packaged CRISPR/VP64–sadCas9 (Kemaladewi et al., 2019).
Potential problems of this approach include off-target activities, regulating CRISPR/VP64–sadCas9 activity so laminin-α1 expression is more precisely controlled, assessing the long-term impacts of CRISPR/Cas9 system in human muscle, potential requirement for multiple dosing, and immune response to the AAV delivery system. Nonetheless, once these limitations are addressed, this approach represents an exciting new frontier for the treatment of LAMA2-CMD.
Future Perspectives
Laminin-211/221 is the main ligand for muscle cell receptors: the dystroglycan complex (DGC) and integrin-α7β1 heterocomplex. Therefore, mutations in laminin-α2 lead to disrupted communication between the basal lamina and muscle cell receptors. These interactions are detrimental for mechanical support during muscle contraction. Overexpression of integrin-α7 in a LAMA2-CMD mouse model was reported to reduce muscle disease progression in a mouse model of LAMA2-CMD. The observations indicate that overexpression of integrin-α7 can reestablish the linkage between muscle cells and laminin isoforms in laminin-211/221-deficient muscle. Possible approaches to restore integrin-α7β1 to the sarcolemma of LAMA2-CMD patients include AAV–ITGA7 expression. These studies were performed in two models of DMD and shown to improve muscle pathology and physiology (Heller et al., 2013, 2015). Problems with an AAV–ITGA7 approach include regulating ITGA7 expression in muscle and immune response to AAV delivery systems. Another approach is using small molecules that target an increase in integrin-α7. A muscle cell-based screen has recently identified SU9516 and a structural analog Sunitinib (an FDA-approved compound), which increase the α7β1 integrin in the muscle of mdx mice (Sarathy et al., 2017; Fontelonga et al., 2019). These small molecules could be used to restore the integrin-α7β1 linkage system to the sarcolemma and reduce muscle disease progression in LAMA2-CMD. However, this approach remains to be explored.
Laminin-111 was shown to be an effective protein replacement therapy for LAMA2-CMD by multiple groups (Table 1). EHS-derived laminin-111 was studied in multiple animal models with different forms of muscular dystrophy. Treatment with laminin-111 in LAMA2-CMD mouse model increased life expectancy, muscle function, and muscle regeneration and repair. Additionally, laminin-111 protein therapy may provide the unique ability to deliver different concentrations of the biological throughout the body. High local concentration of laminin-111 may enhance myoblast differentiation while maintaining their survival through myokine signaling. Low local laminin-111 concentrations may induce myogenic proliferation. However, the effects of laminin-111 on SCs and myogenic proliferation and differentiation require further investigation.
Translation of laminin-111 protein therapy from the bench to the bedside will require production of recombinant human laminin-111 protein. Human laminin-111 has 70% amino acid homology compared to the mouse isoform. Cross-species differences in amino acid sequence and/or glycosylation patterns may trigger an immune response in mice or humans. Treatment using EHS mouse laminin-111 in the GRMD dog model did stimulate an immune response, but appeared not to have any adverse effects in this large animal model of DMD. Potential problems for laminin-111 therapeutics include: (1) production of a 900-kDa recombinant human laminin-111 in sufficient quantity and purity to treat LAMA2-CMD patients; (2) potential immune response since multiple injections will be required throughout the lifespan of the patient; (3) care in administering the recombinant laminin to ensure it does not polymerize during intravenous treatments; and (4) determining dosing requirements and drug efficacy. Once these problems are resolved, then laminin-111 could serve as a potent therapy for LAMA2-CMD and other muscular dystrophies.
Finally, another approach to produce laminin-111 in skeletal muscle is via endogenous upregulation of laminin-α1 protein chain. Since the LAMA1 cDNA is ~10 kb, then delivery using AAV is not feasible. A potential solution may be upregulation via LAMA1 promoter stimulation using CRISPR/sadCas9–VP64 technology. Treatment with CRISPR/sadCas9–VP64 in the more severe laminin-α2 negative mouse model, the dy3k, is needed to assess the efficacy of this approach with congenital onset. Although CRISPR/sadCas9–VP64 may be the strongest potential approach to upregulate laminin-α1 in patients, delivery challenges may include immune response against AAV.
In the development of several approaches based on integrin-α7β1 regulation, laminin- and cell-based therapies, laminin-111 protein replacement therapy, or laminin-α1 enhancement, each represents novel approaches for LAMA2-CMD. Each approach has unique strengths and weaknesses that will require extensive testing and assessment before entering clinical trials. Finally, a combinatorial approach using each of these approaches may serve as the most effective approach for treating this devastating muscle disease.
Author Contributions
PB-F conceptualized and designed the article. PB-F, CB, and AO-S drafted the article. DB critically revised the article.
Funding
This work was supported by grants from NIH/NIAMS R01AR064338-01A1 and MDA628561 to DB. PB-F and CB were supported by Mick Hitchcock scholarships.
Conflict of Interest
The University of Nevada, Reno, has a patent on the therapeutic use of laminin-111 and its derivatives. This patent has been licensed to Prothelia Inc., and the University of Nevada, Reno has a small equity share in this company.
References
Accorsi, A., Mehuron, T., Kumar, A., Rhee, Y., and Girgenrath, M. (2015). Integrin dysregulation as a possible driver of matrix remodeling in laminin-deficient congenital muscular dystrophy (MDC1A). J. Neuromuscul. Dis. 2, 51–61. doi: 10.3233/jnd-140042
Barraza-Flores, P., Fontelonga, T. M., Wuebbles, R. D., Hermann, H. J., Nunes, A. M., Kornegay, J. N., et al. (2019). Laminin-111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of duchenne muscular dystrophy. Hum. Mol. Genet. 28, 2686–2695. doi: 10.1093/hmg/ddz086
Belkin, A. M., Retta, S. F., Pletjushkina, O. Y., Balzac, F., Silengo, L., Fassler, R., et al. (1997). Muscle B1D integrin reinforces the cytoskeleton-matrix link: modulation of integrin adhesive function by alternative splicing. J. Cell Biol. 139, 1583–1595. doi: 10.1083/jcb.139.6.1583
Boppart, M. D., and Mahmassani, Z. S. (2019). Integrin signaling: linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 317, C629–C641. doi: 10.1152/ajpcell.00009.2019
Burkin, D. J., and Kaufman, S. J. (1999). The α7β1 integrin in muscle development and disease. Cell Tissue Res. 296, 183–190. doi: 10.1007/s004410051279
Cohn, R. D., Mayer, U., Saher, G., Herrmann, R., Van Der Flier, A., Sonnenberg, A., et al. (1999). Secondary reduction of α7B integrin in laminin α2 deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J. Neurol. Sci. 163, 140–152. doi: 10.1016/s0022-510x(99)00012-x
Crawley, S., Farrell, E. M., Wang, W., Gu, M., Huang, H. Y., Huynh, V., et al. (1997). The α7β1 integrin mediates adhesion and migration of skeletal myoblasts on laminin. Exp. Cell Res. 235, 274–286. doi: 10.1006/excr.1997.3671
Doe, J. A., Wuebbles, R. D., Allred, E. T., Rooney, J. E., Elorza, M., and Burkin, D. J. (2011). Transgenic overexpression of the α7 integrin reduces muscle pathology and improves viability in the Dy(W) mouse model of merosin-deficient congenital muscular dystrophy type 1A. J. Cell Sci. 124, 2287–2297. doi: 10.1242/jcs.083311
Flintoff-Dye, N. L., Welser, J., Rooney, J., Scowen, P., Tamowski, S., Hatton, W., et al. (2005). Role for the α7β1 integrin in vascular development and integrity. Dev. Dyn. 234, 11–21. doi: 10.1002/dvdy.20462
Fontelonga, T. M., Jordan, B., Nunes, A. M., Barraza-Flores, P., Bolden, N., Wuebbles, R. D., et al. (2019). Sunitinib promotes myogenic regeneration and mitigates disease progression in the mdx mouse model of duchenne muscular dystrophy. Hum. Mol. Genet. 28, 2120–2132. doi: 10.1093/hmg/ddz044
Gawlik, K. I., and Durbeej, M. (2010). Transgenic overexpression of laminin α1 chain in laminin α2 chain-deficient mice rescues the disease throughout the lifespan. Muscle Nerve 42, 30–37. doi: 10.1002/mus.21616
Gawlik, K. I., and Durbeej, M. (2015). Deletion of integrin α7 subunit does not aggravate the phenotype of laminin α2 chain-deficient mice. Sci. Rep. 5:13916. doi: 10.1038/srep13916
Gawlik, K. I., Harandi, V. M., Cheong, R. Y., Petersén, Å., and Durbeej, M. (2018). Laminin α1 reduces muscular dystrophy in dy2J mice. Matrix Biol. 70, 36–49. doi: 10.1016/j.matbio.2018.02.024
Gawlik, K. I., Holmberg, J., and Durbeej, M. (2014). Loss of dystrophin and β-sarcoglycan significantly exacerbates the phenotype of laminin α2 chain-deficient animals. Am. J. Pathol. 184, 740–752. doi: 10.1016/j.ajpath.2013.11.017
Gawlik, K. I., Li, J.-Y., Petersén, A., and Durbeej, M. (2006a). Laminin α1 chain improves laminin α2 chain deficient peripheral neuropathy. Hum. Mol. Genet. 15, 2690–2700. doi: 10.1093/hmg/ddl201
Gawlik, K. I., Mayer, U., Blomberg, K., Sonnenberg, A., Ekblom, P., and Durbeej, M. (2006b). Laminin α1 chain mediated reduction of laminin α2 chain deficient muscular dystrophy involves integrin α7β1 and dystroglycan. FEBS Lett. 580, 1759–1765. doi: 10.1016/j.febslet.2006.02.027
Gawlik, K., Miyagoe-Suzuki, Y., Ekblom, P., Takeda, S., and Durbeej, M. (2004). Laminin α1 chain reduces muscular dystrophy in laminin chain deficient mice. Hum. Mol. Genet. 13, 1775–1784. doi: 10.1093/hmg/ddh190
Gawlik, K. I., Oliveira, B. M., and Durbeej, M. (2011). Transgenic expression of laminin α1 chain does not prevent muscle disease in the mdx mouse model for duchenne muscular dystrophy. Am. J. Pathol. 178, 1728–1737. doi: 10.1016/j.ajpath.2010.12.030
Geranmayeh, F., Clement, E., Feng, L. H., Sewry, C., Pagan, J., Mein, R., et al. (2010). Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 20, 241–250. doi: 10.1016/j.nmd.2010.02.001
Gillies, A. R., and Lieber, R. L. (2011). Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44, 318–331. doi: 10.1002/mus.22094
Goudenege, S., Lamarre, Y., Dumont, N., Rousseau, J., Frenette, J., Skuk, D., et al. (2010). Laminin-111: a potential therapeutic agent for duchenne muscular dystrophy. Mol. Ther. 18, 2155–2163. doi: 10.1038/mt.2010.165
Grounds, M. D., Sorokin, L., and White, J. (2005). Strength at the extracellular matrix-muscle interface. Scand. J. Med. Sci. Sports 15, 381–391. doi: 10.1111/j.1600-0838.2005.00467.x
Guo, L. T., Zhang, X. U., Kuang, W., Xu, H., Liu, L. A., Vilquin, J.-T., et al. (2003). Laminin A2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul. Disord. 13, 207–215. doi: 10.1016/s0960-8966(02)00266-3
Hayashi, Y. K., Ogawa, M., Matsuda, C., Tsukahara, T., Arahata, K., Ozawa, E., et al. (1998). Mutations in the integrin α7 gene cause congenital myopathy. Nat. Genet. 19, 94–97. doi: 10.1038/ng0598-94
Heller, K. N., Montgomery, C. L., Janssen, P. M. L., Clark, K. R., Mendell, J. R., and Rodino-Klapac, L. R. (2013). AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther. 21, 520–525. doi: 10.1038/mt.2012.281
Heller, K. N., Montgomery, C. L., Shontz, K. M., Clark, K. R., Mendell, J. R., and Rodino-Klapac, L. R. (2015). Human α7 integrin gene (ITGA7) delivered by adeno-associated virus extends survival of severely affected dystrophin/utrophin-deficient mice. Hum. Gene Ther. 26, 647–656. doi: 10.1089/hum.2015.062
Hodges, B. L., Hayashi, Y. K., Nonaka, I., Wang, W., Arahata, K., and Kaufman, S. J. (1997). Altered expression of the α7β1 integrin in human and murine muscular dystrophies. J. Cell Sci. 110, 2873–2881.
Holmberg, J., and Durbeej, M. (2013). Laminin-211 in skeletal muscle function. Cell Adh. Migr. 7, 111–121. doi: 10.4161/cam.22618
Ishii, K., Sakurai, H., Suzuki, N., Mabuchi, Y., Sekiya, I., Sekiguchi, K., et al. (2018). Recapitulation of extracellular LAMININ environment maintains stemness of satellite cells in vitro. Stem Cell Rep. 10, 568–582. doi: 10.1016/j.stemcr.2017.12.013
Kemaladewi, D. U., Bassi, P. S., Erwood, S., Al-Basha, D., Gawlik, K. I., Lindsay, K., et al. (2019). A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature 572, 125–130. doi: 10.1038/s41586-019-1430-x
Kölbel, H., Hathazi, D., Jennings, M., Horvath, R., Roos, A., and Schara, U. (2019). Identification of candidate protein markers in skeletal muscle of laminin-211-deficient CMD type 1A-patients. Front. Neurol. 10:470. doi: 10.3389/fneur.2019.00470
Konkay, K., Kannan, M. A., Lingappa, L., Uppin, M. S., and Challa, S. (2016). Congenital muscular dystrophy with inflammation: diagnostic considerations. Ann. Indian Acad. Neurol. 19, 356–359. doi: 10.4103/0972-2327.186814
Kornegay, J. N. (2017). The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 7:9. doi: 10.1186/s13395-017-0124-z13
Kuang, W., Xu, H., Vachon, P. H., and Engvall, E. (1998a). Disruption of the Lama2 gene in embryonic stem cells: laminin α2 is necessary for sustenance of mature muscle cells. Exp. Cell Res. 241, 117–125. doi: 10.1006/excr.1998.4025
Kuang, W., Xu, H., Vachon, P. H., Ling Liu, F. L., Wewer, U. M., and Engvall, E. (1998b). Merosin-deficient congenital muscular dystrophy. Partial genetic correctio in two mouse models. J. Clin. Invest. 102, 844–852. doi: 10.1172/JCI3705
Kuang, W., Xu, H., Vilquin, J. T., and Engvall, E. (1999). Activation of the Lama2 gene in muscle regeneration: abortive regeneration in laminin α2-deficiency. Lab. Invest. 79, 1601–1613.
Lepper, C., Partridge, T. A., and Fan, C.-M. (2011). An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development 138, 3639–3646. doi: 10.1242/dev.067595
Liu, J., Burkin, D. J., and Kaufman, S. J. (2008). Increasing α7β1-integrin promotes muscle cell proliferation, adhesion, and resistance to apoptosis without changing gene expression. Am. J. Physiol. Cell Physiol. 294, C627–C640. doi: 10.1152/ajpcell.00329.2007
Marcinczyk, M., Elmashhady, H., Talovic, M., Dunn, A., Bugis, F., and Garg, K. (2017). Laminin-111 enriched fibrin hydrogels for skeletal muscle regeneration. Biomaterials 141, 233–242. doi: 10.1016/j.biomaterials.2017.07.003
Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–495. doi: 10.1083/jcb.9.2.493
Mayer, U. (2003). Integrins: redundant or important players in skeletal muscle? J. Biol. Chem. 278, 14587–14590. doi: 10.1074/jbc.r200022200
Mayer, U., Saher, G., Fassler, R., Bornemann, A., Echtermeyer, F., von der, M. H., et al. (1997). Absence of integrin α 7 causes a novel form of muscular dystrophy. Nat. Genet. 17, 318–323. doi: 10.1038/ng1197-318
Mohassel, P., Foley, A. R., and Bönnemann, C. G. (2018). Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. 71–72, 188–204. doi: 10.1016/j.matbio.2018.06.005
Morgan, J. E., and Partridge, T. A. (2003). Muscle satellite cells. Int. J. Biochem. Cell Biol. 35, 1151–1156. doi: 10.1016/s1357-2725(03)00042-6
Murphy, M. M., Lawson, J. A., Mathew, S. J., Hutcheson, D. A., and Kardon, G. (2011). Satellite Cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637. doi: 10.1242/dev.064162
Nguyen, Q., Lim, K. R. Q., and Yokota, T. (2019). Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy. Appl. Clin. Genet. 12, 113–130. doi: 10.2147/TACG.S187481
Nissinen, M., Vuolteenaho, R., Boot-Handford, R., Kallunki, P., and Tryggvason, K. (1991). Primary structure of the human laminin A chain. Limited expression in human tissues. Biochem. J. 276, 369–379. doi: 10.1042/bj2760369
Öcalan, M., Goodman, S. L., Kühl, U., Hauschka, S. D., and von der Mark, K. (1988). Laminin alters cell shape and stimulates motility and proliferation of murine skeletal myoblasts. Dev. Biol. 125, 158–167. doi: 10.1016/0012-1606(88)90068-1
Patton, B. L., Miner, J. H., Chiu, A. Y., and Sanes, J. R. (1997). Distribution and function of laminins in the neuromuscular system of developing, adult and mutant mice. J. Cell Biol. 139, 1507–1521. doi: 10.1083/jcb.139.6.1507
Pegoraro, E., Mancias, P., Swerdlow, S. H., Raikow, R. B., Garcia, C., Marks, H., et al. (1996). Congenital muscular dystrophy with primary laminin-A2 (merosin) deficiency presenting as inflammatory myopathy. Ann. Neurol. 40, 782–791. doi: 10.1002/ana.410400515
Perrin, A., Rousseau, J., and Tremblay, J. P. (2017). Increased Expression of Laminin Subunit Alpha 1 Chain by dCas9-VP160. Mol. Ther. Nucleic Acids 6, 68–79. doi: 10.1016/j.omtn.2016.11.004
Rayagiri, S. S., Ranaldi, D., Raven, A., Mohamad Azhar, N. I. F., Lefebvre, O., Zammit, P. S., et al. (2018). Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self-renewal. Nat. Commun. 9:1075. doi: 10.1038/s41467-018-03425-3
Reinhard, J. R., Lin, S., Mckee, K. K., Meinen, S., Crosson, S. C., Sury, M., et al. (2017). Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9:eaal4649. doi: 10.1126/scitranslmed.aal4649
Rooney, J. E., Gurpur, P. B., Yablonka-Reuveni, Z., and Burkin, D. J. (2009). Laminin-111 restores regenerative capacity in a mouse model for α7 integrin congenital myopathy. Am. J. Pathol. 174, 256–264. doi: 10.2353/ajpath.2009.080522
Rooney, J. E., Knapp, J. R., Hodges, B. L., Wuebbles, R. D., and Burkin, D. J. (2012). Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am. J. Pathol. 180, 1593–1602. doi: 10.1016/j.ajpath.2011.12.019
Rooney, J. E., Welser, J. V., Dechert, M. A., Flintoff-Dye, N. L., Kaufman, S. J., and Burkin, D. J. (2006). Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci. 119, 2185–2195. doi: 10.1242/jcs.02952
Ross, J., Benn, A., Jonuschies, J., Boldrin, L., Muntoni, F., Hewitt, J. E., et al. (2012). Defects in glycosylation impair satellite stem cell function and niche composition in the muscles of the dystrophic largemyd mouse. Stem Cells 30, 2330–2341. doi: 10.1002/stem.1197
Sambasivan, R., Yao, R., Kissenpfennig, A., Van Wittenberghe, L., Paldi, A., Gayraud-Morel, B., et al. (2011). Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656. doi: 10.1242/dev.067587
Sanes, J. R. (2003). The basement membrane/basal lamina of skeletal muscle. J. Biol. Chem. 278, 12601–12604. doi: 10.1074/jbc.r200027200
Sarathy, A., Wuebbles, R. D., Fontelonga, T. M., Tarchione, A. R., Mathews Griner, L. A., Heredia, D. J., et al. (2017). SU9516 increases α7β1 integrin and ameliorates disease progression in the mdx mouse model of duchenne muscular dystrophy. Mol. Ther. 25, 1395–1407. doi: 10.1016/j.ymthe.2017.03.022
Sasaki, T., Giltay, R., Talts, U., Timpl, R., and Talts, J. F. (2002). Expression and distribution of laminin α1 and α2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp. Cell Res. 275, 185–199. doi: 10.1006/excr.2002.5499
Silva-Barbosa, S. D., Butler-Browne, G. S., De Mello, W., Riederer, I., Di Santo, J. P., Savino, W., et al. (2008). Human myoblast engraftment is improved in laminin-enriched microenvironment. Transplantation 85, 566–575. doi: 10.1097/tp.0b013e31815fee50
Silva Garcia, J. M., Panitch, A., and Calve, S. (2019). Functionalization of hyaluronic acid hydrogels with ECM-derived peptides to control myoblast behavior. Acta Biomater. 84, 169–179. doi: 10.1016/j.actbio.2018.11.030
Summers, P. J., and Parsons, R. (1981). A quantitative assessment of dystrophic mouse (129 ReJ dy/dy) myogenesis in vitro. Neuropathol. Appl. Neurobiol. 7, 269–277. doi: 10.1111/j.1365-2990.1981.tb00098.x
Summers, P. J., and Parsons, R. (1981). An electron microscopic study of satellite cells and regeneration in dystrophic mouse muscle. Neuropathol. Appl. Neurobiol. 7, 257–268. doi: 10.1111/j.1365-2990.1981.tb00097.x
Taniguchi, M., Kurahashi, H., Noguchi, S., Sese, J., Okinaga, T., Tsukahara, T., et al. (2006). Expression profiling of muscles from fukuyama-type congenital muscular dystrophy and laminin-α2 deficient congenital muscular dystrophy; is congenital muscular dystrophy a primary fibrotic disease? Biochem. Biophys. Res. Commun. 342, 489–502. doi: 10.1016/j.bbrc.2005.12.224
Thomas, K., Engler, A. J., and Meyer, G. A. (2015). Extracellular matrix regulation in the muscle satellite cell niche. Connect. Tissue Res. 56, 1–8. doi: 10.3109/03008207.2014.947369
Timpl, R., and Brown, J. C. (1996). Supramolecular assembly of basement membranes. Bioessays 18, 123–132. doi: 10.1002/bies.950180208
Vachon, P. H., Loechel, F., Xu, H., Wewer, U. M., and Engvall, E. (1996). Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J. Cell Biol. 134, 1483–1497. doi: 10.1083/jcb.134.6.1483
Vachon, P. H., Xu, H., Liu, L., Loechel, F., Hayashi, Y., Arahata, K., et al. (1997). Integrins (α7β1) in muscle function and survival disrupted expression in merosin-deficient congenital muscular dystrophy. J. Clin. Invest. 100, 1870–1881. doi: 10.1172/JCI119716
van Lunteren, E., Moyer, M., and Leahy, P. (2006). Gene expression profiling of diaphragm muscle in α2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiol. Genomics 25, 85–95. doi: 10.1152/physiolgenomics.00226.2005
van Ry, P. M., Minogue, P., Hodges, B. L., and Burkin, D. J. (2014). Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet. 23, 383–396. doi: 10.1093/hmg/ddt428
von der Mark, H., Williams, I., Wendler, O., Sorokin, L., von der Mark, K., and Pöschl, E. (2002). Alternative splice variants of α 7 β 1 integrin selectively recognize different laminin isoforms. J. Biol. Chem. 277, 6012–6016. doi: 10.1074/jbc.M102188200
Yanay, N., Elbaz, M., Konikov-Rozenman, J., Elgavish, S., Nevo, Y., Fellig, Y., et al. (2019). Pax7, Pax3 and mamstr genes are involved in skeletal muscle impaired regeneration of Dy2J/Dy2J mouse model of Lama2-CMD. Hum. Mol. Genet. 28, 3369–3390. doi: 10.1093/hmg/ddz180
Yin, H., Price, F., and Rudnicki, M. A. (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67. doi: 10.1152/physrev.00043.2011
Yurchenco, P. D., McKee, K. K., Reinhard, J. R., and Rüegg, M. A. (2018). Laminin-deficient muscular dystrophy: molecular pathogenesis and structural repair strategies. Matrix Biol. 72, 174–187. doi: 10.1016/j.matbio.2017.11.009
Yurchenco, P. D., and Patton, B. L. (2009). Developmental and pathogenic mechanisms of basement membrane assembly. Curr. Pharm. Des. 15, 1277–1294. doi: 10.2174/138161209787846766
Ziemkiewicz, N., Talovic, M., Madsen, J., Hill, L., Scheidt, R., Patel, A., et al. (2018). Laminin-111 functionalized polyethylene glycol hydrogels support myogenic activity in vitro. Biomed. Mater. 13:065007. doi: 10.1088/1748-605x/aad915
Keywords: LAMA2-CMD, Laminin, α7 integrin, muscular dystrophy, muscle, therapeutic
Citation: Barraza-Flores P, Bates CR, Oliveira-Santos A and Burkin DJ (2020) Laminin and Integrin in LAMA2-Related Congenital Muscular Dystrophy: From Disease to Therapeutics. Front. Mol. Neurosci. 13:1. doi: 10.3389/fnmol.2020.00001
Received: 03 October 2019; Accepted: 06 January 2020;
Published: 11 February 2020.
Edited by:
Markus A. Ruegg, University of Basel, SwitzerlandReviewed by:
Janosch P. Heller, Royal College of Surgeons in Ireland, IrelandRuth Herbst, Medical University of Vienna, Austria
Copyright © 2020 Barraza-Flores, Bates, Oliveira-Santos and Burkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dean J. Burkin, ZGJ1cmtpbkBtZWQudW5yLmVkdQ==